

Abstract
Carbohydrates with high glycemic index are proposed to promote the development of obesity, insulin resistance and fatty liver, but the mechanism by which this occurs remains unknown. High serum glucose concentrations glucose are known to induce the polyol pathway and increase fructose generation in the liver. Here we show that this hepatic, endogenously-produced fructose causes systemic metabolic changes. We demonstrate that mice unable to metabolize fructose are protected from an increase in energy intake and body weight, visceral obesity, fatty liver, elevated insulin levels and hyperleptinemia after exposure to 10% glucose for 14 weeks. In normal mice, glucose consumption is accompanied by aldose reductase and polyol pathway activation in steatotic areas. In this regard, we show that aldose reductase deficient mice were protected against glucose-induced fatty liver. We conclude that endogenous fructose generation and metabolism in the liver represents an important mechanism whereby glucose promotes the development of metabolic syndrome.
Introduction
There is increasing evidence that excessive intake of carbohydrates may have a role in the epidemic of obesity and insulin resistance. Low carbohydrate diets have been found effective at inducing weight loss, often with improvement in hypertriglyceridemia and insulin resistance.1–5 One of the postulated mechanisms revolves around the fructose content of carbohydrate-containing foods.6–8 Fructose, present in added sugars such as sucrose and high fructose corn syrup, can induce all of the features of metabolic syndrome in animals and the metabolic effects occur independently of excessive energy intake.9 In previous studies, when rats are pair-fed either fructose or glucose as part of their diet, the fructose-fed rats show worse features of metabolic syndrome.10–11 Similarly, in humans the administration of beverages containing 25% fructose to overweight individuals also causes more visceral obesity and insulin resistance compared to subjects administered 25% glucose.12 Studies such as these suggest it is the fructose content in added sugars which may account for why sugary soft drinks are so strongly linked with the development of obesity, fatty liver, and insulin resistance.13–18
At present there is a vast literature suggesting that carbohydrates with a high glycemic index increase the risk for obesity and insulin resistance.19–20 The glycemic index relates to the ability of carbohydrates to increase plasma glucose levels following ingestion, for which glucose carries the highest level (1.0), whereas fructose has a low glycemic index (0.2).21 Whether high glycemic index carbohydrates increase the risk for insulin resistance beyond that associated with increased energy intake has been controversial.2,22 However, a recent study reported that a diet low in high glycemic carbohydrates may help maintain weight loss independently of energy intake.23
Many high glycemic foods also contain fructose. For instance, sucrose is a disaccharide of glucose and fructose, and high fructose corn syrup is a mixture of glucose and fructose, raising the question of whether fructose is responsible for the effects of high glycemic foods to increase the risk for metabolic syndrome.24 To further complicate this issue, fructose can be generated endogenously via the polyol pathway from glucose. Specifically, aldose reductase metabolizes glucose to sorbitol, which can then be converted to fructose by sorbitol dehydrogenase. Thus, if aldose reductase were highly expressed or activated in the liver, some of the glucose absorbed might be converted to fructose, and hence possibly provide a mechanism for inducing features of the metabolic syndrome. To test this hypothesis, we provided drinking water with or without glucose (10% (wt/vol)) to wild type mice or fructokinase (ketohexokinase, KHK) knockout mice for 14 weeks. We selected 14 weeks based on our previous studies on mice drinking fructose25. Parallel groups of mice were also maintained on regular chow (containing 60 % carbohydrate and no fructose).
We have previously shown that fructokinase knockout mice (KHK-A/C KO) have a normal phenotype26 and are protected from fructose-induced metabolic syndrome.25 Importantly, fructokinase does not metabolize glucose.27 In this manuscript we show that there is a significant activation of aldose reductase and the polyol pathway in the liver of mice exposed to drinking glucose (10% in water) that leads to the production of "endogenous fructose" and that the blockade of its metabolism in fructokinase deficient mice exerts protection against glucose induced fatty liver and insulin resistance
Results
Similar glucose intake in wild type and KHK-A/C-deficient mice
As shown in Figure 1, both wild type and KHK-A/C KO mice ingested similar amounts of glucose water (cumulative, Fig 1A). Both glucose-fed groups reduced their chow intake in response to the energy intake from the glucose (Fig 1B, left). However, the reduction in chow intake was not enough to compensate for the energy intake from the glucose water and hence both groups remained in positive energy balance (Fig 1B, left). Nevertheless, the KHK-A/C KO mice reduced their chow intake more and hence their total energy intake was lower than that observed in WT mice fed glucose (Fig 1B left). Measurements of urinary fructose and glucose are also included (Supplementary Fig S1A and S1B) since KHK-A/C KO mice are known to excrete increased levels of urinary fructose. However, the amount of energy loss via the urine was minor and did not alter the energy balance results.
Figure 1. Effect of Glucose consumption on Metabolic Parameters in WT and KHK A/C KO Mice.

WT mice and KHK-A/C KO mice were provided drinking water containing 10% glucose or tap water with normal mouse chow ad libitum for 14 weeks (n = 6 per group). (a) Cumulative energy intake of glucose water for 14 weeks. (b) Left. Cumulative total energy intake of chow diet with 10% glucose water or tap water for 14 weeks. Right. Total energy intake per gram body weight per day of normal chow diet with 10% glucose water or tap water for 14 weeks. (c) Growth curves of WT mice and KHK-A/C KO mice. Epididymal fat weight (d). Serum insulin (e). HOMA-R (f). Serum leptin (g). Key: HOMA-R, homeostasis model assessment for insulin resistance. Data represent means ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001 vs. respective water control. #P < 0.05. ### P < 0.001 (ANOVA, Tukey post hoc test).
As animals gain weight, their energy requirements increase thus requiring a re-evaluation of energy intake factored for weight. In this regard, both WT and KHK-A/C KO mice receiving glucose had greater total energy intake than mice drinking only water, but there was no difference between the glucose-fed WT and KHK-A/C KO mice based on calculations of daily intake as well as cumulative energy intake (Fig 1B, right).
KHK deficiency blocks metabolic syndrome in mice
WT mice drinking glucose water showed significant weight gain, visceral (epididymal) fat accumulation, hyperinsulinemia and elevated HOMA-R (homeostasis model assessment ratio), and hyperleptinemia compared to WT mice receiving water (Fig 1 C–G). KHK-A/C KO mice given glucose also gained weight, visceral fat and developed elevated leptin levels compared to KHK-A/C KO mice on regular water, although levels of insulin and HOMA-R were not different (Fig 1 C–G). Nevertheless, KHK-A/C KO mice receiving glucose had significantly less weight gain, visceral obesity, hyperinsulinemia, HOMA-R, and hyperleptinemia compared to glucose-fed WT mice (Fig 1C–1G). Of interest, despite having different insulin levels, no differences were observed between WT and KHK-A/C KO receiving glucose in insulin tolerance and hepatic glucokinase expression (Supplementary Fig S2 and S3)
WT and KHK-A/C KO mice receiving glucose showed higher glycated hemoglobin (Hb) A1C, higher total and LDL cholesterol, and lower BUN levels compared to control mice receiving regular drinking water (Supplementary Table S1). However, there were no differences in these parameters between the glucose-fed WT and KHK-A/C KO mice. Serum creatinine, triglycerides and HDL cholesterol did not differ among the 4 groups of mice.
KHK deficiency blocks glucose-induced fatty liver in mice
WT mice receiving glucose developed enlarged livers that were grossly pale; these changes were not observed in the glucose-fed KHK-A/C KO mouse (Fig 2A and B). Histologically, H&E stained sections and Oil red O staining documented the presence of steatosis (Fig 2C and Fig 2D, Supplementary Table S2). H&E staining of the livers of WT mice fed glucose water showed that macrovesicular steatosis was dominant around the portal triad (zone 1) and into the adjoining midzonal area (zone 2) whereas microvesicular lipid accumulation occured near the central vein (zone 3) and also extended into the adjoining midzonal area (Fig 2 D, Supplementary Table S2). In contrast, KHK-A/C KO animals on the high glucose diet showed neither macro- nor micro-steatosis, and similarly neither types of steatosis were observed in the control WT or KHK-A/C KO mice on tap water. The increased lipid deposition in glucose-fed WT mice was shown to be due to increased intrahepatic triglyceride but not cholesterol content (Fig 2E–F).
Figure 2. Effect of Glucose consumption on Liver histology and function.

WT mice and KHK-A/C KO mice were given 10% glucose water, or tap water for 14 weeks with normal chow diet ad libitum (n = 6 per group). (a) Liver weight. (b) Gross images of liver showing increased liver size and pale color in WT but not KHK-A/C KO mice given glucose (size bar: 1 cm). (c) Representative images of oil red O staining demonstrating increased fat accumulation (red) in WT but not KHK-A/C KO mice given glucose. (size bar 50 µM) (d) Representative images of H and E staining showing areas of macrosteatosis (white vesicles) in zone 2 and microsteatosis in zone 1 (surrounding the central vein, CV) in WT but not KHK-A/C KO mice given glucose, PT: Portal Triad. (e) Intrahepatic triglyceride levels. (size bar 50 µM) (f) Intrahepatic cholesterol levels. (g) Serum aspartate aminotransferase (AST) levels. (h) Serum alanine aminotransferase (ALT) levels. Data represent means ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001 vs. respective water control. #P < 0.05. ##P < 0.01. ### P < 0.001. (ANOVA, Tukey post hoc)
Trichrome stain analysis demonstrated a small amount of collagen deposition in the periportal sinusoids of the high glucose-fed WT animals (Supplementary Figure S4) without evidence for inflammation or other pathological alterations (Supplementary Table S2). Mild elevation in transaminase levels was also present in the glucose-fed WT mice, as noted by elevated serum aspartate aminotransferase, AST and serum alanine aminotransferase, ALT levels (Fig 2G and 2H).
Western blotting of liver tissue documented increased expression of enzymes involved in fat synthesis (ATP citrate lyase (ACL) and Fatty acid synthase (FAS)) in the glucose-fed WT mice compared to control WT mice (Figs 3A, B and Supplementary Fig S5). Glucose-fed KHK-A/C KO mice showed a lower level of ACL compared to glucose fed control mice. In addition, glucose-fed KHK-A/C KO mice had a tendency for lower FAS levels compared to glucose-fed WT mice, and also more evidence for active fatty acid oxidation, as noted by higher intrahepatic β-hydroxybutyrate levels (Fig 3C). Of interest. changes observed at the protein levels of these lipogenic genes correlated with changes of these genes at the mRNA level (Supplementary Fig S6). These studies document that glucose-induced fatty liver is almost entirely mediated by fructokinase.
Figure 3. Effect of Glucose consumption on Liver enzymes.

(a, b) Western blot of ATP citrate lyase (ACL, a) and fatty acid synthase (FAS, b). Relative intensity to β-actin in liver (n = 5), (full length image of representative western blot in Supplementary Fig S5). (c) Serum β-hydroxy butyrate concentration (n = 6). (d) Hepatic Fructose content. (e) Hepatic sorbitol content. (f) Serum fructose concentration. (g–i) Western blot analysis of aldose reductase (AR, g), KHK (h) and sorbitol dehydrogenase (SDH, i). Relative intensity to to β-actin in liver (n = 5).), (full length image of representative western blot in supplementary Fig S5). Data represent means ± s.e.m. *P < 0.05, **P < 0.01 vs. respective water control. (ANOVA, Tukey post hoc) a, P < 0.05 vs WT water by t-test.
Activation of the polyol pathway in glucose-fed mice
The observation that KHK-A/C KO mice were protected from glucose-mediated metabolic effects suggests that some of the glucose was being converted to fructose in the liver by the polyol pathway. Consistent with this hypothesis, we found elevated levels of fructose and sorbitol in the liver of WT mice given glucose water compared to regular water (Fig 3D and 3E). Serum fructose levels were also higher in glucose-fed WT mice compared to WT controls (Fig 3F). This increase in hepatic fructose and sorbitol levels was accompanied by higher expression of aldose reductase and KHK expression in WT mice by western blotting, however sorbitol dehydrogenase was not increased (Fig 3G–I and Supplementary Fig S5). Of interest, KHK-A/C KO mice showed higher aldose reductase expression, perhaps as a compensatory mechanism to the lack of KHK. In addition, KHK-A/C KO mice fed glucose had higher hepatic fructose and sorbitol levels compared to KHK-A/C KO mice on tap water.
To determine whether the injured areas corresponded with the sites of expression of the polyol pathway, we performed immunofluorescence studies employing specific antibodies. As shown in Supplementary figures S7 and S8, both AR and KHK are predominantly expressed in zones 2 and 3 in glucose-fed animals correlating with the areas of severe hepatic steatosis. However, we also observed AR and KHK up-regulation in zone 1 surrounding the portal triad,.
To evaluate the importance of the polyol pathway in the mechanism by which glucose induces hepatic steatosis, we gave 10% glucose to aldose reductase deficient mice (ARKO) for 14 weeks and analyzed intrahepatic levels of sorbitol and fructose. As shown in figure 4, similar to our previous result in WT and KHK-A/C KO mice, ARKO mice drinking glucose had greater total energy intake compared to mice drinking tap water which was no different compared to WT siblings drinking glucose (Fig 4A). Of interest, ARKO mice drinking glucose had reduced body, liver and fat weight compared to WT mice (Fig 4B, Fig4C and Supplementary Fig S9 and S10). As expected, ARKO mice did not accumulate sorbitol (Fig 4D) and fructose (Fig 4E) intrahepatically compared to WT mice receiving water indicating that the polyol pathway was responsible for the generation of endogenous fructose in mice drinking glucose. Consistent with reduced liver weight in ARKO mice drinking glucose, these mice had reduced fatty liver as denoted by lower intrahepatic triglyceride concentration, H&E staining and oil red O determination (Fig 4F and Supplementary Fig S10). Also, serum of AST and ALT, were not up-regulated in the serum of ARKO mice drinking glucose compared to WT siblings (Fig 4G and Fig 4H). Furthemore, exposure of human HepG2 cells to high glucose levels (20 mM) resulted in significant intracellular triglyceride accumulation that was prevented with an aldose reductase inhibitor, sorbinil (10 µM, Supplementary Fig S11). Of interest, and as shown in the supplementary figure, addition back of fructose blocked the inhibitory effect of sorbinil on glucose induced triglyceride accumulation in a dose dependent manner. It is important to note that aldose reducatse has already been implicated in the pathogenesis of fatty liver in diabetic animal models28–29 another condition in which the polyol pathway has been implicated.
Figure 4. Aldose reductase knockout mice do not develop glucose-induced fatty liver.

WT mice and aldose reductase knockout mice (AR KO) given ad libitum normal chow diet with 10% glucose water, or tap water for 14 weeks (n = 3–4). (a) Cumulative total energy intake of chow diet with 10% glucose water or tap water for 14 weeks. (b) Growth curves of WT mice and AR KO mice. (c) Liver weight. (d) Hepatic sorbitol content. (e) Hepatic fructose content. (f) Intrahepatic triglyceride levels. (g) Serum aspartate aminotransferase (AST) levels. (h) Serum alanine aminotransferase (ALT) levels. Data represent means ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001 vs. respective water control. #P < 0.05. ##P < 0.01. ### P < 0.001.(ANOVA, Tukey post hoc)
Role of energy intake in fructose-induced metabolic effects
These studies show that glucose-mediated obesity, visceral fat accumulation, hyperinsulinemia, hyperleptinemia and fatty liver are all dependent in part on the conversion of glucose to fructose in the liver with metabolism of the fructose by KHK. In other words, the mechanism by which glucose induces its metabolic effects is largely dependent on fructose metabolites resulting from the fructose generated from glucose by the polyol pathway.
Some of the protection that occurred in glucose-fed KHK-A/C KO mice can be attributed to better control of overall energy intake, as KHK-A/C KO mice had lower energy intake due to a greater reduction in chow intake in response to similar glucose intake. To address the relative role of energy intake in mediating the KHK-dependent effects, we performed a subset analysis by comparing glucose-fed WT mice and glucose-fed KHK-A/C KO mice that had nearly identical glucose and total energy intake (n= 4 per group, Fig 5A and B). In these mice, significant differences were still noted in the degree of fatty liver and serum insulin levels (Fig 5C–F), whereas no difference was shown in overall weight, visceral fat accumulation, or leptin levels (Supplementary Table S3). These studies suggest that glucose causes fatty liver and insulin resistance almost completely due to the conversion of glucose to fructose and that the effect of fructose to cause these metabolic effects does not require excessive energy intake. These studies are consistent with our observation that we can induce fatty liver, insulin resistance, and frank diabetes in rats with a sucrose (40%) diet even when caloric intake is reduced to 90 percent of normal intake.30
Figure 5. Analysis of Mice Matched for GLucose and Total Energy Intake.
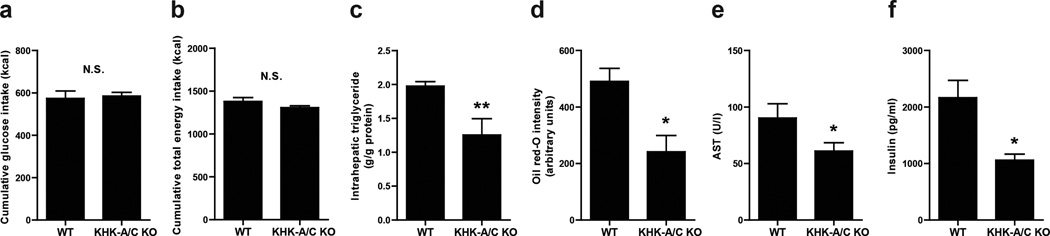
WT mice and KHK-A/C KO mice were give 10% glucose water for 14 weeks with normal chow diet ad libitum (n = 4 per group. (n = 4 per group). (a) Cumulative glucose intake (n = 4). (a) Cumulative total energy intake (n = 4). (c) Intrahepatic triglyceride levels (n = 4). (d) Quantification of oil red O staining (n = 4). (e) Serum aspartate aminotransferase (AST) levels (n = 4). (f) Serum insulin levels (n = 4). Data represent means ± s.e.m. *P < 0.05, **P < 0.01 by t-test. N.S., not significant.(ANOVA Tukey post hoc analysis)
Discussion
These studies suggest that under certain conditions glucose may induce metabolic syndrome in mice via an aldose reductase, fructose-dependent pathway. Whether this is occurring in humans ingesting high glycemic foods remains to be determined. However, as in mice, aldose reductase can be activated in humans by a number of ways, including by high glucose concentrations. Total parenteral nutrition, in which a 35% glucose solution is provided by vein, is associated with the development of fatty liver and experimental studies have shown that it is mediated by the glucose component of the infused fluids.31–32 Soft drinks containing high fructose corn syrup also deliver markedly high glucose solutions (4–6%) to the gut, and sucrose-containing soft drinks provide similar amounts of glucose after the sucrose is degraded in the intestine. Thus, soft drinks are a special problem as they provide both fructose and glucose together, in which the presence of glucose is also known to enhance fructose absorption.33 It is thus not surprising that soft drink intake is associated with increased risk for obesity, fatty liver, and insulin resistance.13–18,34–35 Furthermore, in a randomized study of overweight subjects, the administration of sugary soft drinks (1 L/d) for 6 months was associated with significantly more visceral fat, hepatic fat, and serum triglycerides compared to groups given diet soda, milk or water.36
In conclusion, the mechanism by which glucose causes weight gain, fat accumulation, fatty liver, and insulin resistance is largely due to activation of aldose reductase in the liver with conversion of glucose to fructose. While some of these effects are due to the ability of fructose to increase energy intake, the effect of fructose to induce insulin resistance and fatty liver is independent of energy intake. These studies provide new insights into how carbohydrates may cause obesity, fatty liver, and insulin resistance.
Materials and Methods
Animal study
Ketohexokinase-A and -C knockout (KHK-A/C KO) mice, which were of C57BL/6 background and were lacking both ketohexokinase-A and ketohexokinase-C, were generated as described previously37–38. KHK-A/C knockout homozygous mice and wild-type (WT) litter mates (male, 10 week-old) were used. They were maintained in temperature- and humidity-controlled specific pathogen-free condition on a 14-hour dark/10-hour light cycle, and allowed ad libitum access to normal laboratory chow (Harlan Teklad, #2920X). WT mice and KHK-A/C KO mice were assigned to control group or glucose water group (n = 6), matching mean body weight among the groups. Mice had free access to normal laboratory chow and water containing 10% glucose or tap water for 14 weeks. Glucose water was prepared by dissolving D-(+)-glucose (Sigma-Aldrich) in tap water. Body weight was measured every week, and energy intake from both normal chow and glucose water was measured 2 times per week. Urine samples were collected at 12 weeks. Mice were sacrificed after 6 hours fasting of food and glucose water. Blood were withdrawn, and tissues were taken and frozen in liquid nitrogen.
Aldose reductase knockout mice (ARKO) in the C57BL/E background were obtained from Jackson Laboratory (Bar Harbor, ME) and bred in our institution in temperature- and humidity-controlled specific pathogen-free condition on a 14-hour dark/10-hour light cycle, and allowed ad libitum access to normal laboratory chow (Harlan Teklad, #2920X). The experimental procedure was similar to the one explained above for KHK-A/C KO mice.
All experiments were conducted with adherence to the NIH Guide for the Care and Use of Laboratory Animals. The animal protocol was approved by the Animal Care and Use Committee of the University of Colorado.
Biochemical analysis
Biochemical analysis for alanine aminotransferase, aspartate aminotransferase, urea nitrogen, creatinine, total cholesterol, LDL cholesterol, HDL cholesterol, triglyceride, glucose, HbA1c and uric acid was done with an automated chemistry analyzer (VetACE Clinical Chemistry System, Alfa Wassermann Diagnostic Technologies). The serum level of leptin and insulin was determined using a mouse leptin ELISA kit and Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem) respectively. Liver fructose, sorbitol and β-hydroxybutyrate concentration was measured with EnzyChrom fructose assay kit (BioAssay Systems) and β-hydroxybutyrate and sorbitol assay kits (Biovision) respectively. Briefly, the sorbitol determination assay (biovision) is based on the conversion of sorbitol to fructose with generation of NADH (color) through a recombinant sorbitol dehydrogenase. Therefore, to avoid the effects from endogenous hepatic sorbitol dehydrogenase, liver lysates are protein cleared through a 10 kDa spin column (biovision) before analyzing sorbitol levels with the kit. In contrast, the fructose assay kit is based in its metabolism by fructose dehydrogenase, an enzyme found only in plants but not mammals. For hepatic triglyceride determination, fat was solubilized by homogenization in 1 ml solution containing 5% nonidet P40 (NP-40) in water, slowly samples were exposed to 80–100°C in a water bath for 2–5 minutes until the NP-40 became cloudy, then cooled down to room temperature. Samples were then centrifuged for 2 minutes to remove any insoluble material. Triglyceride determination with the VetAce autoanalyzer consisted in their initial breakdown into fatty acids and glycerol. Glycerol is then oxidized to generate a product which reacts with the probe to generate color at 570 nm.
Histopathology
Paraffin-embedded sections were stained with either hematoxylin and eosin (H & E) or Mallory trichrome. Histological examination was performed using the procedures described by Brunt et al.39 as modified by Kleiner et al.40 and expanded for use in rodents by Orlicky et al41. Briefly, for H & E staining, sections were deparaffined and passed dehydrated with xylenes and ethanol. Slides were thoroughly washed and dipped in hematoxylin, washed again with water and ethanol 95% and incubated with eosin for 30 minutes.nAfter incubation, sections were re-incubated with ethanol and xylenes and covered with mounting medium. For Mallory trichrome, slides were hydrated with xylenes and ethanol and stained with 1% fuchsin acid solution for 2 minutes. After incubation, slides were washed and stained with 1% phosphomolybdic acid solution, washed again and dipped into solution containing 2% Orange G, 0.5% Methyl Blue and 2% oxalic acid for 15 minutes. Slides were then washed thoroughly, dehydrated with ethanol/xylenes and mounted. Images were captured on an Olympus BX51 microscope equipped with a four megapixel Macrofire digital camera (Optronics; Goleta, CA) using the PictureFrame Application 2.3 (Optronics). Composite images were assembled with the use of Adobe Photoshop. All images in each composite were handled identically.
For oil red O staining, 0.3% Oil Red O solution (Sigma-Aldrich) was freshly prepared by mixing 0.5% Oil Red O solution dissolved in isopropanol and distilled water at a ratio of 3:2, and then filtered. Liver sections (4 µm thick) were cut with a cryostat, fixed in 10% formalin and washed with running water. Sections were rinsed with 60% isopropanol, stained with Oil Red O staining solution for 15 minutes, and then rinsed with 60% isopropanol. Finally, sections were counterstained with hematoxylin, and then examined under a light microscope. Immunostaining for AR and KHK was performed from deparaffinized sections. The sections were immunostained with the specific primary antibodies described in the text, followed by the appropriate secondary antibody labeled with either Alexa 488 or Alexa 594 (Molecular Probes, Eugene OR) and nuclei were stained with DAPI (Sigma Chemical Company, St Louis, MO).
Image Analysis and CLD Quantitation
Immunofluorescence images were captured at room temperature on a Nikon Diaphot fluorescence microscope equipped with a Cooke SensiCam CCD camera (Tonawand, NY) using Slidebook software (Intelligent Imaging Innovations Inc., Denver CO) as previously described (20). Fluorescence images were digitally deconvolved using the No Neighbors algorithm (Slidebook) and converted to TIFF files. All images were processed and assembled into montages in Photoshop (Adobe Systems Inc. Mountain View, CA). Images shown in any one montage have been taken and processed similarly to allow visual comparisons of relative protein levels in cells expressing the same protein(s).
Western blotting
Protein lysates were prepared from mouse tissue employing MAP Kinase lysis buffer as previously described42. Briefly, tissues (approx 50 mg) were homogenized in 500 µl of buffer containing 0.5% triton X-100, 2 mM MgCl2, 1 mM EGTA and 1 mM DTT supplemented with protease and phosphatase inhibitors (Roche), samples were then incubated on ice for 30 minutes with occasional vortex and spin it down at 13000 rpm fppr 15 minutes at 4°C. Supernatant was collected and content determined by the BCA protein assay (Pierce). 50 µg of total protein was loaded per lane for SDS-PAGE (10% w/v) analysis and then transferred to PVDF membranes. Membranes were incubated with primary antibodies (all of them at a 1:1000 dilution) (AR, custom made raised in rabbit; SDH Proteintech 15881-1-AP; KHK, Sigma HPA007040; ACL, Cell Signaling 4332S; FAS, Cell signaling 3180S) and visualized using a horseradish peroxidase secondary antibody(1:2000) and the HRP Immunstar® detection kit (Bio-Rad, Hercules, CA). Chemiluminescence was recorded with an Image Station 440CF and results analyzed with the 1D Image Software (Kodak Digital Science, Rochester, NY).
Cultured cells
Human-derived hepatocyte cell line HepG2 cells were grown o 80% confluency in RPMI-1640 medium (invitrogen) supplemented with 10% fetal calf serum and antibiotics. Once, cells reached the confluency they were exposed to glucose (20 mM) for three days with fresh changes of medium twice daily in the presence or absence of sorbinil (10 µM, kindly provided by Pfizer Central Research, New York, NY). After 72 hours, cells were lysed with 5% NP40 and exposed to 80–100°C in a water bath for 2–5 minutes until the NP-40 became cloudy, then cooled down to room temperature. Samples were then centrifuged for 2 minutes to remove any insoluble material and triglyceride concentration determined with the VetAce autoanalyzer.
Statistical analysis
All data are presented as the mean ± s.e.m. Independent replicates for each data point (n) are identified in figure legends. Data graphics and statistical analysis were performed using Prism 5 (GraphPad). Data without indications were analyzed by one-way ANOVA, Tukey post hoc test. P < 0.05 was regarded as statistically significant.
Supplementary Material
Acknowledgments
We thank Erin Genova and Ashley Bassett from the Department of Pathology at the University of Colorado for their work with the histology. This work was supported by Grants HL-68607 and RC4 DK090859-01 (to R.J.J) and Grant 1K01DK095930-01 (to M.A.L.) from National Institutes of Health, Diabetes UK (United Kingdom) Grant RD04/0002833, and startup funds from the University of Colorado.
Conflict of interest: RJ has received research grants from Amway and Danone and is on the Scientific Advisory Board of Amway. Dr Johnson also has authored two lay books on sugar (fructose) and its role in obesity and metabolic syndrome (The Sugar Fix, Rodale, 2008 and The Fat Switch, mercola.com, 2012).
Footnotes
Author contributions
Author contributions: M.A.L., T.I., Y.Y.S. and R.J.J. designed research; M.A.L., T.I., N.L., C.C., D.J.O., P.R., C.R., S.I., C.A.R.J., E.S.B., and T.K., performed research; C.P.D., A.A., J.M.P., T.K., S.M., L.G.S.-L., J.M., and D.T.B. contributed new reagents/analytic tools; M.A.L., T.I, D.O.J.., and R.J.J. analyzed data; and M.A.L., T.I., Y.Y.S., and R.J.J. wrote the paper.
References
- 1.Atkins R. Dr. Atkins’ New Diet Revolution. Avon Books; 1998. [Google Scholar]
- 2.Ebbeling CB, Leidig MM, Feldman HA, Lovesky MM, Ludwig DS. Effects of a low-glycemic load vs low-fat diet in obese young adults: a randomized trial. Jama. 2007;297:2092–2102. doi: 10.1001/jama.297.19.2092. [DOI] [PubMed] [Google Scholar]
- 3.Gardner CD, et al. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA. 2007;297:969–977. doi: 10.1001/jama.297.9.969. [DOI] [PubMed] [Google Scholar]
- 4.Dansinger ML, Gleason JA, Griffith JL, Selker HP, Schaefer EJ. Comparison of the Atkins, Ornish, Weight Watchers, and Zone diets for weight loss and heart disease risk reduction: a randomized trial. JAMA. 2005;293:43–53. doi: 10.1001/jama.293.1.43. [DOI] [PubMed] [Google Scholar]
- 5.Foster GD, et al. Weight and metabolic outcomes after 2 years on a low-carbohydrate versus low-fat diet: a randomized trial. Annals of internal medicine. 2010;153:147–157. doi: 10.1059/0003-4819-153-3-201008030-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tappy L, Le KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev. 2010;90:23–46. doi: 10.1152/physrev.00019.2009. [DOI] [PubMed] [Google Scholar]
- 7.Havel PJ. Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutrition reviews. 2005;63:133–157. doi: 10.1301/nr.2005.may.133-157. [DOI] [PubMed] [Google Scholar]
- 8.Johnson RJ, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. The American journal of clinical nutrition. 2007;86:899–906. doi: 10.1093/ajcn/86.4.899. [DOI] [PubMed] [Google Scholar]
- 9.Johnson RJ, et al. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocr Rev. 2009;30:96–116. doi: 10.1210/er.2008-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagawa T, et al. A causal role for uric acid in fructose-induced metabolic syndrome. American journal of physiology. 2006;290:F625–F631. doi: 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 11.Gersch MS, et al. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. American journal of physiology. 2007;293:F1256–F1261. doi: 10.1152/ajprenal.00181.2007. [DOI] [PubMed] [Google Scholar]
- 12.Stanhope KL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. The Journal of clinical investigation. 2012;119:1322–1334. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludwig DS, Peterson KE, Gortmaker SL. Relation between consumption of sugar-sweetened drinks and childhood obesity: a prospective, observational analysis. Lancet. 2001;357:505–508. doi: 10.1016/S0140-6736(00)04041-1. [DOI] [PubMed] [Google Scholar]
- 14.Schulze MB, et al. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. Jama. 2004;292:927–934. doi: 10.1001/jama.292.8.927. [DOI] [PubMed] [Google Scholar]
- 15.Ouyang X, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. 2008;48:993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu FB, Malik VS. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: epidemiologic evidence. Physiology & behavior. 2010;100:47–54. doi: 10.1016/j.physbeh.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malik VS, Popkin BM, Bray GA, Despres JP, Hu FB. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation. 2010;121:1356–1364. doi: 10.1161/CIRCULATIONAHA.109.876185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malik VS, et al. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes care. 2010;33:2477–2483. doi: 10.2337/dc10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ludwig DS. The glycemic index: physiological mechanisms relating to obesity, diabetes, and cardiovascular disease. JAMA. 2002;287:2414–2423. doi: 10.1001/jama.287.18.2414. [DOI] [PubMed] [Google Scholar]
- 20.Willett W, Manson J, Liu S. Glycemic index, glycemic load, and risk of type 2 diabetes. The American journal of clinical nutrition. 2002;76:274S–280S. doi: 10.1093/ajcn/76/1.274S. [DOI] [PubMed] [Google Scholar]
- 21.Jenkins DJ, et al. Glycemic index of foods: a physiological basis for carbohydrate exchange. The American journal of clinical nutrition. 1981;34:362–366. doi: 10.1093/ajcn/34.3.362. [DOI] [PubMed] [Google Scholar]
- 22.Liese AD, et al. Dietary glycemic index and glycemic load, carbohydrate and fiber intake, and measures of insulin sensitivity, secretion, and adiposity in the Insulin Resistance Atherosclerosis Study. Diabetes care. 2005;28:2832–2838. doi: 10.2337/diacare.28.12.2832. [DOI] [PubMed] [Google Scholar]
- 23.Ebbeling CB, et al. Effects of dietary composition on energy expenditure during weight-loss maintenance. JAMA. 2012;307:2627–2634. doi: 10.1001/jama.2012.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segal MS, Gollub E, Johnson RJ. Is the fructose index more relevant with regards to cardiovascular disease than the glycemic index? European journal of nutrition. 2007;46:406–417. doi: 10.1007/s00394-007-0680-9. [DOI] [PubMed] [Google Scholar]
- 25.Ishimoto T, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4320–4325. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diggle CP, et al. Both isoforms of ketohexokinase are dispensable for normal growth and development. Physiol Genomics. 2010;42A:235–243. doi: 10.1152/physiolgenomics.00128.2010. [DOI] [PubMed] [Google Scholar]
- 27.Van den Berghe G. Fructose: metabolism and short-term effects on carbohydrate and purine metabolic pathways. Progress in biochemical pharmacology. 1986;21:1–32. [PubMed] [Google Scholar]
- 28.Qiu L, et al. Aldose reductase regulates hepatic peroxisome proliferator-activated receptor alpha phosphorylation and activity to impact lipid homeostasis. J Biol Chem. 2008;283:17175–17183. doi: 10.1074/jbc.M801791200. [DOI] [PubMed] [Google Scholar]
- 29.Qiu L, et al. Inhibition of aldose reductase activates hepatic peroxisome proliferator-activated receptor-alpha and ameliorates hepatosteatosis in diabetic db/db mice. Exp Diabetes Res. 2012;2012:789730. doi: 10.1155/2012/789730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roncal-Jimenez CA, et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism: clinical and experimental. 2011;60:1259–1270. doi: 10.1016/j.metabol.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guglielmi FW, et al. Total parenteral nutrition-related gastroenterological complications. Dig Liver Dis. 2006;38:623–642. doi: 10.1016/j.dld.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Nishimura M, Yamaguchi M, Naito S, Yamauchi A. Soybean oil fat emulsion to prevent TPN-induced liver damage: possible molecular mechanisms and clinical implications. Biol Pharm Bull. 2006;29:855–862. doi: 10.1248/bpb.29.855. [DOI] [PubMed] [Google Scholar]
- 33.Ushijima K, Riby JE, Fujisawa T, Kretchmer N. Absorption of fructose by isolated small intestine of rats is via a specific saturable carrier in the absence of glucose and by the disaccharidase-related transport system in the presence of glucose. The Journal of nutrition. 1995;125:2156–2164. doi: 10.1093/jn/125.8.2156. [DOI] [PubMed] [Google Scholar]
- 34.Brownell KD, et al. The public health and economic benefits of taxing sugar-sweetened beverages. The New England journal of medicine. 2009;361:1599–1605. doi: 10.1056/NEJMhpr0905723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vartanian LR, Schwartz MB, Brownell KD. Effects of soft drink consumption on nutrition and health: a systematic review and meta-analysis. American journal of public health. 2007;97:667–675. doi: 10.2105/AJPH.2005.083782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maersk M, et al. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: a 6-mo randomized intervention study. The American journal of clinical nutrition. 2012;95:283–289. doi: 10.3945/ajcn.111.022533. [DOI] [PubMed] [Google Scholar]
- 37.Diggle CP, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem. 2009;57:763–774. doi: 10.1369/jhc.2009.953190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trinh CH, Asipu A, Bonthron DT, Phillips SE. Structures of alternatively spliced isoforms of human ketohexokinase. Acta Crystallogr D Biol Crystallogr. 2009;65:201–211. doi: 10.1107/S0907444908041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunt EM. Grading and staging the histopathological lesions of chronic hepatitis: the Knodell histology activity index and beyond. Hepatology. 31:241–246. doi: 10.1002/hep.510310136. [DOI] [PubMed] [Google Scholar]
- 40.Kleiner DE, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2000;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 41.Orlicky DJ, et al. Chronic ethanol consumption in mice alters hepatocyte lipid droplet properties. Alcohol Clin Exp Res. 2011;35:1020–1033. doi: 10.1111/j.1530-0277.2011.01434.x. 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanaspa MA, et al. The tight junction protein, MUPP1, is up-regulated by hypertonicity and is important in the osmotic stress response in kidney cells. Proc Natl Acad Sci U S A. 2007;104:13672–13677. doi: 10.1073/pnas.0702752104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.