

Abstract
The presence of tangles of abnormally phosphorylated tau is a characteristic of Alzheimer's disease (AD), and the loss of synapses correlates with the degree of dementia. In addition, the overexpression of interleukin-1 (IL-1) has been implicated in tangle formation in AD. As a direct test of the requirement for IL-1 in tau phosphorylation and synaptophysin expression, IL-1 actions in neuron–microglia cocultures were manipulated. Activation of microglia with secreted β-amyloid precursor protein or lipopolysaccharide elevated their expression of IL-1α, IL-1β, and tumor necrosis factor α (TNFα) mRNA. When such activated microglia were placed in coculture with primary neocortical neurons, a significant increase in the phosphorylation of neuronal tau was accompanied by a decline in synaptophysin levels. Similar effects were evoked by treatment of neurons with recombinant IL-1β. IL-1 receptor antagonist (IL-1ra) as well as anti-IL-1β antibody attenuated the influence of activated microglia on neuronal tau and synaptophysin, but anti-TNFα antibody was ineffective. Some effects of microglial activation on neurons appear to be mediated by activation of p38 mitogen-activated protein kinase (p38-MAPK), because activated microglia stimulated p38-MAPK phosphorylation in neurons, and an inhibitor of p38-MAPK reversed the influence of IL-1β on tau phosphorylation and synaptophysin levels. Our results, together with previous observations, suggest that activated microglia may contribute to neurofibrillary pathology in AD through their production of IL-1, activation of neuronal p38-MAPK, and resultant changes in neuronal cytoskeletal and synaptic elements.
Keywords: Alzheimer's disease, β-amyloid precursor protein, cortical neuron, interleukin-1, microglia, mitogen-activated protein kinase, synaptophysin, phosphorylated tau
Introduction
Increased levels of proinflammatory factors such as interleukin (IL)-1 and tumor necrosis factor α (TNFα), as well as other indications of microglial activation, have been correlated with several neurodegenerative situations, including Alzheimer's disease (AD), AIDS-associated dementia, and traumatic brain injury (Griffin and Mrak, 2002). Several lines of evidence indicate that microglia and their products (cytokines, excitatory amino acids, and other neurotoxins) can cause neuronal damage (Giulian et al., 1995; Meda et al., 1995; Barger and Harmon, 1997; Jeohn et al., 1998; Barger and Basile, 2001; Li et al., 2001). These observations, together with previous findings in AD patients, suggest that a chronic inflammatory reaction, driven mainly by activated microglia, may contribute to the process of neuropathological changes in AD brain (Griffin et al., 1989, 1998).
Neurofibrillary changes, in the form of neuritic plaques, neuropil threads, and neurofibrillary tangles, are key histological features of AD. Tau is one of the microtubule-associated proteins that stabilizes growing axons necessary for the development and growth of neurites. However, in AD, for unknown reasons, tau becomes excessively phosphorylated and appears in paired helical filaments, dystrophic neurites, and neurofibrillary tangles (Braak et al., 1994). This neurofibrillary pathology suggests a loss of axonal integrity and an eventual decline in connectivity and synapses, a consistent correlate of dementia in AD (DeKosky and Scheff, 1990; Terry et al., 1991). Previously, we reported that secreted β-amyloid precursor protein (sAPP) can cause neuronal cell damage that can be restricted to functional synapses (Barger and Basile, 2001). This and other sAPP-evoked neurotoxicity occurs via an indirect mechanism that involves microglial activation (Barger and Harmon, 1997; Li et al., 2000a; Barger and Basile, 2001). To further explore these relationships, we used a microglial–neuronal coculture model to (1) assess the effects of microglial activation on synthesis of neuronal synaptophysin and tau, (2) probe the possible link between phosphorylation and the maintenance of synapses, and (3) determine potential signal transduction components mediating phosphorylation events in glial–neuronal interactions.
Materials and Methods
Reagents. sAPP was purified from a recombinant expression system as described previously (Barger and Basile, 2001). Lipopolysaccharide (LPS) and recombinant rat IL-1β were obtained from Sigma (St. Louis, MO). Mouse IL-1ra was from R&D Systems (Minneapolis, MN), monoclonal hamster anti-murine IL-1β antibody was from Genzyme (Cambridge, MA), and anti-murine TNFα antibody was from PeproTech (Rocky Hill, NJ). SB203580-HCl was from Calbiochem (Sunnyvale, CA). Monoclonal anti-phospho-tau antibody AT8 was from RDI (Flanders, NJ); monoclonal anti-Tau1 antibody was kindly provided by Dr. L. I. Binder (Northwestern University); monoclonal anti-synaptophysin antibody SY38 was from ICN Biomedicals (Costa Mesa, CA); antibodies against phospho-p38-MAPK or total p38-MAPK were from New England Biolabs (Beverly, MA). Medium, serum, and B27 supplement for cell cultures were from Invitrogen (Grand Island, NY).
Cell cultures. Primary neuronal cultures were derived from the cerebral cortex of fetal Sprague Dawley rats (embryonic day 18), as described previously (Li et al., 1998). Experiments using primary neuronal cell cultures were performed after 10–14 d in culture. Primary cultures of rat microglia were generated from the cortical tissue of neonatal (0–3 d) Sprague Dawley rats, as described previously (Barger and Basile, 2001). The N9 mouse microglial cell line (Corradin et al., 1993) was maintained in DMEM (Invitrogen) supplemented with 10% with fetal bovine serum. Coculture of N9 cells or primary microglia with primary neuronal cells was performed as described previously (Li et al., 2000a). Either microglial cell type was grown on semi-permeable membranes of basket-type cell culture inserts (Falcon; pore size 0.4 μm; Fisher Scientific, Houston, TX), treated with activation stimuli (LPS or sAPP), and then washed before placement into culture wells containing neurons. When applied to cocultures, IL1-ra, anti-IL-1β antibody, anti-TNFα antibody, or SB203580-HCl (aqueous solution) was added to the neuronal culture medium before placement of the microglial inserts into the neuronal culture wells.
Viability assays. Cell survival was assessed using a previously described 3-(4-5-dimethythiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay (Li et al., 1998).
RT-PCR amplification. Total RNA was extracted from cultured cells using TriReagent RNA (Molecular Research Center, Cincinnati, OH) according to the manufacturer's instructions. RT-PCR was performed as described previously (Li et al., 2001). Briefly, for comparisons of mRNA levels among different RNA samples, RT was performed simultaneously using reagents from a single master mix. PCR was performed using reagents from Clontech (Palo Alto, CA). The sequences of primers for both mouse and rat IL-1α, IL-1β, and TNFα and glyceraldehyde 3-phosphate dehydrogenase (G3PDH) were used in this study; the mouse sequences were given previously (Li et al., 2000a, 2001). The following are the sequences of primers used for rat IL-1α: upstream 5′-CT AAG AAC TAC TTC ACA TCC GCA GC-3′; downstream 5′-CTG GAA TAA AAC CCA CTG AGG TAG G-3′; for rat IL-1β: upstream 5′-TGA CTC GTG GGA TGA TGA CG-3′; downstream 5′-CTG GAG ACT GCC CAT TCT CG-3′; for rat TNFα: upstream 5′-GCA CAG AAA GCA TGA TCC GAG-3′; downstream 5′-CCT GGT ATG AAG TGG CAA ATC G-3′. PCR amplification was performed through 26 cycles (for IL-1α and IL-1β) or 28 cycles (for TNFα) at 94°C for 45 sec, 60°C for 45 sec, and 72°C for 45 sec. For G3PDH, amplification was performed through 26 cycles at 94°C for 45 sec, 55°C for 45 sec, and 72°C for 45 sec. The PCR reaction was stopped by final extension for 10 min at 72°C. The PCR cycle for each target gene was determined by sampling 5 μl aliquots every third cycle from cycle 21 on. The indicated cycles were verified to be within the linear range of product accumulation for the specific PCR reaction. Equal volumes of reaction mixture from each sample were loaded onto 1.5% agarose gels, and fluorescent images were digitally captured for analysis of intensity with NIH Image software. Levels of IL-1α, IL-1β, and TNFα mRNA were normalized relative to G3PDH mRNA in the same sample.
Western immunoblot assay. Proteins were extracted using lysis buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, and 0.1% SDS and quantified using a Micro BCA assay reagent kit (Pierce, Rockford, IL) as described previously (Li et al., 1998). Aliquots (40 μg each) were loaded onto a 10% SDS-polyacrylamide gel, subjected to electrophoresis at 90 V for 1.5 hr, and transferred to Immobilon-P membranes. Membranes were incubated overnight at 4°C with primary antibody and for 2 hr at room temperature with secondary antibody and visualized using the Western-Light Chemiluminescent Detection System (Tropix, Bedford, MA). Films were digitized and analyzed using NIH Image software, version 1.60.
Statistical analysis. Data were analyzed using an unpairedt test, and values were considered significantly different when the two-tailed p value was <0.05. Results are expressed as mean ± SEM.
Results
Two strategies were used to assess the capacity of IL-1 to directly regulate neuronal tau phosphorylation and synaptophysin synthesis and to assess the possibility that p38-MAPK mediates these events. For the first, we directly applied IL-1 to neuronal cultures, and for the second, we used a coculture paradigm that allows one cell type to be treated and analyzed independently of the other, in this case, activated microglia and neurons. Studies were initiated with the N9 microglial cell line for the sake of homogeneity (i.e., independence from astrocytes or other contaminants of primary glial cultures); additional experiments were performed with rat primary microglia. Both N9 cells and primary microglia were grown on semi-permeable membranes of basket-type cell culture inserts and stimulated with either sAPP (10 nm) or LPS (30 ng/ml). Both agonists caused an elevation of IL-1α, IL-1β, and TNFα mRNA levels in N9 microglia (Fig.1A). To confirm whether primary microglial cells have similar responses to sAPP, and whether there is a chronic production of cytokine expression by primary microglia after a transient exposure to sAPP, primary microglia were incubated with sAPP (10 nm) for 2 hr, washed twice with fresh medium, and then cultured in fresh medium for 6 or 12 hr and harvested for assay of cytokine expression. As shown in Figure1B, in primary microglia, IL-1α and IL-1β and TNFα remain elevated for 12 hr after a 2 hr treatment with sAPP.
Fig. 1.

Time course of LPS or sAPP induction of IL-1α, IL-1β, and TNFα mRNA expression in microglia. A, N9 microglial cells were treated with either LPS (30 ng/ml) or sAPP (10 nm) for the indicated times [hours (h)]. Total RNA was extracted and subjected to RT-PCR of sequences specific to IL-1α, IL-1β, or TNFα; G3PDH was amplified to assess equivalency of loading. B, Primary microglia were treated with sAPP (10 nm) for 2 hr, followed by two washes with fresh medium. These microglia were then cultured in fresh medium for up to 12 hr and harvested for cytokine expression assay. Controls (Con) were untreated sister cultures. Data are representative of at least two separate experiments.
There was a significant decrease in the steady-state levels of synaptophysin and a significant increase in the steady-state levels of phosphorylated tau in primary neocortical neurons cocultured for 24 hr with baskets containing LPS- or sAPP-activated microglia (Fig.2). These changes were accompanied by an increase in phosphorylation, i.e., activation, of p38-MAPK (Fig. 2), a signal transduction protein that can phosphorylate tau at five sites that are phosphorylated in paired helical filaments (Reynolds et al., 2000). As early as 30 min after application of recombinant IL-1β, there was a marked elevation of tau phosphorylation, increasing to approximately twofold after 2 hr of incubation, compared with untreated sister cultures (Fig.3A,C). However, total neuronal tau was unchanged by treatment with IL-1β for up to 12 hr (Fig. 3A). Treatment of neuronal cells with IL-1β also resulted in a marked, time-dependent decrease in steady-state levels of synaptophysin, which was first apparent after a 1 hr incubation (Fig. 3B). After 2 hr of treatment with IL-1β, the synaptophysin level decreased by 55% compared with untreated sister cultures (Fig.3B,C).
Fig. 2.

Effects of activated microglia on synaptophysin, tau, and p38-MAPK in neocortical neurons. N9 microglia were stimulated with LPS (30 ng/ml) for 2 hr (LPS), washed with fresh medium, and then placed in coculture with primary neocortical neurons for 24 hr; untreated sister cultures of microglia were also cocultured with neurons (Con). Proteins were extracted from the neurons, and Western blot analysis was performed for the detection of synaptophysin (Syn), phosphorylated tau (Tau-phos), or phosphorylated p38-MAPK (p38-phos). A, Western immunoblots. B, Relative levels of Syn, Tau-phos, and p38-phos in treated versus control cultures. Values are expressed as mean ± SEM (n = 4). **p< 0.01.
Fig. 3.

IL-1β induces tau phosphorylation and decreases synaptophysin in cortical neurons. A, Illustrations of phosphorylated tau (Tau-phos) and total tau (Total-tau) in cortical neurons treated with IL-1β (30 ng/ml) for up to 12 hr. B, Synaptophysin (Syn) in cultures treated for up to 12 hr.C, Quantification of protein levels of Syn and Tau-phos in neuronal cultures treated with IL-1β (30 ng/ml) for 2 hr. Values are expressed as mean ± SEM of four samples. *p< 0.05; **p < 0.01.
To test whether IL-1 and TNFα play essential roles in the interactions between activated microglia and neurons, IL-1ra, anti-IL-1β antibodies, or anti-TNFα antibodies were used to block the actions of these two activated microglia-derived cytokines on neurons. The presence of IL-1ra in primary neurons cocultured with N9 cells inhibited the IL-1-induced portion of the increase in phosphorylated tau as well as the decrease in synaptophysin to levels similar to those in control sister cultures (Fig.4A,B). However, addition of anti-TNFα antibody to the cocultures did not show significant protective effects on synaptophysin (Fig.4A,B). Cocultures of primary microglia with primary neurons pretreated with IL-1ra yielded similar results (Fig.5A,B). In addition, IL-1ra blocked the IL-1-induced activation of p38-MAPK (Fig. 5A,B). As confirmation that sAPP activation of microglia results in similar changes in synaptophysin, tau, and p38-MAPK, coculture of sAPP-activated primary microglia with primary neurons resulted in a 50% decrease in synaptophysin and a 50% increase in tau phosphorylation. These were concomitant with a ∼250% increase in activation of p38-MAPK. All of these changes in neuronal proteins were suppressed by pretreating the neuronal cultures with either anti-IL-β antibody or IL-1ra (Fig.6A–C). As in cocultures with LPS-activated microglia, pretreatment of the neuronal cultures with anti-TNFα antibody did not show these salutary effects, suggesting that TNFα does not play a role in these particular deleterious effects of microglial activation on neurons (Fig.6A–C). However, compared with control sister cultures, neither total p38-MAPK nor total tau levels were changed in neuronal cultures by 24 hr coculture with sAPP-activated microglia (Fig. 6A,B).
Fig. 4.
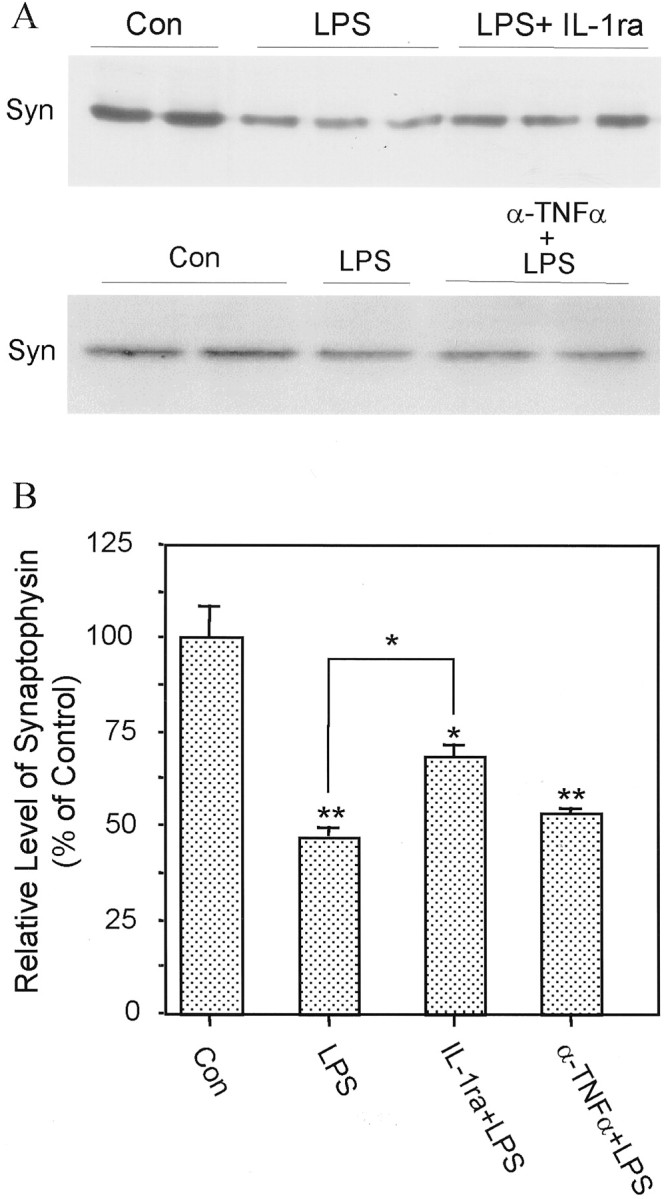
Protective effect of IL-1ra on neuronal synaptophysin loss induced by activated microglia. A, Western immunoblots illustrating proteins from cortical neurons cocultured for 24 hr with naive N9 cells (Con), LPS-stimulated N9 cells (LPS), LPS-stimulated N9 cells in the presence of IL-1ra (LPS+ IL-1ra), or LPS-stimulated N9 cells in the presence of anti-TNFα antibody (α-TNFα+LPS). B, Quantification of protein level of synaptophysin. Values are expressed as mean ± SEM of four to six samples. *p < 0.05; **p < 0.01.
Fig. 5.
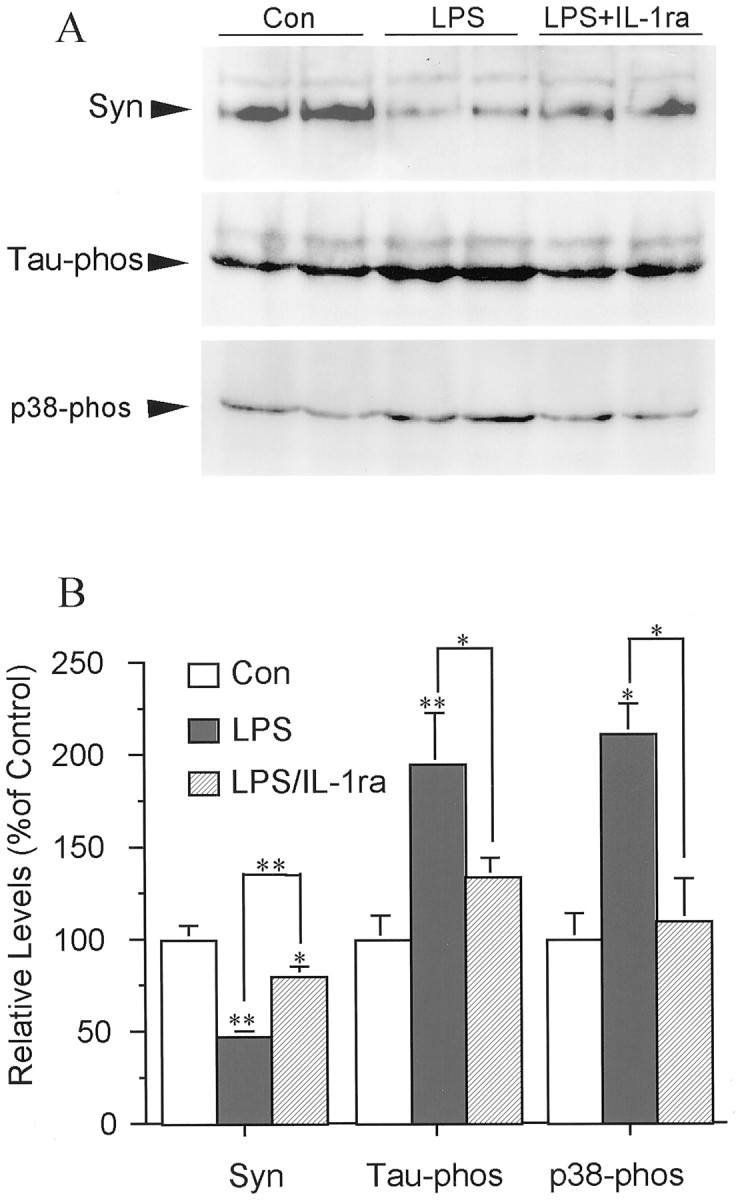
Effects of activated primary cortical microglia on synaptophysin, tau phosphorylation, and p38-MAPK activation in primary neurons. A, Western blot of phosphorylated tau (Tau-phos), synaptophysin (Syn), and p38-MAPK (p38-phos) in cortical neurons cocultured with naive primary microglia (Con), LPS-stimulated microglia (LPS), or LPS-stimulated microglia in the presence of IL-1ra [LPS + IL-1ra (LPS/IL-1ra)]. Results are representative of two independent experiments. B, Quantification of protein levels of Syn, Tau-phos, and p38-phos. Values are expressed as mean ± SEM (n = 4). *p < 0.05; **p < 0.01.
Fig. 6.
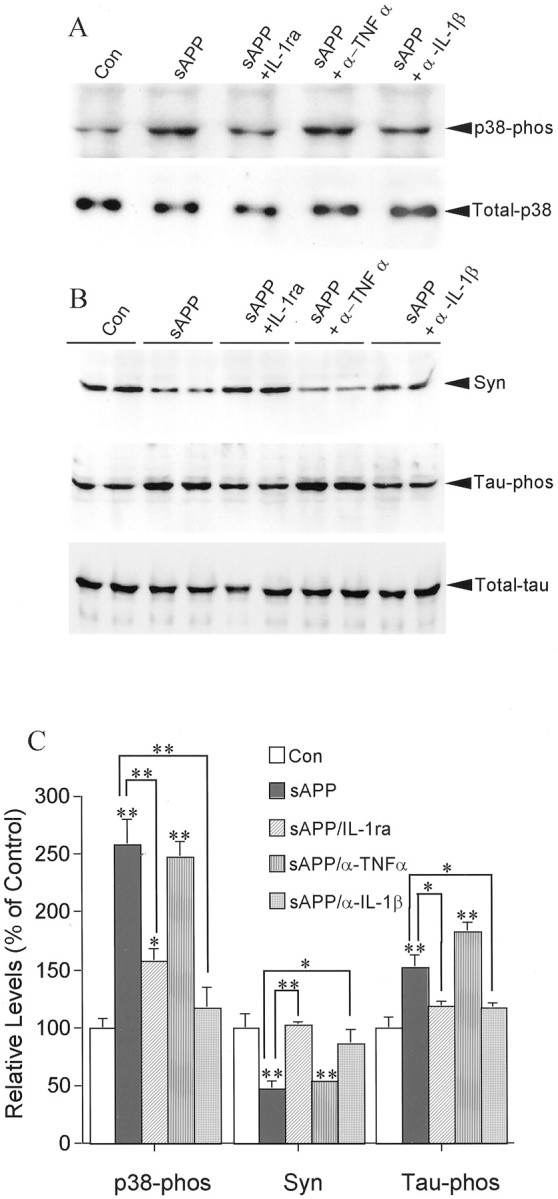
Effects of sAPP-activated primary microglia on the levels of neuronal p38-MAPK and tau phosphorylation and of synaptophysin. A, Western immunoblots of phosphorylated p38-MAPK (p38-phos), total p38-MAPK (Total-p38); B, synaptophysin (Syn), phosphorylated tau (Tau-phos), and total tau (Total-tau) in cortical neurons cocultured with naive primary microglia (Con), sAPP-stimulated microglia (sAPP), sAPP-stimulated microglia in the presence of IL-1ra (sAPP+IL-1ra), of anti-TNFα (sAPP + α-TNFα), or of anti-IL-1β antibody (sAPP+α-IL-1β). C, Quantification of protein levels of p38-phos, Syn, and Tau-phos. Values are expressed as mean ± SEM of three to four samples from two independent experiments. *p < 0.05; **p < 0.01.
We next sought to determine whether microglial neuronal toxicity could account for loss of synaptophysin, increase in tau phosphorylation, and activation of p38-MAPK. In the present coculture models, no significant loss in neuronal viability was detected at 24 hr, suggesting that the loss of synaptophysin, the phosphorylation of tau, and the activation of p38-MAPK shown here was not caused by cell loss, but in fact preceded neuron cell death. As further evidence and in agreement with other studies (Strijbos and Rothwell, 1995), blocking IL-1 activity with IL-1ra resulted in substantially higher neuronal viability. Blockade of TNFα was somewhat effective in maintaining viability, but less than blockade of IL-1 (Fig. 7).
Fig. 7.

IL-1ra or anti-TNFα antibody attenuates the neurotoxicity of activated microglia. Survival was assessed by MTT assay in cortical neurons cocultured with naive N9 cells (Con), LPS-stimulated N9 cells (LPS), LPS-stimulated N9 cells in the presence of IL-1ra (LPS+IL-1ra), or LPS-stimulated N9 cells in the presence of anti-TNFα antibody (LPS+α-TNFα) for 24 or 48 hr. Values are expressed as mean ± SEM of four to eight samples. **p < 0.001.
Activation of p38-MAPK in neuronal cells has been associated with responses to stress (Xia et al., 1995; Kawasaki et al., 1997) and, more specifically, with IL-1 and hyperphosphorylated tau in AD (Sheng et al., 2001). Because an elevation of phosphorylated p38-MAPK appeared in coculture of neocortical neurons with microglia (Figs. 2, 5,6A,C), we tested whether activation of p38-MAPK participates in IL-1-induced tau phosphorylation and decreased synaptophysin in neocortical neurons. In case IL-1-induced activation of astrocytes results in phosphorylation of p38-MAPK, we carefully prepared our primary neuronal cell cultures and maximally limited glial contamination using serum-free Neurobasal medium supplemented with B27 and transiently using 10−5m cytosine arabinoside as described previously (Li et al., 1998, 2000b). In the current neuronal cultures, astrocytes comprised 3% of the total cells, suggesting that the observed elevation of p38-MAPK phosphorylation was neuronal in origin. At 30 and 60 min after application of IL-1β, neurons showed an elevation of activated p38-MAPK, which returned to basal levels after 2 hr (Fig.8), whereas the total neuronal p38-MAPK did not show marked differences after IL-1β treatment (Fig.8A). Pretreatment of neocortical neurons with p38-MAPK inhibitor SB203580 significantly suppressed IL-1β-induced changes in tau and synaptophysin (Fig.8B,C), indicating that activation of p38-MAPK was involved in the IL-1 signal transduction pathway regulating these proteins in neurons.
Fig. 8.
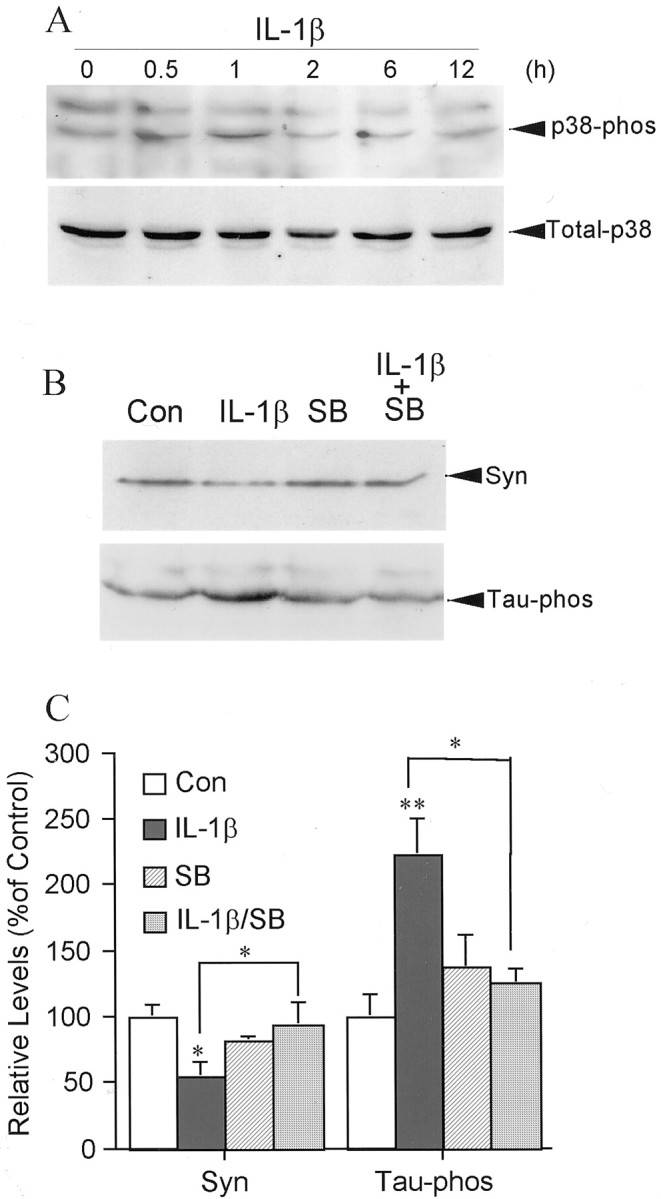
IL-1β induction of tau phosphorylation in cortical neurons is mediated by phosphorylation of p38-MAPK.A, Time course of IL-1β (30 ng/ml) induction of p38-MAPK phosphorylation compared with the level of total p38-MAPK (Total-p38) in the same neuronal cultures;B, an illustration of inhibition of activation of p38-MAPK with SB203580 on IL-1β-induced suppression of synaptophysin (Syn) and on IL-1β-induced phosphorylation of tau protein (Tau-phos) after 2 hr treatment.C, Quantification of protein levels of synaptophysin (Syn) and phosphorylated tau protein (Tau-phos). Values are expressed as mean ± SEM (n = 3–4). *p < 0.05; **p < 0.01.
Discussion
In this study, the effects of glial–neuronal interactions on phosphorylation of tau protein and maintenance of synaptophysin levels were dissected in vitro in a glial–neuronal coculture model. The results demonstrate that (1) activated microglia increase tau phosphorylation and decrease steady-state levels of synaptophysin through release of IL-1; (2) these IL-1-induced changes occur before significant neuronal cell loss associated with microglial neurotoxicity is detectable; and (3) the effects of IL-1 on tau and synaptophysin are mediated, at least in part, through activation of p38-MAPK. These experimental results, together with previous observations (Griffin et al., 1989; Sheng et al., 2001; Griffin and Mrak, 2002), suggest that microglial activation with overexpression of IL-1 contributes to the neurodegenerative consequences of AD.
The cognitive alterations in patients with AD are closely associated with synaptic loss and neurofibrillary pathology in the limbic system and neocortex (Arriagada et al., 1992). Loss of synaptic proteins such as synaptophysin, syntaxin, and SNAP-25 (solubleN-ethylmaleimide-sensitive factor attachment protein) in the frontal neocortex has provided the best neurobiological correlates of cognitive impairment (DeKosky and Scheff, 1990; Terry et al., 1991;Mukaetova-Ladinska et al., 2000). However, the molecular and cellular processes responsible for loss of synapses and the formation of hyperphosphorylated tau in the early stages of AD, before significant neuronal cell death, remain undefined. Three possibilities have been proposed on the basis of the neuropathology: (1) loss of synapses is a nonspecific consequence of global neurodegenerative changes that include neuronal loss (Scheff et al., 1996); (2) loss of synapses results from the direct neurotoxicity of amyloid β-peptide (Aβ) (Mattson, 1997); and (3) loss of synapses results from cytoskeletal changes caused either actively by tau aggregates or passively by loss of tau function (Lee et al., 1991; Wischik et al., 1996). Here we provide evidence that temporal overexpression of cytokines contributes to a cascade of events resulting in specific patterns of tau pathology and neurodegeneration.
IL-1 promotes neuronal production of β-amyloid precursor protein and its derivatives (Buxbaum et al., 1992), but our current findings suggest that IL-1-mediated proinflammatory sequelae could damage neuronal connectivity via mechanisms beyond neurotoxic effects of Aβ production. Previously, we found that activated microglia can influence properties of neurotransmitters, such as cholinergic or glutamatergic systems (Li et al., 2000a; Barger and Basile, 2001). A role for microglia in neuronal cell death is now widely accepted (Griffin et al., 1989; Giulian et al., 1995; Barger and Harmon, 1997; Li et al., 2001). Furthermore, activation of microglia by sAPP can have effects on neuronal function short of outright cell death, including reduction of the density of functional synapses (Barger and Basile, 2001). The effects of activated microglia and IL-1 on tau phosphorylation and synaptophysin expression noted here approach biochemical explanations for this phenomenon.
In the present study, decreased synaptophysin synthesis was accompanied by elevation of phosphorylated tau and preceded significant neuronal cell death, suggesting that phosphorylation of tau and loss of synaptic proteins are early events in the process of neuronal cell death. This is consistent with the hypothesis that altered expression of synaptic proteins occurs early in the neurodegenerative sequelae of AD (Masliah et al., 2001). The induction of tau phosphorylation by IL-1 shown herein vitro and previously in vivo (Sheng et al., 2001) indicates that IL-1 might potentially contribute to the reorganization of the cytoskeleton, interrupt normal microtubule assembly and axon stabilization, and eventually result in loss of synaptic proteins and synapses. This is supported by the observations in AD that a loss of synaptophysin is observed in tangle-bearing neurons (Callahan and Coleman, 1995) and that activated microglia correlate with neurofibrillary pathology (DiPatre and Gelman, 1997), including intracellular tau pathology (Sheng et al., 1997). Furthermore, in a study comparing neuropathology in AD patients with that in cognitively normal individuals with abundant Aβ deposits, the latter group was found to have normal synapse densities and scant evidence of inflammation (Lue et al., 1996), suggesting that inflammatory events contribute to neuronal abnormalities that lead to clinical symptoms. This suggestion is consistent with our finding that preincubation of neuronal cells with IL-1ra suppressed the effect of activated microglia on synthesis of neuronal synaptophysin. Although blocking IL-1 or TNFα enhanced neuronal viability, TNFα seemed to have no significant influence on synaptophysin synthesis or tau phosphorylation, suggesting that not all cytokines released by activated microglia use the same pathways to achieve their deleterious effects on neurons.
Activation of p38-MAPK is involved in neuronal responses to various stresses (Xia et al., 1995; Kawasaki et al., 1997), and this kinase is closely related to hyperphosphorylated tau protein in AD (Sheng et al., 2001). Like several other members of the MAPK family, p38-MAPK is activated by dual phosphorylation; cytokines, including IL-1, can effect this activation. Here phosphorylation of p38-MAPK was significantly increased in neuronal cells either treated with recombinant IL-1β or cocultured with activated microglia, indicating that activation of p38-MAPK is involved in IL-1 signal transduction in neurons. The finding that inhibition of p38-MAPK significantly suppressed IL-1β-induced tau phosphorylation and IL-1β-decreased synaptophysin adds to the evidence for a role for this kinase in neural physiology and connectivity. Interestingly, treatment of neuronal cells with recombinant IL-1β resulted in transient increases in activated p38-MAPK, whereas neuronal cells cocultured with activated microglia showed a sustained increase that could be blocked by IL-1ra. This suggests that persistent microglial activation such as that proposed for AD may result in continued release of IL-1 with neurodegenerative consequences. This observation further confirms our previous finding that chronic activation of microglia causes significant and prolonged microglial production of cytokines, which leads to neuronal cell damage and further microglia activation (Li et al., 2000a; Barger and Basile, 2001). All of this points to the existence of neurodegenerative cycles that are dependent on neuronal–microglial interactions such as those proposed in AD and other neurodegenerative conditions (for review, seeGriffin et al., 2000).
The IL-1-mediated interactions shown here provide experimental support for the idea that microglial–neuronal relationships and the concomitant overexpression of IL-1 observed in AD brain contribute to the neuronal dysfunction and loss characteristic of AD, particularly those involved in formation of neurofibrillary tangles and loss of synapses. Our findings regarding the involvement of IL-1 and p38-MAPK in microglial activation-induced neuronal tau phosphorylation and synaptic function suggest that pharmacological approaches more directly targeting excessive expression of cytokines, e.g., cytokine-suppressant anti-inflammatory drugs, should be developed against brain inflammatory conditions, which are neuropathogenic not only in AD but also in early Down's syndrome, in AIDS, in head injury, and in demyelinating diseases such as multiple sclerosis.
Footnotes
This research was supported in part by National Institutes of Health Grant AG12411 and the Donald W. Reynolds Foundation. We thank Pam Free for secretarial support.
Correspondence should be addressed to Prof. W. Sue T. Griffin, Donald W. Reynolds Center on Aging, Room 3103, 629 South Elm Street, Little Rock, AR 72205. E-mail: griffinsuet@uams.edu.
References
- 1.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 2.Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- 3.Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E, Strothjohann M. Abnormally phosphorylated tau protein related to the formation of neurofibrillary tangles and neuropil threads in the cerebral cortex of sheep and goat. Neurosci Lett. 1994;171:1–4. doi: 10.1016/0304-3940(94)90589-4. [DOI] [PubMed] [Google Scholar]
- 5.Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc Natl Acad Sci USA. 1992;89:10075–10078. doi: 10.1073/pnas.89.21.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callahan LM, Coleman PD. Neurons bearing neurofibrillary tangles are responsible for selected synaptic deficits in Alzheimer's disease. Neurobiol Aging. 1995;16:311–314. doi: 10.1016/0197-4580(95)00035-d. [DOI] [PubMed] [Google Scholar]
- 7.Corradin SB, Mauel J, Donini SD, Quattrocchi E, Ricciardi-Castagnoli P. Inducible nitric oxide synthase activity of cloned murine microglial cells. Glia. 1993;7:255–262. doi: 10.1002/glia.440070309. [DOI] [PubMed] [Google Scholar]
- 8.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 9.DiPatre PL, Gelman BB. Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol. 1997;56:143–149. doi: 10.1097/00005072-199702000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D, Li X, Kirkpatrick JB. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int. 1995;27:119–137. doi: 10.1016/0197-0186(95)00067-i. [DOI] [PubMed] [Google Scholar]
- 11.Griffin WS, Mrak RE. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer's disease. J Leukoc Biol. 2002;72:233–238. [PMC free article] [PubMed] [Google Scholar]
- 12.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffin WS, Nicoll JA, Grimaldi LM, Sheng JG, Mrak RE. The pervasiveness of interleukin-1 in Alzheimer pathogenesis: a role for specific polymorphisms in disease risk. Exp Gerontol. 2000;35:481–487. doi: 10.1016/s0531-5565(00)00110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeohn GH, Kong LY, Wilson B, Hudson P, Hong JS. Synergistic neurotoxic effects of combined treatments with cytokines in murine primary mixed neuron/glia cultures. J Neuroimmunol. 1998;85:1–10. doi: 10.1016/s0165-5728(97)00204-x. [DOI] [PubMed] [Google Scholar]
- 16.Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. doi: 10.1074/jbc.272.30.18518. [DOI] [PubMed] [Google Scholar]
- 17.Lee VM, Balin BJ, Otvos L, Trojanowski JQ., Jr A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Wang J, Sheng JG, Liu L, Barger SW, Jones RA, Van Eldik LJ, Mrak RE, Griffin WS. S100 beta increases levels of beta-amyloid precursor protein and its encoding mRNA in rat neuronal cultures. J Neurochem. 1998;71:1421–1428. doi: 10.1046/j.1471-4159.1998.71041421.x. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Liu L, Kang J, Sheng JG, Barger SW, Mrak RE, Griffin WS. Neuronal–glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J Neurosci. 2000a;20:149–155. doi: 10.1523/JNEUROSCI.20-01-00149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Barger SW, Liu L, Mrak RE, Griffin WS. S100β induction of the proinflammatory cytokine interleukin-6 in neurons. J Neurochem. 2000b;74:143–150. doi: 10.1046/j.1471-4159.2000.0740143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Liu L, Barger SW, Mrak RE, Griffin WS. Vitamin E suppression of microglial activation is neuroprotective. J Neurosci Res. 2001;66:163–170. doi: 10.1002/jnr.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lue LF, Brachova L, Civin WH, Rogers J. Inflammation, Aβ deposition, and neurofibrillary tangle formation as correlates of Alzheimer's disease neurodegeneration. J Neuropathol Exp Neurol. 1996;55:1083–1088. [PubMed] [Google Scholar]
- 23.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 24.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 25.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 26.Mukaetova-Ladinska EB, Garcia-Siera F, Hurt J, Gertz HJ, Xuereb JH, Hills R, Brayne C, Huppert FA, Paykel ES, McGee M, Jakes R, Honer WG, Harrington CR, Wischik CM. Staging of cytoskeletal and beta-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer's disease. Am J Pathol. 2000;157:623–636. doi: 10.1016/s0002-9440(10)64573-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J Neurochem. 2000;74:1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- 28.Scheff SW, Sparks DL, Price DA. Quantitative assessment of synaptic density in the outer molecular layer of the hippocampal dentate gyrus in Alzheimer's disease. Dementia. 1996;7:226–232. doi: 10.1159/000106884. [DOI] [PubMed] [Google Scholar]
- 29.Sheng JG, Mrak RE, Griffin WS. Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol. 1997;56:285–290. [PubMed] [Google Scholar]
- 30.Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WS. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strijbos PJ, Rothwell NJ. Interleukin-1 β attenuates excitatory amino acid-induced neurodegeneration in vitro: involvement of nerve growth factor. J Neurosci. 1995;15:3468–3474. doi: 10.1523/JNEUROSCI.15-05-03468.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 33.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]