

Abstract
Aims
Traumatic brain injury is a significant cause of morbidity and mortality worldwide. An epidemiological association between head injury and long-term cognitive decline has been described for many years and recent clinical studies have highlighted functional impairment within 12 months of a mild head injury. In addition chronic traumatic encephalopathy is a recently described condition in cases of repetitive head injury. There are shared mechanisms between traumatic brain injury and Alzheimer’s disease, and it has been hypothesised that neuroinflammation, in the form of microglial activation, may be a mechanism underlying chronic neurodegenerative processes after traumatic brain injury.
Methods
This study assessed the microglial reaction after head injury in a range of ages and survival periods, from <24 hours survival through to 47 years survival. Immunohistochemistry for reactive microglia (CD68 and CR3/43) was performed on human autopsy brain tissue and assessed “blind” by quantitative image analysis. Head injury cases were compared to age matched controls, and within the traumatic brain injury group cases with diffuse traumatic axonal injury were compared to cases without diffuse traumatic axonal injury.
Results
A major finding was a neuroinflammatory response which develops within the first week and persists for several months after TBI, but has returned to control levels after several years. In cases with diffuse traumatic axonal injury the microglial reaction is particularly pronounced in the white matter.
Conclusions
These results demonstrate that prolonged microglial activation is a feature of traumatic brain injury, but that the neuroinflammatory response returns to control levels after several years.
Keywords: neurotrauma, microglia, neuroinflammation, traumatic axonal injury
Introduction
There is a considerable retrospective epidemiological literature suggesting that traumatic brain injury (TBI) is associated with an increased risk of developing Alzheimer’s disease (AD) in later life [1, 2] although not all studies have confirmed this association [3]. Data from prospective studies has also been conflicting with some studies showing an association [4] while others show no association [5]. Meta-analysis of 7 case-control studies [6] calculated a relative risk of developing AD of 1.82 for head injury with loss of consciousness, only reaching statistical significance for males. Fleminger et al. [7] studied 15 case-control studies and calculated an odds-ratio (OR) of 1.58. Again, however, this study showed that the association between head injury and AD was only statistically significant for males (males OR 2.26, females OR 0.92) the group that form the majority of the head-injured population.
Follow up of patients 10–20 years after admission to hospital with TBI provided further evidence of late stage neurodegeneration [8]. Even mild head injury (acute Glasgow Coma Score [GCS] 13–15) is associated with a higher than expected incidence of disability (Glasgow Outcome Score: moderate or severe disability) at one year post injury [9].
The data relating to repetitive head injury is more secure. Dementia pugilistica has been known for many years [10], although is a rare condition, with cerebellar and parkinsonian phenotypes being seen more commonly than overt dementia. Tau deposition is described although in a distribution that differs from AD [11], and the tau isoform is the same as seen in AD [12]. Recent studies have focused on the idea of a specific neurodegenerative condition after repetitive head injury, chronic traumatic encephalopathy (CTE) [13]. The concept of CTE has been extended beyond cognitive problems to motor disorders, with pathological TDP-43 deposition being described in 3 CTE cases and linked to an amyotrophic lateral sclerosis (ALS)- type disorder [14], although many researchers have objected to this term [15,16]. Studies looking at outcomes after a single head injury using this Glasgow cohort could not demonstrate tau deposition in acute head injury with a survival time of up to 1 month [17], but a higher incidence of tau and β-amyloid (Aβ) deposition has recently been described with survivals ranging from 1–47 years, compared with age-matched controls [18], providing further evidence of an association between TBI and AD.
Previous studies have focused attention on neuroinflammation in the form of microglial activation, as a mechanism of potential relevance to neurodegeneration both in (AD) and in the response to brain injury [19,20,21,22]. Microglial phenotypes may be modified by an external or intrinsic stimulus, and undergo morphological changes and release proteins which may be detrimental or beneficial to the surrounding brain tissue [23]. When activated the microglia can change their morphology and can express new cell surface markers, or alter expression of pre-existing markers, including the MHC class II antigen [24] and a marker of phagocytic activity, CD68 [25].
After brain injury cytokines are released activating microglia, the degree of activation reflecting the severity of the injury [26]. Microglial activation will lead to further cytokine release, including IL-1, possibly secondary to elevated levels of ATP released from damaged cells [27], with activation of purinergic P2X7 receptors on microglia [28]. IL-1 is expressed in increased quantities in the cerebral cortex within hours of TBI [29], and chronic overexpression of IL-1 is found in AD [30]. Griffin et al. [21] have proposed a “Cytokine Cycle” in which traumatic brain injury, or other forms of brain injury, can, in susceptible individuals, initiate an over-exuberant sustained inflammatory response which can result in neurodegeneration. IL-1 positive microglial cells lie in close relation to β-APP positive neurons and dystrophic neurites in the brains of head-injured patients [29] and are also found in close apposition to neurofibrillary tangle-containing neurons in AD [30]. β-APP is up-regulated in response to increased IL-1 levels, and is known to be up-regulated in AD [31,32].
In an attempt to test the hypothesis that the microglial response to head injury may persist and provoke chronic neurodegeneration, we have characterised in detail the time course and magnitude of the microglial reaction in human post mortem cases.
Materials and methods
Case selection
The study was approved by the Research Ethics Committee of the Southern General Hospital, Glasgow, Scotland. Cases were selected from the paraffin-embedded tissue archive of the Neuropathology department, Institute of Neurological Sciences, Glasgow. This is a unique archive of more than 1500 well-characterised TBI cases, with a range of survival periods. The acute (less than 12 months survival) cases used in the current study were selected from a cohort accrued between 1987–1999, and the long-term survivors were selected from the entire cohort (1968–1999). 57 cases were selected from the 1987–1999 cohort to represent the <12 months survival group, cases being selected based on completeness of clinical information and available tissue blocks. Ages ranged from 1.5–89 years, and there were 39 males and 18 females. 42 long-term survivor cases were identified from the archive again based on available clinical information and tissue availability. Ages ranged from 19–89 years and there were 37 males and 5 females. This resulted in a total of 99 TBI cases with varying survival times ranging from <24 hours up to 47 years being included in this study (Tables 1 and 2). Of the 57 cases that survived < 1 year, 45 cases had experienced a severe head injury, 34 cases defined by their admission GCS (GCS 8 or less) and 11 defined on review of the clinical history (patients died rapidly before hospital admission and pathology suggested a head injury was a major feature of the autopsy findings). In 3 cases there had been a moderate injury based on admission GCS (GCS 9- 12) and 3 cases had had a mild head injury (GCS 13–15) and died of pathology not directly related to the brain injury. In 6 of the longer survivors in the 12 month survival group there was no data relating to the GCS at time of admission although review of the clinical data again suggested a severe head injury with coma since the time of injury (survival ranging from 5 weeks to 1 year in these cases). For the 42 cases with survival beyond 1 year, admission GCS data was not available for the majority of cases. The mechanisms of injury varied with road traffic accidents (RTA) being more common in the <20 year old group, and falls more common in the >50 year old group. Post mortem interval data was available for all cases and ranged from 24–60 hours.
Table 1.
Details of cases used
| Group | n | Age range (years) | Admission GCS range | Survival range |
|---|---|---|---|---|
| Control | 20 | 18–71 | NA | NA |
| Trauma age <20 | 15 | 1.5- 19 | 3–6 | 7 hours- 5 years |
| Trauma age 20–49 | 37 | 21–49 | 3–15 | 8 hours- 22 years |
| Trauma age >50 | 47 | 50–89 | 3–14 | 7 hours- 47 years |
GCS= Glasgow Coma Scale
NA= Not Applicable
Table 2.
TBI survival times in different age groups (n=99)
| Age | 0–19 years | 20–49 years | 50+ years | |
|---|---|---|---|---|
| Survival | ||||
| <24 hours | 4 | 4 | 5 | |
| 1 day- 1 week | 4 | 5 | 4 | |
| 1 week- 1 month | 5 | 4 | 4 | |
| 1month- 1 year | 1 | 5 | 12 | |
| 1 year- 5 years | 1 | 13 | 9 | |
| >5 years | 0 | 6 | 13 | |
20 cases without TBI and with no other significant neurological impairment or neuropathological abnormality were used as controls (Table 1). Ages ranged from 18–71 years and there were 12 males and 8 females. These patients all had hospital post mortem examinations which were fully consented. Tissue was retained for diagnostic purposes with full neuropathological examination including neurohistology. The cause of death in the control cases is detailed in Table 3.
Table 3.
Details of control cases used in the neuroinflammatory study
| <20 years | 20–50 years | >50 years | ||||
|---|---|---|---|---|---|---|
| Age | Cause of death | Age | Cause of death | Age | Cause of death | |
| Non-head injured controls with no neurological disease | 18 | Leukaemia | 20 | Drug overdose | 50 | Metastatic carcinoma |
| 18 | Systemic Hodgkin’s disease | 21 | Septic shock | 55 | Gastric lymphoma PTE | |
| 24 | Drug overdose | 59 | Disseminated Langerhan’s histiocytosis | |||
| 28 | Drug overdose | 60 | Bronchial carcinoma | |||
| 33 | Ischaemic heart disease | 64 | Sarcoidosis | |||
| 35 | Malignant teratoma | 69 | Acute pyelonephritis | |||
| 43 | Hodgkin’s lymphoma | 69 | Congestive cardiac failure | |||
| 44 | Myocardial infarct | 70 | Breast carcinoma | |||
| 47 | T cell lymphoma | 71 | Pneumonia | |||
PTE= pulmonary thrombo-embolus
All cases were coded such that assessment was “blind”.
Macroscopic brain examination and sampling for histology
The brains had been fixed in 10% formal saline for 3 weeks prior to dissection after which a brain cut and histological sampling was undertaken. No brains were subjected to prolonged formalin fixation. The brains were cut at a 1-cm thickness in the coronal plane. Full macroscopic and microscopic examination was undertaken in each case. The histological sections examined from each case in this study were the parasagittal cortex including corpus callosum, internal capsule and hippocampus, all sampled at the level of the lateral geniculate body; the cerebellum including dentate nucleus; and the pons including cerebellar peduncles. This represents the minimum recommended sampling when assessing diffuse traumatic axonal injury (TAI) [43]. The tissue was processed in a VIP tissue processor (Bayer Diagnostics, Newbury, UK) using a 60-hour cycle and embedded in paraffin wax. 8 μm thick sections of the paraffin blocks were cut and stained with haematoxylin and eosin (H&E). For the assessment of TAI a monoclonal antibody against the N-terminus of the human APP molecule (clone 22C11, Böehringer, dilution 1:50) was used, and TAI was documented as being absent or present.
Immunohistochemistry for activated microglia
Immunohistochemistry was undertaken for markers of microglial activation. Sections were immunostained with anti-CD68 which binds lysosomes and therefore identifies microglia with phagocytic functions (mouse monoclonal antibody to a macrophage-specific 110 kDa glycoprotein -Dako, 1:1000) and CR3/43 which labels activated microglia (mouse monoclonal antibody to class II MHC: HLA-DR, -DQ and -DP β chains - Dako, 1:800). The antibodies were detected using the ABC kit (Vecta Stain, Vector Laboratories, Peterborough, UK) and developed with DAB. In this study counterstaining with haematoxylin was weak to allow greater sensitivity of image analysis as discussed below.
Image analysis
Image analysis was undertaken to assess the immunostaining load within defined anatomical regions in cases of TBI and to compare these with control cases. The regions of interest were the hippocampus, the inferior temporal gyrus, and the corpus callosum and cingulate gyrus at the level of the lateral geniculate body. These areas were selected as they frequently show pathological changes in TBI cases.
The morphometric study used an image analysis system consisting of a digital CCD-Camera (CoolSnap-Pro®) linking an Olympus BX 40 Light Microscope to a PC with the image analysis software (Image-Pro® Plus, Media Cybernetics). Multiple non-overlapping colour images of the area of interest were captured using x10 stage objective lens and the images were tiled together automatically to give a large composite image. Lamp intensity, digital camera set-up and calibration parameters were kept constant throughout the capture of images.
The image analysis software (Image-Pro® Plus, Media Cybernetics) used in this study allowed the definition of immunoreactive profiles based on a defined threshold (segmentation). Segmentation is a process which allows the isolation of certain colours from an image as a whole. In this study the immunoreaction was developed by diaminobenzidine (DAB) which produces a brown precipitate. A manual segmentation technique was used to isolate the brown immunoreaction in the captured images. The sections were all weakly counterstained with haematoxylin to allow greater differentiation and to permit neuroanatomical orientation. The stored images were magnified to allow greater sensitivity during the segmentation process. The number generated by image analysis (immunoload) is a reflection of the number of pixels with a colour determined by the threshold within the image analysed against the number of pixels not showing that colour pattern.
While this programme did allow the threshold setting to be applied as a constant to all images this was not done due to immunostaining intensity variation between batches of immunostaining. Therefore each slide had unique parameters set for segmentation based on the intensity of immunostaining. While this was more labour intensive it allowed greater sensitivity in the segmentation process. All results were generated by one analyst blind to clinical data to remove any inter-observer variation. To assess intra-observer variation, the same field of the same slide was analysed at the start of each session, and the load scores checked to see if they were similar. This showed less than 5% variation over a ten day period.
Data analysis
The project was designed as an observational study. For the purpose of analysis an individual’s overall immunoload was obtained for each case by averaging the available values for each anatomical region. Immunostaining load is presented as a percentage of the total area multiplied by 10. Box and whisker plots showing the minimum, lower quartile, median, upper quartile, maximum and outliers(*) were used to display the data graphically. Actual data points were superimposed (°) on the boxplots. Overall immumoloads for groups were compared using a Kruskal Wallis test with Mann Whitney pairwise comparisons if differences were identified. In the pairwise comparisons a Bonferroni correction was applied to take account of multiple comparisons. Summary statistics for the individual areas are presented. All tests were two sided. Statistical significance was taken as p<0.05. Minitab 16 was used for data analysis.
Results
CD68 and CR3/43 immunohistochemistry was undertaken on all 99 pre-selected cases for the neuroinflammation study. Microglial cells were seen with differing morphological appearances; these ranged from resting ramified cells to rounded phagocytosing amoeboid cells (Figure 1). There was considerable variation in CD68 and CR3/43 immunoreactivity within cases (Figure 2). Immunostaining was seen throughout the areas of interest. The pattern of staining was diffuse, with no specific anatomical pattern. There was no accentuation of immunostaining in association with regions described as being most susceptible to neurodegenerative pathology in CTE (subpial region, periventricular region, sulcal depths, or perivascular distribution) [13]. While post mortem interval and period of formalin fixation can influence immunohistochemical staining, no significant differences were seen in the groups studied with relation to these variables. All cases were handled in a standardised protocol with good post mortem refrigeration and relatively consistent post mortem interval of around 40 hours (range 24–60 hours). The cases with longer post mortem intervals (towards 60 hours) showed no less immunostaining compared to those cases with a shorter post mortem interval. No cases had a period of prolonged formalin fixation.
Figure 1.
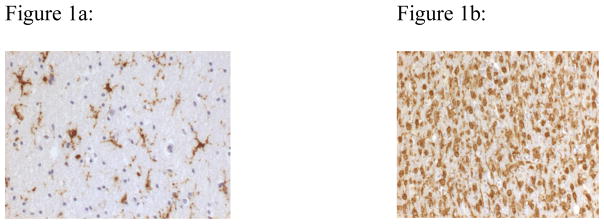
Changes in the morphological appearances of microglia, in response to stimuli. Figure 1a shows ramified microglial cells (CR3/43 immunostaining x40), while figure 1b shows amoeboid (phagocytosing) microglial cells (CD 68 immunostaining x40).
Figure 2.

Examples of the variation of immunostaining seen between cases. The left column (figures 2a, c and e) was stained with CD68; the right column (figures 2b, d and f) was stained with CR3/43. All figures are ×10.
Effects of age on immunoload in control cases (Figure 3)
Figure 3.
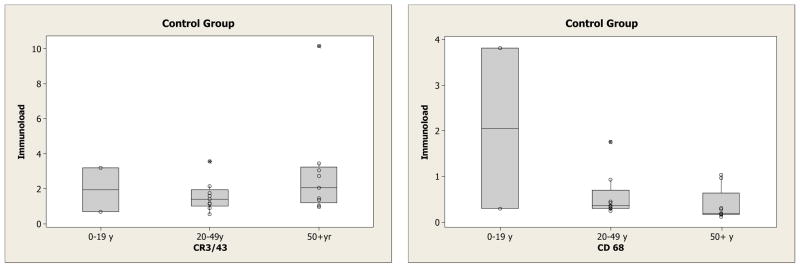
Box and whisker plots of the overall immunoloads (CR3/43 and CD68) in the control group for those <20 years (n = 2), 20–49 years (n = 9) and 50+ years (n = 9)
To examine the effect of age on overall immunoload all control cases under the age of 50 (n=11) and control cases over the age of 50 (n=9) were compared. There was no significant difference observed between the two groups for CR3/43 (median 1.41 vs 2.01, p=0.27). The median immunoload for each separate region is given in supplementary material and shows the values were higher in all regions including the temporal lobe and hippocampus, sites commonly involved in Alzheimer’s disease. For CD68 the immunoload was less pronounced although there was a significant reduction in the overall immunoload with ageing (median 0.36 vs 0.19, p=0.04).
Immunoload in Controls and TBI cases in relation to survival time (Figures 4 & 5)
Figure 4.
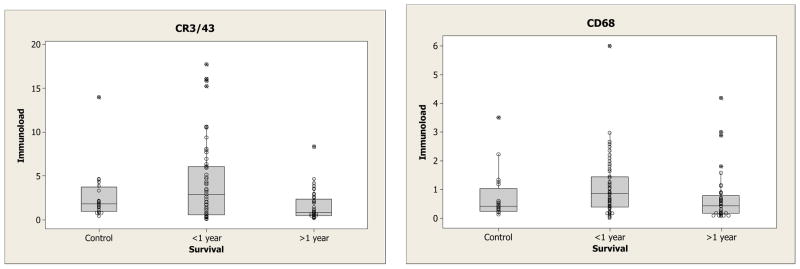
Box and whisker plots of the overall immunoload in the controls, those who survived less than one year and those who survived more than 1 year.
Figure 5.
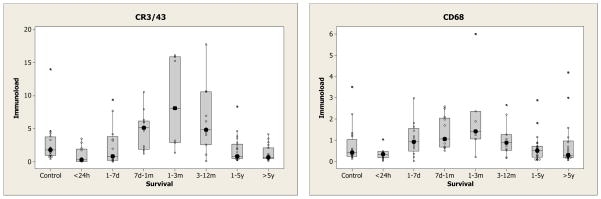
Box and whisker plots of the overall immunoload for differing survival intervals. The median immunoload is emphasised with a large solid dot to illustrate the pattern over survival time.
Firstly, inflammation markers were compared in the controls, those who survived less than one year and those who survived more than 1 year (Figure 4). Both CR3/43 (p = 0.02) and CD68 (p = 0.01) showed statistically significant differences amongst the 3 groups. With CR3/43, those who survived >1 year (median 0.8) had lower levels than control cases (median 1.8) (p = 0.05) and those who survived <1 year (median 2.9) (p = 0.04). However, there was no statistically significant difference between controls and those surviving <1 year. With CD68, those who survived >1 year had lower levels (median 0.43) than those who survived <1 year (median 0.85) (p = 0.02) but neither was significantly different from control (median 0.42). Secondly a more detailed relationship with survival is shown in Figure 5. In these boxplots the median is highlighted by large solid dot. Broadly speaking the levels go down from control in the first 24 hours then rise with a maximum around 3 months, and then fall again. This is particularly noticeable in the corpus callosum (which has the highest levels of response of all 4 areas) but is also seen in the cingulate white matter (supplementary material).
Effect of the presence of traumatic axonal injury (TAI) on immunoload (Figure 6)
Figure 6.
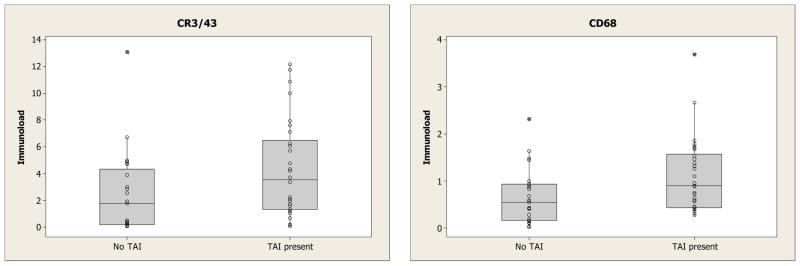
Box and whisker plots of the overall immunoload in those TBI cases who survived less than 1 year showing those with and without TAI.
Within the group of 57 TBI cases with survival <1 year, 30 had a pathological diagnosis of TAI. Image analysis of CR3/43 of TAI cases when compared to non-TAI cases showed a higher overall immunoload (median 3.5 vs 1.8, p = 0.03). Immunoload was higher in all regions (supplementary material), but particularly in regions related to the white matter. CD68 also showed a significant increase in cases of TAI (median 0.90 vs 0.55, p= 0.02) although this was less pronounced than that seen for CR3/43. Again the median immunoload was higher in all regions (supplementary material). The findings were not related to focal pathologies such as contusions, haematomas and infarcts.
Discussion
This study has shown that there is a significant up-regulation of the neuroinflammatory response as identified with CD68 immunoreactivity after TBI when compared to non-trauma controls, and that this response peaks around 3 months survival but returns to control levels after several years. Within the group of TBI cases a difference was seen between cases of TAI and cases of non-TAI with both CD68 and CR3/43 being up-regulated in the cases of TAI. As with all autopsy based studies the majority of cases were from the severe TBI group with death being an end-point. It must be recognised that this group of patients may not necessary be representative of TBI patient groups, and it is difficult to draw a direct correlation with long-term survivors of mild head injury.
Neuroinflammation in controls
Assessing the resting levels of MHC class II expression in human brains is difficult as even control (non-trauma) brains will have been subjected to some form of agonal event which may have resulted in microglial activation. A further confounding factor is the increasing realisation that inflammation within the CNS can be modified by systemic inflammatory responses [33,34]. Clinical studies of AD patients have demonstrated that impairment of cognitive function persists for up to 2 months after systemic infection and is associated with elevated serum levels of IL-1β [35]. Delerium is a well recognised consequence of systemic infection, and delirium is associated with increased microglial activation [36]. TNFα in particular may have a role in activating microglia during systemic sepsis [37]. The cases used as controls in this study were defined as having no neurological disease during life and no significant neuropathology demonstrated at autopsy. However, on review some of these cases died with pneumonia and others had systemic haematological malignancies or inflammatory conditions. It is therefore possible that some of the control cases had a neuroinflammatory response to systemic disorders such as infection and malignancy. Systemic disorders did not appear to affect the CNS in a predictable fashion such that some cases of systemic haematological malignancy were associated with increased microglial reaction while others were not. Future studies assessing the neuroinflammatory response may need to apply more stringent definitions to their control tissue.
Despite these limitations the age-matched control cases used in this study have results which suggest that MHC class II expression is normally low in the human brain and that while there is an age-related increase in MHC class II expression in temporal and hippocampal regions this did not reach statistical significance. These are regions frequently affected in AD and MHC class II expressing microglia have been described in relation to neuritic plaques [38,39] in AD, a condition predominantly associated with ageing. Phagocytic activity, as demonstrated by CD 68 showed a significant reduction with ageing.
Microglial activation in TBI
The most pronounced increase in MHC class II expression after TBI was seen in the central white matter regions (corpus callosum and cingulate gyrus) of cases which were diagnosed pathologically as having TAI. As expected, given that there would be Wallerian degeneration secondary to axonal disruption, there was an increase in phagocytic activity in these regions, although the phagocytic response was less pronounced than the MHC class II expression. These findings again focus interest on the white matter as a region of great importance in the long-term response to TBI. TUNEL positive cells, both oligodendrocytes [40] and macrophages/microglia [41], have been detected in the white matter of TBI cases many months after the injury. One interpretation of these findings is that axonal disruption may continue for many months after the initial forces associated with TBI have been applied, and that there is little, if any, axonal recovery; the neuroinflammatory response may contribute to or be secondary to this. Sulaiman et al [42], using the guinea pig stretch optic nerve model, showed on-going axonal and neuronal cell body degeneration over a 3-month period after a single stretch.
The distribution of tau pathology has been described in CTE [13], and after a single head injury with survival <12 months [17] and survival 1–47 years [18]. Tau pathology after TBI is described as involving predominantly superficial cortical layers, accumulating in the depths of sulci, and in a perivascular distribution. The distribution of activated microglia was diffuse and did not show any accentuation in these regions. This suggests that there is no direct spatial link between activated microglia and neurons expressing abnormal tau, although any potential interplay between activated microglia and neurodegenerative mechanisms that ultimately result in abnormal neuronal tau accumulation are likely to be much more complex and this observation in itself does not exclude a role for microglia in on-going TBI-related neurodegeneration.
Potential mechanisms and future studies
Microglia show considerable plasticity, and morphology may tell the investigator little about function [43]. Activated microglia may have a range of phenotypes, and these phenotypes are not fixed but will be modified by exposure to a changing local environment. Three broad macrophage phenotypes have been proposed; classically activated, wound healing and regulatory [44]. Microglia can bind amyloid fibrils via a number of receptors [45], although they are not effective at phagocytosing the fibrils, possibly as a result of co-expression of serum amyloid P (SAP) [46].
Activation of microglial cells can be produced by a variety of mechanisms. In a study of patients with a severe head injury IL-8 was noted to be markedly up-regulated acutely, with a x1000 increase in CSF when compared to peripheral blood levels [47]. The authors, therefore, postulated a role of this cytokine in initiating the neuroinflammatory response.
Cultured microglial cells have been shown to express α7 nicotinic acetylcholinergic receptors [48] and acetyl choline and nicotine pre-treatment can inhibit the lipopolysaccharide-induced microglial response. An α7 selective nicotinic antagonist can attenuate this inhibitory effect. Therefore, the intrinsic cholinergic system within the CNS may modulate the neuroinflammatory response. This system is frequently damaged in severe TBI and may result in loss of inhibition of the neuroinflammatory response [49]. A second receptor system which may modulate the microglial response is the purinergic receptor group P2X. Activation of the ionotrophic P2X7 microglial receptor by extracellular ATP increases diacylglycerol lipase activity and inhibits monoacylglycerol lipase [50]. This may result in increased microglial production of 2-arachidonoylglycerol. This molecule is currently thought to have a major role in co-ordinating the neuroinflammatory response. 2-arachidonoylglycerol, via cannabinoid receptors, can reduce excitotoxic damage by reducing glutamate release [51], limiting cerebral oedema by reducing cerebral blood flow [52], and inhibiting the production of neurotoxic agents by microglia [53]. Recently modulation of interactions between the neuronal, glial and vascular compartments of the brain by adenosine has been studied in rodent models of neuroinflammation, with inhibition of the A2A receptors (A2AR) limiting the neuroinflammatory response [54]. Clearly there are a number of potential molecular pathways that may be activated after TBI, and therefore a number of potential pharmacological targets to modulate the neuroinflammatory response. Our studies would indicate that any detrimental effect of neuroinflammation after TBI has a maximal effect around 3 months post-TBI, suggesting a time limited window for intervention if neuroinflammation is an important mechanism in long-term neurodegeneration.
β-APP is not only up-regulated in acute TBI, but there is increased intraneuronal processing of the molecule [55] potentially resulting in Aβ production and deposition. Rapid Aβ deposition has been described in fatal head injury [56], and deposition of pathological Aβ is described in long-term survivors of a single episode of TBI [18]. It has been suggested that acute brain injury may damage synapses resulting in damage associated molecular pattern molecules (DAMPs) being released, producing a pro-inflammatory phenotype, whereas neuronal apoptosis may induce an anti-inflammatory phenotype [43].
Other potential functions of MHC class II up-regulation secondary to brain injury are speculative. Microglia are usually activated prior to astrocyte activation and gliosis although both cellular responses are commonly seen in response to brain injury. Microglia may act to control the astrocyte response and to limit the degree of gliosis [57] and may be involved in regulating synaptogenesis [58]. Microglia are important to the long-term re-organisation of neuronal synaptic connections after TBI. TBI results in primary neuronal loss (ischaemia, excitotoxicity) with substantial re-organisation of the residual tissue including synaptic sprouting and synaptogenesis. Eyupoglu et al. [58] studied microglial roles in synaptogenesis in both in vivo entorhinal cortex lesion and complex organotypic entorhino-hippocampal slice cultures. Pharmacological blocking of microglial activation protected neurons from microglia-induced secondary dendritic modification and promoted useful re-innervation. Synaptic degeneration is important in neurodegeneration, although whether this is primary or secondary pathology is unknown. Microglial activation and synaptic degeneration have been described in animal models [59] although a direct causative link between these two processes has not been firmly established.
As stated above, human neuropathological studies are limited by the human tissue available. In studies of TBI most cases are at the severe end of TBI and provide little information regarding the long-term effects of mild TBI. The development of ligands which bind to the peripheral benzodiazepine receptor on up-regulated microglia will allow the study of microglial activation state in living subjects. PK11195 labels activated microglia during life [60], and expression correlates inversely with cognitive function [61]. To date no studies have been undertaken in post-TBI patients, although a single paper studying a rat model of TBI demonstrated the effectiveness of both PK11195 and DAA1106, although suggested DAA1106 had a higher specificity [62]. Such ligands are now being used to study TBI and allow a direct comparison with the current post mortem based study [63]. These studies may guide future neuropathological work and may target different anatomical sites, such as subcortical areas.
Future studies will be required to assess the potential mechanisms whereby microglia may have deleterious effects on cognition. This may be by direct effects on neuron and axons resulting in increased neuronal and axonal damage, or may be more subtle, affecting synaptic organisation and structure. Our studies suggest microglial responses are maximal around 3 months, but that they return to control levels gradually over several years. It may be that by limiting microglial activation early on after traumatic brain injury, using agents such as nicotinic or adenosine antagonists, or cannabinoids, some TBI patients may have a better long-term outcome with regard to cognitive function.
In summary
There was no significant increase in neuroinflammation with ageing in control cases.
Increased microglial activity was seen after TBI, peaking around 3 months but returning to control levels after several years.
There was increased expression of MHC class II and increased phagocytic activity in TAI cases.
Supplementary Material
Acknowledgments
This work was funded by NIH grant AG12411.
Dr C Smith was supported by a Clinical Research Fellow grant from the Scottish Council for Postgraduate Medical and Dental Education.
CS, PDL and SMG undertook the assessment and interpretation. LSM undertook all statistical analyses.
Abbreviations
- TBI
traumatic brain injury
- TAI
traumatic axonal injury
- AD
Alzheimer’s disease
Footnotes
All authors contributed to the manuscript, with CS being the lead author.
Conflict of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of Alzheimer's disease in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 2.Mayeux R, Ottman R, Tang MX, Noboa-Bauza L, Marder K, Gurland B, Stern Y. Genetic susceptability and head injury as risk factors for Alzheimer's disease among community-dwelling elderly persons and their first degree relatives. Ann Neurol. 1993;33:494–501. doi: 10.1002/ana.410330513. [DOI] [PubMed] [Google Scholar]
- 3.Broe GA, Henderson AS, Creasey H, McCusker E, Korten AE, Jorm AF, Longley W, Anthony JC. A case-control study of Alzheimer’s disease in Australia. Neurology. 1990;40:1698–1707. doi: 10.1212/wnl.40.11.1698. [DOI] [PubMed] [Google Scholar]
- 4.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JCS. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 5.Launer LJ, Anderson K, Dewey ME, Letenneur L, Ott A, Amaducci LA, Brayne C, Copeland JR, Dartigues JF, Kragh-Sorensen P, Lobo A, Martinez-Lage JM, Stijnen T, Hofman A. Rates and risk factors for dementia and Alzheimer’s disease: results from EURODEM incidence research group and work groups. European studies of dementia. Neurology. 1999;52:78–84. doi: 10.1212/wnl.52.1.78. [DOI] [PubMed] [Google Scholar]
- 6.Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Rocca WA. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM risk factors research group. Int J Epidemiol. 1991;20 (Suppl 2):S28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- 7.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74:857–62. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millar K, Nicoll JA, Thornhill S, Murray GD, Teasdale GM. Long term neuropsychological outcome after head injury: relation to APOE genotype. J Neurol Neurosurg Psychiatry. 2003;74:1047–1052. doi: 10.1136/jnnp.74.8.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thornhill S, Teasdale GM, Murray GD, McEwen J, Roy CW, Penny KI. Disability in young people and adults one year after head injury: prospective cohort study. BMJ. 2000;320:1631–1635. doi: 10.1136/bmj.320.7250.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 11.Geddes JF, Vowles GH, Nicoll JA, Révész T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt ML, Zhukareva V, Newell KL, Lee VM-Y, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001;101:518–524. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- 13.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C, Morin P, Lee HS, Kubilus CA, Daneshvar DH, Wulff M, Budson AE. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armon C, Miller RG. Correspondence regarding: TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–29. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]; J Neuropathol Exp Neurol. 2011;70:97–98. doi: 10.1097/01.JNEN.0000392910.86750.32. [DOI] [PubMed] [Google Scholar]
- 16.Bedlack RS, Genge A, Amato AA, Shaibani A, Jackson CE, Kissel JT, Wall C, King WM, Cupler E, Lou JS, Ensrud E, Tan E, Goldstein JM, Katz J, Dimachkie MM, Barohn RJ, Mozaffar T. Correspondence regarding: TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2011;70:96–97. doi: 10.1097/NEN.0b013e318204782b. [DOI] [PubMed] [Google Scholar]
- 17.Smith C, Graham DI, Murray LS, Nicoll JAR. Tau immunohistochemistry in acute brain injury. Neuropath Applied Neurobiol. 2003;29:496–502. doi: 10.1046/j.1365-2990.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- 18.Johnson VE, Stewart W, Smith DH. Widespread Tau and Amyloid-Beta Pathology Many Years After a Single Traumatic Brain Injury in Humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engel S, Schluesener H, Mittelbronn M, Seid K, Adjodah D, Wehner HD, Meyermann R. Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta Neuropathologica. 2000;100:313–322. doi: 10.1007/s004019900172. [DOI] [PubMed] [Google Scholar]
- 20.Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WS, Nicoll JA. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 21.Griffin WST, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicoll JA, Mrak RE, Graham DI, Stewart J, Wilcock G, MacGowan S, Esiri MM, Murray LS, Dewar D, Love S, Moss T, Griffin WS. Associations of Interleukin-1 gene polymorphisms with Alzheimer’s disease. Ann Neurol. 2000;47:365–368. [PMC free article] [PubMed] [Google Scholar]
- 23.Kettenmann H, Hanisch U-K, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 24.Streit WJ, Graeber MB, Kreutzberg GW. Peripheral nerve lesion produces increased levels of major histocompatibility complex antigens in the central nervous system. J Neuroimmunol. 1989;21:117–123. doi: 10.1016/0165-5728(89)90167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holness CL, Simmons DL. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood. 1993;81:1607–1613. [PubMed] [Google Scholar]
- 26.Igarashi T, Potts MB, Noble-Haeusslein LJ. Injury severity determines Purkinje cell loss and microglial activation in the cerebellum after cortical contusion injury. Exp Neurol. 2007;203:258–268. doi: 10.1016/j.expneurol.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 27.Di Virgilio F. The P2Z purinoceptor: an intriguing role in immunity, inflammation and cell death. Immunol Today. 1995;16:524–528. doi: 10.1016/0167-5699(95)80045-X. [DOI] [PubMed] [Google Scholar]
- 28.Ferrari D, Chiozzi P, Falzoni S, Hanau S, DiVirgilio Purinergic modulation of interleukin-1β release from microglial cells stimulated with bacterial endotoxin. J Exp Med. 1997;185:579–582. doi: 10.1084/jem.185.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffin WST, Sheng JG, Gentleman SM, Graham DI, Mrak RE, Roberts GW. Microglial interleukin-1α expression in human head injury: correlations with neuronal and neuritic β-amyloid precursor protein expression. Neurosci Lett. 1994;176:133–136. doi: 10.1016/0304-3940(94)90066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheng JG, Mrak RE, Griffin WST. Glial-neuronal interactions in Alzheimer’s disease: progressive association of IL-1α+ microglia and S100β + astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol. 1997;56:285–290. [PubMed] [Google Scholar]
- 31.Mrak RE, Sheng JG, Griffin WST. Correlation of astrocytic S100β expression with dystrophic neurites in amyloid plaques of Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55:273–279. doi: 10.1097/00005072-199603000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffin WST, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CD, III, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer’s disease. Proc Natl Acad Sci. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 34.Semmler A, Hermann S, Mormann F, Weberpals M, Paxian SA, Okulla T, Schäfers M, Kummer MP, Klockgether T, Heneka MT. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflammation. 2008;5:38. doi: 10.1186/1742-2094-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:788–789. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munster BC, Aronica E, Zwinderman AH, Eikelenboom P, Cunningham C, Rooij SE. Neuroinflammation in Delirium: A Postmortem Case-Control Study. Rejuvenation Res. 2011;14:615–622. doi: 10.1089/rej.2011.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Gool WA, van de Beek D, Eikelenboom P. Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet. 2010;375:773–775. doi: 10.1016/S0140-6736(09)61158-2. [DOI] [PubMed] [Google Scholar]
- 38.Haga S, Akai K, Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol. 1989;77:569–575. doi: 10.1007/BF00687883. [DOI] [PubMed] [Google Scholar]
- 39.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 40.Williams S, Raghupathi R, MacKinnon M-A, McIntosh TK, Saatman KE, Graham DI. In-situ DNA fragmentation occurs in white matter up to 12 months after head injury in man. Acta Neuropathol. 2001;102:581–590. doi: 10.1007/s004010100410. [DOI] [PubMed] [Google Scholar]
- 41.Wilson S, Raghupathi R, Saatman KE, MacKinnon MA, McIntosh TK, Graham DI. Continued in situ DNA fragmentation of microglia/macrophages in white matter weeks and months after traumatic brain injury. J Neurotrauma. 2004;21:239–250. doi: 10.1089/089771504322972031. [DOI] [PubMed] [Google Scholar]
- 42.Mohammed Sulaiman A, Denman N, Buchanan S, Porter N, Vesi S, Sharpe R, Graham DI, Maxwell WL. Stereology and ultrastructure of chronic phase axonal and cell soma pathology in stretch-injured central nerve fibers. J Neurotrauma. 2011;28:383–400. doi: 10.1089/neu.2010.1707. [DOI] [PubMed] [Google Scholar]
- 43.Perry VH, Nicoll JAR, Holmes C. Microglia in neurodegenerative disease. Nat Reviews Neurosci. 2010;6:1–9. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 44.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer's disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. 2009;87:181–194. doi: 10.1016/j.pneurobio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 46.Kalaria RN, Grahovac I. Serum amyloid P immunoreactivity in hippocampal tangles, plaques and vessels: implications for leakage across the blood-brain barrier in Alzheimer's disease. Brain Res. 1990;516:349–353. doi: 10.1016/0006-8993(90)90941-4. [DOI] [PubMed] [Google Scholar]
- 47.Kushi H, Saito T, Makino K, Hayashi N. IL-8 is a key mediator of neuroinflammation in severe traumatic brain injuries. Acta Neurochir Suppl. 2003;86:347–350. doi: 10.1007/978-3-7091-0651-8_74. [DOI] [PubMed] [Google Scholar]
- 48.Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- 49.Murdoch I, Nicoll JAR, Graham DI, Dewar D. Nucleus basalis of Meynert pathology in the human brain after fatal head injury. J Neurotrauma. 2002;19:279–284. doi: 10.1089/08977150252807018. [DOI] [PubMed] [Google Scholar]
- 50.Witting A, Walter L, Wacker J, Moller T, Stella N. P2X7 receptors control 2-arachidonoylglycerol production by microglial cells. Proc Natl Acad Sci. 2004;101:3214–3219. doi: 10.1073/pnas.0306707101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt M, López-Rodriguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz Bl. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- 52.Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci. 2002;22:9771–9775. doi: 10.1523/JNEUROSCI.22-22-09771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klegeris A, Bissonnette CJ, McGeer PL. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br J Pharmacol. 2003;139:775–786. doi: 10.1038/sj.bjp.0705304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rebola N, Simões AP, Canas PM, Tomé AR, Andrade GM, Barry CE, Agostinho PM, Lynch MA, Cunha RA. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J Neurochem. 2011;117:100–111. doi: 10.1111/j.1471-4159.2011.07178.x. [DOI] [PubMed] [Google Scholar]
- 55.Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer βA4 amyloid protein precursor. Proc Natl Acad Sci. 1992;89:10075–10078. doi: 10.1073/pnas.89.21.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's dis ease. J Neurol Neurosurg Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagata K, Takei N, Nakajima K, Saito H, Kohsaka S. Microglial conditioned medium promotes survival and development of cultured mesencephalic neurons from embryonic rat brain. J Neurosci Res. 1993;34:357–363. doi: 10.1002/jnr.490340313. [DOI] [PubMed] [Google Scholar]
- 58.Eyupoglu IY, Bechmann I, Nitsch R. Modification of microglia function protects from lesion-induced neuronal alterations and promotes sprouting in the hippocampus. FASEB J. 2003;17:1110–1111. doi: 10.1096/fj.02-0825fje. [DOI] [PubMed] [Google Scholar]
- 59.Mandolesi G, Grasselli G, Musumeci G, Centonze D. Cognitive deficits in experimental autoimmune encephalomyelitis: neuroinflammation and synaptic degeneration. Neurol Sci. 2010;31(Suppl 2):S255–259. doi: 10.1007/s10072-010-0369-3. [DOI] [PubMed] [Google Scholar]
- 60.Banati RB. Visualising microglial activation in vivo. Glia. 2002;40:206–217. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- 61.Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer's disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 62.Venneti S, Wagner AK, Wang G, Slagel SL, Chen X, Lopresti BJ, Mathis CA, Wiley CA. The high affinity peripheral benzodiazepine receptor ligand DAA1106 binds specifically to microglia in a rat model of traumatic brain injury: implications for PET imaging. Exp Neurol. 2007;207:118–127. doi: 10.1016/j.expneurol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.