

Abstract
Human pluripotent stem cells (hPSCs) have an unparalleled potential to generate limitless quantities of any somatic cell type. However, current methods for producing populations of various somatic cell types from hPSCs are generally not standardized and typically incorporate undefined cell culture components often resulting in variable differentiation efficiencies and poor reproducibility. To address this, we have developed a defined approach for generating epithelial progenitor and epidermal cells from hPSCs. In doing so, we have identified an optimal starting cell density to maximize yield and maintain high purity of K18+/p63+ simple epithelial progenitors. In addition, we have shown that the use of synthetic, defined substrates in lieu of Matrigel and gelatin can successfully facilitate efficient epithelial differentiation, maintaining a high (>75%) purity of K14+/p63+ keratinocyte progenitor cells and at a two to threefold higher yield than a previously reported undefined differentiation method. These K14+/p63+ cells also exhibited a higher expansion potential compared to cells generated using an undefined differentiation protocol and were able to terminally differentiate and recapitulate an epidermal tissue architecture in vitro. In summary, we have demonstrated the production of populations of functional epithelial and epidermal cells from multiple hPSC lines using a new, completely defined differentiation strategy.
Introduction
Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), have the unique capability to indefinitely self-renew and to generate somatic cell types from all three embryonic germ layers.1–3 Because of these characteristics, hPSCs possess a tremendous potential for numerous tissue engineering applications, including, but not limited to, regenerative medicine, in vitro model systems to study tissue morphogenesis and disease, and pharmaceutical or toxicity screens. To meet the industrial and clinical demands for the various cell types that can be generated from hPSCs, it will be necessary to employ reproducible hPSC differentiation systems that exclusively incorporate defined cell culture components. Such standardized and well-characterized differentiation systems should be amenable for scale-up, reduce variability observed in hPSC differentiation systems, and be compatible with good manufacturing practice (GMP).4,5 Since the initial derivation of hESCs, researchers have engineered such defined culture systems for both the maintenance6–14 and differentiation of hPSCs to certain cell lineages, including but not limited to retinal pigment epithelial cells, neural precursor cells, and cardiomyocytes.5,15,16
Epithelial cells have been derived and characterized from hPSCs using a variety of methods.17–22 One method, developed by Hewitt et al., involves culturing hESCs in a serum-containing normal human keratinocyte medium, supplementing cells with bone morphogenetic protein 4 (BMP4), and subculturing cells on type I collagen. The resulting epithelial cells express the simple epithelial cell marker cytokeratin 18 (K18) and can further differentiate, indicated by expression of cytokeratin 12 (K12) and loss of K18.18 Another method for generating epithelial cells involves coculturing hESCs with PA6 stromal cells and supplementing media with BMP4 followed by culture in a serum-containing medium. The resulting cells express K18 and can acquire expression of basal keratinocyte marker cytokeratin 14 (K14) if cultured on a keratinocyte-derived extracellular matrix.17 A robust method for engineering high purity populations of non-neural epithelia from hPSCs, illustrated in Figure 1, utilizes retinoic acid (RA) to initiate hPSC commitment to an epithelial cell fate, while the cells are plated in a feeder-free system on a Matrigel substrate.23 While BMP4 can be exogenously added to the medium to synergize with RA, endogenous BMP4 activity in the unconditioned differentiation medium (UM+RA) has shown to be sufficient for the epithelial commitment.20,24,25 After 1 week of culture in UM+RA, the resulting epithelial progenitor cells coexpressed K18 and the transcription factor, p63, which has been found necessary for epidermal development in vivo and for simple epithelial cell differentiation to K14+ keratinocytes in vitro.17,26 These K18+/p63+ cells were coerced into maturing towards an epidermal cell fate when subcultured on a gelatin substrate in a defined keratinocyte medium. While this differentiation method has been shown to generate functional K14+/p63+ epidermal keratinocyte cells with the capacity to stratify, cornify, and recapitulate an epidermal-like architecture when cultured at the air-liquid interface (ALI),27 the effect of cell density on epithelial cell purity and yield is not yet clear. In addition, this differentiation system incorporates two undefined substrates, Matrigel and gelatin, in which lot-to-lot variability of these substrates can have a potential impact on differentiation reproducibility.
FIG. 1.
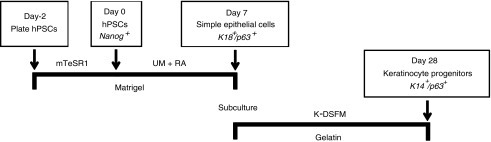
Method for generating simple epithelial cells and keratinocyte progenitors from human pluripotent stem cells (hPSCs). hPSCs are passaged onto Matrigel and expanded in an mTeSR1 medium for 2 days before differentiation. Cells in this study were characterized on day 0, 7, and 28 for marker proteins to assess pluripotency or epithelial commitment. UM, unconditioned hESC medium; RA, retinoic acid; K-DSFM, keratinocyte defined serum-free medium.
Numerous synthetic, defined substrates have been developed to support the maintenance and growth of hPSCs and, in some cases, differentiation. Two of these substrates include StemAdhere (Stemcell Technologies, Vancouver, Canada) and Synthemax (Corning, Corning, NY).28,29 To date, no method exists to generate epithelial and epidermal cell populations from hPSCs using a defined, feeder-free culture system. Here we demonstrate efficient, completely defined differentiation of hPSCs epithelial and epidermal progenitors.
Materials and Methods
Cell culture and differentiation
H9 hESCs and IMR90 clone 4 hiPSCs were cultured in feeder-free conditions on Matrigel (BD Biosciences, San Jose, CA), StemAdhere (Stemcell Technologies), or Synthemax in mTeSR1 medium (Stemcell Technologies) and passaged every 4–5 days using Versene (Life Technologies, Grand Island, NY). Singularized hPSCs were passaged using ROCK inhibitor Y27632 (Sigma-Aldrich, St. Louis, MO). Before differentiation, cell counts were made from one well of a culture plate to determine cell density at the onset of differentiation. For differentiation, hPSCs cultured in mTeSR1 were switched to unconditioned hESC medium (UM) supplemented with retinoic acid (UM+RA): DMEM/F12 containing 20% knockout serum replacer, 1× MEM nonessential amino acids, 1 mM L-glutamine (all from Life Technologies), 0.1 mM β-mercaptoethanol, and 1 μM all-trans RA (Sigma-Aldrich). Cells were cultured in UM+RA for 7 days, changing medium daily. RA-treated cells were detached using 2 mg/mL Dispase (Life Technologies) and passaged onto Synthemax, StemAdhere, or gelatin-coated plates in Keratinocyte Defined Serum-free Medium (K-DSFM; Life Technologies). After 3–4 weeks, differentiated cells were subcultured using trypsin to obtain high purity epithelial cell populations.
Flow cytometry
Cells were detached from culture plates using Trypsin-EDTA, fixed in 1% paraformaldehyde for 10 min at 37°C, and permeabilized in FACS buffer (phosphate-buffered saline [PBS] with 0.5% bovine serum albumin, 0.1% NaN3, and 0.1% Triton X-100). Primary antibodies were incubated overnight in FACS buffer; control samples were included using no primary antibody. After a 1 h secondary stain cells were analyzed on a FACScalibur using CellQuest software or a BD LSRII flow cytometer using BD FACSDiva software.
Antibodies and immunostaining
Cells in monolayer were fixed in 4% paraformaldehyde for 20 min, rinsed in PBS, and incubated in 5% milk with 0.4% (v/v) Triton X-100 in PBS for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C in blocking buffer, and after subsequent washes in PBS, fluorophore-conjugated secondary antibodies were applied for 1 h at room temperature. Nuclear staining was accomplished using Hoechst dye (Sigma-Aldrich). Primary antibodies used for immunostaining and flow cytometry were rabbit anti-K14 polyclonal (Lab Vision, Fremont, CA), mouse anti-p63 monoclonal, rabbit anti-K18 polyclonal (Santa Cruz Biotechnology, Santa Cruz, CA), AlexaFluor 700-conjugated mouse anti-K18 monoclonal (Abcor, Holland, MI), and AlexaFluor 488-conjugated mouse anti-Ki67 (BD Biosciences). Species-specific secondary antibodies were obtained from Life Technologies and were conjugated to AlexaFluor 488 and 594 dyes. Mouse IgG1 and Rabbit IgG isotype controls were obtained from BD Biosciences and Lab Vision, respectively. Immunofluorescence images were acquired on an Olympus IX70 microscope using MetaVue imaging software.
Expansion potential/proliferation experiments
K14+/p63+ cells were passaged onto either Synthemax or gelatin-coated 6-well plates and plated at about 1–5× 104 cells per well. Cultures were trypsinized at various times points when there was sufficient cell growth and the number of cells per well was determined using a hemacytometer.
Terminal differentiation and organotypic skin culture
Terminal differentiation in monolayer culture was induced by adding 0.66 mM Ca2+ to cells in K-DSFM at day 28 of differentiation. Cells were cultured in high K-DSFM+Ca2+ for 7–10 days before analysis. Organotypic skin culture was performed based on protocols by Egles et al. and Metallo et al.27,30 Briefly, dermal constructs were prepared by combining 240 μL 10× EMEM (Lonza, Walkersville, MD), 21.6 μL L-glutamine (Life Technologies), 270 μL fetal bovine serum (FBS; Hyclone, Logan, UT), 74.8 μL NaHCO3, 1.33 mL 1× PBS, and 667 μL rat tail collagen (3 mg/mL; Life Technologies) on ice. Two hundred microliters of this acellular matrix was added to each of twelve 0.4 μm pore size inserts and cultured in a 12-well plate. After gelation of the acellular matrix, the cellular dermal construct was prepared on ice consisting of 300 μL 10× EMEM, 27.1 μL L-glutamine, 337 μL FBS, 93.3 μL NaHCO3, 1.67 mL 1× PBS, 833 μL rat tail collagen (3 mg/mL), and 200 μL of 3×105 human foreskin fibroblasts. Two hundred fifty microliters of the cellular dermal construct mixture was added to each insert. After gelation of the cellular dermal construct, wells were flooded with prewarmed flavinoid adenine dinucleotide (FAD) medium, which consisted of 3:1 F12/DMEM basal medium with 2.5% FBS (Life Technologies) and 0.4 μg/mL hydrocortisone, 8.4 ng/mL cholera toxin, 5 μg/mL insulin, 24 μg/mL adenine (all from Sigma-Aldrich), 10 ng/mL epidermal growth factor, and 1× antibiotic/antimycotic. The dermal constructs were gently detached from the sides of the inserts using a Pasteur pipet to facilitate contraction over a few days. About 5×105 hPSC-derived K14+/p63+ cells were seeded on contracted gels in 100 μL of FAD medium and allowed to attach for 3–4 h before flooding the wells with FAD medium. After 2 days of submerged culture in FAD, cells were submerged in FAD medium+0.66 mM Ca2+ for an additional 2 days. Cells were then maintained for 2–3 weeks at the ALI where the bottom of the insert just contacts the interface with the FAD+Ca2+ media.
Histology and immunohistochemistry
Organotypic skin cultures were removed from well inserts and fixed overnight in 10% formalin at 4°C and stored in 70% ethanol before embedding in paraffin. Sections were cut at 5 μm and mounted on charged slides. The slides were deparaffinized in three changes of xylene and dehydrated in graded alcohols to water. Endogenous peroxidase was blocked with 0.3% hydrogen peroxide in methanol for 20 min. Antigen retrieval was completed according to the manufacturer's instructions for each antibody. After antigen retrieval, the slides were blocked in 5% goat serum and incubated for 1 h with titered primary antibodies. After washing, slides were incubated with fluorescent secondary antibodies for 1 h or stained with hematoxylin and eosin, and mounted with antifade reagent.
Results
Effect of initial cell density
The effect of initial cell density on differentiation efficiency is a phenomenon that has been observed in hPSC differentiation approaches to generate neuroepithelia and non-neural epithelia.19,31 For example, Ji et al. reported that the plating density of embryoid bodies (EBs) had a profound effect on the number of keratinocyte progenitors formed where a lower plating density of EBs resulted in a greater yield of keratinocytes.19 In the study of neural differentiation of hESCs, it was found that the plating density of hESCs had an effect on the ratio of Pax6+ cells to Pax6− neural crest-like cells where high hESC plating densities resulted in mostly Pax6+ cells and low densities promoted primarily neural crest differentiation.31 Given these observations in other epithelial and neural differentiation approaches, we determined the effect of initial hPSC density on our previously reported epithelial differentiation method and given this effect, identified an optimal density to maximize cell purity and yield of our hPSC-derived epithelial cells.
We previously reported a directed differentiation method for generating epithelial cells and epidermal progenitors from hESCs.23 This approach, illustrated in Figure 1, uses RA to induce simple epithelial differentiation and subsequently involves subculturing simple epithelial cells in a defined keratinocyte growth medium to generate highly enriched populations of K14+/p63+ epidermal keratinocyte progenitor cells. To identify how initial cell density affects RA-induced epithelial differentiation, we passaged H9 hESCs onto a Matrigel substrate at varying cell densities. After 1–2 days of expansion, we induced epithelial differentiation at starting cell densities of 1000, 3000, 6500, and 30,000 cells/cm2 for 1 week and found a striking difference in the resulting cellular morphologies (Fig. 2A). Initial hPSC densities below 6500 cells/cm2 resulted in cell populations primarily containing cells with a mesenchymal morphology on day 7 (Fig. 2Ai, ii, v). In contrast, cultures at cell densities at or above 6500 cells/cm2 upon initiation of differentiation resulted in cell populations containing colonies of cells possessing an epithelial morphology by day 7 (Fig. 2Aiii, iv, vi). In addition, we found that cultures where differentiation initiated at a cell density of 6500 cells/cm2 yielded cells that uniformly expressed E-cadherin by day 7 of epithelial differentiation (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/tec), whereas cultures differentiated from a cell density 1000 cells/cm2 failed to express E-cadherin by day 7 of differentiation (Supplementary Fig. S1B). These results suggest that the initial cell density of hPSCs has a profound effect on the ability of hPSCs to form simple epithelial cell populations when cultured in the presence of RA.
FIG. 2.

Effect of initial hPSC density on epithelial differentiation. (A) Phase contrast images of H9 human embryonic stem cells (hESCs) at day 0 (top panels) and day 7 (bottom panels) of epithelial differentiation at starting cell densities of (i) 1000, (ii) 3000, (iii) 6500, and (iv) 30,000 cells/cm2. High-resolution images showing representative cells from (v) 1000 cells/cm2 day 7 culture exhibiting a mesenchymal-like morphology and from (vi) 6500 cells/cm2 day 7 culture exhibiting an epithelial morphology. Scale bar in panels i-iv is 200 μm and scale bar in panels v-vi is 100 μm. (B) Representative flow cytometry dot plots showing expression of K18 in day 7 cell populations at various starting cell densities compared to an isotype control. Color images available online at www.liebertpub.com/tec
To quantify the effect of initial cell density on epithelial commitment of hPSCs, we investigated expression of the simple epithelial marker K18 in H9 hESC-derived epithelial cell populations using flow cytometry and found that the starting densities of 6500 and 30,000 cells/cm2 at day 0 resulted in >95% pure K18+ epithelial cell populations by day 7 (Fig. 2B). We also found that these cells coexpressed K18 and p63 in culture (Fig. 3C, D). In contrast, cultures at cell densities of 1000 cells/cm2 at day 0 resulted in populations of <20% K18+ epithelial cells and representative cell colonies were found to not express p63 (Figs. 2B and 3A). At a starting density of 3000 cells/cm2, we found that 75% of cells at day 7 expressed K18 (Fig. 2B) and some of these K18+ cells also expressed p63 (Fig. 3B). No cells at day 7 at any starting density expressed the pluripotency marker Nanog (Supplementary Fig. S2).
FIG. 3.

Expression of K18 and p63 in H9-derived simple epithelial cells at day 7 of differentiation at various starting densities. Representative colonies are shown stained with antibodies against K18 and p63 for day 0 hPSC densities of (A) 1000, (B) 3000, (C) 6500, and (D) 30,000 cells/cm2. Insets show higher magnification images of representative cells that coexpress K18 and p63. Scale bar in all panels is 50 μm. Color images available online at www.liebertpub.com/tec
To identify the threshold of initial hPSC density required to achieve highly enriched K18+ simple epithelial cell populations, we expanded our analysis by investigating a range of densities from 500 to 40,000 cells/cm2. We found that a cell density of at least 4500 cells/cm2 was necessary to achieve >90% pure populations of K18+ simple epithelial cells (Fig. 4A). Samples with initial cell densities lower than 4500 cells/cm2 resulted in populations that had not only low purities of K18+ cells, but were also highly variable among biological replicates. For each starting hPSC density, we calculated the yield of K18+ simple epithelial by normalizing the absolute number of K18+ cells in culture at day 7 to the number of Nanog+ cells present at day 0. These results, shown in Figure 4B, indicate a maximum yield of K18+ epithelial cells, 20 K18+ cells at day 7 per undifferentiated cell at day 0, was achieved at a starting density of 6500 cells/cm2. In addition, we demonstrated successful differentiation of these simple epithelial cells at this starting density to validate the potential of these cells to form K14+ epidermal keratinocyte progenitors (Supplementary Fig. S3). Our analysis of the effects of hPSC density upon initiation of epithelial differentiation has identified (1) a lower limit of initial cell density to achieve high K18+ simple epithelial cell purities and (2) an initial cell density for maximizing epithelial cell yield, while maintaining high epithelial cell purity. We observed no difference in initial cell density dependence when passaging hPSCs as either single cells or colonies before differentiation (Supplementary Fig. S4). For the remainder of this study, we used the optimal cell density of 6500 cells/cm2 to achieve highly enriched (>90%) simple epithelial cell populations and maximize our epithelial cell yield.
FIG. 4.

Yield and purity of K18+ simple epithelial cells as a function of day 0 cell density. (A) Purity of H9-derived simple epithelial cells at day 7 of differentiation at various starting densities as measured by flow cytometry. (B) Yield of simple epithelial cells at day 7 of differentiation calculated by normalizing the absolute number of K18+ epithelial cells at day 7 to the starting number of Nanog+ cells at day 0. Error bars indicate standard deviation of three independent replicates. Color images available online at www.liebertpub.com/tec
Epithelial commitment using defined substrates
In addition to optimizing epithelial cell yield and purity by altering hPSC density, we also sought to completely define all cell culture components of this epithelial differentiation system. First, we investigated possibilities to replace Matrigel with a defined, xeno-free substrate. We identified StemAdhere and Synthemax, two synthetic and defined substrates, as potential candidates to replace Matrigel given their ability to promote hPSC growth.28,29 StemAdhere is functionalized with the extracellular portion of E-cadherin, a protein present in epithelial cells that modulates cell-cell contact.29 Synthemax is functionalized with an RGD-containing peptide to facilitate cell adhesion via integrin binding.28 Given the importance of both cell-cell interactions and cell-matrix interactions on epithelial and epidermal differentiation, we chose StemAdhere and Synthemax as potentially suitable candidates to facilitate the epithelial differentiation of hPSCs in lieu of Matrigel.32,33
We investigated the ability of each of these defined substrates to promote epithelial commitment of hPSCs following culture in UM containing RA (Fig. 5A). We analyzed cells via flow cytometry after 7 days of differentiation and found that StemAdhere and Synthemax both successfully facilitated the differentiation of H9 hESCs to highly enriched (>90%) epithelial populations, similar to those produced on Matrigel in terms of K18 or p63 expression throughout the cell population (Fig. 5B–D and Supplementary Fig. S3). We have demonstrated that cell populations generated on these defined substrates coexpressed K18 and p63 epithelial markers by day 7 of differentiation, similar to cell populations generated on Matrigel substrates (Fig. 5E). We have also demonstrated the ability of StemAdhere and Synthemax to efficiently promote simple epithelial differentiation of IMR90 clone 4 hiPSCs (Supplementary Fig. S5)
FIG. 5.

Epithelial commitment of hPSCs on defined substrates. (A) Method of generating K18+ simple epithelial cells by culturing hPSCs on a StemAdhere or Synthemax substrate in lieu of a Matrigel substrate. (B) Quantification of flow cytometry data exhibiting highly enriched K18+ or p63+ cell populations generated on Matrigel, StemAdhere, and Synthemax substrates. Error bars indicate standard deviation of three independent replicates for each condition (*, # indicates p>0.05 compared to Matrigel condition). (C) Representative flow cytometry dot plots showing homogenous K18+ or (D) p63+ populations generated from H9 hESCs cultured on defined substrates compared to Matrigel on day 7 of differentiation. Isotype controls are shown for each condition. (E) Immunofluorescence images of K18 expression in H9 hESC-derived cell populations at day 7 of differentiation. Scale bar in all panels is 100 μm. Color images available online at www.liebertpub.com/tec
Generation of epidermal keratinocyte progenitors in defined conditions
Our previously reported method for generating epidermal keratinocyte progenitors from hPSCs involved subculture of hPSC-derived simple epithelial cells on a gelatin substrate in a keratinocyte defined, serum-free medium (K-DSFM) for 3 weeks (Fig. 1). To develop a completely defined differentiation system to generate K14+/p63+ epidermal keratinocyte progenitors, we investigated two approaches, both outlined in Figure 6A. The first approach involved simple epithelial commitment of hPSCs on a StemAdhere or Synthemax substrate followed by subculturing these simple epithelial cells onto StemAdhere or Synthemax in K-DSFM for 3 weeks (Fig. 6A-i). The second approach also involved the simple epithelial commitment of hPSCs on either a StemAdhere or Synthemax substrate, but rather than enzymatically removing the cells and subculturing, the cells remained plated on the initial substrate and were cultured in K-DSFM for a 3 week period (Fig. 6A-ii). In both cases, cell populations were analyzed for expression of keratinocyte markers at day 28.
FIG. 6.

Development of a completely defined, standardized epithelial differentiation system. (A) Method for generating epithelial and epidermal progenitor cells from hPSCs using a completely defined system (i) with or (ii) without a subculture step. (B) Representative flow cytometry dot plots showing K14 expression in H9 hESC-derived cell populations at day 28 of differentiation generated using conventional substrates or defined substrates (top panels) with a subculture step or (bottom panels) without a subculture step compared to an isotype control. (C) Quantification of flow cytometry data measuring purity of K14+ cells at day 28 of differentiation from each condition shown in (B). N=3 independent replicates for each condition. (D) Yield of K14+ cells at day 28 of differentiation of conditions incorporating a subculture step. Yield measured by absolute number of K14+ cells at day 28 relative to the starting number of Nanog+ cells at day 0 of differentiation. Proliferative fraction of K14+ cells shown as measured by Ki67 coexpression as well as the nonproliferative (Ki67-) fraction of K14+ cells. Error bars indicate standard deviation of three independent replicates for each condition (* indicates p<0.05 compared to Matrigel/gelatin condition). (E) Immunofluorescence images of K14 expression in day 28 differentiated cell populations generated on Matrigel/gelatin or defined substrates. Scale bar in all panels is 100 μm. Color images available online at www.liebertpub.com/tec
In investigating both of these approaches to completely define our differentiation system, we made a few key observations. First, simple epithelial cell colonies derived from H9 hESCs on either a StemAdhere or Synthemax substrate did not survive beyond a few days when subcultured onto a new StemAdhere surface indicating that the StemAdhere surface does not provide the cues needed differentiation or even survival of the hPSC-derived simple epithelial cells (data not shown). Second, H9 hESCs differentiated without a subculture step, using the approach outlined in Figure 6A-ii, failed to produce high purity K14+ keratinocyte progenitor cell populations at day 28 of differentiation regardless of the substrate used (Fig. 6B, C). Specifically, differentiation on a StemAdhere surface yielded a 19.8%±7.1% K14+ cell population at day 28, while differentiation on a Synthemax surface yielded a 13.3%±3.0% K14+ cell population. These cultures were found not to be enriched in K10+ cells indicating that elimination of the subculture step did not induce terminal differentiation to epidermal keratinocytes (Supplementary Fig. S6). These results indicate that eliminating the subculture step in this differentiation system will result in low purity K14+ epidermal keratinocyte progenitor populations. Third, simple epithelial cells differentiated on either a StemAdhere or Synthemax substrate could be subcultured onto a Synthemax substrate and successfully produced highly enriched (>75%) cell populations of K14+ epidermal keratinocyte progenitor cells similar to those cell populations generated using Matrigel and gelatin substrates (Fig. 6B, C). We found that these cell populations generated on defined substrates also coexpressed p63 similar to those derived on Matrigel and gelatin (Fig. 6E). In addition, we observed the generation of high purity K14+ cell populations from IMR90 clone 4 hiPSCs using this same system outlined in Figure 6A-i (Supplementary Fig. S7A). Together, these results demonstrate that in replacing Matrigel with either StemAdhere or Synthemax and replacing the gelatin with Synthemax thus creating a completely defined culture system, one can successfully generate high purity populations of K14+/p63+ cells from a hESC or a hiPSC source.
Keratinocyte progenitor yield and expansion potential
In addition to developing a defined, efficient epithelial differentiation method by incorporating StemAdhere or Synthemax to replace Matrigel and Synthemax to replace gelatin, we surprisingly found that this system produced higher yields of K14+ keratinocyte progenitors. In fact, we determined that this defined epithelial differentiation system yielded of 20–25 K14+ cells per undifferentiated cell, two- to three-fold higher than the original epithelial differentiation system using Matrigel and gelatin substrates (Fig. 6D). We also observed a three to fourfold greater yield when using the defined culture system to generate K14+ epidermal keratinocyte progenitors from the IMR90 clone 4 hiPSC line and achieved approximately 30 K14+ cells per undifferentiated cell (Supplementary Fig. S7B). In addition, K14+ cell populations generating using the defined culture system had a greater proliferative fraction of cells, quantified by Ki67 staining, compared to the original culture system by measuring Ki67 coexpression in these K14+ cell populations (Fig. 6D).
Given the higher fraction of Ki67+ cells in our K14+ cell populations derived using a defined system compared to our original system, we sought to further investigate the expansion potential of our epidermal keratinocyte progenitor cells. We have previously determined that K14+ epidermal keratinocyte progenitor cells derived using our original differentiation system, outlined in Figure 1, have a very limited expansion potential.20,23 A higher expansion potential would, in fact, make these cells more attractive for tissue engineering applications. We therefore, investigated the proliferative capacity of our K14+ keratinocyte progenitor cell populations derived in our defined differentiation system by quantifying the amount of cell doublings possible. By plating H9-derived K14+ epidermal keratinocyte progenitors differentiated using our defined culture system on a Synthemax substrate and counting doublings for a period of 3–4 weeks, we found that these K14+ cell populations underwent twice as many population doublings before senescence compared to differentiation on Matrigel and gelatin (Fig. 7A). All cell populations maintained high K14 expression after senescence as measured by flow cytometry (Fig. 7B). This higher proliferative capacity of the K14+ keratinocyte progenitor population generated using the defined differentiation system could be advantageous for engineering skin tissues from hPSCs in the future.
FIG. 7.

Expansion potential of epidermal keratinocyte progenitors generated using defined, standardized substrates. (A) K14+/p63+ cell populations at day 28 of differentiation were subcultured onto the same substrate (gelatin or Synthemax) on which the cells were generated and measurements of cell counts were taken until senescence was observed. (B) Flow cytometry data taken of populations after senescence to confirm the cell populations remained enriched with K14+ cells compared to an isotype control. Color images available online at www.liebertpub.com/tec
Terminal differentiation to epidermal keratinocytes
To confirm that these K14+/p63+ keratinocyte progenitor cells differentiated using our defined culture system are fit to be incorporated into skin tissue models and other tissue engineering applications, we investigated the ability of these cells to terminally differentiate to epidermal keratinocytes in culture. We incorporated H9-derived K14+/p63+ keratinocyte progenitor cells differentiated on a Synthemax substrate into an organotypic culture system and observed stratification and terminal-differentiation of these keratinocyte progenitors to epidermal keratinocytes (Fig. 8A–F). Specifically, while we observed the presence of K14 and p63 in the basal and the immediate suprabasal layers (Fig. 8B, C), the terminal differentiation markers filaggrin, involucrin, and cytokeratin 10 (K10) were exclusively present in the suprabasal layers of the stratified tissue (Fig. 8D–F) similar to what is observed in human skin.27 These results indicate that these cells have the ability to recapitulate an epidermal-like architecture in vitro and expressed markers of terminal epidermal differentiation.
FIG. 8.

Stratification and terminal differentiation of H9 hESC-derived keratinocyte progenitors to epidermal keratinocytes. (A) H&E stain of a cross-section of H9 hESC-derived K14+/p63+ cell populations generated using the defined epithelial differentiation system on Synthemax substrates after culture for in an organotypic culture system. Inset shows multiple cell layers. Immunohistochemical stains were performed on cross-sections of these tissues showing the presence of (B) K14 (red), (C) p63 (red), and terminal-differentiation markers (D) filaggrin (green), (E) involucrin (green), and (F) K10 (green). Scale bar in all panels is 50 μm.
Discussion
Research efforts on engineering hPSCs to generate populations of specific somatic cell types have focused on designing culture systems that are efficient, robust, and scalable, often using completely defined cell culture systems. We have previously demonstrated that we can guide hESCs towards an epithelial and, subsequently, an epidermal cell fate by treating hESCs with RA on a Matrigel substrate followed by subculture in K-DSFM on a gelatin substrate.20,23 In the present study, we report that cell density upon initiation of differentiation affects purity and yield of epithelial progenitors. Specifically, we found that commencing epithelial differentiation of singularized hPSCs at a density of 6500 cells/cm2 is optimal considering the balance between purity and yield of cells. This cell density maximizes epithelial cell yield while maintaining high purity (>90%) populations of K18+/p63+ cells. The intercellular and/or intracellular mechanisms that give rise to this phenomenon have not specifically been investigated in this study, but elucidation of these mechanisms may provide insight as to how to further optimize this system of epithelial and epidermal differentiation of hPSCs. In addition, such knowledge might be essential for the future scale-up of this differentiation system to provide large quantities of epithelial or epidermal cells to meet industrial or clinical demands for various tissue engineering applications.
In this study, we have also designed a completely defined culture system to facilitate epithelial and epidermal differentiation of hPSCs by replacing the two undefined culture components, Matrigel and gelatin, with defined substrates. Using this defined protocol, we demonstrated high epidermal progenitor cell purities (>80%) as measured by K14 expression, in both hESCs and hiPSCs. In addition, we have demonstrated that this defined epithelial differentiation system can yield two to threefold more K14+/p63+ cells compared to differentiation on Matrigel and gelatin, and that cells differentiated via the defined protocol also exhibit a greater expansion potential. The improvement in yield in the defined system compared to the original differentiation protocol may be attributed to the improved plating efficiency of K18+/p63+ simple epithelial cell colonies subcultured on Synthemax compared to those cells plated on gelatin (data not shown). However, this does not necessarily explain why a larger proliferative fraction and the higher expansion potential of the K14+/p63+ keratinocyte progenitor cells resulted from the defined system. Differences in cell-matrix interactions between the two differentiation systems could account for this difference in the growth and maturation of K18+/p63+ simple epithelial cells derived from hPSCs. One key difference between gelatin and Synthemax is that Synthemax has been reported to be functionalized specifically with the binding domains of bone sialoprotein and vitronectin.28,34 Given that epidermal keratinocytes express high levels of αvβ5 integrin,33 a vitronectin receptor, it is conceivable that Synthemax, upon stimulation of this integrin, may actually promote either more potent differentiation and/or facilitate improved growth of epidermal keratinocytes derived from hPSCs accounting for the improvements in cell yield and expansion potential demonstrated in this study. Elucidating this mechanism by which Synthemax promotes a greater yield and expansion potential of K14+/p63+ cells compared to gelatin would be advantageous to further improve epithelial or epidermal keratinocyte cell yield.
Surprisingly, we found that StemAdhere did not facilitate growth or even survival of subcultured K18+/p63+ simple epithelial colonies. A study by Charest et al. demonstrates that keratinocyte differentiation is mediated by cell-cell contact via E-cadherin.32 Therefore, we predicted that StemAdhere, which is functionalized with extracellular E-cadherin, would be a promising candidate for facilitating epithelial cell growth and differentiation. While this demonstrated to be true with respect to StemAdhere successfully facilitating epithelial commitment of hPSCs, it failed to support growth and maturation of K18+/p63+ simple epithelial cells to K14+/p63+ epidermal keratinocyte progenitors. We speculate that while cell-cell contact via E-cadherin has been shown to be necessary for keratinocyte differentiation, it has demonstrated so in the context of simultaneous cell-matrix interaction, as reported by Charest et al., and that this synergistic effect is necessary for successful keratinocyte differentiation of hPSCs.32 Again, the mechanisms by which the matrix influences the cell fate decisions of this cell population would be useful for designing improvements in this cell culture system to maximize output of these epithelial or epidermal keratinocyte cells for tissue engineering applications.
While we have contributed to efforts in defining hPSC epithelial differentiation processes, future studies should not only focus on elucidating mechanisms to improve differentiation, but also seek to replace xenogeneic cell culture constituents. Many researchers have developed methods for maintaining, differentiating, and even freezing hPSCs in xeno-free conditions.5,35–40 Generating somatic cell populations from hPSCs using xeno-free culture conditions is essential for not only regenerative therapies to eliminate introduction of potential pathogens from animal-derived components into patients, but also for in vitro model systems that are designed to emulate a human system. Matrigel and gelatin are not ideal substrates for scale-up and application of hPSC-based culture systems given that there is lot-to-lot variability in the composition of these products and these substrates are not suitable for human tissue engineering applications as a result of their xenogeneic origin. Therefore, the advantage of replacing of these animal-derived matrices with synthetic, defined substrates is twofold for the purpose of generating epithelial cells for human tissue engineering applications. Efforts should continue to standardize and define hPSC cell culture maintenance and differentiation systems for making such systems more conducive for scale-up and for reproducibly generating somatic cells that can be utilized for in vitro model systems to study tissue morphogenesis and disease or for regenerative therapies. In this paper, we have shown that RA treatment of hPSCs at an optimal density and subsequent culture of cells in K-DSFM on defined substrates can efficiently generate populations of functional epithelial and epidermal cells, which can be used for various human tissue engineering applications.
Supplementary Material
Acknowledgments
The authors thank Joe Hardin and colleagues in Experimental Pathology with the UW-Madison Carbone Cancer Center (N.C.I. No. P30CAO14520) for the histological processing and immunohistochemistry of organ. This work was supported by National Science Foundation Grant CBET-1066311 (S.P.P.) and a University of Wisconsin Stem Cell and Regenerative Medicine Predoctoral Fellowship (J.A.S.) and a National Institutes of Health training grant NIDCD T32 DC009401 (J.A.S.). Human foreskin fibroblasts were kindly provided by the Garlick Laboratory (Tufts University, Boston, MA).
Disclosure Statement
No competing financial interests exist.
References
- 1.Thomson J. Itskovitz-Eldor J. Shapiro S. Waknitz M. Swiergiel J. Marshall V., et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Yu J. Vodyanik M. Smuga-Otto K. Antosiewicz-Bourget J. Frane J. Tian S., et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi K. Tanabe K. Ohnuki M. Narita M. Ichisaka T. Tomoda K., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 4.Ausubel L.J. Lopez P.M. Couture L.A. GMP scale-up and banking of pluripotent stem cells for cellular therapy applications. Methods Mol Biol. 2011;767:147. doi: 10.1007/978-1-61779-201-4_11. [DOI] [PubMed] [Google Scholar]
- 5.Vaajasaari H. Ilmarinen T. Juuti-Uusitalo K. Rajala K. Onnela N. Narkilahti S., et al. Toward the defined and xeno-free differentiation of functional human pluripotent stem cell-derived retinal pigment epithelial cells. Mol Vis. 2011;17:558. [PMC free article] [PubMed] [Google Scholar]
- 6.Amit M. Carpenter M.K. Inokuma M.S. Chiu C.P. Harris C.P. Waknitz M.A., et al. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev Biol. 2000;227:271. doi: 10.1006/dbio.2000.9912. [DOI] [PubMed] [Google Scholar]
- 7.Xu C. Inokuma M.S. Denham J. Golds K. Kundu P. Gold J.D., et al. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol. 2001;19:971. doi: 10.1038/nbt1001-971. [DOI] [PubMed] [Google Scholar]
- 8.Amit M. Shariki C. Margulets V. Itskovitz-Eldor J. Feeder layer- and serum-free culture of human embryonic stem cells. Biol Reprod. 2004;70:837. doi: 10.1095/biolreprod.103.021147. [DOI] [PubMed] [Google Scholar]
- 9.Chen G. Gulbranson D.R. Hou Z. Bolin J.M. Ruotti V. Probasco M.D., et al. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8:424. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valamehr B. Tsutsui H. Ho C.M. Wu H. Developing defined culture systems for human pluripotent stem cells. Regen Med. 2011;6:623. doi: 10.2217/rme.11.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu H.F. Narayanan K. Lim S.X. Gao S. Leong M.F. Wan A.C. A 3D microfibrous scaffold for long-term human pluripotent stem cell self-renewal under chemically defined conditions. Biomaterials. 2012;33:2419. doi: 10.1016/j.biomaterials.2011.11.077. [DOI] [PubMed] [Google Scholar]
- 12.Frank S. Zhang M. Schöler H.R. Greber B. Small molecule-assisted, line-independent maintenance of human pluripotent stem cells in defined conditions. PLoS One. 2012;7:e41958. doi: 10.1371/journal.pone.0041958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng G. Liu S. Rancourt D.E. Synergistic effect of medium, matrix, and exogenous factors on the adhesion and growth of human pluripotent stem cells under defined, xeno-free conditions. Stem Cells Dev. 2012;21:2036. doi: 10.1089/scd.2011.0489. [DOI] [PubMed] [Google Scholar]
- 14.Heng B.C. Li J. Chen A.K. Reuveny S. Cool S.M. Birch W.R., et al. Translating human embryonic stem cells from 2-dimensional to 3-dimensional cultures in a defined medium on laminin- and vitronectin-coated surfaces. Stem Cells Dev. 2012;21:1701. doi: 10.1089/scd.2011.0509. [DOI] [PubMed] [Google Scholar]
- 15.Lian X. Hsiao C. Wilson G. Zhu K. Hazeltine L.B. Azarin S.M., et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109:E1848. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozhich O.A. Hamilton R.S. Mallon B.S. Standardized generation and differentiation of neural precursor cells from human pluripotent stem cells. Stem Cell Rev. 2012 doi: 10.1007/s12015-012-9357-8. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aberdam E. Barak E. Rouleau M. de LaForest S. Berrih-Aknin S. Suter D., et al. A pure population of ectodermal cells derived from human embryonic stem cells. Stem Cells. 2008;26:440. doi: 10.1634/stemcells.2007-0588. [DOI] [PubMed] [Google Scholar]
- 18.Hewitt K.J. Shamis Y. Carlson M.W. Aberdam E. Aberdam D. Garlick J.A. Three-dimensional epithelial tissues generated from human embryonic stem cells. Tissue Eng Part A. 2009;15:3417. doi: 10.1089/ten.tea.2009.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji L. Allen-Hoffmann B. de Pablo J. Palecek S. Generation and differentiation of human embryonic stem cell-derived keratinocyte precursors. Tissue Eng. 2006;12:665. doi: 10.1089/ten.2006.12.665. [DOI] [PubMed] [Google Scholar]
- 20.Metallo C.M. Ji L. de Pablo J.J. Palecek S.P. Retinoic acid and bone morphogenetic protein signaling synergize to efficiently direct epithelial differentiation of human embryonic stem cells. Stem Cells. 2008;26:372. doi: 10.1634/stemcells.2007-0501. [DOI] [PubMed] [Google Scholar]
- 21.Iuchi S. Dabelsteen S. Easley K. Rheinwald J.G. Green H. Immortalized keratinocyte lines derived from human embryonic stem cells. Proc Natl Acad Sci U S A. 2006;103:1792. doi: 10.1073/pnas.0510953103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dabelsteen S. Hercule P. Barron P. Rice M. Dorsainville G. Rheinwald J. Epithelial cells derived from human embryonic stem cells display p16INK4A senescence, hypermotility, and differentiation properties shared by many P63+ somatic cell types. Stem Cells. 2009;27:1388. doi: 10.1002/stem.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metallo C. Ji L. de Pablo J. Palecek S. Directed differentiation of human embryonic stem cells to epidermal progenitors. Methods Mol Biol. 2010;585:83. doi: 10.1007/978-1-60761-380-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu C. Rosler E. Jiang J. Lebkowski J. Gold J. O'Sullivan C., et al. Basic fibroblast growth factor supports undifferentiated human embryonic stem cell growth without conditioned medium. Stem Cells. 2005;23:315. doi: 10.1634/stemcells.2004-0211. [DOI] [PubMed] [Google Scholar]
- 25.James D. Levine A.J. Besser D. Hemmati-Brivanlou A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132:1273. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- 26.Mills A.A. Zheng B. Wang X.J. Vogel H. Roop D.R. Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 27.Metallo C. Azarin S. Moses L. Ji L. de Pablo J. Palecek S. Human embryonic stem cell-derived keratinocytes exhibit an epidermal transcription program and undergo epithelial morphogenesis in engineered tissue constructs. Tissue Eng Part A. 2010;16:213. doi: 10.1089/ten.tea.2009.0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber J.L. Dolley-Sonneville P. Weber D.M. Fadeev A.G. Zhou Y. Yang J., et al. Corning synthemax surface: a tool for feeder-free, xeno-free culture of human embryonic stem cells. Nat Methods. 2010;7:an6. [Google Scholar]
- 29.Monsma S.A. Garcia II B.H. Marty A. Information from Primorigen Biosciences. Madison, WI: Stemcell Technologies; 2011. StemAdhere Defined Matrix for hPSC. [Google Scholar]
- 30.Egles C. Garlick J.A. Shamis Y. Three-dimensional human tissue models of wounded skin. Methods Mol Biol. 2010;585:345. doi: 10.1007/978-1-60761-380-0_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chambers S.M. Fasano C.A. Papapetrou E.P. Tomishima M. Sadelain M. Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charest J.L. Jennings J.M. King W.P. Kowalczyk A.P. García A.J. Cadherin-mediated cell-cell contact regulates keratinocyte differentiation. J Invest Dermatol. 2009;129:564. doi: 10.1038/jid.2008.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watt F.M. Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 2002;21:3919. doi: 10.1093/emboj/cdf399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melkoumian Z.K. Soni M. Dolley-Sonneville P.J. Martin A.W. Zhou Y. Pal S., et al. Synthetic peptide-acrylate surface (PAS) for culture of human mesenchymal stem cells in serum-free defined medium. www.selectscience.net. Mar 13, 2011. www.selectscience.net
- 35.Bergström R. Ström S. Holm F. Feki A. Hovatta O. Xeno-free culture of human pluripotent stem cells. Methods Mol Biol. 2011;767:125. doi: 10.1007/978-1-61779-201-4_9. [DOI] [PubMed] [Google Scholar]
- 36.Holm F. Ström S. Inzunza J. Baker D. Strömberg A.M. Rozell B., et al. An effective serum- and xeno-free chemically defined freezing procedure for human embryonic and induced pluripotent stem cells. Hum Reprod. 2010;25:1271. doi: 10.1093/humrep/deq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marinho P.A. Vareschini D.T. Gomes I.C. Paulsen B.S. Furtado D.R. Castilho L.D., et al. Xeno-free production of human embryonic stem cells in stirred microcarrier systems using a novel animal/human-component-free medium. Tissue Eng Part C Methods. 2013;19:146. doi: 10.1089/ten.TEC.2012.0141. [DOI] [PubMed] [Google Scholar]
- 38.Tannenbaum S.E. Turetsky T.T. Singer O. Aizenman E. Kirshberg S. Ilouz N., et al. Derivation of xeno-free and GMP-grade human embryonic stem cells—platforms for future clinical applications. PLoS One. 2012;7:e35325. doi: 10.1371/journal.pone.0035325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q. Mou X. Cao H. Meng Q. Ma Y. Han P., et al. A novel xeno-free and feeder-cell-free system for human pluripotent stem cell culture. Protein Cell. 2012;3:51. doi: 10.1007/s13238-012-2002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swistowski A. Zeng X. Scalable production of transplantable dopaminergic neurons from hESCs and iPSCs in xeno-free defined conditions. Curr Protoc Stem Cell Biol. 2012;Chapter 2(Unit2D.12) doi: 10.1002/9780470151808.sc02d12s22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.