

Abstract
MEKK3 is a conserved Ser/Thr protein kinase belonging to the MAPK kinase kinase (MAP3K) family. MEKK3 is constitutively expressed in T cells, but its function in T cell immunity has not been fully elucidated. Using Mekk3 T cell conditional knockout (T-cKO) mice, we show that MEKK3 is required for T cell immunity in vivo. Mekk3 T-cKO mice had reduced T cell response to bacterial infection and were defective in clearing bacterial infections. The Ag-induced cytokine production, especially IFN-γ production, was impaired in Mekk3-deficient CD4 T cells. The TCR-induced ERK1/2, JNK, and p38 MAPKs activation was also defective in Mekk3-deficient CD4 T cells. In vitro, MEKK3 is not required for Th1 and Th2 cell differentiation. Notably, under a nonpolarizing condition (Th0), Mekk3 deficiency led to a significant reduction of IFN-γ production in CD4 T cells. Furthermore, the IL-12/IL-18–driven IFN-γ production and MAPK activation in Mekk3-deficient T cells was not affected suggesting that MEKK3 may selectively mediate the TCR-induced MAPK signals for IFN-γ production. Finally, we found that MEKK3 activation by TCR stimulation requires Rac1/2. Taken together, our study reveals a specific role of MEKK3 in mediating the TCR signals for IFN-γ production.
Mitogen-activated protein kinases, including the ERK, JNK, and p38 MAPKs, are key intracellular signaling molecules used by eukaryotic cells to transduce a wide spectrum of extracellular signals (1–3). MAPKs play important roles in immune responses and regulate immune cell development, activation, differentiation, and survival. MAPK is activated through a cascade that also includes a MAPK kinase (MAP2K) and a MAP2K kinase (MAP3K). Upon stimulation of immune receptors or proinflammatory cytokine receptors, the MAPK pathways are rapidly induced, leading to the expression of genes that are essential for both the innate and adaptive immune responses (4–7). However, precisely how MAPKs are specifically induced and the physiological roles of individual MAPK cascades in T cell immunity remain to be fully elucidated.
MEKK3 is a member of the MAP3K superfamily and shares substantial homology in the kinase domain with other members in this family (8–10). Biochemical studies showed that MEKK3 is able to activate multiple MAPKs including the JNKs, ERK1/2, p38, and ERK5 MAPKs in vitro under certain conditions (11, 12). Mouse gene knockout studies showed that MEKK3 is essential for embryonic cardiovascular development, a function that cannot be compensated by its closest homologue MEKK2 (10, 13). Activation of MEKK3, like MEKK2, requires a dimerization-induced autophosphorylation on a conserved serine residue in its activation loop (14, 15). MEKK3 is involved in the proinflammatory cytokine- and TLR-induced JNK and p38 MAPK activation and is also required for IKK–NF-κB activation (16–19). Several studies, including two recent reports, suggest that MEKK3 may cooperate with another MAP3K, TAK1, and act downstream of E3 ubiquitin ligase TRAF6 in mediating the proinflammatory signals for MAPK and IKK–NF-κB activation (19–22). MEKK3 is constitutively expressed in both innate and adaptive immune cells, but little is known about the role of MEKK3 in adaptive immune responses.
Using a Mekk3 T cell conditional knockout (T-cKO) mouse line generated recently in our laboratory, we reported that MEKK3 is dispensable for thymic T cell development on the C57BL/6 background. However, the peripheral T cell homeostasis is impaired in the Mekk3 T-cKO mice (23). Similar phenotype in peripheral T cells homeostasis was also reported by another group with independently generated Mekk3 T-cKO mice (24).
To understand further the physiological role of MEKK3 in T cell-mediated adaptive immunity, we studied the function of Mekk3-deficient T cells and examined how MEKK3 deficiency affects the Ag-induced cytokine production, anti-bacteria immunity, and signal transduction pathways. Our results demonstrate that MEKK3 is required for mounting optimal T cell responses in vivo and is involved in mediating the TCR-dependent Rac1/2 signals for IFN-γ production through the MAPK pathways.
Materials and Methods
Mice
Mekk3 T-cKO mice were described previously (23). All mice were on C57BL/6 background and used between 6 and 12 wk of age. Our mice were housed in a pathogen-free animal care facility at M.D. Anderson Cancer Center (Houston, TX) or Yale University (New Haven, CT). All mouse experiments were approved by the Institutional Animal Care and Use Committees of the University of Texas M.D. Anderson Cancer Center and Yale University.
Immunization
Eight-week-old mice were used for the immunization. A mixture of 1 mg/ml OVA (Sigma-Aldrich, St. Louis, MO) and 1 mg/ml CFA (Pierce, Rockford, IL) in PBS was vortexed for 4 h at room temperature before being used for injection. Each mouse received one single s.c. injection of 100 μl OVA–CFA mixture at the base of the tail. Nine days later, total spleen cells were restimulated with different concentrations of OVA257–264, SIINFEKL (OT1p), or OVA323–339 (ISQAVHAAHAEINEAGR) (OT2p) peptides. Twenty-four hours after restimulation, total cells were analyzed by intracellular FACS staining, and the supernatants were collected to measure the production of IL-4, IL-10, IFN-γ, and IL-2 by ELISA.
Listeria monocytogenes infection
To measure bacteria counts in infected mice, 8- to 10-wk-old mice were infected i.v. with 5 × 105 CFU L. monocytogenes per mouse in 100 μl PBS at day 1 and sacrificed at day 10. Livers were homogenized and resuspended in 5 ml PBS, and lysates were then diluted and inoculated onto brain heart infusion agar plate (BD Biosciences). L. monocytogenes-OVA (a gift from Kimberly Schluns, M.D. Anderson Cancer Center) (25) were used to test Ag-specific T cell response. Each mouse was injected with 50,000 CFU L. monocytogenes-OVA in 100 μl PBS. Seven days after injection, blood was taken from the mice by orbital bleeding. Blood lymphocytes were stained with an OT1-tetramer Ab for the CD8 response (26). Mice were sacrificed at day 10, and total spleen cells were restimulated with different concentrations of OVA, OT1p, OT2p, or 12-O-tetradecanoyl phorbol-13-acetate (TPA) plus ionomycin. Intracellular staining of IL-4 and IFN-γ was performed at 6 h (for TPA stimulation) and 24 h (for OVA stimulation) after restimulation. GolgiStop (BD Biosciences, San Jose, CA) was added during the last 4 h of stimulation. Twenty-four hours after restimulation, supernatant was collected for assaying the production of IL-4, IL-10, IFN-γ, and IL-2 by ELISA.
Cell preparation, staining, and flow cytometry
Lymphocytes were isolated from thymi, spleens, and lymph nodes from 6-to 12-wk-old mice. Tissues were mechanically disrupted to obtain single-cell suspension. RBCs in the spleen were lysed in RBC Lysis Buffer (Sigma-Aldrich) at room temperature for 3 min. Cells were then labeled with anti-CD4–coated magnetic microbeads (Miltenyi Biotec, Auburn, CA). The labeled cells were separated by Auto-MACS (Miltenyi Biotec) according to the manufacturer’s instructions. The enriched CD4+ cells were stained with anti-CD4–FITC, anti-CD62L–allophycocyanin, and anti-CD44–PE (BD Biosciences). CD4+CD62L+CD44low naive T cells were sorted on a BD FACSAria cell sorter. For intracellular staining of cytokines, in vitro-differentiated CD4 T cells were stimulated with TPA (50 ng/ml)/ionomycin (500 ng/ml) or anti-CD3 Ab (1 μg/ml) for 6 h in the presence of GolgiStop (BD Biosciences). Cells were first stained for cell surface markers, followed by fixation and permeabilization using Fix/Perm Kit (BD Biosciences). Cells were then stained with anti–IL-4–PE (1:100; eBioscience, San Diego, CA), and/or anti–IFN-γ–allophycocyanin (1:200; eBioscience) thereafter, washed two more times with FACS buffer, and fixed in 1% paraformaldehyde in PBS before being analyzed with an LSR II FACS machine (BD Biosciences). Data were collected on an LSR II Flow Cytometer (BD Biosciences) using FACSDiva software (BD Biosciences). Postacquisition analysis was performed with FlowJo software (Tree Star, Ashland, OR).
T cell differentiation
Total splenocytes were incubated with anti-CD4–coated magnetic microbeads, and the CD4+ cells were then separated by Auto-MACS (Miltenyi Biotec) according to the manufacturer’s instructions. The enriched CD4+ cells were stained with anti-CD4–FITC, anti-CD62L–allophycocyanin, and anti-CD44–PE Abs (BD Biosciences), respectively. CD4+CD62L+CD44low naive T cells were sorted by a BD FACSAria cell sorter. Purified naive CD4 T cells were then stimulated with plate-bound anti-CD3 (1 μg/ml) plus 1 μg/ml anti-CD28 in 24-well plates in the presence of 50 U/ml rIL-2 (Peprotech, Rocky Hill, NJ) in T cell medium (RPMI 1640, 10% FBS, 1× antibiotics [Sigma, St. Louis, MO], and 50 μM 2-ME). For Th1 differentiation, anti–IL-4 (5 μg/ml; Pierce) and IL-12 (10 ng/ml; Peprotech) were added to the culture. For Th2 differentiation, anti–IFN-γ (5 μg/ml; Pierce) and IL-4 (10 ng/ml; Peprotech) were added to the culture. Cells were cultured for 7 d before harvesting for immunoblotting analysis.
T cell stimulation and proliferation
Thymocytes or purified naive CD4 T cells were stimulated with different concentrations of plate-bound anti-CD3 (clone 145-2C11). Cells were stimulated in 96-well plates at 2 × 105 cells/well in T cell medium (RPMI 1640, 10% FBS, 1× antibiotics [Sigma], and 50 μM 2-ME). Anti-CD28 (clone 37.51) was added to the medium at 1 μg/ml when indicated. The plates were incubated in a 37°C CO2 incubator. Supernatant was collected at 24 h to measure the production of IL-2 by ELISA using an ELISA kit (Pierce) according to the manufacturer’s protocol. For proliferation assay, cells were pulsed with 1 μCi [3H]thymidine (PerkinElmer, San Jose, CA) per well during the last 8 h of a 72-h culture period. The cells were then harvested with a Tomtec cell harvester and counted with a β-plate liquid scintillation counter. All results are expressed as mean ± SD of triplicate cultures.
Immunoblot analysis
Purified naive or effector CD4 T cells were stimulated with plate-bound anti-CD3 (5 μg/ml), or TPA (20 ng/ml)/ionomycin (100 ng/ml), or IL-12 (2 ng/ml)/IL-18 (20 ng/ml) for the indicated time periods. To inhibit the activity of Rac1/2, T cells were pretreated with NSC23766 or its analogue (Raci-analogue) (Dr. Yi Zheng, Children’s Hospital Research Foundation, Cincinnati, OH) at 50 μM for 2 h before stimulation. Cells were then lysed in lysis buffer (25 mM Tris-HCI pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM PMSF, 1 mM Na3VO4, and protease inhibitor mixture [Roche, Nutley, NJ]). Total cell lysates were resolved by 8 or 10% SDS-PAGE gels and transferred to an Immobilon-P membrane (Millipore, Billerica, MA). Membrane was blocked in 5% nonfat dry milk in TBS with 0.1% Tween 20, followed by blotting with anti-MEKK3 Ab (BD Biosciences), anti–phospho-p38, anti–phospho-ERK1/2, anti–phospho-JNK, anti–phospho-IKKα/β, anti–phospho-IκBα (all from Cell Signaling, Beverly, MA), or blotted with anti-ERK2, anti-p38, anti–T-bet, anti-IκBα (Santa Cruz Biotechnology, Santa Cruz, CA). Integrated OD values of bands were measured by ImagePro Plus 5.0 software.
Results
MEKK3 is required for IFN-γ production in vitro and in vivo in T cells
To investigate the in vivo role of MEKK3 in T cell-mediated immune responses, we immunized Lck-Cre-Mekk3flox/+, designated as normal control littermate (NCL) or f/+ mice, and Lck-Cre-Mekk3flox/−, designated as Mekk3 T-cKO or f/− mice, with OVA plus CFA. The Mekk3 T-cKO mice and NCL mice were described in our recent studies (23). Nine days after immunization, total splenocytes were isolated and restimulated with OVA or with OT2p (for CD4 T cells) peptide to determine the Ag-specific T cell responses. As shown in Fig. 1A, stimulation of T cells with OVA or OT2p induced robust production of IFN-γ, IL-2, and IL-10 in the control splenocytes from NCL mice. However, the Ag-induced IFN-γ production was dramatically reduced in the splenocytes from the Mekk3 T-cKO mice. Although it was less dramatic compared with the IFN-γ production, the Ag-induced production of IL-2 and IL-10 was also significantly reduced in the splenocytes from the Mekk3 T-cKO mice compared with that from the control mice. These data show that the Mekk3 T-cKO mice have a defective T cell Ag recall response.
FIGURE 1.

MEKK3 is required for CD4 T cell IFN-γ production. A, NCL (f/+) and Mekk3 T-cKO (f/−) mice (three mice per group) were s.c. immunized with OVA plus CFA. Nine days after immunization, spleen cells were stimulated with the indicated concentration of OVA or OVA323–339 (OT2p) peptide. Cytokine production was measured by ELISA. B and C, Naive CD4 T cells sorted from NCL (f/+) and Mekk3 T-cKO (f/−) mice were stimulated with plate-bound anti-CD3 (B) or together with 1 μg/ml soluble anti-CD28 (C). Cell proliferation was measured by [3H]thymidine incorporation. Production of IL-2 and IFN-γ was measured by ELISA. D and E, Thymocytes from NCL (f/+) and Mekk3 T-cKO (f/−) mice were stimulated with plate-bound anti-CD3 at the indicated concentration (D) or together with 1 μg/ml soluble anti-CD28 (E). Cell proliferation was measured by [3H]thymidine incorporation. Production of IL-2 and IFN-γ was measured by ELISA. Data shown are representative of three independent experiments.
We showed previously that the T cell number in the spleens of Mekk3 T-cKO mice was reduced compared with that in control mice (23). Therefore, the deficiency in cytokine production observed above could be due to either the reduced T cell number or an intrinsic T cell defect in response to Ag stimulation, or both. Consistently, there were fewer CD4 and CD8 T cells in the spleens of immunized Mekk3 T-cKO mice than that in the control mice (data not shown). To determine if the reduction of cytokine production in the splenocytes of Mekk3 T-cKO mice might also be due to an intrinsic defect of Mekk3-KO T cells in response to Ag stimulation, we isolated naive CD4 T cells (CD44lowCD62Lhigh) by FACS sorting from NCL and Mekk3 T-cKO mice. The same number of purified T cells was stimulated with an anti-CD3 Ab alone or together with an anti-CD28 Ab (Fig. 1B, 1C). As expected, stimulation of the NCL T cells resulted in robust T cell proliferation and IFN-γ and IL-2 production. Although the TCR-induced T cell proliferation was unaffected in Mekk3-KO T cells, which is consistent with the results from our recent report (23), the IFN-γ production by Mekk3-KO T cells was dramatically reduced (Fig. 1B, 1C). The IL-2 production by Mekk3-KO T cells was also reduced but was not as severe as that of IFN-γ production. These results indicate that MEKK3 is required not only for T cell homeostasis but also for Ag-induced IFN-γ production.
To confirm further the cell intrinsic role of MEKK3 in IFN-γ production, we also stimulated Mekk3 T-cKO thymocytes with a plate-bound anti-CD3 Ab alone or together with an anti-CD28 (Fig. 1D, 1E). As expected, no cell proliferation defect was observed in Mekk3-deficient thymocytes. However, the IFN-γ production was still defective in Mekk3-deficient thymocytes.
MEKK3 is required for the clearance of Listeria infection
To investigate the in vivo role of MEKK3 in T cell-mediated immune responses, we examined whether the Mekk3 T-cKO mice were able to mount an efficient anti-bacteria immune response. Previous studies have established a critical role of IFN-γ in anti-Listeria immune responses in mice (27). Therefore, we examined whether Mekk3 T-cKO mice were more or less susceptible to L. monocytogenes infection. When NCL and Mekk3 T-cKO mice were infected with 5 × 105 CFU L. monocytogenes per mouse, a lethal dose for around 50% of the infected mice, we found that the same percentages of mice were killed in both groups and the dynamics of lethality was also similar between the NCL and Mekk3 T-cKO mice (data not shown). These results suggest that MEKK3 in T cells was not required for the protection of early bacterial infection-induced lethality. However, when we immunized NCL and Mekk3 T-cKO mice with a sublethal dose (5 × 104 CFU per mouse) of a recombinant L. monocytogenes strain that expressed OVA (L. monocytogenes-OVA), we found that whereas 80% of the infected NCL mice (7 of 9) cleared bacteria in their livers 10 d postinfection, only 33% of Mekk3 T-cKO mice (2 of 6) were able to clear the bacteria (Fig. 2A). Importantly, the number of bacteria in the livers of Mekk3 T-cKO mice was two-log higher than that in the NCL mice. These results indicate that MEKK3 is important for the T cell-mediated anti-bacteria immunity.
FIGURE 2.

MEKK3 is required for clearance of L. monocytogenes-OVA infection. A, Nine NCL (f/+) and six Mekk3 T-cKO (f/−) mice were immunized with 5 × 104 CFU L. monocytogenes-OVA per mouse by tail vein injection. Livers from Listeria-infected mice were homogenized in 5 ml PBS. The lysates were series diluted and inoculated onto brain heart infusion agar plate for calculation of bacteria concentration. The bacteria numbers in the livers are shown as numbers of bacteria per milligram of liver tissue and plotted on a log scale. Data represent a summary of three independent experiments. B, NCL (f/+) and Mekk3 T-cKO (f/−) mice (three mice per group) were immunized with 5 × 104 CFU L. monocytogenes-OVA per mouse by tail vein injection. Ten days after immunization, spleen cells from infected mice were restimulated with OVA, SIINFEKL (OT1p), or OVA323–339 (OT2p) peptides, and then the cytokine production was assayed by ELISA. C, Representative FACS plots of CD4 and CD8 staining of splenocytes of Listeria-infected mice. D and E, Spleen cells from infected NCL (f/+) and Mekk3 T-cKO (f/−) mice were restimulated with TPA plus ionomycin (T+I) or OVA. Expression of IFN-γ was analyzed on CD4+ (D) or CD8+ (E) T cells by intracellular cytokine staining. Data shown are representative of two independent experiments. N.S., non-stimulated.
To analyze the Ag-specific T cell responses during anti-bacteria immunity, we rechallenged the spleen T cells from the infected mice with OVA or OVA peptides OT1p or OT2p. Consistent with the results shown above from the OVA plus CFA immunization experiments, the IFN-γ production induced by OVA and OT2p was impaired in splenic T cells from Mekk3 T-cKO mice (Fig. 2B). The IFN-γ production induced by OT1p in CD8 T cells was also reduced. In addition, IL-2 and IL-10 production was also modestly reduced in splenic T cells from the Mekk3 T-cKO mice. Because the numbers of splenic CD4 and CD8 T cells were reduced in the Mekk3 T-cKO mice (Fig. 2C), these results indicate that MEKK3 may play a cell intrinsic role for IFN-γ production in CD4 T cells but may be dispensable for the production of other cytokines such as IL-2 or IL-10.
To determine whether the decrease in Ag-induced IFN-γ production defects in Mekk3-deficient CD4 and CD8 T cells was due to the cell intrinsic defects to differentiate into IFN-γ–producing cells or due to the total cell number reduction, or to both, we examined the frequency of IFN-γ+ T cells within total CD4 or CD8 T cells, respectively, from the L. monocytogenes-OVA infected mice by using an intracellular cytokine staining assay after restimulation with TPA/ionomycin or OVA. As shown in Fig. 2D, there were fewer Ag-specific IFN-γ–producing CD4 T cells (reduced frequency) in the spleens of Mekk3 T-cKO mice than that in NCL mice. However, the frequency of the OVA-induced IFN-γ–producing CD8 T cells was comparable in NCL and Mekk3 T-cKO mice, whereas a moderate decrease of IFN-γ production by TPA/ionomycin stimulation was observed (Fig. 2E). We also measured the OT1p tetramer-positive CD8 T cells before or 7 d after immunization and found that both NCL and Mekk3 T-cKO mice had similar percentages of tetramer-positive CD8 cells (Supplemental Fig. 1). Given that the percentage of CD8 T cells in the spleens of Mekk3 T-cKO mice was decreased, although the frequency of the OVA-induced IFN-γ–producing CD8 T cells was the same, the total number of the OVA-induced IFN-γ–producing CD8 T cells was still decreased, which explained the decrease of IFN-γ production in total splenocytes as measured by ELISA in Fig. 2B. These results suggest that MEKK3 may be more critical for CD4 T cells than for CD8 T cells in inducing IFN-γ producing cells. To further confirm these results, we purified WT and MEKK3-deficient naive CD8 T cells and found that there was no significant difference in IFN-γ production when stimulated with anti-CD3/anti-CD28 (Supplemental Fig. 2). However, MEKK3-deficient naive CD8 T cells produced less IFN-γ when stimulated with anti-CD3 only. This could be due to a survival defect of MEKK3-deficient CD8 T cells in the absence of CD28 signal. Together, these results further demonstrate that MEKK3 plays a cell intrinsic role for CD4 T cell-mediated IFN-γ production.
MEKK3 is not required for naive CD4 T cells to differentiate in vitro into Th1 and Th2 lineages
Because the Mekk3-KO CD4 T cells were defective in IFN-γ production, this prompted us to examine if MEKK3 is required for the differentiation of Th1 cells, which are the main IFN-γ–producing CD4 T cells. Naive CD4 T cells (CD62LhighCD44low) from NCL and Mekk3 T-cKO mice were isolated by FACS sorting and cultured under the Th1 polarizing condition in vitro for 6 d. The IFN-γ production and the number of IFN-γ–producing cells were evaluated thereafter. To our surprise, Mekk3-KO T cells were only marginally affected in total IFN-γ production and in the induction of IFN-γ–producing cells under the Th1 condition (Fig. 3A, 3B). Consistent with these results, we found that the induction of T-bet, a key transcription factor required for IFN-γ production by T cells, was comparable in WT T cells and Mekk3-KO T cells (Fig. 3C). In addition, Th2, Th17, and regulatory T cell differentiation in vitro from the naive Mekk3-KO T cells was also comparable with that from the control CD4 T cells (Fig. 3A, 3B, and data not shown).
FIGURE 3.

MEKK3 is not required for CD4 T cell Th1 and Th2 differentiation. A and B, Naive CD4 T cells were sorted from NCL (f/+) and Mekk3 T-cKO (f/−) mice and differentiated under either Th1 or Th2 condition for 6 d. The in vitro-differentiated cells were washed and restimulated with anti-CD3 Ab for 24 h. IFN-γ production was measured by ELISA (A). The differentiated cells were also restimulated with TPA plus ionomycin for 6 h, and the percentage of IFN-γ– and IL-4–producing CD4 T cells was measured by intracellular cytokine staining (B). C, Naive CD4 T cells were sorted from NCL (f/+) and Mekk3 T-cKO (f/−) mice and differentiated under either nonpolarizing condition (Th0) or Th1 or Th2 condition for 6 d. The in vitro-differentiated cells were washed and restimulated with vehicle (−) or TPA plus ionomycin (T/I) (+) for 6 h. T-bet expression was measured by immunoblotting. The p38 level was used as a loading control. D and E, Naive CD4 T cells were sorted from NCL (f/+) and Mekk3 T-cKO (f/−) mice and then stimulated with plate-bound anti-CD3 plus anti-CD28 and IL-2 for 5 d. The in vitro-cultured cells were washed, restimulated with TPA plus ionomycin for 6 h, and the percentage of IFN-γ–producing CD4 T cells was measured by intracellular cytokine staining (D). The in vitro-cultured cells were also washed, then restimulated with anti-CD3 Ab for 24 h to measure the cytokine production by ELISA (E). Data shown are representative of five independent experiments. Each experiment used one pair of NCL and T-cKO mice.
Notably, when Mekk3-KO naive CD4 T cells were cultured under nonpolarizing (Th0) conditions (anti-CD3/anti-CD28 plus IL-2), the number of IFN-γ–producing cells and the total amount of IFN-γ produced were significantly reduced compared with those of the control cells when restimulated with TPA/ionomycin treatment followed by intracellular cytokine staining (Fig. 3D) or with plate-bound anti-CD3 followed by ELISA analysis (Fig. 3E). Similar intracellular staining results were also found when WT and MEKK3-KO Th0 CD4 T cells were restimulated with an anti-CD3 Ab (Supplemental Fig. 3). This defect appeared to be IFN-γ specific, as induction of IL-2, IL-4, and IL-10 was the same in control and Mekk3-KO CD4 T cells. These results suggest that in the absence of polarizing cytokines, the TCR-mediated IFN-γ production by CD4 T cells requires MEKK3.
MEKK3 is required for the TCR-induced MAPK activation
To understand the molecular mechanism of MEKK3-mediated signal transduction in CD4 T cells, we stimulated naive Mekk3-KO CD4 T cells with an anti-CD3 Ab or by treatment with TPA plus ionomycin. Activation of the ERK1/2 and p38 MAPKs by the anti-CD3 stimulation was severely impaired in the Mekk3-KO naive T cells (Fig. 4A). The JNK was only weakly activated in either control or Mekk3-KO T cells with no major difference (data not shown). However, the TPA/ionomycin-induced JNK activation, as well as ERK1/2 and p38 MAPK activation, were all markedly reduced in the Mekk3-KO T cells. Surprisingly, the IKKβ activation induced by TPA/ionomycin was not significantly impaired in Mekk3-KO T cells (Fig. 4B).
FIGURE 4.

MEKK3 is required for TCR-induced MAPK activation. A and B, Naive CD4 T cells sorted from NCL (f/+) and Mekk3 T-cKO (f/−) mice were stimulated with 5 μg/ml plate-bound anti-CD3 (A) or TPA (20 ng/ml) plus ionomycin (100 ng/ml) (T+I) (B) for the indicated time periods. Total cell lysates were prepared for immunoblotting analysis with the indicated Abs. The integrated OD (IOD) values of bands are shown in the tables below the bands. C, In vitro-expanded Th0 CD4 T cells from NCL (f/+) and Mekk3 T-cKO (f/−) mice were stimulated with TPA plus ionomycin (T+I) for the indicated time periods. Total cell lysates were prepared for immunoblotting analysis with the indicated Abs. The IOD values of bands are shown in the tables below the bands. Data shown are representative of four independent experiments. Each experiment used one pair of NCL and T-cKO mice.
In addition to naive CD4 T cells, the in vitro-expanded CD4 T cells under the Th0 conditions were also used for the above study. As shown in Fig. 4C, the induction of the ERK1/2, p38, and JNK MAPKs was also impaired in the Mekk3-KO effector T cells compared with that in control cells. Again, the IκBα degradation and induction of p-IκBα was not significantly affected suggesting that MEKK3 might not be involved in IKKβ activation or IκBα degradation in response to TCR/CD28 or TPA/ionomycin stimulation.
MEKK3 is not required for IL-12/IL-18–induced IFN-γ production
We were intrigued by the results showing that the IFN-γ production in Mekk3-KO T cells was defective under the Th0 but not the Th1 culture condition. Because the main difference between these two culture conditions was the presence of IL-12 and anti–IL-4 Ab in Th1 condition but not Th0 condition, Mekk3-KO T cells were also cultured under the Th0 plus IL-12 or plus anti–IL-4 Ab, then assayed for their ability to produce IFN-γ. In the presence of anti–IL-4 Ab, Mekk3-KO T cells remained defective in IFN-γ production (data not shown). However, addition of IL-12 almost restored the ability of Mekk3-KO T cells to produce IFN-γ similar to that by control T cells (Fig. 5A, Supplemental Fig. 4, Supplemental Fig. 5). Furthermore, the in vitro-cultured Mekk3-KO T cells under the Th0 or Th0 plus IL-12 condition were able to produce an almost identical amount of IFN-γ as that of the control T cells in response to IL-12/IL-18 restimulation (Fig. 5A, Supplemental Fig. 6).
FIGURE 5.
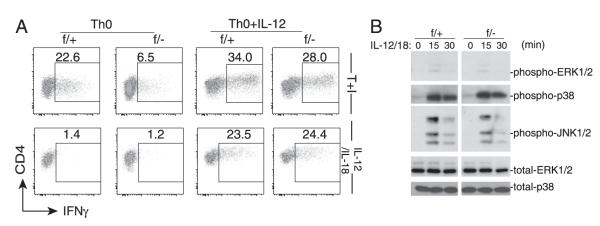
MEKK3 is not required for IL-12/IL-18–induced IFN-γ production. A, Naive CD4 T cells sorted from NCL (f/+) and Mekk3 T-cKO (f/−) mice were cultured under Th0 condition or Th0 plus IL-12 for 6 d. The in vitro-cultured cells were washed and then restimulated with either TPA plus ionomycin (T+I) or with IL-12 plus IL-18 for 6 h. Percentage of IFN-γ–producing cells was determined by intracellular cytokine staining. B, Naive CD4 T cells sorted from NCL (f/+) and Mekk3 T-cKO (f/−) mice were cultured under the Th0 condition for 6 d. The in vitro-cultured cells were washed and then restimulated with IL-12 plus IL18 (IL-12/18) for the indicated time points. Total cell lysates were analyzed by immunoblotting assays for p-ERK1/2, p-p38, and p-JNK1/2. Total ERK1/2 and p38 levels were used as loading controls. Data shown are representative of three independent experiments. Each experiment used one pair of NCL and T-cKO mice.
These results suggest that MEKK3 is not required for the IL-12–induced IFN-γ production. Consistently, we found that the cytokine IL-12/IL-18–induced ERK1/2 and p38 MAPK activation was not affected in Mekk3-KO T cells (Fig. 5B), in contrast to the TPA- or TCR-induced activation of these kinases (Fig. 4). It is noteworthy that IL-12/IL-18 induced weaker ERK1/2 activation but much stronger JNK and p38 MAPK activation than the TCR-induced MAPK activation.
MEKK3 requires Rac1/2 for its activation by TCR stimulation
To understand how MEKK3 is involved in mediating the TCR signals for IFN-γ production, we examined MEKK3 activation by TCR stimulation. As shown in Fig. 6A, stimulation of WT CD4 T cells by an anti-CD3 Ab led to quick and transient activation of MEKK3 as evident by its mobility up-shift on an SDS-PAGE gel. This mobility up-shift was due to phosphorylation, as it was reversed by a phosphatase treatment (Fig. 6A). IL-12/IL-18 seemed unable to activate MEKK3, as very small fractions of MEKK3 were up-shifted. These results suggest that the TCR-mediated T cell activation signals may induce MEKK3 activation, which in turn activates the p38 MAPK to regulate IFN-γ production in T cells, as several studies in the past show that p38 activation is crucial for IFN-γ expression by T cells (28, 29).
FIGURE 6.

MEKK3 requires Rac1/2 for its activation upon TCR stimulation. A, CD4 T cells were either untreated (−) or pretreated (+) with calf intestine phosphatase (CIP) for 15 min as indicated before being stimulated with IL-12/IL-18 or anti-CD3 Ab for the indicated time points. Total cell lysates were prepared and analyzed for MEKK3 by immunoblotting. The p-MEKK3 is indicated. B, CD4 T cells were either untreated or pretreated with 50 μM NSC23766 for 2 h before stimulation with anti-CD3 Ab for the indicated time points. Total cell lysates were prepared for immunoblotting analysis of the indicated molecules. The integrated OD (IOD) value of each band was calculated as shown in the tables below the bands. Data shown are representative of three independent experiments with two NCL mice used in each experiment. C, Naive CD4 T cells sorted from NCL (f/+) and Mekk3 T-cKO (f/−) mice CD4 T cells were either untreated (DMSO) or treated with 50 mM NS23766 for 6 d under Th0 condition. Cells were then restimulated with TPA/ionomycin for 6 h to check the percentage of IFN-γ–producing cells by intracellular staining. Data shown are representative of three independent experiments. Each experiment used one pair of NCL and T-cKO mice. D, An illustration showing the role of MEKK3 in TCR-mediated T cell signal pathways and responses.
How MEKK3 is activated after TCR stimulation has not been studied. Previously, it was shown that small GTPase Rac1/2 is involved in MEKK3 activation in response to osmotic stress (30). Because Rac1/2 play an important role in IFN-γ production in T cells (28) and are known upstream activators of the JNK and p38 MAPKs, this prompted us to examine whether Rac1/2 are upstream activators of MEKK3 in T cells. We thus pretreated WT T cells with a Rac1/2-specific inhibitor, NSC23766 (31, 32), before subjecting these cells to TCR stimulation, and then determined MEKK3 activation. As shown in Fig. 6B, inhibition of Rac1/2 almost completely blocked the TCR-mediated MEKK3 activation in T cells. Consistently, the downstream MAPK activation was also inhibited (Fig. 6B, Supplemental Fig. 9).
To examine further the involvement of Rac1/2 in CD4 T cell differentiation, we cultured WT or Mekk3-KO CD4 T cells under either Th0 or Th0 plus IL12 condition in the presence of 50 μM NSC23766. At this inhibitor concentration, neither T cell survival nor T cell proliferation was significantly affected (Supplemental Fig. 7, Supplemental Table I). Notably, NSC23766 significantly inhibited IFN-γ production in WT CD4 T cells but not Mekk3-KO CD4 T cells, which already had reduced IFN-γ production (Fig. 6C). However, under Th0 plus IL-12 conditions, treatment of NSC23766 can moderately decrease IFN-γ production through restimulation by TPA/ionomycin or IL-12/IL-18; this is consistent with previously published data on RAC2 (28) (Supplemental Fig. 8). Similar results were obtained by using a different Rac1/2 inhibitor newly developed by Dr. Yi Zheng’s laboratory (Raci-analogue) (Supplemental Fig. 9). Together, these results suggest that MEKK3 may be a critical signaling molecule for the TCR-mediated, Rac1/2-dependent MAPK activation that plays a critical role in IFN-γ production and homeostasis (Fig. 6D).
Discussion
T cell activation and function are controlled by complex cell surface signals after stimulation of multiple receptors such as TCR, costimulatory receptors, cytokine receptors, as well as many growth factor receptors like insulin, EGF, PDGF, etc. A common feature for the response to the above receptor stimulation in T cells is rapid and transient activation of the MAPK pathways and the IKK–NFκB pathway. How the limited numbers of MAPK pathways are being used by such diverse T cell stimuli to confer specific physiological responses is still largely unknown, and answering this question is central for our understanding of MAPK function and regulation in T cells and other cell types. It is believed that the MAPK specificity is controlled by members of upstream MAP3Ks that act either alone or together with associated adapter molecules to organize unique MAPK modules in response to different signal inputs.
We recently reported that the MEKK3–MAPK module plays an important role in T cell homeostasis. Notably, MEKK3-deficient T cells did not exhibit defects in Ag-induced T cell proliferation despite the fact that the number of peripheral T cells in Mekk3 T-cKO mice was reduced (23). We show that the TCR-mediated homeostatic signals is impaired when MEKK3 is deleted from T cells.
In this study, we further explored the roles of MEKK3 in T cell-mediated immune responses. We showed that the Ag-specific T cell immunity was impaired in the Mekk3 T-cKO mice. After immunization, the Mekk3 T-cKO mice had fewer Ag-specific CD4 and CD8 T cells than that in the control mice and produced significantly less T cell cytokines after Ag restimulation. The defects in T cell cytokine production, especially the IFN-γ production, were due to the reduced T cell numbers as well as the cell intrinsic defect of peripheral CD4 T cells in Mekk3 T-cKO mice. Compared with CD4 T cells, the Mekk3-deficient CD8 T cells appeared to be less affected in IFN-γ production suggesting that MEKK3 may play a more important role in CD4 T cells than in CD8 T cells.
MEKK3 also plays a role in T cell immunity against bacterial infection. Although Mekk3 T-cKO mice had a similar survival curve to acute Listeria infection, which is known to be independent of T cell function, the T cell-mediated adaptive immune responses against bacteria infection were impaired in Mekk3 T-cKO mice. For instance, the Ag-induced T cell cytokine production (e.g., IL-2, IL-4, and IFN-γ, etc.) was markedly reduced in the spleens of Mekk3 T-cKO mice infected with Listeria compared with that of WT mice. Again, the Mekk3-KO T cells were intrinsically defective in IFN-γ production in response to bacterial infection. In addition, the Mekk3 T-cKO mice had impaired ability to clear the infected bacteria compared with that of the control mice. These data are consistent with the findings that T cells are not the major cell types in response to acute Listeria infection. Macrophages and NK cells play crucial roles in the early phases of bacterial infection, whereas T cells and IFN-γ are required for bacteria clearance (33, 34).
Although MEKK3 is required for IFN-γ production, it is dispensable for Th1 cell differentiation in vitro. Notably, MEKK3 is required for IFN-γ production by the effector CD4 T cells differentiated under a nonpolarizing condition (Th0). This result suggests that IL-12, which is required for Th1 polarization, may mediate a MEKK3-independent signal and partially compensate for the reduced TCR signals in MEKK3-KO T cells, as both the TCR- and cytokine-generated signals are critical for IFN-γ production (35). Indeed, we found that the cytokine IL-12/IL-18–mediated IFN-γ production in effector CD4 T cells was MEKK3 independent, a process that might use a different MAP3K such as MEKK4 to initiate the downstream signaling (36). Furthermore, we found that the induction of the ERK1/2, JNK, and p38 MAPKs by TCR stimulation differed from that induced by cytokine IL-12/IL-18 (Figs. 4, 5). Whereas TCR stimulation led to a strong ERK1/2 activation but weak p38 and JNK activation, IL-12/IL-18 induced a weak ERK1/2 activation but a much stronger JNK and p38 MAPK activation than that of the TCR stimulation. Because the p38 MAPK is well characterized for its role in IFN-γ production by T cells, the strong induction of p38 MAPK in Mekk3-KO T cells by IL-12/IL-18 may explain why the defect in IFN-γ production was restored under this condition.
Nevertheless, the induction of IFN-γ–producing CD4 T cells in vivo was impaired in both OVA immunization and Listeria infection models. As discussed above, such a defect is a combinatorial deficiency of reduced CD4 T cell number and TCR signaling. Notably, Mekk3-KO CD8 T cells were not cell intrinsically defective in IFN-γ production. In vivo, OVA immunization or Listeria infection were able to induce similar frequency of IFN-γ–producing CD8 T cells, although the total number of such cells was still reduced due to homeostasis defect. These results suggest that there is cell-type specificity of MEKK3 function in T cells.
MEKK3 activity is rapidly and transiently induced after TCR stimulation indicating that MEKK3 is involved in transducing the TCR signals. Indeed, in the absence of MEKK3, the TCR-mediated MAPK activation is partially blocked. Surprisingly, not only was the JNK and p38 MAPK activation impaired in Mekk3-KO T cells, but also the ERK1/2 activation was defective. MEKK3 was found previously to mediate JNK1/2 and p38 MAPK through activating their respective activating kinases JNKK1/2 and MKK3/6 (10, 18, 37). Although in vitro MEKK3 was able to activate MEK1/2, which in turn led to ERK1/2 activation, it is unclear at the moment how MEKK3 may influence the ERK1/2 activation in vivo. Because the Ras–Raf–ERK1/2 pathway is the major cascade for ERK1/2 activation in various cell types including T cells, MEKK3 may indirectly affect this pathway to modulate ERK1/2 activation in T cells.
MEKK3 is found in a large protein complex that contains proteins encoded by ccm1, ccm2, and ccm3, whose mutations have been linked to a human disease called cerebral cavernous malformation (CCM). CCM1, the product of ccm1 gene, has been identified previously from a yeast two-hybrid screen as Krit1, which binds to a small GTPase Rap1 (38). Rap1 has been shown to control various cellular responses by influencing the Ras function. Many early studies showed that Rap1 participates in T cell signaling (39, 40). Therefore, it is possible that MEKK3 could indirectly modulate ERK1/2 activity through the Rap1-mediated Ras regulation.
It was recently reported that independently generated Mekk3 T-cKO mice had decreased NF-κB signaling in CD4SP thymocytes (24). We observed a similar defect in thymocytes after stimulation (data not shown). However, the TCR-induced IKKβ activation and IκBα degradation was not defective in MEKK3-deficient peripheral naive or effector T cells. In the periphery, it is possible that IKKβ activation may be mediated by multiple MAP3Ks.
Finally, we show that MEKK3 activation in T cells requires Rac1/2. These data are consistent with previous findings in non-T cells showing that in response to osmotic stress, MEKK3 was activated by Rac1/2 in association with adapter protein OSM1/CCM2 in the CCM complex (30, 41). Rac1/2 are potent activators of MAPKs including the JNK and p38 MAPKs (42–44). It is thus likely that the Rac1/2 activating signals to MAPKs are mediated by MEKK3. The function of Rac1/2 in IFN-γ production in T cells, especially during Th1 differentiation, has been well documented (28, 45). Consistently, we found that the IFN-γ production in T cells was blocked in the presence of Rac1/2-specific inhibitors.
Given the fact that p38 MAPK is critical for IFN-γ production in T cells, our study thus suggests that one of the MEKK3 functions in T cell immunity may be to link the TCR-induced Rac1/2 signal to p38 MAPK to control IFN-γ production. Notably, MEKK3 appears to be required only for the TCR-mediated but not IL-12/IL-18–mediated IFN-γ production, whereas Rac1/2 appears to regulate both the TCR and IL-12/IL-18–induced IFN-γ production. It is possible that in response to IL-12/IL-18 stimulation, Rac1/2 may form a different signaling complex with other MAP3Ks (e.g., MEKK4) (36) to stimulate the downstream pathway.
Although it remains to be determined if OSM1/CCM2 is also involved in MEKK3 activation in T cells, the key proteins in the CCM complex, namely, CCM1, CCM2, and CCM3, are all expressed in T cells (data not shown). Future studies to determine their roles in the regulation of MEKK3 will be necessary to understand fully the physiological roles of MEKK3 in T cell immunity.
Supplementary Material
Figure S1. Normal CD8 response after LM-OVA infection: NCL (f/+) and Mekk3 T-cKO (f/−) mice were infected with 5×104 CFU of LM-OVA. Blood were collected at day 7 after infection. Percentages of OT1p (OVA257-264) tetramer positive CD8 cells in the CD8+CD3+ population were determined by flow cytometry. Data are representative of two individual experiments. Blood from non-infected wild type mice (non-infection) was used as negative controls. Data shown are representative of three independent experiments.
Figure S2. IFNγ production of MEKK3 deficient CD8 T cell: Naïve CD8 T cells sorted from NCL (f/+) and Mekk3 T-CKO (f/−) mice were stimulated with plate-bound anti-CD3 alone, or with 1 μg/ml of soluble anti-CD28. Cell proliferation was measured by [3H] thymidine incorporation. Production of IL-2 and IFNγ were measured by ELISA. Data shown are representative of three independent experiments.
Figure S3. MEKK3 is required for TCR induced IFNγ production by CD4 T cells: Naive CD4 T cells were stimulated with plate-bound anti-CD3 plus anti-CD28 antibodies in the presence of IL-2 (Th0 condition) for 5 days. The in vitro cultured cells were washed and re-stimulated with plate-coated anti-CD3 (1 μg/ml) for 6 h, the percentages of IFNγ producing CD4 T cells was measured by intracellular cytokine staining. Data shown are representative of three independent experiments.
Figure S4. MEKK3 is not required for IL-12 induced IFNγ production by CD4 T cells: Naive CD4 T cells were stimulated with plate-bound anti-CD3 plus anti-CD28 antibodies in the presence of IL-2 and IL-12 (Th0+IL-12 condition) for 5 days. The in vitro cultured cells were washed and re-stimulated with plate-coated anti-CD3 antibody (1 μg/ml) for 6 h, the percentages of IFNγ producing CD4 T cells was measured by intracellular cytokine staining. Data shown are representative of three independent experiments.
Figure S5. IL-12 partially restores IFNγ production in MEKK3 deficient CD4 T cells: Naïve CD4 T cells were differentiated for 6 days under Th0 or Th0+IL-12 conditions. Cells were washed then re-stimulated with plate-bound anti-CD3 antibody for 24 h, followed by ELISA analysis of IFNγ production. Data shown are representative of three independent experiments.
Figure S6. MEKK3 is not required for the IL-12/IL-18 induced IFNγ production: Naïve CD4 T cells were differentiated for 6 days under Th0 or Th0+IL-12 conditions. Cells were washed then re-stimulated with IL-12/IL-18 for 24 h, followed by ELISA analysis of IFNγ production. Data shown are representative of three independent experiments.
Figure S7. RAC1/2 inhibitor NS23766 doesn’t affect T cell survival: Naïve CD4 T cells were treated with NS23766 (50 μM) for 12 or 24 h, before being analyzed for cell survival by 7-AAD and Annexin-V staining as indicated. Numbers to the gated boxes show percentages of viable cells, which are 7-AAD and Annexin-V double negative. Data shown are representative of three independent experiments.
Figure S8. Inhibition of RAC1/2 with NS23766 decreases the differentiation of IFNγ producing CD4 T cells under Th0+IL-12 condition: Naïve CD4 T cell from NCL mice were either untreated (DMSO) or treated with 50 μM of NS23766 for 5 days under Th0+IL-12 condition. Cells were washed then re-stimulated with TPA/ionomycin or IL-12/IL18 for 6 h as indicated, Numbers next to the gated boxes show the percentage of IFNγ producing cells determined by intracellular cytokine staining. Data shown are representative of three independent experiments.
Figure S9. Inhibition of RAC1/2 with NS23766-Analog (Raci-Analog) decreases the differentiation of IFNγ producing CD4 T cells and MEKK3 phosphorylation: Upper panel, CD4 T cells from NCL mice were either untreated (DMSO) or pre-treated with 50 μM of NSC23766 for 2 h before stimulation with anti-CD3 antibody for 5 min. Total cell lysates were prepared for immunoblotting analysis of the indicated molecules. Data shown are representative of three independent experiments. Bottom panel, Naïve CD4 T cell from NCL mice were either untreated (DMSO) or treated with 50 μM of Raci-Analog for 5 days under Th0 condition. Cells were washed then re-stimulated with TPA/ionomycin for 6 h. Numbers next to the gated boxes show the percentage of IFNγ producing cells determined by intracellular cytokine staining.
Table S1. RAC1/2 inhibitor NS23766 doesn’t affect cell proliferation: Half million of naïve CD4 T cells from wild type mice were either untreated (DMSO) or treated with 50 μM NS23766 under the Th0 or Th0+IL-12 conditions. The total viable cells were determined 5 days later and shown in the table. Data shown are pooled from three independent experiments.
Abbreviations used in this article
- CCM
cerebral cavernous malformation
- NCL
normal control littermate
- T-cKO
T cell conditional knockout
- TPA
12-O-tetradecanoyl phorbol-13-acetate
- WT
wild-type
Footnotes
Disclosures The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 2.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 3.Avruch J. MAP kinase pathways: the first twenty years. Biochim. Biophys. Acta. 2007;1773:1150–1160. doi: 10.1016/j.bbamcr.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- 5.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu. Rev. Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 6.Rincón M, Davis RJ. Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 2009;228:212–224. doi: 10.1111/j.1600-065X.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- 7.Huang G, Shi LZ, Chi H. Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine. 2009;48:161–169. doi: 10.1016/j.cyto.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blank JL, Gerwins P, Elliott EM, Sather S, Johnson GL. Molecular cloning of mitogen-activated protein/ERK kinase kinases (MEKK) 2 and 3. Regulation of sequential phosphorylation pathways involving mitogen-activated protein kinase and c-Jun kinase. J. Biol. Chem. 1996;271:5361–5368. doi: 10.1074/jbc.271.10.5361. [DOI] [PubMed] [Google Scholar]
- 9.Ellinger-Ziegelbauer H, Brown K, Kelly K, Siebenlist U. Direct activation of the stress-activated protein kinase (SAPK) and extracellular signal-regulated protein kinase (ERK) pathways by an inducible mitogen-activated protein Kinase/ERK kinase kinase 3 (MEKK) derivative. J. Biol. Chem. 1997;272:2668–2674. doi: 10.1074/jbc.272.5.2668. [DOI] [PubMed] [Google Scholar]
- 10.Yang J, Boerm M, McCarty M, Bucana C, Fidler IJ, Zhuang Y, Su B. Mekk3 is essential for early embryonic cardiovascular development. Nat. Genet. 2000;24:309–313. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 11.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 12.Su B. Linking stress to immunity? Nat. Immunol. 2005;6:541–542. doi: 10.1038/ni0605-541. [DOI] [PubMed] [Google Scholar]
- 13.Deng Y, Yang J, McCarty M, Su B. MEKK3 is required for endothelium function but is not essential for tumor growth and angiogenesis. Am. J. Physiol. Cell Physiol. 2007;293:C1404–C1411. doi: 10.1152/ajpcell.00058.2007. [DOI] [PubMed] [Google Scholar]
- 14.Cheng J, Yu L, Zhang D, Huang Q, Spencer DM, Su B. Dimerization through the catalytic domain is essential for MEKK2 activation. J. Biol. Chem. 2005;280:13477–13482. doi: 10.1074/jbc.M414258200. [DOI] [PubMed] [Google Scholar]
- 15.Zhang D, Facchinetti V, Wang X, Huang Q, Qin J, Su B. Identification of MEKK2/3 serine phosphorylation site targeted by the Toll-like receptor and stress pathways. EMBO J. 2006;25:97–107. doi: 10.1038/sj.emboj.7600913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Q, Yang J, Lin Y, Walker C, Cheng J, Liu ZG, Su B. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat. Immunol. 2004;5:98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- 17.Blonska M, Shambharkar PB, Kobayashi M, Zhang D, Sakurai H, Su B, Lin X. TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappaB activation. J. Biol. Chem. 2005;280:43056–43063. doi: 10.1074/jbc.M507807200. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu Z, Su B. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat. Immunol. 2001;2:620–624. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- 19.Qin J, Yao J, Cui G, Xiao H, Kim TW, Fraczek J, Wightman P, Sato S, Akira S, Puel A, et al. TLR8-mediated NF-kappaB and JNK activation are TAK1-independent and MEKK3-dependent. J. Biol. Chem. 2006;281:21013–21021. doi: 10.1074/jbc.M512908200. [DOI] [PubMed] [Google Scholar]
- 20.Blonska M, Shambharkar PB, Kobayashi M, Zhang D, Sakurai H, Su B, Lin X. TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappa B activation. J. Biol. Chem. 2005;280:43056–43063. doi: 10.1074/jbc.M507807200. [DOI] [PubMed] [Google Scholar]
- 21.Yamazaki K, Gohda J, Kanayama A, Miyamoto Y, Sakurai H, Yamamoto M, Akira S, Hayashi H, Su B, Inoue J. Two mechanistically and temporally distinct NF-kappaB activation pathways in IL-1 signaling. Sci. Signal. 2009;2:ra66. doi: 10.1126/scisignal.2000387. [DOI] [PubMed] [Google Scholar]
- 22.Di Y, Li S, Wang L, Zhang Y, Dorf ME. Homeostatic interactions between MEKK3 and TAK1 involved in NF-kappaB signaling. Cell. Signal. 2008;20:705–713. doi: 10.1016/j.cellsig.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Chang X, Facchinetti V, Zhuang Y, Su B. MEKK3 is essential for lymphopenia-induced T cell proliferation and survival. J. Immunol. 2009;182:3597–3608. doi: 10.4049/jimmunol.0803738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinohara H, Yamasaki S, Maeda S, Saito T, Kurosaki T. Regulation of NF-kappaB-dependent T cell activation and development by MEKK3. Int. Immunol. 2009;21:393–401. doi: 10.1093/intimm/dxp007. [DOI] [PubMed] [Google Scholar]
- 25.Pope C, Kim SK, Marzo A, Masopust D, Williams K, Jiang J, Shen H, Lefrançois L. Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J. Immunol. 2001;166:3402–3409. doi: 10.4049/jimmunol.166.5.3402. [DOI] [PubMed] [Google Scholar]
- 26.Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 27.Shedlock DJ, Whitmire JK, Tan J, MacDonald AS, Ahmed R, Shen H. Role of CD4 T cell help and costimulation in CD8 T cell responses during Listeria monocytogenes infection. J. Immunol. 2003;170:2053–2063. doi: 10.4049/jimmunol.170.4.2053. [DOI] [PubMed] [Google Scholar]
- 28.Li B, Yu H, Zheng W, Voll R, Na S, Roberts AW, Williams DA, Davis RJ, Ghosh S, Flavell RA. Role of the guanosine triphosphatase Rac2 in T helper 1 cell differentiation. Science. 2000;288:2219–2222. doi: 10.1126/science.288.5474.2219. [DOI] [PubMed] [Google Scholar]
- 29.Lu B, Yu H, Chow C, Li B, Zheng W, Davis RJ, Flavell RA. GADD45gamma mediates the activation of the p38 and JNK MAP kinase pathways and cytokine production in effector TH1 cells. Immunity. 2001;14:583–590. doi: 10.1016/s1074-7613(01)00141-8. [DOI] [PubMed] [Google Scholar]
- 30.Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE, Horne EA, Dell’Acqua ML, Johnson GL. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 2003;5:1104–1110. doi: 10.1038/ncb1071. [DOI] [PubMed] [Google Scholar]
- 31.Akbar H, Cancelas J, Williams DA, Zheng J, Zheng Y. Rational design and applications of a Rac GTPase-specific small molecule inhibitor. Methods Enzymol. 2006;406:554–565. doi: 10.1016/S0076-6879(06)06043-5. [DOI] [PubMed] [Google Scholar]
- 32.Nassar N, Cancelas J, Zheng J, Williams DA, Zheng Y. Structure-function based design of small molecule inhibitors targeting Rho family GTPases. Curr. Top. Med. Chem. 2006;6:1109–1116. doi: 10.2174/156802606777812095. [DOI] [PubMed] [Google Scholar]
- 33.Pamer EG. Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 34.Edelson BT, Unanue ER. Immunity to Listeria infection. Curr. Opin. Immunol. 2000;12:425–431. doi: 10.1016/s0952-7915(00)00112-6. [DOI] [PubMed] [Google Scholar]
- 35.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 36.Chi H, Lu B, Takekawa M, Davis RJ, Flavell RA. GADD45beta/GADD45gamma and MEKK4 comprise a genetic pathway mediating STAT4-independent IFNgamma production in T cells. EMBO J. 2004;23:1576–1586. doi: 10.1038/sj.emboj.7600173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deacon K, Blank JL. Characterization of the mitogen-activated protein kinase kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38 pathways regulated by MEK kinases 2 and 3. MEK kinase 3 activates MKK3 but does not cause activation of p38 kinase in vivo. J. Biol. Chem. 1997;272:14489–14496. doi: 10.1074/jbc.272.22.14489. [DOI] [PubMed] [Google Scholar]
- 38.Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, Touchman JW, Gallione CJ, Lee-Lin SQ, Kosofsky B, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum. Mol. Genet. 1999;8:2325–2333. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- 39.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 40.Morton AM, McManus B, Garside P, Mowat AM, Harnett MM. Inverse Rap1 and phospho-ERK expression discriminate the maintenance phase of tolerance and priming of antigen-specific CD4+ T cells in vitro and in vivo. J. Immunol. 2007;179:8026–8034. doi: 10.4049/jimmunol.179.12.8026. [DOI] [PubMed] [Google Scholar]
- 41.Hilder TL, Malone MH, Bencharit S, Colicelli J, Haystead TA, Johnson GL, Wu CC. Proteomic identification of the cerebral cavernous malformation signaling complex. J. Proteome Res. 2007;6:4343–4355. doi: 10.1021/pr0704276. [DOI] [PubMed] [Google Scholar]
- 42.Kang ZB, Ge Y, Chen Z, Cluette-Brown J, Laposata M, Leaf A, Kang JX. Adenoviral gene transfer of Caenorhabditis elegans n—3 fatty acid desaturase optimizes fatty acid composition in mammalian cells. Proc. Natl. Acad. Sci. USA. 2001;98:4050–4054. doi: 10.1073/pnas.061040198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 44.Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J. Biol. Chem. 1996;271:27225–27228. doi: 10.1074/jbc.271.44.27225. [DOI] [PubMed] [Google Scholar]
- 45.Guo F, Cancelas JA, Hildeman D, Williams DA, Zheng Y. Rac GTPase isoforms Rac1 and Rac2 play a redundant and crucial role in T-cell development. Blood. 2008;112:1767–1775. doi: 10.1182/blood-2008-01-132068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Normal CD8 response after LM-OVA infection: NCL (f/+) and Mekk3 T-cKO (f/−) mice were infected with 5×104 CFU of LM-OVA. Blood were collected at day 7 after infection. Percentages of OT1p (OVA257-264) tetramer positive CD8 cells in the CD8+CD3+ population were determined by flow cytometry. Data are representative of two individual experiments. Blood from non-infected wild type mice (non-infection) was used as negative controls. Data shown are representative of three independent experiments.
Figure S2. IFNγ production of MEKK3 deficient CD8 T cell: Naïve CD8 T cells sorted from NCL (f/+) and Mekk3 T-CKO (f/−) mice were stimulated with plate-bound anti-CD3 alone, or with 1 μg/ml of soluble anti-CD28. Cell proliferation was measured by [3H] thymidine incorporation. Production of IL-2 and IFNγ were measured by ELISA. Data shown are representative of three independent experiments.
Figure S3. MEKK3 is required for TCR induced IFNγ production by CD4 T cells: Naive CD4 T cells were stimulated with plate-bound anti-CD3 plus anti-CD28 antibodies in the presence of IL-2 (Th0 condition) for 5 days. The in vitro cultured cells were washed and re-stimulated with plate-coated anti-CD3 (1 μg/ml) for 6 h, the percentages of IFNγ producing CD4 T cells was measured by intracellular cytokine staining. Data shown are representative of three independent experiments.
Figure S4. MEKK3 is not required for IL-12 induced IFNγ production by CD4 T cells: Naive CD4 T cells were stimulated with plate-bound anti-CD3 plus anti-CD28 antibodies in the presence of IL-2 and IL-12 (Th0+IL-12 condition) for 5 days. The in vitro cultured cells were washed and re-stimulated with plate-coated anti-CD3 antibody (1 μg/ml) for 6 h, the percentages of IFNγ producing CD4 T cells was measured by intracellular cytokine staining. Data shown are representative of three independent experiments.
Figure S5. IL-12 partially restores IFNγ production in MEKK3 deficient CD4 T cells: Naïve CD4 T cells were differentiated for 6 days under Th0 or Th0+IL-12 conditions. Cells were washed then re-stimulated with plate-bound anti-CD3 antibody for 24 h, followed by ELISA analysis of IFNγ production. Data shown are representative of three independent experiments.
Figure S6. MEKK3 is not required for the IL-12/IL-18 induced IFNγ production: Naïve CD4 T cells were differentiated for 6 days under Th0 or Th0+IL-12 conditions. Cells were washed then re-stimulated with IL-12/IL-18 for 24 h, followed by ELISA analysis of IFNγ production. Data shown are representative of three independent experiments.
Figure S7. RAC1/2 inhibitor NS23766 doesn’t affect T cell survival: Naïve CD4 T cells were treated with NS23766 (50 μM) for 12 or 24 h, before being analyzed for cell survival by 7-AAD and Annexin-V staining as indicated. Numbers to the gated boxes show percentages of viable cells, which are 7-AAD and Annexin-V double negative. Data shown are representative of three independent experiments.
Figure S8. Inhibition of RAC1/2 with NS23766 decreases the differentiation of IFNγ producing CD4 T cells under Th0+IL-12 condition: Naïve CD4 T cell from NCL mice were either untreated (DMSO) or treated with 50 μM of NS23766 for 5 days under Th0+IL-12 condition. Cells were washed then re-stimulated with TPA/ionomycin or IL-12/IL18 for 6 h as indicated, Numbers next to the gated boxes show the percentage of IFNγ producing cells determined by intracellular cytokine staining. Data shown are representative of three independent experiments.
Figure S9. Inhibition of RAC1/2 with NS23766-Analog (Raci-Analog) decreases the differentiation of IFNγ producing CD4 T cells and MEKK3 phosphorylation: Upper panel, CD4 T cells from NCL mice were either untreated (DMSO) or pre-treated with 50 μM of NSC23766 for 2 h before stimulation with anti-CD3 antibody for 5 min. Total cell lysates were prepared for immunoblotting analysis of the indicated molecules. Data shown are representative of three independent experiments. Bottom panel, Naïve CD4 T cell from NCL mice were either untreated (DMSO) or treated with 50 μM of Raci-Analog for 5 days under Th0 condition. Cells were washed then re-stimulated with TPA/ionomycin for 6 h. Numbers next to the gated boxes show the percentage of IFNγ producing cells determined by intracellular cytokine staining.
Table S1. RAC1/2 inhibitor NS23766 doesn’t affect cell proliferation: Half million of naïve CD4 T cells from wild type mice were either untreated (DMSO) or treated with 50 μM NS23766 under the Th0 or Th0+IL-12 conditions. The total viable cells were determined 5 days later and shown in the table. Data shown are pooled from three independent experiments.