

Abstract
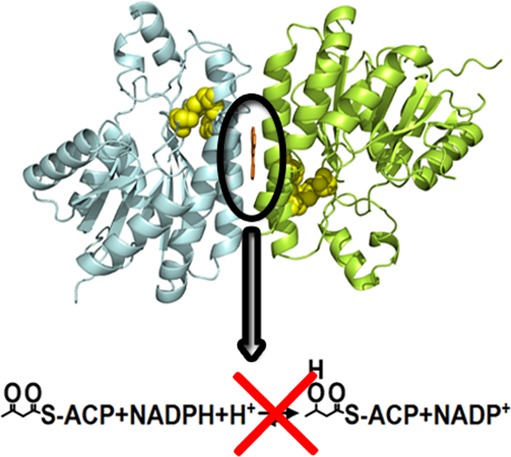
3-Oxo-acyl-acyl carrier protein (ACP) reductase (FabG) plays a key role in the bacterial fatty acid synthesis II system in pathogenic microorganisms, which has been recognized as a potential drug target. FabG catalyzes reduction of a 3-oxo-acyl-ACP intermediate during the elongation cycle of fatty acid biosynthesis. Here, we report gene deletion experiments that support the essentiality of this gene in P. aeruginosa and the identification of a number of small molecule FabG inhibitors with IC50 values in the nanomolar to low micromolar range and good physicochemical properties. Structural characterization of 16 FabG-inhibitor complexes by X-ray crystallography revealed that the compounds bind at a novel allosteric site located at the FabG subunit–subunit interface. Inhibitor binding relies primarily on hydrophobic interactions, but specific hydrogen bonds are also observed. Importantly, the binding cavity is formed upon complex formation and therefore would not be recognized by virtual screening approaches. The structure analysis further reveals that the inhibitors act by inducing conformational changes that propagate to the active site, resulting in a displacement of the catalytic triad and the inability to bind NADPH.
Pseudomonas aeruginosa is a ubiquitous free-living Gram-negative bacterium that often causes opportunistic infections, mainly in patients with immunosuppression, burns, or cystic fibrosis. P. aeruginosa is able to adapt to diverse environmental conditions, and consequently, the range of pathologies associated with this microorganism is broad, including respiratory tract, skin, and blood infections.1,2 The treatment of P. aeruginosa infections is complicated due to its high intrinsic resistance to antibiotics and capability of developing/acquiring new mechanisms of resistance.3,4 The spread of drug-resistant strains underlines the need to identify novel drug leads/hit compounds.5 Recent efforts toward this objective are directed to better understand the biology of P. aeruginosa, to characterize different aspects of pathologies associated with this bacterium, and to improve containment of P. aeruginosa infections.6−10
Fatty acid synthesis type II (FAS II) exists in bacteria, plants, and parasites.11−13 FAS II consists of several proteins that catalyze individual reactions in fatty acid biosynthesis. The FAS II system has been identified as an attractive drug target, and several antibiotics targeting this pathway are in use, such as triclosan or isoniazid.14−18
3-Oxo-acyl-ACP reductase (FabG; EC 1.1.1.100) catalyzes the first reduction step that leads to the conversion of 3-oxo-acyl-ACP to 3-D-hydroxyacyl-ACP intermediates during the elongation cycle of the FAS II system11,13 (Figure 1A). FabG belongs to the short-chain dehydrogenase/reductase (SDR) family of NAD(P)(H)-dependent oxidoreductases.19 The members of this family share a Rossmann fold motif that is involved in cofactor binding and are engaged in a broad range of dehydrogenation and reduction reactions. FabG is a promising drug target due to its essentiality, high conservation in bacteria, and presence of a single isoform in many bacterial species.18 Although several potential inhibitors of FabG have been identified,20−24 these are largely natural product extracts and pose significant drug development challenges. So far, none have reached the clinic.
Figure 1.

Enzymatic reactions catalyzed by FabG. (A) In fatty acid biosynthesis FabG uses NADPH to reduce 3-oxoacyl-ACP substrate (represented here by the shortest substrate, acetoacetyl-ACP) to respective 3-D-hydroxyacyl-ACP. (B and C) FabG is also able to reduce non-natural substrates, such as acetoacetyl-CoA (B) and 3-oxodecanoyl-N-acetylcysteamine (C). These two activities were employed in this study, and the directions of the individual reactions monitored are indicated by thick arrows.
Here, we validate FabG from P. aeruginosa as a drug target by gene deletion experiments and present a series of novel small-molecule FabG inhibitors with nanomolar to low micromolar IC50 values and good physicochemical properties. Some of these molecules have phenotypic activity against a Gram-positive bacterium, Staphylococcus aureus, but none were active against P. aeruginosa. We also report the crystal structures of FabG, the binary complex of FabG-NADPH, and FabG-inhibitor complexes. In all complexes the inhibitors bind at a novel allosteric site located at the intersubunit interface, inducing structural changes within FabG incompatible with NADPH binding and catalysis.
Results and Discussion
Target Validation: fabG Is an Essential Gene in P. aeruginosa PAO1
Existence of fabG as a single isoform in most bacteria suggests its potential use as a drug target; however, experimental evidence for gene essentiality has been reported only for Escherichia coli, Salmonella enterica, and Mycobacterium tuberculosis.25,26 In order to examine the essentiality of fabG and thus its suitability as a drug target in P. aeruginosa, we attempted to construct a P. aeruginosa PAO1 ΔfabG mutant using the pEX18Ap suicide vector.27 In this vector (LEXYB122ΔPA2967), a gentamicin resistance cassette replaces the fabG gene, and the cassette is flanked at both ends by 400 bp fragments of homologous DNA. After several conjugations and counter-selection utilizing the sacB gene in the vector, several hundred gentamicin-resistant colonies were isolated and analyzed. They were all found to be carbenicillin-resistant, indicating the presence of the plasmid backbone and a single crossover event in all isolated colonies. The presence of the gentamicin cassette and the fabG gene in these clones was confirmed by PCR. All these suspected mutants were sucrose-sensitive. Spontaneous sucrose- and gentamicin-resistant mutants, which had also lost the carbenicillin resistance, indicated a possible double crossover event by loss of the vector backbone. However, genotypic characterization of the isolated DNA of these suspected mutants showed the presence of the wild type fabG sequence, thus representing only single crossover events. Disruption of the chromosomal fabG gene using the knockout procedure with different supplementation of the culture media, e.g., with palmitic acid or a fatty acid cocktail, was also unsuccessful.
We therefore constructed a strain carrying a second chromosomal copy of fabG under the control of its native promoter and attempted the deletion of the native copy of fabG at the PA2967 locus. The suicide mini-CTX2 plasmid based method28 was utilized for site-specific integration of the second copy of fabG. We successfully obtained a PAO1 strain with a dual copy of fabG (PAO1-LEXYB141). The presence of the two fabG copies was confirmed by genotypic characterization and sequencing. The PAO1-LEXYB141 strain was then used to delete the native fabG copy by the same methods as described above. We were able to replace the fabG gene at the chromosomal PA2967 locus by the gentamicin resistance casette in the presence of the second copy at the attB site. These clones were gentamicin-resistant, carbenicillin-sensitive, and sucrose-resistant. The replacement of the fabG sequence at the PA2967 locus and the presence of the second fabG copy at the attB site were confirmed by PCR (see Supporting Information and Figure S1) and DNA sequencing. The successful deletion of wild type fabG at the PA2967 locus in the P. aeruginosa mutant PAO1-LEXYB141 carrying two copies of this gene demonstrates that the employed protocol works, and we therefore conclude that fabG is an essential gene for growth in this organism.
Structures of apo-FabG and the FabG-NADPH Complex
The crystal structures of apo-FabG and the FabG-NADPH complex were determined to 2.3 Å and 1.8 Å resolution, respectively, using molecular replacement (Supplementary Tables S1 and S2). FabG adopts a canonical Rossmann fold commonly found in tyrosine-dependent oxidoreductases19 with a seven-stranded parallel β-sheet surrounded by α helices, forming together a three-layer (α/β/α) sandwich (Figure 2). The central β-sheet has β7β6β5β4β1β2β3 topology and packs against helices α1, α2, and α8 on one side and against helices α3, α4, and α5 on the other side. The overall structure of FabG from P. aeruginosa is similar to the structures of FabG from other organisms (Supplementary Figure S2), including FabG from other pathogenic bacteria such as E. coli,29M. tuberculosis,30Bacillus anthracis,31Rickettsia prowazekii,32 and Staphylococcus aureus.33
Figure 2.
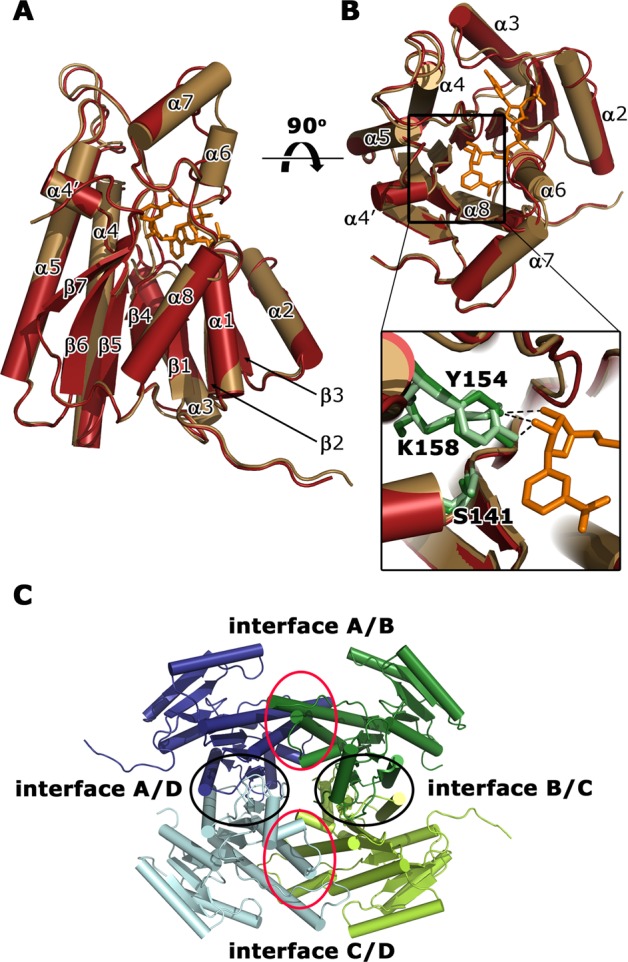
Overall crystal structure of a subunit of apo-FabG (yellow) and FabG-NADPH complex (red). Side (A) and top (B) views of the superimposed crystal structures in cartoon representation. NADPH (orange) and three catalytic residues (S141, Y154, and K158) in apo-FabG (light green) and the FabG-NADPH complex structure (dark green) are shown in stick representation. The important hydrogen-bond interaction between the catalytic residues and NADPH are represented by dashed lines. (C) Quaternary structure of FabG from P. aeruginosa. Cartoon representation of four apo-FabG subunits (dark blue, dark green, light green, light blue), which are related by D2 symmetry. The four subunit–subunit interfaces are encircled.
The conformation of FabG in the binary complex with NADPH is very similar to apo-FabG with an rmsd of 0.5–1.6 Å for Cα atoms between individual protein chains. The only significant structural changes are ordering and decreased mobility for residues located in helices α6 and α7 and a loop β3−α3 that are involved in NADPH binding (Figure 2). In the structure of the FabG-NADPH complex the electron density for the cofactor is observed in three protein molecules of the asymmetric unit (Supplementary Figure S3A). The absence of NADPH in the fourth FabG molecule is most likely an artifact of the soaking experiment with accessibility of this NADPH binding site limited by crystal packing.
In the complex, NADPH binds in a deep cleft formed by the C-terminus of the parallel β-sheet and helices α4, α5, α6, and α7 (Figure 2) burying ∼650 Å2 of solvent accessible area. Overall, the recognition of NADPH by FabG is typical for this class of enzymes (Supplementary Figure S3B), and the structure of the FabG-NADPH complex from P. aeruginosa superimposes with an rmsd of 1.2–2.0 Å for Cα atoms to FabG-NAD(P)H complexes from other bacteria (Supplementary Figure S2B).
Quaternary Structure of P. aeruginosa FabG
The asymmetric units of the crystals of apo-FabG and the FabG-NADPH complex contain four protein subunits that form a tetramer (Figure 2C). The subunits within the tetramer are related by D2 symmetry, which can be described as a dimer of dimers. One dimer is formed by chains A and D, and the second dimer by chains B and C. The dimer interface (interface A/D) and its equivalent chains B and C (interface B/C) (Figure 2C) involves the C-terminal part of helices α5 and α8, strand β7, loops β6−α6, α7−α8, and α8−β7, and the C-termini, burying ∼1440 Å2 of solvent-accessible area in each subunit. The interactions are mainly hydrophobic with ∼10 stabilizing intersubunit hydrogen bonds.
The dimer–dimer interface, formed between chains A and B (interface A/B) and between chains C and D (interface C/D) (Figure 2C), involves helices α4 and α5 and loop β5−α4 with a short α4′ helix from each subunit, burying ∼1600 Å2 of solvent-accessible area per monomer. This interface is also mainly hydrophobic and is further stabilized by ∼20 intersubunit hydrogen-bond interactions at the edge of the interface.
Characterization of FabG
We have employed two different enzymatic assays to facilitate identification and evaluation of potential inhibitors of FabG. In the first assay, we monitored NADPH consumption by FabG as it performs the biologically relevant reduction of the 3-oxo group of the acetoacetyl-coenzyme A (AcAcCoA) substrate analogue (Forward Assay, Figure 1B). In the second assay, we have taken advantage of the reversibility of the FabG-catalyzed reaction and monitored NADPH production using the product mimic 3-hydroxydecanoyl-N-acetylcysteamine (3-OH decanoyl-NAC), which is oxidized to 3-oxodecanoyl-N-acetylcysteamine (Back Assay, Figure 1C).
In the Forward Assay, FabG catalyzes reduction of AcAcCoA with a KM = 1.0 ± 0.2 mM and kcat/KM = (12 ± 3) × 103 M–1 s–1 in the presence of 0.8 mM NADPH (Supplementary Figure S4A). The KM for NADPH was 0.29 ± 0.03 mM in the presence of 4 mM AcAcCoA. In the Back Assay, the KM of 3-OH decanoyl-NAC could not be determined due to limited solubility of the substrate. However, the response was linear at least to 0.3 mM, indicating that the KM was above this concentration. The KM of NADP+ in the presence of 0.3 mM 3-OH decanoyl-NAC was 0.29 mM (Supplementary Figure S4B).
Identification of FabG Inhibitors: NMR Fragment Screen
FabG was screened against a fragment library of 436 compounds at a single concentration (1 mM) using an NMR-based binding assay. Compounds consistently identified in STD and waterLOGSY experiments were considered as potentially binding to FabG, resulting in 48 hits from the screen (11% hit rate). Due to compound availability and compatibility with the biochemical assay, only 32 of these molecules were screened at a single concentration (1 mM) using the Forward Assay with AcAcCoA as substrate. Twenty-five molecules displayed >50% inhibition at 1 mM concentration (Supplementary Figure S5A). The most potent compounds were selected for crystallography, but a crystal structure was obtained only for compound 498 (see below). This compound was profiled in more detail in the Forward Assay giving an IC50 of 138 μM (Table 1).
Table 1. IC50 and Hill Coefficient Values Determined for Compounds with Crystal Structures.
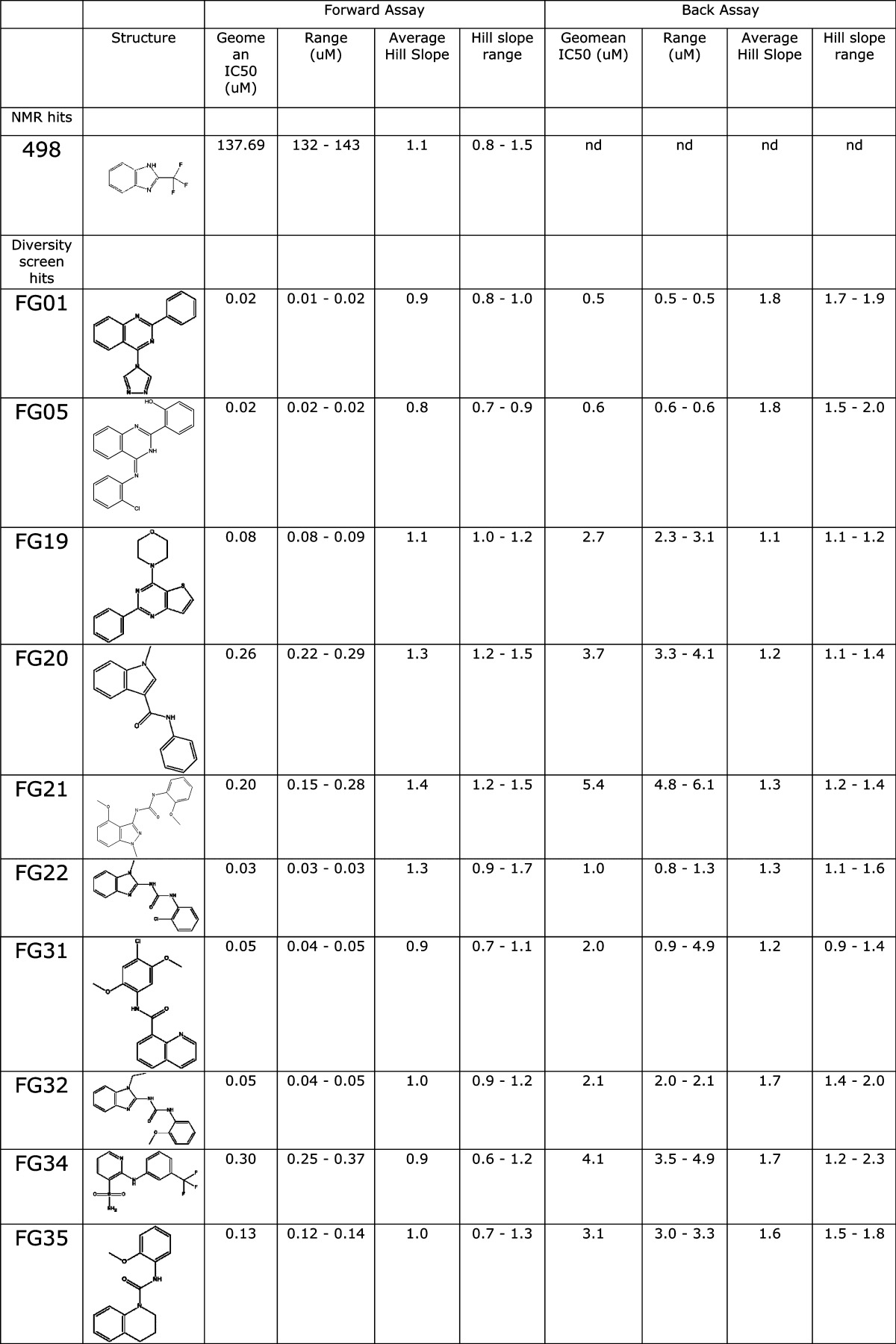
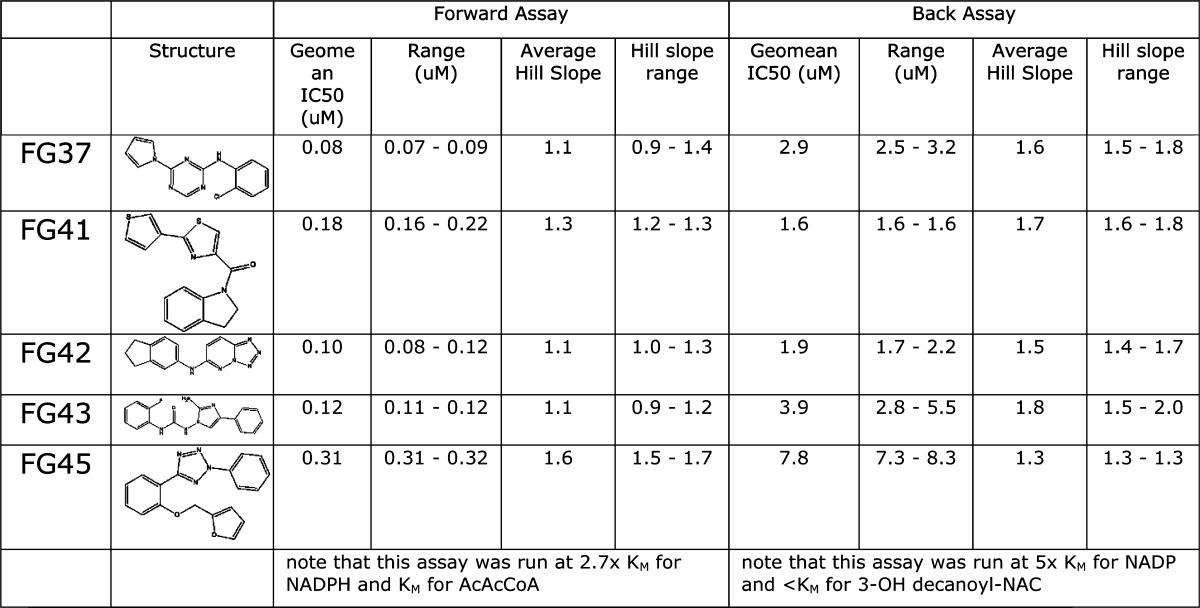
nd = not determined.
Identification of FabG Inhibitors: Diversity Biochemical Screen
FabG was screened against a diverse library of 15667 compounds at a single concentration (10 μM) using the Back Assay with 3-OH decanoyl-NAC as substrate. Compounds with activity greater than 3x the standard deviation around the baseline were considered to be active. This resulted in 101 compounds with >25% activity. Limits on compound availability and a pragmatic raise of the cutoff to 50% resulted in 30 compounds being selected for concentration/effect curves in the Back Assay. IC50 values are presented in Table 1 and range from 500 nM to 11.5 μM. The Hill slopes were steep with a median of 1.5 for all compounds tested and 28 of 30 compounds >1.0, suggesting cooperativity in the interaction between the enzyme, substrates (NADP+ and 3-OH decanoyl-NAC), and compounds. Negative cooperativity in binding of NADPH and ACP has also been observed previously in FabG from E. coli.29
Twenty-one compounds were also evaluated in the Forward Assay, giving >10% inhibition at single concentration (0.1 μM) (Supplementary Figure S5B). Concentration/effect curves were obtained for these compounds in the Forward Assay, giving IC50 values in the range 0.02–0.31 μM. Strikingly, although the compounds had broadly the same rank order of potency as in the Back Assay (correlation coefficient R2 = 0.78 following the removal of a single outlier), the IC50’s were, on average, 30-fold lower and the Hill slopes median was 1.1 with 10 of 21 compounds tested giving Hill slopes ≤1 (Table 1 and Supplementary Table S3). We also verified by UV–vis spectrophotometry that protein aggregation triggered by the addition of the hit compounds as the cause for FabG inhibition can be excluded (Supplementary Figure S6).
Although the two assays are set up differently with respect to the ratio between the concentration of substrates and their respective KM values, this accounts for less than 2-fold of the observed difference in potencies. The detailed enzymology of this dimer of dimer enzyme is clearly complex, and the explanation for this difference may lie in the different α cooperativity factors underlying the complex cooperativity interactions between the substrates (NADPH, H+, AcAcCoA for the Forward Assay, and NADP+ and 3-OH decanoyl-NAC for the Back Assay).
Finally, the 30 diversity hit molecules were profiled for their ability to inhibit bacterial cell growth in 10-point concentration/effect curves against P. aeruginosa and the Gram-positive bacterium S. aureus. No compounds showed a significant effect against P. aeruginosa, but FG48 completely inhibited the growth of S. aureus, with a pEC50 of 4.7 (Supplementary Figure S7). In addition, FG39 and FG43 inhibited cell growth by 35% and 64%, respectively, at a concentration of 100 μM. A comparison shows that the crystal structures of FabG from P. aeruginosa and S. aureus(33) are very similar and that the inhibitor binding pockets at the A/B and C/D interfaces (see below) could potentially also be formed in the S. aureus enzyme.
Crystal Structures of Inhibitor Complexes
To further understand the structural basis for the compound-mediated inhibition of FabG activity, we determined the crystal structures of several FabG-hit complexes. The structures of FabG complexed with NMR hit 498 and with HTS hits FG01, FG05, FG19, FG20, FG21, FG22, FG31, FG32, FG34, FG35, FG37, FG41, FG42, FG43, and FG45 could be obtained in resolution ranges of 1.8–2.9 Å (Supplementary Tables S1 and S2).
Remarkably, despite a lack of structural similarity between the hit compounds present in the complexes (Tanimoto score34 of ≤0.81), they all bind in a similar conformation at the same allosteric site, located at the dimer–dimer interface, about 14 Å away from the catalytic center (Figure 3A). Two inhibitor molecules are bound per FabG tetramer and in several cases one or two orientations of the compounds were modeled at each binding site to fit the experimental electron density maps (Supplementary Figure S8). Pairwise superimposition of the individual subunits of the FabG-hit compound complexes results in rmsd values in the range of 0.2–2.4 Å for Cα atoms. In the following, we use the structure of the complex of FabG with FG01, which is one of the most potent inhibitors, to describe the protein–ligand interactions.
Figure 3.

Inhibitor binding to the FabG protein. A) Subunits C (light green) and D (light blue) of the structure of FabG in the complex with hit FG01. The compound (orange, stick representation) binds at the allosteric site located at the interface between subunit C from the B/C dimer and subunit D from the A/D dimer. The catalytic triad residues (S141, Y154, K158) of each protein subunit are shown in space-fill representation (yellow). (B) Stereo view of the inhibitor binding site with bound FG01 included. The residues forming the hydrophobic cavity are shown in stick representation. (C) Superimposed hit compounds 498, FG01, FG05, FG19, FG20, FG21, FG22, FG31, FG32, FG34, FG35, FG37, FG41, FG42, FG43, and FG45 (stick representation) at the interface C/D demonstrating the almost planar conformation of the bound hits.
The two equivalent binding cavities at the dimer–dimer interface are mainly hydrophobic and are formed by residues belonging to helices α4 (W106, F107, V110, N111, L114, and N115) and α5 (A156, A159, G160, G163, F164, and A167) from each subunit (Figure 3B). Binding excludes ∼180 Å2 of solvent-accessible area in each protein subunit and completely buries the inhibitor within the protein (buried surface area of 440 Å2). The hydrophobic nature of the cavities limits the number of possible hydrogen-bond donors and acceptors. Nevertheless, some of the ligands form specific hydrogen bonds to FabG (Supplementary Figure S9) with the most striking example of the FG34 complex, where the side chain of N111 adopts a different conformation to mediate this interaction. Interestingly, the FabG-inhibitor interactions impose strong constraints on the conformation of the bound ligands, which despite having 2–4 rings connected by rotatable bonds, are almost planar in all 16 structures (Figure 3C).
The cavity is absent in the apo-FabG structure and is induced by the ligand; moreover the size of the cavity is dependent on the size of the inhibitor (Supplementary Figure S10), a striking example of induced fit by an inhibitor. Absence of this cavity in the noninhibited enzyme reveals a severe limitation of most present-day docking-based virtual screening methods for drug identification, as such approaches will miss such a cryptic binding site.35 Cryptic binding cavities also introduce a bias in computational druggability assessments as they will remain undetected in the absence of ligand-bound templates. The set of 16 FabG-inhibitor structures determined in this study represents a useful study-case that could help to improve the algorithms dealing with protein flexibility.
The binding site has to be temporarily solvent-exposed to allow for inhibitor binding. This can be achieved either by local rearrangements in helices α4 or by dissociation of the tetramer into dimers. Both scenarios imply that FabG should exhibit dynamic behavior. The differences observed between the structures of the apo-form and inhibitor-complexes of FabG show that the protein can adopt distinct conformations and therefore likely undergoes extensive motions in solution. Furthermore, some regions of the protein have high B factors or are completely disordered in the crystals, emphasizing the dynamic nature of the enzyme.
Structural Basis of Inhibition
The consequence of ligand binding is a profound perturbation of the structure of FabG. The conformational rearrangement primarily occurs at subunit interfaces involved in ligand recognition (dimer–dimer interface A/B and C/D), leading to a ∼15° rotation of the B/C dimer with respect to the A/D dimer (Figure 4A). Thus, the structures of the dimers are practically unchanged upon inhibitor binding and align with an rmsd of about 2.0 Å to the equivalent dimers in apo-FabG while alignment of A and B or C and D subunit pairs from the ligand complexes superpose with an rmsd in the order of 3.2 Å to corresponding pairs in the apo-FabG structure.
Figure 4.
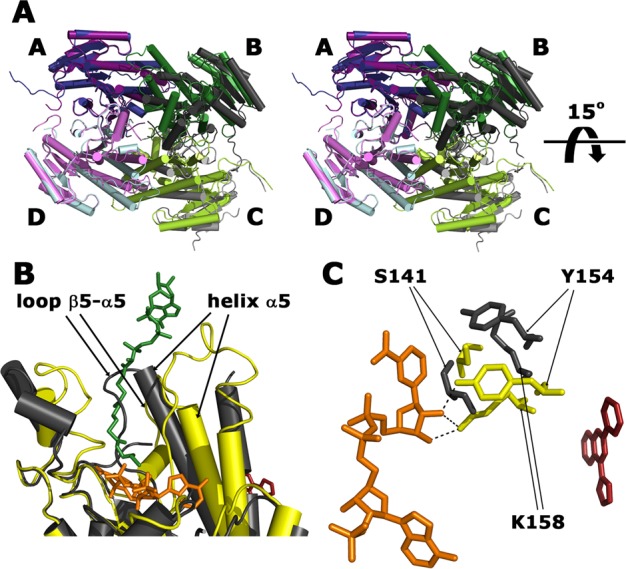
Conformational changes in FabG upon inhibitor binding. (A) Stereo view of superimposed structures of tetrameric apo-FabG (dark blue, dark green, light green, light blue) and the FabG-FG01 complex (dark purple, dark gray, light gray, light purple). The structures are aligned on the A/D dimer illustrating the rotation of the B/C dimer. (B) Superimposed structures of the FabG-NADPH and FabG-FG01 complexes showing the reorientation of the catalytic triad upon FG01 binding. Formation of the FabG-inhibitor complex displaces the catalytic triad (S141, Y154, K158) from the NADPH-binding compatible conformation (yellow) to a novel orientation (dark gray). NADPH and FG01 are displayed as orange and red stick models, respectively. Hydrogen bonds between Y154 and K158 and NADPH are shown as dashed lines. (C) A section of the superimposed structures of subunits of FabG-NADPH (yellow) and FabG-FG01 (dark gray). NADPH and hit FG01 are shown as orange and red stick models, respectively. Hexanoyl-coenzyme A substrate (green stick) is modeled based on the structure of FabG4-NAD+-hexanoyl-coenzyme A complex from M. tuberculosis (PDB accession code 3v1u). Structural elements that are displaced upon FG01 binding and result in a conformation incompatible with substrate binding are labeled.
At the subunit level, the structural changes are localized to the proximity of the inhibitor binding site and involve the N-terminal parts of the helices α4 (residues 103–124) and α5 (residues 152–166), loops β4−α4 (residues 90–102) and β5−α5 (residues 140–151), which connect the N-termini of helices α4 and α5 to the core of the protein, as well as the four C-terminal residues of the monomer (residues 244–247). These movements (Figure 4B), in combination with head-to-tail arrangement of the helices at the intersubunit interface, lead to the formation of a cavity where the inhibitors bind (Figure 3B). Simultaneously, the two dimers in the tetramer reorient by ∼15° with respect to each other, thus decreasing the solvent-accessible area buried in the interfaces A/B and C/D from ∼1440 Å2 in the apo structure to ∼1030 Å2 in the inhibitor-bound structures, also accounting for the area excluded by ligand binding.
Crucially, these conformational changes propagate to the catalytic triad (residues S141, Y154, and K158), resulting in an active site conformation that is incompatible with NADPH binding (Figure 4B). S141, Y154 and K158 are displaced and form hydrogen-bond contacts with the backbone carbonyl groups of P184 and M246, thus preventing interactions for binding of the ribose moiety of NADPH as seen in the holoenzyme (Figure 2B) and shown to be critical for its efficient binding by mutational studies of E. coli FabG.36 The loop containing S141 adopts a conformation that would cause steric hindrance with NADPH binding (Figure 4C). Overall, we conclude that the inhibitor acts by preventing the binding of NADPH and disrupting the catalytically competent orientation of the catalytic triad.
Mode of Inhibition
To gain further insights into the inhibitory mechanism of identified hits, a kinetic analysis of inhibition was performed for one of the most potent inhibitors (FG01). Within the limits of experimental error, the Lineweaver–Burk plot analysis shows a noncompetitive mode of inhibition with respect to NADPH (Figure 5), in agreement with our conclusion from the structural analysis. Comparison of the structures of the FabG-FG01 complex with that of the 3-oxo-acyl-acyl carrier protein reductase from M. tuberculosis in complex with NAD+ and hexanoyl-coenzyme A37 shows that reorientation of helix α5 caused by FG01 binding displaces loop β5−α5, thus also blocking the fatty acid binding site (Figure 4C).
Figure 5.
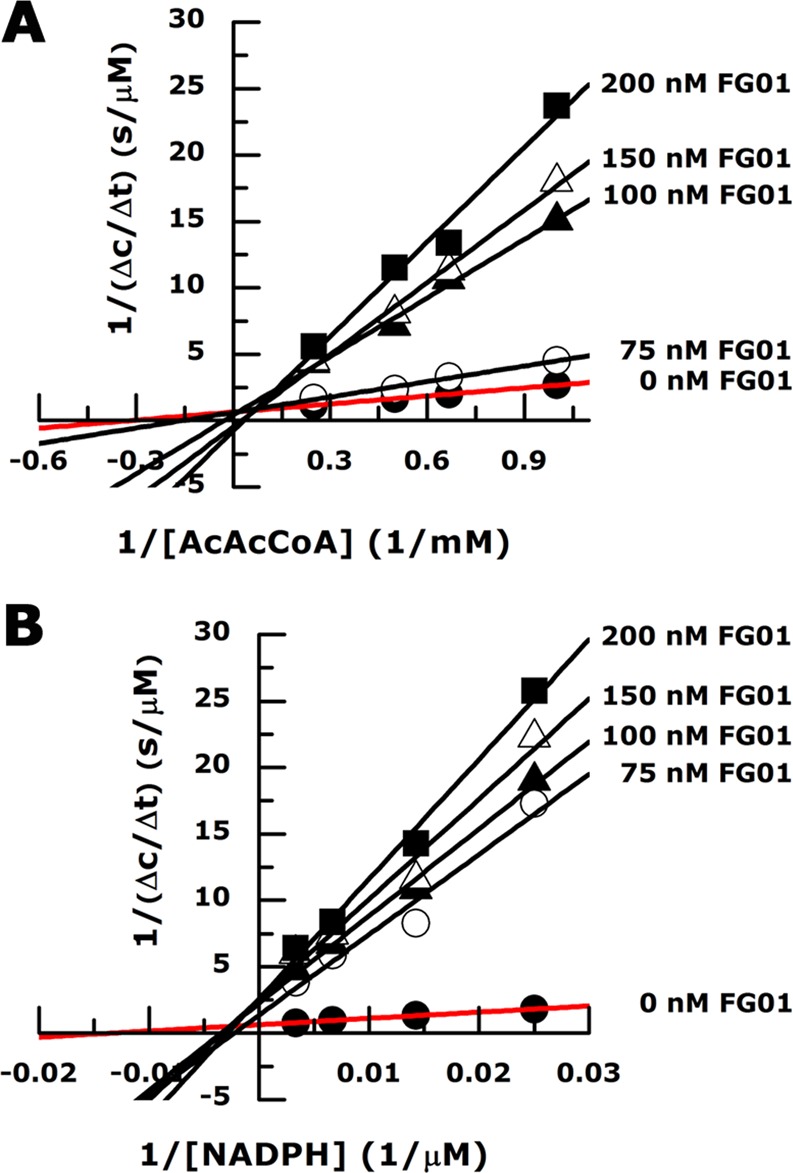
Lineweaver–Burk plots illustrating the effect of FG01 on FabG activity. Within the limits of experimental error, FG01 shows a mixed inhibition pattern with respect to AcAcCoA (A) and a noncompetitive inhibition pattern with NADPH (B).
To test experimentally the proposed mechanism of inhibition, we evaluated the influence of the inhibitors on NADPH binding by FabG enzyme. A fluorescence-based binding assay was used to analyze binding of the dinucleotide to the enzyme. The sensitivity of the assay required a FabG concentration of 3 μM, limiting the assay conditions to the nonoptimal concentrations of the compounds (≥IC50) that do not allow measurement of the dose-dependent response at equilibrium. Nevertheless, we could demonstrate that at 1.5 μM all compounds inhibited the binding of NADPH to FabG (Supplementary Figure S11).
Structure–Activity Relationship for Lead Development
The small-molecule inhibitors described represent attractive start points for drug discovery with ligand efficiencies in the range 0.37–0.53 (Supplementary Table S3).38 Although lipophilic lipid efficiency may be more appropriate to drive lead optimization, the initial range of 2.9–5.3 represents excellent starting values.39 The compounds show no phenotypic response in the Gram-negative P. aeruginosa. However, the observation of activity of some compounds in the Gram-positive bacterium S. aureus suggests that this is either due to poor penetration of the compounds through the Gram-negative cell wall or, alternatively, rapid efflux of the compounds. Although the physicochemical properties to penetrate Gram-negative and Gram-positive bacteria are poorly defined,40 the diversity of chemotypes described gives a number of options for optimization to increase intrabacterial concentrations.
The analysis of the FabG-inhibitor complexes suggests that the optimal scaffold for optimization of potency should be symmetrical and planar. The binding cavity is formed within a four-helix bundle structure at the intersubunit interface, with two helices provided by each monomer. The subunits pack in head-to-tail orientation, leading to a symmetric binding cavity, reflected in two binding modes for the bound ligands in several cases. Therefore symmetrization of a chemical scaffold should allow for effective exploitation of the available space and interactions at the binding site. The optimal shape of the lead molecule could be based on the alternative orientations of some hit compounds observed in protein–ligand structures, e.g., for hit FG01 in the binding site located at the A/B interface.
The hit compounds in the FabG-inhibitor structures adopt almost planar conformations, which are not representative of one of their low energy conformers.41 Such an observation implies that the free energy of ligand binding could potentially be decreased by increasing the planarity of the lead molecule. This could be achieved by introducing condensed aromatic rings and other functional chemical groups with sp2 hybridization.
Finally, the designed chemical scaffold for a drug lead has to be supplemented with hydrogen-bond acceptor and/or donor groups at certain positions to acquire the desired specificity by interactions with the side chains of N111 and N115 and backbone amide and carbonyl groups.
Conclusions
Fatty acid biosynthesis, and in particular FabG, has been considered as an attractive drug target for treatment of infectious diseases.17,18 Our studies validate FabG as a drug target in P. aeruginosa, identify a series of novel FabG inhibitors, and provide important insight into mechanism of inhibition from the structural and biochemical characterization. A major, and unexpected, finding was the discovery of a cryptic binding site at the dimer–dimer interface. Binding of ligands at this site induces conformational changes that propagate to the active site and result in disturbance of the catalytic triad and loss of the ability to bind the cosubstrate NADPH. The results provide a framework for future rational design of potent small molecule inhibitors that target FabG.
Methods
Materials
3-Hydroxydecanoyl-N-acetylcysteamine (3-OH decanoyl-NAC) was synthesized using a published procedure42,43 by coupling (±)-3-hydroxydecanoic acid (Wako Chemicals) with NAC (Sigma-Aldrich). See Supporting Information for a complete list of the inhibitory compounds used.
fabG Gene Deletion
Construction of a P. aeruginosa PAO1 containing two chromosomal copies of fabG and construction of a suicide vector and the gene knockout at the native fabG locus were carried out using protocols as described in the Supporting Information.
Protein Production
Recombinant FabG was produced and purified as described in detail in the Supporting Information.
Enzyme Assays
Two independent assays, using either 3-OH decanoyl-NAC or acetoacetyl-coenzyme A (AcAcCoA) as substrate, were established to monitor enzymatic activity of FabG. The first assay (Forward Assay as it leads to NADP+ formation) utilizes the FabG-catalyzed reduction of AcAcCoA with NADPH as an electron donor. The reaction was followed spectrophotometrically by monitoring the consumption of NADPH at 340 nm. The second assay (Back Assay as it leads to NADPH formation) is based on the FabG-catalyzed oxidation of 3-OH decanoyl-NAC with NADP+ as cosubstrate. NADPH production was measured as an increase in fluorescence using an excitation wavelength of 340 nm and emission wavelength of 455 nm. For details see Supporting Information.
Enzymatic HTS Assay
The FabG high-throughput screen was performed using the Back Assay and a library of 15667 compounds (Dundee Drug Discovery Unit in-house diverse compound collection).44 Compounds were screened at a single concentration of 10 μM in the presence of NADP+ (1.5 mM) and 3-OH decanoyl-NAC (450 μM). Compounds exhibiting a percentage inhibition (PI) of >25% were considered as hits. For details see Supporting Information.
NMR Binding Assay
One-dimensional (1D) saturation transfer difference (STD) and water ligand observation with gradient spectroscopy (waterLOGSY) NMR experiments45 were used to screen a fragment library of 436 compounds (derived from Maybridge Ro3 library) for binding of FabG following the procedure applied previously for FabA.46 Compounds identified as FabG ligands in both experiments were considered as hits. For more details see Supporting Information.
FabG Inhibitor Studies
The identified hit compounds from both screens were either cherry-picked from the corresponding screening libraries or purchased from external suppliers. To evaluate the inhibitory properties of identified hit compounds in the Forward Assay, the enzymatic reaction was performed in the presence of NMR hit compounds at 1 mM concentration or HTS hits at 0.1 μM concentration.
To determine IC50 values 10-point (0.5 nM to 10 μM) (Back Assay) or 7-point (14 nM to 10 μM) (Forward Assay), dose–response curves were recorded using 3-fold serial dilution of each hit compound. For details see Supporting Information.
Fluorescence based NADPH binding studies, aggregation assay, and phenotypic growth assay are described in the Supporting Information.
Structure Determination
Three-dimensional structures of apo-FabG, the FabG-NADPH complex, and 16 enzyme–inhibitor complexes were determined using protein crystallography as outlined in the Supporting Information.
Acknowledgments
We gratefully acknowledge access to synchrotron radiation at the ESRF, Grenoble, France and at MAX IV Laboratory, Lund, Sweden. We also acknowledge access to equipment at the Protein Science Facility at Karolinska Institutet and the NMR Facility at the University of St. Andrews. We want to thank A. Holmgren for access to an EnSpire 2300 Multilabel reader. This work was funded by the Seventh Framework Program of the European Commission (FP7/2007-2013) under grant agreement no 223461, the Swedish Research Council, and the Wellcome Trust (grant no 083481/Z/07/Z).
Supporting Information Available
Methods details; structural overlays; KM determinations; unbiased electron density maps for 16 ligands; enzyme-NADPH and enzyme–inhibitor interaction schemes; fluorescence binding assays for NADPH; growth inhibition curve for S. aureus; data and refinement statistics; chemical and physicochemical properties and kinetic data for the small-molecule inhibitors. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and crystallographic data have been deposited under PDB accession codes 4afn (apo-FabG), 4ag3 (FabG-NADPH complex) and 4bnt, 4bnu, 4bnx, 4bnv, 4bny, 4bnz, 4bo0, 4bo1, 4bo2, 4bo3, 4bo4, 4bo5, 4bo6, 4bo7, 4bo8 and 4bo9 for the FabG–ligand complexes.
The authors declare no competing financial interest.
There were errors present in Table 1 of the version that was published September 23, 2014. The corrected version was re-posted on September 30, 2013.
Supplementary Material
References
- Kerr K. G.; Snelling A. M. (2009) Pseudomonas aeruginosa: a formidable and ever-present adversary. J. Hosp. Infect. 73, 338–344. [DOI] [PubMed] [Google Scholar]
- Mesaros N.; Nordmann P.; Plesiat P.; Roussel-Delvallez M.; Van Eldere J.; Glupczynski Y.; Van Laethem Y.; Jacobs F.; Lebecque P.; Malfroot A.; Tulkens P. M.; Van Bambeke F. (2007) Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infect. 13, 560–578. [DOI] [PubMed] [Google Scholar]
- Breidenstein E. B.; de la Fuente-Nunez C.; Hancock R. E. (2011) Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 19, 419–426. [DOI] [PubMed] [Google Scholar]
- Strateva T.; Yordanov D. (2009) Pseudomonas aeruginosa - a phenomenon of bacterial resistance. J. Med. Microbiol. 58, 1133–1148. [DOI] [PubMed] [Google Scholar]
- Hirsch E. B.; Tam V. H. (2010) Impact of multidrug-resistant Pseudomonas aeruginosa infection on patient outcomes. Expert Rev. Pharmacoecon. Outcomes Res. 10, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fothergill J. L.; Winstanley C.; James C. E. (2012) Novel therapeutic strategies to counter Pseudomonas aeruginosa infections. Expert Rev. Anti Infect. Ther. 10, 219–235. [DOI] [PubMed] [Google Scholar]
- Moynie L.; Schnell R.; McMahon S. A.; Sandalova T.; Boulkerou W. A.; Schmidberger J. W.; Alphey M.; Cukier C.; Duthie F.; Kopec J.; Liu H.; Jacewicz A.; Hunter W. N.; Naismith J. H.; Schneider G. (2013) The AEROPATH project targeting Pseudomonas aeruginosa: crystallographic studies for assessment of potential targets in early-stage drug discovery. Acta Crystallogr. F69, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Krause A.; Worgall S. (2012) Recent developments for Pseudomonas vaccines. Human Vaccines 7, 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsor G. L.; Lam D. K.; Fleming L.; Lo R.; Whiteside M. D.; Yu N. Y.; Hancock R. E.; Brinkman F. S. (2011) Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39, D596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Su S.; Bhatnagar R. K.; Hassett D. J.; Lu L. J. (2012) Prediction and analysis of the protein interactome in Pseudomonas aeruginosa to enable network-based drug target selection. PLoS One 7, e41202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D. I.; Vogel H. J. (2010) Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem. J. 430, 1–19. [DOI] [PubMed] [Google Scholar]
- Goodman C. D.; McFadden G. I. (2008) Fatty acid synthesis in protozoan parasites: unusual pathways and novel drug targets. Curr. Pharm. Des. 14, 901–916. [DOI] [PubMed] [Google Scholar]
- Rock C. O.; Jackowski S. (2002) Forty years of bacterial fatty acid synthesis. Biochem. Biophys. Res. Commun. 292, 1155–1166. [DOI] [PubMed] [Google Scholar]
- Campbell J. W.; Cronan J. E. Jr. (2001) Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu. Rev. Microbiol. 55, 305–332. [DOI] [PubMed] [Google Scholar]
- Heath R. J.; Rock C. O. (2004) Fatty acid biosynthesis as a target for novel antibacterials. Curr. Opin. Invest. Drugs 5, 146–153. [PMC free article] [PubMed] [Google Scholar]
- Pan P.; Tonge P. J. (2012) Targeting InhA, the FASII enoyl-ACP reductase: SAR studies on novel inhibitor scaffolds. Curr. Top. Med. Chem. 12, 672–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. B.; Rock C. O. (2011) Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery?. Curr. Opin. Microbiol. 14, 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. M.; Lu Y. J.; Rock C. O. (2004) The reductase steps of the type II fatty acid synthase as antimicrobial targets. Lipids 39, 1055–1060. [DOI] [PubMed] [Google Scholar]
- Kavanagh K. L.; Jornvall H.; Persson B.; Oppermann U. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 65, 3895–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn M. J.; Zheng C. J.; Kim W.G . (2008) Macrolactin S, a new antibacterial agent with FabG-inhibitory activity from Bacillus sp. AT28. J. Antibiot. (Tokyo) 61, 687–691. [DOI] [PubMed] [Google Scholar]
- Tasdemir D.; Lack G.; Brun R.; Ruedi P.; Scapozza L.; Perozzo R. (2006) Inhibition of Plasmodium falciparum fatty acid biosynthesis: evaluation of FabG, FabZ, and FabI as drug targets for flavonoids. J. Med. Chem. 49, 3345–3353. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe S. R.; Inglis K. A.; Urch J. E.; Muller S.; van Aalten D. M.; Fairlamb A. H. (2006) Kinetic, inhibition and structural studies on 3-oxoacyl-ACP reductase from Plasmodium falciparum, a key enzyme in fatty acid biosynthesis. Biochem. J. 393, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Luo S. Y.; Ye Y. B.; Zhao W. H.; Sun X. G.; Wang Z. Q.; Li R.; Sun Y. H.; Tian W. X.; Zhang Y. X. (2008) The antibacterial efficacy of an aceraceous plant [Shantung maple (Acer truncatum Bunge)] may be related to inhibition of bacterial beta-oxoacyl-acyl carrier protein reductase (FabG). Biotechnol. Appl. Biochem. 51, 73–78. [DOI] [PubMed] [Google Scholar]
- Zhang Y. M.; Rock C. O. (2004) Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty-acid synthase. J. Biol. Chem. 279, 30994–31001. [DOI] [PubMed] [Google Scholar]
- Lai C. Y.; Cronan J. E. (2004) Isolation and characterization of beta-ketoacyl-acyl carrier protein reductase (fabG) mutants of Escherichia coli and Salmonella enterica serovar Typhimurium. J. Bacteriol. 186, 1869–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish T.; Roberts G.; Laval F.; Schaeffer M.; Daffe M.; Duncan K. (2007) Functional complementation of the essential gene fabG1 of Mycobacterium tuberculosis by Mycobacterium smegmatis fabG but not Escherichia coli fabG. J. Bacteriol. 189, 3721–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T. T.; Karkhoff-Schweizer R. R.; Kutchma A. J.; Schweizer H. P. (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86. [DOI] [PubMed] [Google Scholar]
- Hoang T. T.; Kutchma A. J.; Becher A.; Schweizer H. P. (2000) Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43, 59–72. [DOI] [PubMed] [Google Scholar]
- Price A. C.; Zhang Y. M.; Rock C. O.; White S. W. (2001) Structure of beta-ketoacyl-[acyl carrier protein] reductase from Escherichia coli: negative cooperativity and its structural basis. Biochemistry 40, 12772–12781. [DOI] [PubMed] [Google Scholar]
- Cohen-Gonsaud M.; Ducasse S.; Hoh F.; Zerbib D.; Labesse G.; Quemard A. (2002) Crystal structure of MabA from Mycobacterium tuberculosis, a reductase involved in long-chain fatty acid biosynthesis. J. Mol. Biol. 320, 249–261. [DOI] [PubMed] [Google Scholar]
- Zaccai N. R.; Carter L. G.; Berrow N. S.; Sainsbury S.; Nettleship J. E.; Walter T. S.; Harlos K.; Owens R. J.; Wilson K. S.; Stuart D. I.; Esnouf R. M. (2008) Crystal structure of a 3-oxoacyl-(acylcarrier protein) reductase (BA3989) from Bacillus anthracis at 2.4-A resolution. Proteins 70, 562–567. [DOI] [PubMed] [Google Scholar]
- Subramanian S.; Abendroth J.; Phan I. Q.; Olsen C.; Staker B. L.; Napuli A.; Van Voorhis W. C.; Stacy R.; Myler P. J. (2011) Structure of 3-ketoacyl-(acyl-carrier-protein) reductase from Rickettsia prowazekii at 2.25 A resolution. Acta Crystallogr. F67, 1118–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D.; Bhattacharyya S.; Das A. K. (2012) Crystal structure and fluorescence studies reveal the role of helical dimeric interface of staphylococcal fabg1 in positive cooperativity for NADPH. Proteins 80, 1250–1257. [DOI] [PubMed] [Google Scholar]
- Willett P.; Barnard J. M.; Downs G. M. (1998) Chemical similarity searching. J. Chem. Inf. Comput. Sci. 38, 983–996. [Google Scholar]
- Cheng T.; Li Q.; Zhou Z.; Wang Y.; Bryant S. H. (2012) Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 14, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A. C.; Zhang Y. M.; Rock C. O.; White S. W. (2004) Cofactor-induced conformational rearrangements establish a catalytically competent active site and a proton relay conduit in FabG. Structure 12, 417–428. [DOI] [PubMed] [Google Scholar]
- Dutta D.; Bhattacharyya S.; Roychowdhury A.; Biswas R.; Das A. K. (2013) Crystal structure of hexanoyl-CoA bound to β-ketoacyl reductase FabG4 of Mycobacterium tuberculosis. Biochem. J. 450, 127–39. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. (2004) Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Keserü G. M. (2012) Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discovery 11, 355–365. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Bickerton R. (2010) Drug discovery: Know your chemical space. Nat. Chem. Biol. 6, 482–483. [DOI] [PubMed] [Google Scholar]
- Bolton E. E., Wang Y., Thiessen P. A., Bryant S. H., Ralph A. W. and David C. S. (2008) PubChem: Integrated Platform of Small Molecules and Biological Activities, in Annual Reports in Computational Chemistry Vol. 4, 12, pp 217–241, Elsevier, New York. [Google Scholar]
- Kim M. I.; Kwon S. J.; Dordick J. S. (2009) In vitro precursor-directed synthesis of polyketide analogues with coenzyme a regeneration for the development of antiangiogenic agents. Org. Lett. 11, 3806–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapeyre C.; Delomenede M.; Bedos-Belva l. F.; Duran H.; Negre-Salvayre A.; Baltas M. (2005) Design, synthesis, and evaluation of pharmacological properties of cinnamic derivatives as antiatherogenic agents. J. Med. Chem. 48, 8115–8124. [DOI] [PubMed] [Google Scholar]
- Brenk R.; Schipani A.; James D.; Krasowski A.; Gilbert I. H.; Frearson J.; Wyatt P. G. (2008) Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem. 3, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B.; Peters T. (2003) NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem., Int. Ed. 42, 864–890. [DOI] [PubMed] [Google Scholar]
- Moynie L.; Leckie S. M.; McMahon S. A.; Duthie F. G.; Koehnke A.; Taylor J. W.; Alphey M. S.; Brenk R.; Smith A. D.; Naismith J. H. (2013) Structural insights into the mechanism and inhibition of the beta-hydroxydecanoyl-acyl carrier protein dehydratase from Pseudomonas aeruginosa. J. Mol. Biol. 425, 365–377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.