

Abstract
Fas-mediated apoptosis plays an important role in normal tissue homeostasis, and disruption of this death pathway contributes to many human diseases. Induction of apoptosis via Fas activation has been associated with reactive oxygen species (ROS) generation and down-regulation of FLICE inhibitory protein (FLIP); however, the relationship between these two events and their role in Fas-mediated apoptosis are unclear. We show herein that ROS are required for FLIP down-regulation and apoptosis induction by Fas ligand (FasL) in primary lung epithelial cells. ROS mediate the down-regulation of FLIP by ubiquitination and subsequent degradation by proteasome. Inhibition of ROS by antioxidants or by ectopic expression of ROS-scavenging enzymes glutathione peroxidase and superoxide dismutase effectively inhibited FLIP down-regulation and apoptosis induction by FasL. Hydrogen peroxide is a primary oxidative species responsible for FLIP down-regulation, whereas superoxide serves as a source of peroxide and a scavenger of NO, which positively regulates FLIP via S-nitrosylation. NADPH oxidase is a key source of ROS generation induced by FasL, and its inhibition by dominant-negative Rac1 expression or by chemical inhibitor decreased the cell death response to FasL. Taken together, our results indicate a novel pathway of FLIP regulation by an interactive network of reactive oxygen and nitrogen species that provides a key mechanism of apoptosis regulation in Fas-induced cell death and related apoptosis disorders.
The widely expressed protein Fas (CD95) is a member of a family of death receptors known to be involved in various forms of physiologic and pathologic cell death (1, 2). Activation of Fas receptor by Fas ligand (FasL)3 triggers a complex cascade of intracellular events that require Fas-associated death domain (FADD/Mort1) adaptor protein and the formation of death-inducing signaling complex, leading to caspase-8 activation and apoptosis (3, 4). Fas-mediated apoptosis is an essential mechanism for the maintenance of normal tissue homeostasis, and disruption of this death pathway has been associated with several human diseases, including autoimmune diseases (5), lymphoproliferative disorders (6), diabetes (7), and inflammation and fibrosis (8, 9). The Fas death system also plays important roles in various apoptosis conditions such as those evoked by irradiation, chemotherapeutic agents, and viral infections (10-13).
Although Fas activation can trigger apoptotic cell death, the expression of Fas receptor does not necessarily render cells susceptible to death by its ligand (2, 5), indicating that inhibitors of the apoptosis signaling pathway exist and play a role in the apoptotic process. A key apoptosis regulatory protein of the Fas death pathway is FLICE inhibitory protein (FLIP). FLIP has been shown to inhibit apoptosis by preventing the binding of procaspase-8 to FADD or interfering with the autocatalytic activation of caspase-8 at the death-inducing signaling complex (14, 15). Although the antiapoptotic role of FLIP has been well documented (reviewed in Ref. 16), several studies have shown that FLIP may also act as an inducer of apoptosis depending on its expression level and cell type (17, 18). To date, the role of FLIP has been exclusively studied in transformed cell lines. Because these cell lines may exhibit defective apoptosis machinery, its role in normal cells remains unclear. In this study, we sought to determine the role of FLIP in normal lung epithelial cells, which are a known target for Fas-induced apoptosis (8, 9), and to elucidate its mechanisms of Fas death regulation.
Activation of Fas receptor has been shown to be associated with rapid generation of reactive oxygen species (ROS) (19-22). However, the significance of this finding and the role of specific ROS involved in the apoptotic process are unclear. Vercammen et al. (19) demonstrated a dual pathway of apoptotic and necrotic cell death resulting from massive ROS generation and caspase activation. Gulbins et al. (20) and Sato et al. (21) implicated superoxide anion ( ) as a functional mediator of Fas-induced apoptosis in (Jurkat cells, whereas Medan et al. (22) reported hydrogen peroxide (H2O2) and hydroxyl radicals (OH•) as key mediators in macrophages. In contrast, Hug et al. (23) found no requirement of ROS in fibroblasts stably expressing human Fas. The present study was undertaken to further clarify the role of ROS in Fas-induced apoptosis and to determine the specific ROS involved, their cellular sources, and the mechanisms by which they regulate Fas apoptosis. We also tested the hypothesis that FLIP is a key cellular target of ROS that determines cell sensitivity to Fas-induced apoptosis. Using various molecular and cellular approaches, we found that FLIP plays a crucial role as a negative regulator of Fas-induced apoptosis in lung epithelial cells. FLIP is down-regulated by FasL via an ROS-dependent ubiquitin-proteasomal degradation process. Our data also suggest the interaction between reactive oxygen and nitrogen species, which cooperatively regulate FLIP and Fas signaling, thus revealing a novel mechanism of cell death regulation in death receptor-mediated apoptosis.
Materials and Methods
Reagents and Abs
Recombinant FasL (SuperFasLigand), mAb to FLIP (Dave-2), and caspase-8 inhibitor (Z-IETD-FMK) were obtained from ALEXIS Biochemicals. Abs for Fas, FADD, caspase-8, and peroxidase-labeled secondary Abs and protein A-agarose were obtained from Santa Cruz Biotechnology. Abs for ubiquitin, S-nitrosocysteine, and β-actin were from Sigma-Aldrich, as were N-acetyl cysteine, rotenone, diphenylene iodonium, lactacystin, 2-(4-carboxyphenyl)-tetramethylimidazoline-1-oxy-3-oxide, and superoxide dismutase. Catalase was from Roche Diagnostics. The ROS and NO probes, dihydroethidine, dichlorofluorescein diacetate, and diaminofluorescein diacetate were from Molecular Probes. The transfecting agents Lipofectamine and Oligofectamine were from Invitrogen.
Cell isolation and culture
Alveolar epithelial type II cells were isolated from male Sprague-Dawley rats (100–150 g; Jackson Laboratories) as previously described (24). The animals were handled according to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication No. 85-23, 1985). The animals were sacrificed with pentobarbital sodium (150 mg/kg body wt, i.p.) and the lungs were removed. They were perfused with 0.9% NaCl to remove blood cells and lavaged with PBS solution to remove free alveolar macrophages. The lungs were then excised and filled with PBS containing elastase (40 U/ml, type I; Worthington Biochemical) and DNase (0.006%; Sigma-Aldrich) and incubated at 37°C for 20 min. After enzymatic digestion, the lungs were finely minced and the digestion was arrested by incubation for 5 min in PBS containing 25% FBS and 0.006% DNase. The crude cell extract was sequentially filtered through 160- and 45-μm screens and then centrifuged. The resulting cell pellet was spun on a sterile Percoll density gradient, and the type II cell band was collected and resuspended in 1:1 (v/v) F12 and Eagle’s modified essential medium for further use.
Alveolar epithelial A549 cells were obtained from American Type Culture Collection. They were grown in F-12K medium containing 5% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
siRNA preparation and transfection
siRNA oligonucleotides against FLIP (siFLIP) and its control sequences (siCONTROL) were synthesized and obtained from Dharmacon using their custom SMARTpool and siDESIGN technology. The SMARTpool siRNA contains four sets of oligonucleotides specific for the sequences 1093–1111, 1103–1120, 1206–1224, and 1455–1473 of the long isoform of FLIP (FLIPL) gene (accession no. U97074). The oligonucleotides were annealed and suspended to a concentration of 10 μM. Alveolar epithelial type II (AE2) cells were seeded for transfection in a low serum culture medium (1% FBS) without antibiotics. The cells were transfected with siFLIP or siCONTROL oligonucleotides (100 nM) using Oligofectamine according to the manufacturer’s instructions. The cells were incubated with the oligonucleotide-liposome complexes for 6 h, and then low serum medium was replaced with normal medium (10% FBS) without antibiotics and incubated for an additional 48 h before further analysis.
Plasmids and stable transfection
The plasmids used in this study were generously provided by Dr. Christian Stehlik (Northwestern University, School of Medicine, Chicago, IL). Authenticity of all plasmid constructs was verified by DNA sequencing. Stable transfectants of FLIPL, superoxide dismutase 1 (SOD1), glutathione peroxidase (GPx), and dominant-negative Rac1 (Rac1N17) were generated by culturing A549 cells in a 6-well plate until they reached 80–90% confluence. One microgram of cytomegalovirus-neo vector and 15 μl of Lipofectamine reagent with 2 μg of the described plasmid or control pcDNA3 plasmid were used to transfect the cells in the absence of serum. After 10 h, the medium was replaced with culture medium containing 5% FBS. Approximately 36 h after the beginning of the transfection, the cells were digested with 0.03% trypsin and the cell suspensions were plated onto 75-ml culture flasks and cultured for 24–28 days with G418 selection. Resistant clones were isolated using cloning cylinders (Bellco Biotechnology) and transferred for expansion and analysis by Western blotting. Stable transformants were grown in G418-free RPMI medium for at least two passages before each experiment.
ROS and NO detection
Cellular superoxide and peroxide formation were determined by flow cytometry using dihydroethidine (DHE) and 2′,7′-dichlorofluorescein diacetate (DCF-DA) as fluorescent probes, respectively. For detection of NO, 4,5-diaminofluorescein diacetate (DAF-DA) was used as a probe. Cells were incubated with the probes (10 μM) for 30 min at 37°C in the dark. The cells were washed, resuspended in PBS, and analyzed for fluorescence intensity using FACSCalibur (BD Biosciences) at the excitation/emission wavelengths of 488/610 nm for DHE and 488/538 nm for DCF and DAF fluorescence measurements. The median fluorescence intensity was quantified by CellQuest (BD Biosciences) software analysis of the recorded histograms.
Apoptosis and caspase assays
Apoptosis was determined by Hoechst 33342 assay and by ELISA-based DNA fragmentation assay using a kit from Roche Molecular Biochemicals. For Hoechst assay, cells were incubated with 10 μg/ml Hoechst 33342 for 30 min, and the percentage of cells having intensely condensed chromatin and/or fragmented nuclei by fluorescence microscopy (Carl Zeiss Axiovert) were scored using Pixera software. Approximately 1000 nuclei from random fields were analyzed for each sample. The apoptotic index was calculated as (apoptotic nuclei/total nuclei) × 100 (%). For ELISA, cells were lysed with DNA lysis buffer (200 μl), and the cell lysate (20 μl) was mixed with an Ab solution provided by the supplier (80 μl) in 96-well plates at room temperature for 2 h. After washing with the incubation buffer, the substrate buffer (100 μl) was added to each well and incubated for 10 min at 37°C. OD was then measured using a microplate reader at a wavelength of 405 nm.
Caspase-8 activity was determined by fluorometric assay using the enzyme substrate IETD-AMC (Alexis Biochemicals), which is specifically cleaved by the enzymes at the Asp residue to release the fluorescent leaving group, 7-amido-4-methyl coumarin (AMC). Cell extracts containing 20 μg protein were incubated with 100 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid containing 10% sucrose, 10 mM DTT, 0.1% CHAPS, and 50 μM caspase substrate in a total reaction volume of 250 μl. The reaction mixture was incubated for 2 h at 37°C. At the end of incubation, the liberated fluorescent group AMC was determined fluorometrically (RF-531PC spectrofluorometer, Shimadzu) at the excitation and emission wavelengths of 380 and 460 nm, respectively.
Western blotting
Cells were harvested, washed twice with cold PBS, and lysed in cold lysis buffer containing 50 mM Tris-HCl (pH 7.4), 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM NaF, 1 mM EGTA, 1 mM PMSF, 1 mM sodium orthovanadate, and 1% protease inhibitor (Roche Molecular Biochemicals) for 30 min. After cellular debris was precipitated by centrifugation at 14,000 × g for 15 min at 4°C, the supernatants were collected and analyzed for protein content by bicinchoninic acid assay. Equal amount of proteins per sample (20 μg) were resolved on a 10% SDS-PAGE and transferred to a nitrocellulose membrane. The transferred membrane was blocked for 1 h in 5% nonfat dry milk in TBST and incubated at 4°C overnight with primary Abs, including rat monoclonal anti-human c-FLIP Ab (804127, 1:500, ALEXIS Biochemicals), rabbit polyclonal anti-human caspase-8 Ab (E20, 1:1000, Santa Cruz Biotechnology), rabbit polyclonal anti-human Fas Ab (C20, 1:500, Santa Cruz Biotechnology), rabbit polyclonal anti-human FADD Ab (H181, 1:500, Santa Cruz Biotechnology), mouse monoclonal anti-ubiquitin Ab (U0508, 1:1000, Sigma-Aldrich), and rabbit polyclonal anti-S-nitrosocysteine Ab (N5411, 1:500, Sigma-Aldrich). After three washes with TBST, the membrane was incubated with peroxidase-conjugated isotype-specific secondary Abs (1: 500, Santa Cruz Biotechnology) for 1 h at room temperature and washed with 0.05% Tween 20 in PBS. Immunoreactive proteins were detected by chemiluminescence (SuperSignal West Pico, Pierce Biotechnology) and quantified by densitometry using UN-SCAN-IT digitizing software (Silk Scientific). Mean densitometry data from independent experiments were normalized to the control.
Immunoprecipitation
Cells were washed with cold PBS and lysed in lysis buffer at 4°C for 30 min. Cell lysates were centrifuged at 14,000 × g for 15 min, and the supernatants were collected and analyzed for protein content. Samples containing 60 μg of protein were incubated with 12 μl of anti-FLIP agarose beads diluted with 12 μl of G-protein beads for 6 h at 4°C. The immune complexes were washed three times with 20 volumes of lysis buffer, resuspended in 2× Laemmli sample buffer, and heated at 95°C for 5 min. Immunoprecipitates containing 20 μg of protein were separated by 10% SDS-PAGE and analyzed by Western blotting as described.
Statistical analysis
The data represent means ± SD from three or more independent experiments. Statistical analysis was performed by Student’s t test at a significance level of p < 0.05.
Results
Induction of apoptosis and caspase activation by FasL in lung epithelial cells
To study the regulatory mechanisms of Fas-induced apoptosis, we first characterized the apoptotic response to FasL treatment in primary AE2 cells from rats. The cells were treated with varying concentrations of FasL (0–100 ng/ml), and apoptosis was examined by nuclear DNA fragmentation and caspase activation assays. As shown in Fig. 1A, FasL treatment caused a dose-dependent increase in apoptotic cell death over nontreated control, as determined by Hoechst assay. The apoptotic cells exhibited condensed and/or fragmented nuclei with intense nuclear fluorescence (Fig. 1B). ELISA-based assay of apoptosis similarly showed a dose-dependent effect of FasL on DNA fragmentation (Fig. 1C). Caspase-8 activation, which is indicative of death receptor-mediated apoptosis (25, 26), was also induced by the FasL treatment, as evidenced by the cleavage of its inactive precursor form into active fragments (Fig. 1D). Addition of caspase-8 inhibitor (Z-IETD-FMK) potently inhibited apoptosis and caspase-8 activation induced by FasL (Fig. 1, E and F), indicating the role of caspase-8 in epithelial cell death induced by FasL. Treatment of the cells with N-acetylcysteine (NAC), a general antioxidant, also inhibited FasL-induced cell death and caspase-8 activation (Fig. 1, E and F), suggesting the role of ROS in the apoptotic process.
FIGURE 1.
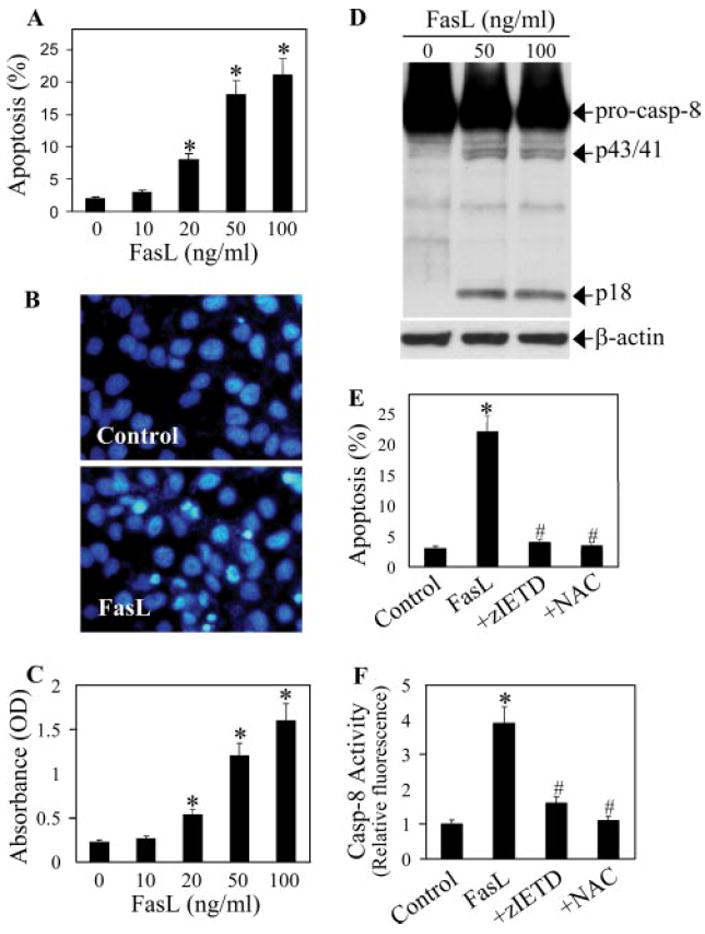
FasL-induced apoptosis and caspase activation in lung epithelial cells. A, Primary AE2 cells (1 × 106 cells/ml) were treated with varying concentrations of FasL (0–100 ng/ml) for 12 h, and apoptosis was determined by Hoechst 33342 assay. B, Fluorescence micrographs of cells treated with FasL (100 ng/ml) for 12 h. Apoptotic cells exhibited shrunken and fragmented nuclei with bright nuclear fluorescence. C, Cells were treated with FasL as described in A and analyzed for DNA nucleosomal fragmentation by ELISA. D, Western blot analysis of caspase-8 at 6 h after FasL treatment. The blots were reprobed with an Ab specific for β-actin as a loading control. E, Cells were treated with FasL (100 ng/ml) in the presence or absence of caspase-8 inhibitor Z-IETD-FMK (10 μM) or NAC (100 μM), and apoptosis was determined by Hoechst assay at 12 h posttreatment. F, Cells were similarly treated as in E for 12 h, and cell lysates (50 μg protein) were prepared and analyzed for caspase-8 activity as described in Materials and Methods. Plots are means ± SD (n = 4). *, p < 0.05 vs nontreated control. #, p < 0.05 vs FasL-treated control.
Role of ROS in FasL-induced apoptosis
To determine the role of specific ROS in FasL-induced apoptosis of lung epithelial cells, AE2 cells were treated with FasL in the presence or absence of SOD ( scavenger), catalase (H2O2 scavenger), or sodium formate (OH• scavenger), and apoptosis was examined by Hoechst assay. Fig. 2A shows that all test scavengers were able to inhibit FasL-induced apoptosis in a dose-dependent manner, with catalase and SOD being the most effective. These results suggest that multiple ROS are involved in the apoptotic process and that H2O2 and play a major role. To confirm the formation of ROS and their inhibition by the antioxidant enzymes in the treated cells, cellular ROS levels were determined using DHE and DCF-DA as fluorescent probes. Fig. 2B shows that FasL was able to induce superoxide and peroxide formation, as indicated by the increase in cellular DHE and DCF fluorescence intensities, respectively. Addition of SOD strongly inhibited FasL-induced DHE fluorescence, whereas catalase and sodium formate were ineffective, indicating the specificity of superoxide detection in the experiment. Analysis of peroxide formation by DCF fluorescence indicates inhibition of FasL-induced peroxide formation by catalase, but not by SOD or sodium formate. Interestingly, SOD increased DCF fluorescence intensity in FasL-treated cells, suggesting increased peroxide formation via dismutation of superoxide in the treated cells.
FIGURE 2.
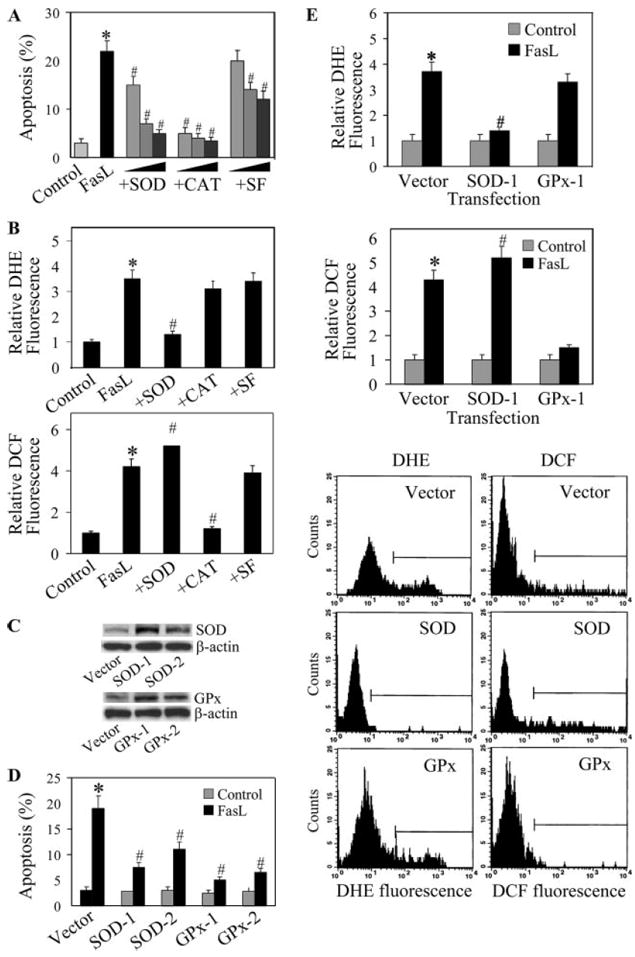
Effects of antioxidants on FasL-induced apoptosis and ROS generation. A, AE2 cells (1 × 106 cells/ml) were either left untreated or pretreated for 1 h with varying concentrations of SOD (500, 1000, 5000 U/ml), catalase (500, 1000, 5000 U/ml), or sodium formate (5, 10, 20 mM). The cells were then treated with FasL (100 ng/ml) for 12 h and analyzed for apoptosis by Hoechst assay. B, Cells were treated with FasL (100 ng/ml) in the presence or absence of SOD (1000 U/ml), catalase (1000 U/ml), or sodium formate (10 mM), and they were analyzed for ROS production by measuring DHE and DCF fluorescence intensities. Plots show relative fluorescence intensity over nontreated control at the peak response time of 1 h after treatment. C, A549 cells were stably transfected with SOD, GPx, or control plasmid. Clonal lines expressing varying degrees of antioxidant enzymes were selected by immunoblotting using Abs specific for SOD and GPx. D, Transfectant lines were treated with FasL (100 ng/ml) for 12 h and analyzed for apoptosis. E, Transfectant lines were treated with FasL (100 ng/ml) and analyzed for DHE and DCF fluorescence intensities at 1 h posttreatment. Representative histograms of DHE and DCF measurements in the treated cells are shown. Plots are mean ± SD (n > 3). *, p < 0.05 vs nontreated control. #, p < 0.05 vs FasL-treated control. CAT indicates catalase.
To confirm the role of superoxide and peroxide in FasL-induced cell death, we analyzed apoptotic and ROS responses to FasL treatment in SOD- and GPx-transfected cells. Because primary AE2 cells were refractory to gene transfection, we used alveolar epithelial A549 cells to aid gene transfer study. The cells were stably transfected with SOD, GPx, or control plasmid, and they were analyzed for antioxidant enzyme expression by Western blotting. Two clonal lines from each of the SOD and GPx transfections were selected based on their relative antioxidant enzyme expression (Fig. 2C). The clonal lines were treated with FasL and analyzed for apoptosis. Fig. 2D shows that the SOD- and GPx-over-expressing lines exhibited significantly reduced apoptosis in response to FasL treatment as compared with the control line. The clonal lines that exhibited higher SOD and GPx expression also showed a more substantial reduction in apoptotic response to FasL treatment, consistent with our earlier data. Flow cytometric analysis of ROS generation also showed a substantial reduction in FasL-induced DHE and DCF fluorescence intensities in the SOD- and GPx-overexpressing lines as compared with the control line (Fig. 2E). Together, these results indicate the role of and H2O2 in FasL-induced apoptosis of lung epithelial cells.
NADPH oxidase is a key source of ROS generation induced by FasL
To determine the source of ROS generation induced by FasL, cells were treated with FasL in the presence or absence of diphenylene iodonium (DPI), a known inhibitor of NADPH oxidase (27, 28), or rotenone, an inhibitor of mitochondrial electron transport chain (28, 29), and their effects on apoptosis and ROS generation were examined. The results show that DPI strongly inhibited FasL-induced apoptosis and ROS generation, whereas rotenone showed weak inhibitory effects (Fig. 3, A and B). Addition of Z-IETD-FMK, which was shown to inhibit FasL-induced caspase-8 activation and apoptosis (Fig. 1), had no significant effect on FasL-induced ROS generation (Fig. 3B). This result is consistent with a previous study showing that caspase inhibition was unable to inhibit ROS generation induced by death receptor activation (30). Together, our results indicate that NADPH oxidase is the primary source of ROS generation and that these ROS act upstream of the caspase-8 activation pathway.
FIGURE 3.
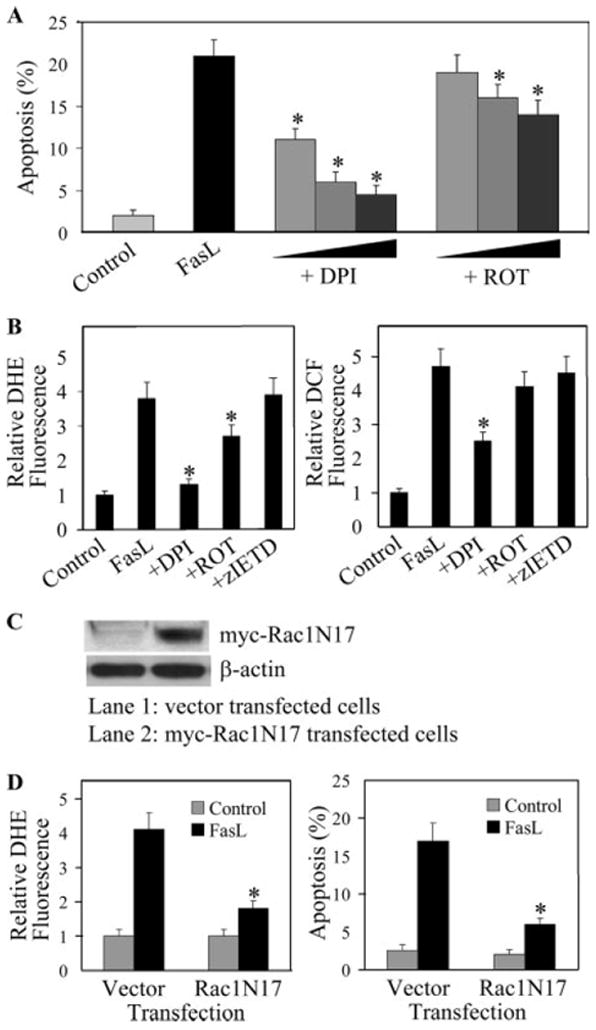
Effects of diphenylene iodonium, rotenone, and Rac1N17 on FasL-induced apoptosis and ROS generation. A, AE2 cells were either left untreated or treated with varying concentrations of DPI (0.1, 0.5, 1 μM) or rotenone (0.1, 0.5, 1 μM) for 1 h, after which they were treated with FasL (100 ng/ml) and analyzed for apoptosis after 12 h. B, Flow cytometric measurements of DHE and DCF fluorescence intensities. Cells were pretreated for 1 h with DPI (1 μM), rotenone (1 μM), or Z-IETD-FMK (10 μM), followed by FasL treatment (100 ng/ml) for 3 h. C, A549 cells were stably transfected with myc-tagged Rac1N17 or control plasmid. Cell lysates were prepared, separated, and probed with anti-myc Ab. D, Transfected cells were treated with FasL (100 ng/ml) and analyzed for DHE fluorescence and apoptosis after 1 and 12 h, respectively. Data are means ± SD (n > 3). *, p < 0.05 vs FasL-treated control. ROT indicates rotenone.
To further determine the role of NADPH oxidase in FasL-induced ROS generation and apoptotic cell death, A549 cells were stably transfected with Rac1N17 or control plasmid, and their effects on FasL-induced ROS generation and apoptosis were determined. Rac1 is a small GTP-binding protein known to be involved in the activation of NADPH oxidase and generation of in various cell types (31-33). Fig. 3C shows that cells transfected with the Rac1N17 plasmid expressed a high level of the protein as compared with vector-transfected control cells. ROS generation, as analyzed by DHE fluorescence, was substantially reduced in the dominant-negative cells as compared with control cells after FasL treatment (Fig. 3D). Likewise, the apoptotic response to FasL treatment was substantially reduced in the dominant-negative cells relative to control cells (Fig. 3D). These results indicate the role of Rac1-mediated ROS generation in FasL-induced apoptosis of lung epithelial cells.
Effect of FasL treatment on apoptosis-regulatory proteins
To determine the mechanism of apoptosis regulation by ROS in FasL-treated cells, we examined by immunoblotting the expression levels of key proteins known to be involved in Fas death signaling, including Fas, FADD, and FLIP. Among these, only the level of FLIP was affected by the FasL treatment in a dose- and time-dependent manner (Fig. 4). Only FLIPL could be detected in this study, whereas the short isoform (FLIPS) was undetectable.
FIGURE 4.
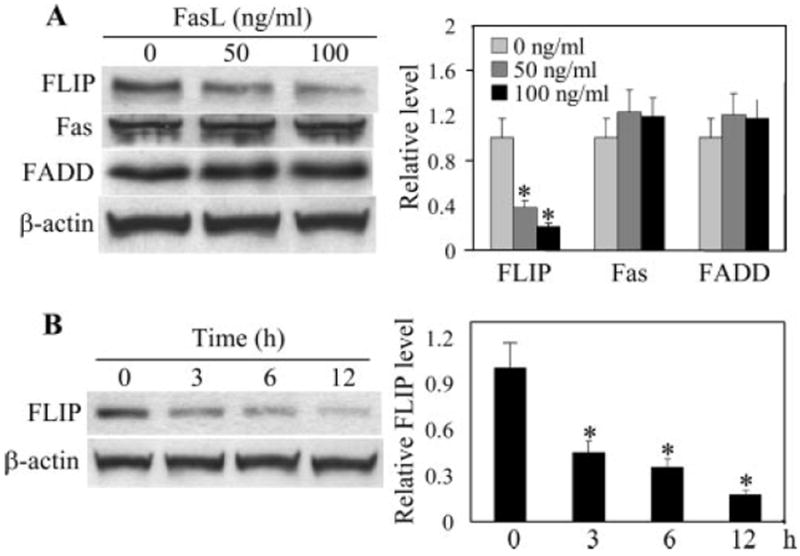
Effect of FasL treatment on apoptosis-regulatory proteins. A, AE2 cells were treated with FasL (0–100 ng/ml) for 12 h. The cells were then washed with ice-cold PBS and extracted with lysis buffer. The cell extracts were separated on polyacrylamide-SDS gels, transferred, and probed with Abs specific for Fas, FADD, and FLIP. Blots were reprobed with β-actin Ab to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry, and mean data from independent experiments (one of which is shown here) were normalized to the results obtained in cells in the absence of FasL (control). B, Time-dependent down-regulation of FLIP by FasL. Cells were treated with FasL (100 ng/ml) for various times and analyzed for FLIP by Western blotting. Values are means ± SD (n > 3). *, p < 0.05 vs nontreated control.
To determine the role of FLIP in FasL-induced apoptosis of lung epithelial cells, AE2 and A549 cells were transfected with siFLIP and full-length FLIP plasmid, respectively, and their effects on FLIP protein expression and cell death response to FasL were examined. Fig. 5, A and B shows that siFLIP-transfected cells exhibited reduced expression of FLIP and were more susceptible to FasL-induced apoptosis than were siCONTROL-transfected cells. Transfection of cells with FLIP plasmid increased FLIP protein expression and reduced cell death response to FasL treatment as compared with vector-transfected control (Fig. 5, C and D). Transfection of the cells with FLIP and siFLIP also resulted in a corresponding decrease and increase in cellular ROS levels following FasL treatment as compared with their respective controls (Fig. 5E). Taken together, these results indicate a negative regulatory role of FLIP in FasL-induced apoptosis and ROS generation in lung epithelial cells.
FIGURE 5.
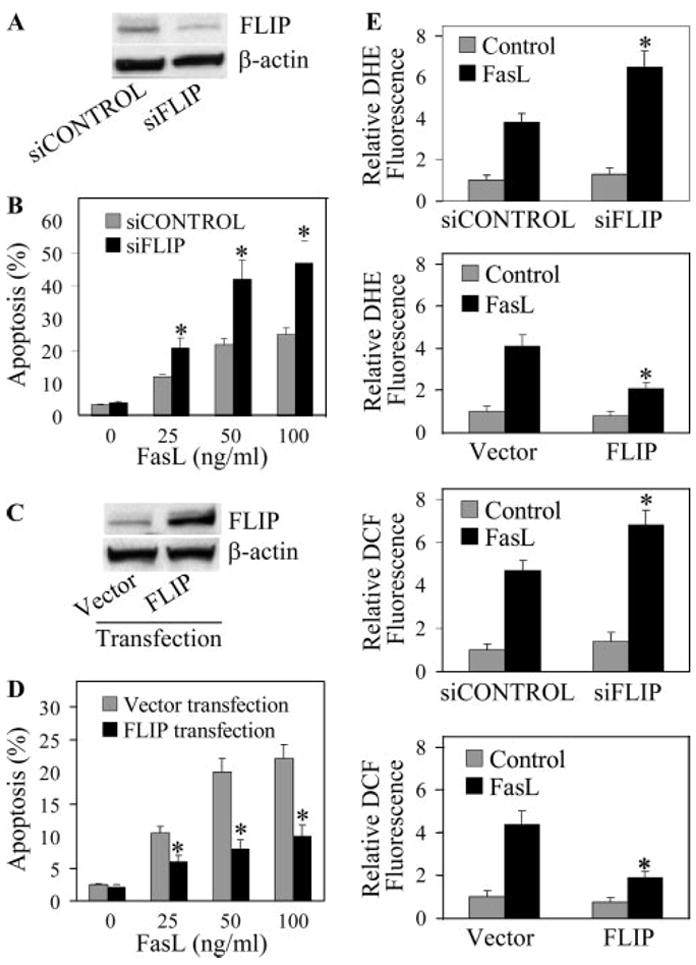
Effects of FLIP siRNA knockdown and gene overexpression on FasL-induced apoptosis and ROS generation. A, AE2 cells were transfected with siFLIP or siCONTROL oligonucleotides as described in Materials and Methods. The cells were characterized for FLIP protein expression by Western blotting. β-actin was used as a loading control. B, Transfected cells were treated with varying doses of FasL (0–100 ng/ml) for 12 h and analyzed for apoptosis by Hoechst assay. C, A549 cells were stably transfected with FLIP or control plasmid. The cells were analyzed for FLIP protein levels by immunoblotting. D, Transfected cells were treated with varying doses of FasL (0–100 ng/ml) for 12 h and analyzed for apoptosis. E, siFLIP, FLIP, and control transfectants were treated with FasL (100 ng/ml) and analyzed for DHE and DCF fluorescence after 1 h. Plots are means ± SD (n = 4). *, p < 0.05 vs FasL-treated controls.
Regulation of FLIP by ROS
FLIP has been shown to be regulated by the ubiquitin-proteasome pathway under different conditions (34-36). To determine whether this pathway is involved in the down-regulation of FLIP by FasL in the test cell system, cells were treated with lactacystin, a highly specific proteasome inhibitor, and its effect on FasL-induced FLIP down-regulation was determined by Western blotting. Fig. 6A shows that lactacystin completely inhibited FLIP down-regulation, indicating the role of proteasomal degradation in FasL-induced down-regulation of FLIP in lung epithelial cells.
FIGURE 6.
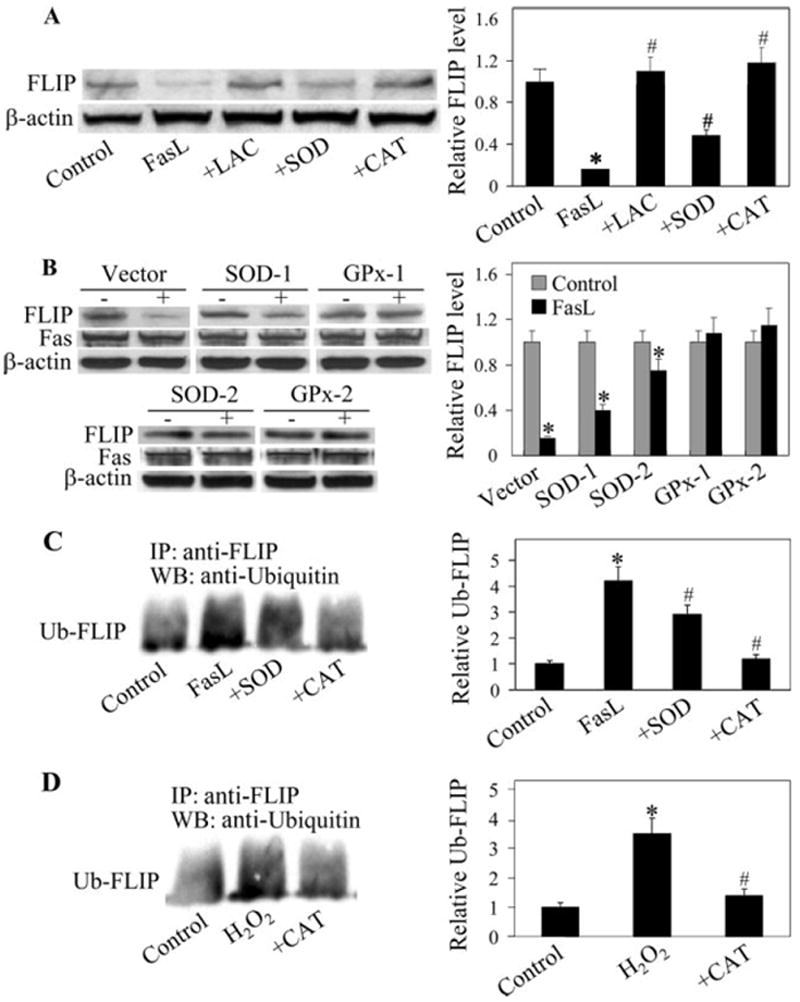
Effects of FasL and antioxidants on FLIP expression and ubiquitination. A, AE2 cells were either left untreated or pretreated with lactacystin (10 μM), SOD (1000 U/ml), or catalase (1000 U/ml) for 1 h, followed by FasL treatment (100 ng/ml) for 12 h. Cell lysates were prepared and analyzed for FLIP protein expression by immunoblotting. Densitometry was performed to determine the relative expression of FLIP in the treated cells compared with nontreated cells. B, SOD-, GPx-, and vector-transfected A549 clonal lines were treated with FasL (100 ng/ml) for 12 h, and FLIP and Fas expressions were determined. C, AE2 cells were pretreated with SOD or catalase as described above and then treated with FasL (100 ng/ml) for 3 h in the presence of lactacystin (10 μM) to prevent proteasomal degradation of FLIP. Cell lysates were immunoprecipitated with anti-FLIP Ab, and the immune complexes were analyzed for ubiquitin by Western blotting. D, Cells were treated with H2O2 (100 μM) for 3 h, with or without catalase (1000 U/ml), and FLIP ubiquitination was similarly determined. Plots are means ± SD (n > 3). *, p < 0.05 vs nontreated control. #, p < 0.05 vs FasL-treated control. LAC indicates lactacysin; CAT, catalase.
To determine the role of ROS in FLIP down-regulation, cells were treated with FasL in the presence or absence of SOD and catalase, and their effect on FLIP protein expression was determined. Fig. 6A shows that treatment of the cells with catalase completely inhibited FasL-induced FLIP down-regulation, whereas SOD treatment showed a partial inhibitory effect. These results were confirmed in GPx- and SOD-overexpressing lines, which exhibited a relatively constant level of Fas expression in the presence or absence of FasL activation (Fig. 6B). The results indicate that H2O2 is a major oxidative species responsible for FLIP down-regulation by FasL. Ubiquitination studies further showed that catalase strongly inhibited FasL-induced ubiquitination of FLIP (Fig. 6C), supporting the role of H2O2 in ubiquitin-mediated down-regulation of FLIP through the proteasome pathway. Direct treatment of the cells with H2O2 similarly induced FLIP ubiquitination, the effect that was inhibited by catalase (Fig. 6D), further supporting the role of this oxidative species in FLIP down-regulation.
Regulation of FLIP by superoxide
The observation that SOD inhibited FLIP down-regulation is somewhat surprising because SOD treatment had no inhibitory effect on peroxide formation, but instead it increased the peroxide level in FasL-treated cells (Fig. 2C). Because previous studies have shown that FLIP ubiquitination is negatively regulated by NO, which is induced by FasL (37), and because NO is known to interact with (38, 39), we investigated this potential interaction as a possible mechanism of FLIP regulation by . Cells were treated with FasL in the presence or absence of SOD, and cellular NO levels were determined by flow cytometry using DAF-DA as a probe. Fig. 7A shows that FasL treatment induced NO production and that cotreatment of the cells with scavenger SOD further increased the NO production. Addition of 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO), a known scavenger of NO, inhibited the NO induction, indicating the specificity of NO detection in the test cell system. Similar results were obtained when NO production was analyzed by Griess assay, which measures nitrite byproduct of NO (data not shown). Western blot analysis of FLIP protein expression shows that PTIO blocked the inhibitory effect of SOD on FasL-induced FLIP down-regulation (Fig. 7B), suggesting that SOD may exert its effect on FLIP through NO modulation. This result was confirmed by the observation that another NO inhibitor, aminoguanidine, also inhibited the effect of SOD on FasL-induced FLIP down-regulation (data not shown). Because previous studies have shown that NO regulates FLIP through S-nitrosylation, which inhibits its ubiquitination and degradation by the proteasome (37), we examined the effect of SOD on FLIP S-nitrosylation in FasL-treated cells by immunoprecipitation and Western blot using anti-S-nitrosocysteine Ab. Treatment of the cells with FasL, as expected, induced S-nitrosylation of FLIP (Fig. 7C). Cotreatment of the cells with SOD further increased this nitrosylation, whereas addition of the NO scavenger PTIO inhibited this effect (Fig. 7C). Taken together, these results indicate that SOD was able to inhibit NO production and S-nitrosylation of FLIP in FasL-treated cells. This finding indicates a novel pathway of FLIP regulation by , which may serve as an additional layer of apoptosis regulation in Fas-induced apoptosis of lung epithelial cells.
FIGURE 7.
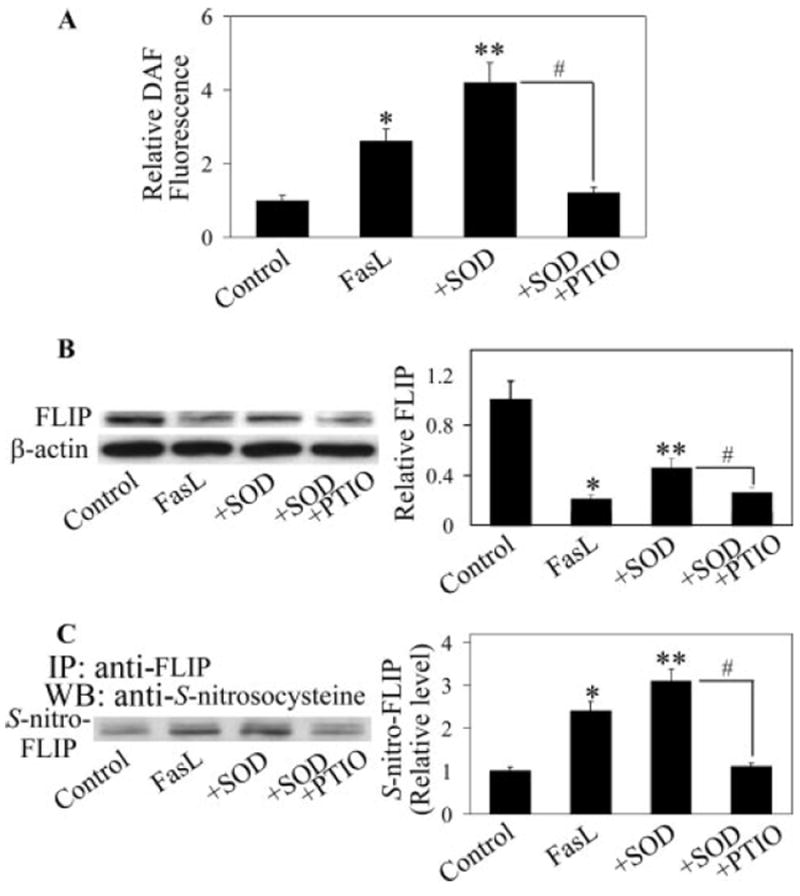
Effects of SOD treatment on NO production and FLIP nitrosylation. A, AE2 cells were either left untreated or pretreated for 1 h with SOD (1000 U/ml) in the presence or absence of NO inhibitor PTIO (100 μM). The cells were then treated with FasL (100 ng/ml) for 3 h and analyzed for NO levels by flow cytometry using DAF-DA as a probe. B, Cells were similarly treated with the test agents and analyzed for FLIP protein levels by Western blotting after 12 h. C, Cells were similarly treated and analyzed for FLIP S-nitrosylation by immunoprecipitation at 3 h posttreatment. Cell lysates were immunoprecipitated with anti-FLIP Ab, and the immune complexes were analyzed by Western blot using S-nitrosocysteine Ab. The density of nitrosylated FLIP bands was normalized against nontreated control band. Plots are means ± SD (n = 3). *, p < 0.05 vs nontreated control. **, p < 0.05 vs FasL-treated control. #, p < 0.05 vs FasL/SOD-treated control.
Discussion
ROS have long been known to be involved in the regulation of apoptosis induced by various pathologic and physiologic stimuli. However, their role in Fas-induced apoptosis and its regulatory mechanisms are unclear. In the present study, we showed that FasL-induced apoptosis of lung epithelial cells is associated with the generation of multiple ROS and that inhibition of these ROS by antioxidant enzymes catalase and SOD effectively inhibited the apoptotic effect of FasL (Fig. 2A). Overexpression of GPx and SOD by gene transfection similarly inhibited FasL-induced apoptosis (Fig. 2D), supporting the role of H2O2 and in the death signaling process. These results are consistent with previous reports showing the proapoptotic role of H2O2 and in various cell types (40-42). In Jurkat T cells, Gulbins et al. (20) and Sato et al. (21) suggested as a mediator of Fas death signaling, whereas Medan et al. (22) showed H2O2 and OH· as key mediators in macrophages. In contrast, Hug et al. (23) found no requirement of ROS in fibroblasts overexpressing Fas, while Aronis et al. (43) showed an antiapoptotic role of ROS in Fas-activated Jurkat cells. These studies suggest that the apoptotic response to Fas activation of ROS is cell type specific and may be determined by the expression levels of antioxidant enzymes. could be the early ROS produced by cells in response to Fas activation, and depending on the expression level of its scavenging enzyme, SOD determines the degree of sensitivity to Fas-induced apoptosis. The expression of SOD has been shown to be up-regulated following death receptor activation through an NF-κB-dependent pathway (44, 45). In NF-κB-deficient cells, ROS generation is enhanced upon treatment with the death ligand TNF-α, and ectopic expression of SOD inhibits this effect (44, 45), suggesting the role of NF-κB in SOD-mediated inhibition of ROS. The induction of ROS by TNF-α has also been shown to be dependent on JNK activation (46), which is negatively regulated by FLIP (47). Because FLIP is known to activate NF-κB (48, 49), its increased expression could lead to SOD up-regulation and decreased formation. Thus, in addition to being affected by ROS, FLIP also regulates ROS through JNK and NF-κB activation, forming a feedback loop that controls the death signaling. Because ROS generation induced by FasL was shown to be mediated through NADPH oxidase, it is also possible that FLIP may have a direct effect on this enzyme and thereby regulate the ROS and apoptotic responses to Fas activation. It is likely that the difference in the expression levels of SOD and other ROS regulatory proteins described herein determine the variable ROS responses to Fas activation observed in the previous studies. Should the level of SOD be high, most of the radicals generated would be rapidly converted to H2O2, thereby putting pressure on the catalase and peroxidase systems. It is also conceivable that potential suboptimal expression levels of catalase and peroxidase could have resulted in increased H2O2 levels. Such elevated levels, detected in our experiments, could in turn initiate a cascade of cellular signaling events, including caspase activation and apoptosis.
The results of this study also show that NADPH oxidase is a key source of ROS generation induced by FasL. Supporting this notion is the evidence that DPI, a known inhibitor of NADPH oxidase (27, 28), strongly inhibited ROS generation and apoptosis induction by FasL, whereas rotenone, a known inhibitor of mitochondrial ROS (28, 29), had minimal effects (Fig. 3). The mechanism by which FasL activates NADPH oxidase is unclear but was shown to involve Rac1 activation because its inhibition by Rac1N17 expression inhibited the induction of by FasL (Fig. 3D). Rac1 protein is known to be involved in the assembly of the NADPH oxidase system, which is responsible for transferring electrons from NADPH to molecular oxygen with the subsequent production of (50). The activation of Rac1 by Fas has also been shown to be mediated through Ras signaling (20), which is activated upon Fas receptor ligation (51). We have further shown that inhibition of Rac1 by dominant-negative expression inhibited apoptotic cell death induced by FasL (Fig. 3D). As indicated earlier, can be dismutated by SOD to form H2O2, which could serve as a key mediator of FasL-induced apoptosis. This was demonstrated by the ability of the peroxide-scavenging enzymes catalase and GPx to inhibit FasL-induced apoptosis (Fig. 2).
The results of this study also show that ROS mediate the apoptotic effect of FasL, at least in part through down-regulation of FLIP. FasL induced down-regulation of FLIP without having a significant effect on FADD and Fas protein expression (Fig. 4). FLIP is known to render cells resistant to death receptor-mediated apoptosis in various cell types (13, 14, 52-54), and its elevated expression has been associated with tumor cells that escape from immune surveillance (55). Consistent with these earlier reports, our gene overexpression and siRNA knockdown studies indicate the inhibitory role of FLIP in FasL-induced apoptosis of lung epithelial cells (Fig. 5). Down-regulation of FLIP by FasL was shown to be dependent on ROS because this effect was inhibited by the antioxidant enzymes catalase, GPx, and SOD (Fig. 6).
The mechanism by which ROS mediates the down-regulation of FLIP by FasL was shown to involve ubiquitin-proteasomal degradation because inhibition of ROS by antioxidants inhibited the ubiquitination and proteasomal degradation of FLIP by FasL (Fig. 6). Direct treatment of the cells with H2O2 similarly induced ubiquitination of FLIP, which was also inhibited by catalase (Fig. 6), supporting the role of ROS in FLIP down-regulation via ubiquitination. Thus, FLIP is a redox-sensitive protein that is subjected to regulation by ROS. Several cytotoxic agents known to increase cellular oxidative stress have been shown to sensitize cells to Fasinduced apoptosis (56-59), supporting the general role of ROS in Fas death signaling through FLIP down-regulation.
In addition to ROS, FLIP has also been shown to be under the regulation of NO. However, unlike ROS, NO plays a protective role in FLIP down-regulation through its ability to stabilize the protein via S-nitrosylation, which prevents its degradation through the ubiquitin-proteasome pathway (36). Our results show that this process is linked to the ROS regulatory mechanism because inhibition of ROS by the antioxidant SOD increased the cellular levels of NO in FasL-treated cells (Fig. 7A). SOD treatment also increased NO-mediated S-nitrosylation of FLIP, which was reversed by cotreatment with the NO inhibitor PTIO (Fig. 7C). PTIO also inhibited the SOD effect on FLIP protein expression in FasL-treated cells (Fig. 7B). Taken together, these results indicate the interactive role of NO and in the regulation of FLIP, thus revealing a novel pathway of apoptosis regulation in Fas-induced cell death.
In conclusion, we have shown that ROS play an important role in the regulation of apoptosis induced by FasL in lung epithelial cells. The mechanism by which ROS regulate apoptosis involves down-regulation of FLIP, which functions as a suppressor of Fas death signaling in these cells. H2O2 is a major oxidative species responsible for FLIP down-regulation via ubiquitin-proteasomal degradation, while serves as a source of H2O2 and as a negative regulator of NO. Because aberrant expression of ROS and NO has been associated with several human disorders, the results of this study could have important implications in the understanding of disease pathogenesis and treatment of associated apoptosis disorders.
Footnotes
This work was supported by National Institutes of Health Grants HL071545 and HL763401.
Abbreviations used in this paper: FasL, Fas ligand; AE2, alveolar epithelial type II; CAT, catalase; DAF-DA, 4,5-diaminofluorescein diacetate; DCF-DA, 2′,7′-dichlorofluorescein diacetate; DHE, dihydroethidine; DPI, diphenylene iodonium; FADD, Fas-associated death domain; FLIP, FLICE inhibitory protein; GPx, glutathione peroxidase; H2O2, hydrogen peroxide; LAC, lactacystin; NAC, N-acetylcysteine; , superoxide anion; OH•, hydroxyl radical; PTIO, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide; Rac1N17, dominant-negative Rac1; ROS, reactive oxygen species; ROT, rotenone; SOD, superoxide dismutase.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 2.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 3.Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis: immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- 4.Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999;33:29–55. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
- 5.Hsu HC, Matsuki Y, Zhang HG, Zhou T, Mountz JD. The Fas signaling connection between autoimmunity and embryonic lethality. J Clin Immunol. 2001;21:1–14. doi: 10.1023/a:1006730112726. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 7.Chervonsky AV, Wang Y, Wong FS, Visintin I, Flavell RA, Janeway CA, Jr, Matis LA. The role of Fas in autoimmune diabetes. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 8.Kuwano K, Hagimoto N, Kawasaki M, Yatomi T, Nakamura N, Nagata S, Suda T, Kentaku R, Maeyama T, Miyazaki H, Hara N. Essential roles of the Fas-Fas-ligand pathway in pulmonary fibrosis. J Clin Invest. 1999;104:13–19. doi: 10.1172/JCI5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: implications for acute pulmonary inflammation. Am J Pathol. 2001;158:153–161. doi: 10.1016/S0002-9440(10)63953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 11.Friesen C, Herr I, Krammer PH, Debatin KM. Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat Med. 1996;2:574–577. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- 12.Muller M, Strand S, Hug H, Heinemann EM, Walczak H, Hofmann WJ, Stremmel W, Krammer PH, Galle PR. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clin Invest. 1997;99:403–413. doi: 10.1172/JCI119174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rehemtulla A, Hamilton CA, Chinnaiyan AM, Dixit VM. Ultraviolet radiation-induced apoptosis is mediated by activation of CD-95 (Fas/APO-1) J Biol Chem. 1997;272:25783–25786. doi: 10.1074/jbc.272.41.25783. [DOI] [PubMed] [Google Scholar]
- 14.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 15.Tschopp J, Irmler M, Thome M. Inhibition of fas death signals by FLIPs. Curr Opin Immunol. 1998;10:552–558. doi: 10.1016/s0952-7915(98)80223-9. [DOI] [PubMed] [Google Scholar]
- 16.Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247–8254. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang DW, Xing X, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X. c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siegel RM, Martin DA, Zheng L, Ng SY, Bertin J, Cohen J, Lenardo MJ. Death-effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. J Cell Biol. 1998;141:1243–1253. doi: 10.1083/jcb.141.5.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vercammen D, Brouckaert G, Denecker G, Van den Craen M, Declercq W, Fiers W, Vandenabeele P. Dual signaling of the Fas receptor: inhibition of both apoptotic and necrotic pathways. J Exp Med. 1998;188:919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gulbins E, Brenner B, Schlottmann K, Welsch J, Heinle H, Koppenhoefer U, Linderkamp O, Coggeshall KM, Lang F. Fas-induced programmed cell death is mediated by a Ras-regulated O2− synthesis. Immunology. 1996;89:205–212. doi: 10.1046/j.1365-2567.1996.d01-743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato T, Machida T, Takahashi S, Iyama S, Sato Y, Kuribayashi K, Takada K, Oku T, Kawano Y, et al. Fas-mediated apoptosome formation is dependent on reactive oxygen species derived from mitochondrial permeability transition in Jurkat cells. J Immunol. 2004;173:285–296. doi: 10.4049/jimmunol.173.1.285. [DOI] [PubMed] [Google Scholar]
- 22.Medan D, Wang L, Toledo D, Lu B, Stehlik C, Shi X, Rojanasakul Y. Regulation of Fas (CD95/APO-1)-mediated apoptosis and necrosis by reactive oxygen species in macrophages. J Cell Physiol. 2005;203:78–84. doi: 10.1002/jcp.20201. [DOI] [PubMed] [Google Scholar]
- 23.Hug H, Enari M, Nagata S. No requirement of reactive oxygen intermediates in Fas-mediated apoptosis. FEBS Lett. 1994;351:311–313. doi: 10.1016/0014-5793(94)00852-3. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Toledo-Velasquez D, Schwegler-Berry D, Ma JKH, Rojanasakul Y. Transport and hydrolysis of enkephalins in cultured alveolar epithelial monolayers. Pharm Res. 1993;10:1662–1667. doi: 10.1023/a:1018941223967. [DOI] [PubMed] [Google Scholar]
- 25.Salvesen GS, V, Dixit M. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 26.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 27.Freeman B, Crapo J. Biology of disease: free radicals and tissue injury. Lab Invest. 1982;47:412–426. [PubMed] [Google Scholar]
- 28.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 29.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 30.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. NF-κB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Claermont P, Finkel T. Rac1 regulates a cytokine-stimulated redox-dependent pathway necessary for NF-κB activation. Mol Cell Biol. 1996;16:7115–7121. doi: 10.1128/mcb.16.12.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sulciner DJ, Yu ZX, Ferrans VJ, Gutkindm K, Irani K, Goldschmidt-Clermont P, Finkel T. Rac1 regulates reactive oxygen species generation in fibroblasts. Biochem J. 1996;318:379–382. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deshpande SS, Angkeow P, Huang J, Ozaki M, Irani K. Rac1 inhibits TNF-α-induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB J. 2000;14:1705–1714. doi: 10.1096/fj.99-0910com. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y, Suh N, Sporn M, Reed JC. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J Biol Chem. 2002;277:22320–22329. doi: 10.1074/jbc.M202458200. [DOI] [PubMed] [Google Scholar]
- 35.Perez D, White E. E1A sensitizes cells to tumor necrosis factor alpha by downregulating c-FLIPs. J Virol. 2003;77:2651–2662. doi: 10.1128/JVI.77.4.2651-2662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poukkula M, Kaunisto A, Hietakangas V, Denessiouk K, Katajamaki T, Johnson MS, Sistonen L, Eriksson JE. Rapid turnover of c-FLIP-short is determined by its unique C-terminal tail. J Biol Chem. 2005;280:27345–27355. doi: 10.1074/jbc.M504019200. [DOI] [PubMed] [Google Scholar]
- 37.Chanvorachote P, Nimmannit U, Wang L, Stehlik C, Lu B, Azad N, Rojanasakul Y. Nitric oxide negatively regulates Fas (CD95)-induced apoptosis through inhibition of ubiquitin-proteasome mediated degradation of FLIP. J Biol Chem. 2005;280:42044–42050. doi: 10.1074/jbc.M510080200. [DOI] [PubMed] [Google Scholar]
- 38.Borutaite V, Brown GC. Nitric oxide induces apoptosis via hydrogen peroxide, but necrosis via energy and thiol depletion. Free Radical Biol Med. 2003;35:1457–1468. doi: 10.1016/j.freeradbiomed.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 39.Wenzel U, Kuntz S, De Sousa UJ, Daniel H. Nitric oxide suppresses apoptosis in human colon cancer cells by scavenging mitochondrial superoxide anions. Int J Cancer. 2003;106:666–675. doi: 10.1002/ijc.11294. [DOI] [PubMed] [Google Scholar]
- 40.Conde de la Rosa L, Schoemaker MH, Vrenken TE, Buist-Homan M, Havinga R, Jansen PL, Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2005;44:918–929. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 41.Malassagne B, Ferret PJ, Hammoud R, Tulliez M, Bedda S, Trébéden H, Jaffray P, Calmus Y, Weill B, Batteux F. The superoxide dismutase mimetic MnTBAP prevents Fas-induced acute liver failure in the mouse. Gastroenterology. 2001;121:1451–1459. doi: 10.1053/gast.2001.29590. [DOI] [PubMed] [Google Scholar]
- 42.Rayner BS, Duong TT, Myers SJ, Witting PK. Protective effect of a synthetic anti-oxidant on neuronal cell apoptosis resulting from experimental hypoxia re-oxygenation injury. J Neurochem. 2006;97:211–221. doi: 10.1111/j.1471-4159.2006.03726.x. [DOI] [PubMed] [Google Scholar]
- 43.Aronis A, Andr’s Melendez J, Golan O, Shilo S, Dicter N, Tirosh O. Potentiation of Fas-mediated apoptosis by attenuated production of mitochondria-derived reactive oxygen species. Cell Death Differ. 2003;10:335–344. doi: 10.1038/sj.cdd.4401150. [DOI] [PubMed] [Google Scholar]
- 44.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, et al. Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 45.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 46.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS, Jr, Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004;18:2905–2915. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakajima A, Komazawa-Sakon S, Takekawa M, Sasazuki T, Yeh W-C, Yagita H, Okumura K, Nakano H. An antiapoptotic protein, c-FLIPL, directly binds to MKK7 and inhibits the JNK pathway. EMBO J. 2006;25:5549–5559. doi: 10.1038/sj.emboj.7601423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M, Tschopp J. The caspase-8 inhibitor FLIP promotes activation of NF-κB and Erk signaling pathways. Curr Biol. 2000;10:640–648. doi: 10.1016/s0960-9822(00)00512-1. [DOI] [PubMed] [Google Scholar]
- 49.Golks A, Brenner D, Krammer PH, Lavrik IN. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-κB activation. J Exp Med. 2006;203:1295–1305. doi: 10.1084/jem.20051556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bokoch GM. Regulation of the human neutrophil NADPH oxidase by the Rac GTP-binding proteins. Curr Opin Cell Biol. 1994;6:212–218. doi: 10.1016/0955-0674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 51.Gulbins E, Bissonette R, Mahboubi A, Nishioka W, Brunner T, Baier G, Baier-Bitterlich G, Byrd C, Lang F, Kolesnick R, et al. FAS-induced apoptosis is mediated via a ceramide-initiated RAS signaling pathway. Immunity. 1995;2:341–351. doi: 10.1016/1074-7613(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 52.Korkolopoulou P, Goudopoulou A, Voutsinas G, Kapralos EP, Patsouris E, Saetta AA. c-FLIP expression in bladder urothelial carcinomas: its role in resistance to Fas-mediated apoptosis and clinicopathologic correlations. Urology. 2004;63:1198–1204. doi: 10.1016/j.urology.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 53.Sharp DA, Lawrence DA, Ashkenazi A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem. 2005;280:19401–19409. doi: 10.1074/jbc.M413962200. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka T, Yoshimi M, Maeyama T, Hagimoto N, Kuwano K, Hara N. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur Respir J. 2002;20:359–368. doi: 10.1183/09031936.02.00252602. [DOI] [PubMed] [Google Scholar]
- 55.Medema JP, de Jong J, van Hall T, Melief CJ, Offringa R. Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J Exp Med. 1999;190:1033–1038. doi: 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinoshita H, Yoshikawa H, Shiki K, Hamada Y, Nakajima Y, Tasaka K. Cisplatin (CDDP) sensitizes human osteosarcoma cell to Fas/CD95-mediated apoptosis by down-regulating FLIP-L expression. Int J Cancer. 2000;88:986–991. doi: 10.1002/1097-0215(20001215)88:6<986::aid-ijc23>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe K, Okamoto K, Yonehara S. Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Differ. 2005;12:10–18. doi: 10.1038/sj.cdd.4401507. [DOI] [PubMed] [Google Scholar]
- 58.Day TW, Najafi F, Wu C-H, Safa AR. Cellular FLICE-like inhibitory protein (c-FLIP): a novel target for Taxol-induced apoptosis. Biochem Pharmacol. 2006;71:1551–1561. doi: 10.1016/j.bcp.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 59.Rogers KMA, Thomas M, Galligan L, Wilson TR, Allen WL, Sakai H, Johnston PG, Longley DB. Cellular FLICE-inhibitory protein regulates chemotherapy-induced apoptosis in breast cancer cells. Mol Cancer Ther. 2007;6:1544–1551. doi: 10.1158/1535-7163.MCT-06-0673. [DOI] [PubMed] [Google Scholar]