

Abstract
The term epigenetics refers to the study of a number of biochemical modifications of chromatin that have an impact on gene expression regulation. Aberrant epigenetic lesions, in particular DNA methylation of promoter associated CpG islands, are common in acute lymphocytic leukemia (ALL). Recent data from multiple laboratories indicates that several hundred genes, involving dozens of critical molecular pathways, are epigenetically suppressed in ALL. Because these lesions are potentially reversible, the reactivation of these pathways using, for instance, hypomethylating agents may have therapeutic potential in this disease. Furthermore, the analysis of epigenetic alterations in ALL may allow: 1) the identification of subsets of patients with poor prognosis when treated with conventional therapy; 2) development of new techniques to evaluate minimal residual disease; 3) better understanding of the differences between pediatric and adult ALL; and 4) new therapeutic interventions by incorporating agents with hypomethylating activity to conventional chemotherapeutic programs. In this review, we desribe the role of epigenetic alterations in ALL from a translational perspective.
Keywords: Acute lymphocytic leukemia, DNA methylation, epigenetics
Introduction
Epigenetics is the study of biochemical modifications of chromatin. These modifications do not alter the primary sequence of DNA but have an impact on gene expression regulation, most frequently gene suppression. The field of epigenetics is rapidly expanding from DNA methylation1 to the realization that histone modifications2 cross talk with DNA methylation3, and the most recent discovery of microRNA4 as having a role in DNA methylation control5. Data from multiple laboratories and for all types of human malignancies has clearly demonstrated that epigenetic alterations, or at least aberrant DNA methylation, are very prevalent in cancer6. Epigenetic lesions complement genetic alterations in oncogenesis. Epigenetic lesions are reversible and it is postulated that a number of chemotherapeutic agents (DNA hypomethylating agents7,8, histone deacetylase inhibitors9) act by reversing aberrant epigenetic marks and inducing physiologic gene expression. In this short review, we focus on current knowledge of the epigenetics of acute lymphocytic leukemia (ALL) from a translational perspective and on the use of this information to develop biomarkers and new therapeutic alternatives. At the present time, the field mainly relates to aberrant DNA methylation, and little is known in terms of abnormal histone code modifications or microRNA gene expression profiles.
Aberrant DNA methylation in ALL
The analysis of DNA methylation in ALL has paralleled the continuous development of simpler and more powerful techniques to assess multiple promoters and many samples in parallel. Most current techniques use bisulfite treatment of DNA10. This method has allowed the development of simple PCR assays to study methylation, including the more recent development of pyrosequencing assays11. Initial studies in ALL consisted of the analysis of single genes in limited number of samples; these investigations (Table 1) focused on genes such as calcitonin12–15, p1516–18, p7319,20, E-cadherin21, ER22. Other single gene studies have included Dkk-323, LATS2/KPM24, Hck25, DBC126, BNIP327, among many others. By the time of these reports, it became apparent that human cancer was characterized by the concomitant methylation of multiple genes28. The studies in solid tumors28 were confirmed in leukemia 29, prompting experiments in ALL30. In the initial profiling study of adult ALL, ten genes (MDR1, THSBS2, THSBS1, MYF3, ER, P15, CD10, c-ABL, p16, p73) were analyzed in a cohort of 80 patients with ALL (Table 1). Results of this profiling analysis were reminiscent of those described in acute myelogenous leukemia (AML)29 or colon cancer28. First, close to 85% of patients had methylation of at least 1 gene and 40% of them of 3 or more genes. Distribution of DNA methylation followed a bimodal pattern, with a group of patients with significant increase in the number of methylated genes.; these patients also had “denser” methylation. There was a strong correlation between methylation of most of these genes indicating the presence of a molecular defect (ie hypermethylator phenotype) leading to the concomitant aberrant methylation of multiple genes. Expression of CD10 was inversely associated with methylation of CD10. At the genetic level, methylation of c-ABL was only observed in patients with Philadelphia chromosome alterations (Ph), and only in those patients with the p210 isoform, a reproducible result31. In the exploratory study, an association was observed between methylation and worse prognosis (for p73 and p15). These data indicated that aberrant DNA methylation of multiple promoter CpG islands is a common feature of adult ALL. A number of large profile studies were subsequently reported 32 (Table 1). When 15 genes were analyzed in more than 250 patients in both adult and pediatric ALL, methylation of at least 1 gene was observed in 77% of patients and 35% of them had methylation of 4 or more genes. Importantly, increased number of methylated genes was associated with a worse outcome. These results confirmed the prevalence and clinical relevance of aberrant DNA methylation of multiple promoter CpG islands in ALL.
Table 1. A list of methylated genes in ALL.
Table 1 summarizes a partial list of genes reported to be methylated in ALL, as well as their chromosomal location, potential function (when known) and the number and percentage of metyhylation. It should be noted that a number of techniques were used to determine methylation and that criteria for “methylation positivity” may difer from study to study. This table is therefore only orientative.
| Gene | Chromosomal location | Function | Number methylated/Number analyzed | % (Range) | References |
|---|---|---|---|---|---|
| GIPC2 | 1p31 | Prostanoid signalling | 31/31 | 100 | 33 |
| RSPO1 | 1p34 | Wnt signalling | 46/46 | 100 | 33 |
| P73 | 1p36 | Transcription factor | 11/35, 17/80, 45/251 | 20 (18–31) | 19,20,30,32 |
| KCNK2 | 1q41 | K+ channel | 14/16 | 87 | 34 |
| LRP1B | 2q21 | LDL complex | 15/16 | 93 | 34 |
| DCL-1 | 2q24 | GTPase negative regulator | 11/16 | 68 | 34 |
| CAST1 | 3p14 | CNS, synapsis | 49/57 | 86 | 33 |
| MAGI1 | 3p14 | Guanylate kinase | 27/45 | 60 | 33 |
| WntA5 | 3p21 | Wnt signaling | 132/307 | 43 | 35 |
| ADCY5 | 3q13 | 38/56 | 68 | 33 | |
| CD10 | 3q25 | Peptidase | 8/80 | 10 | 30 |
| HSPA4L | 4q28 | Heat shock protein | 24/35 | 69 | 33 |
| sFRP2 | 4q31 | Wnt signaling | 42/261 | 16 | 35 |
| OCLN | 5q13 | Tight junction | 31/41 | 76 | 33 |
| EFNA5 | 5q21 | Ephrin signalling | 44/58 | 76 | 33 |
| MSX2 | 5q34 | Transcriptional repressor | 54/55 | 98 | 33 |
| GFPT2 | 5q34 | Aminotransferase | 8/35 | 23 | 33 |
| LATS1 | 6q24 | Kinase | 100/251 | 40 | 32 |
| ER | 6q25 | Estrogen receptor | 17/18, 29/80 | 47 (36–94) | 22, 30 |
| PARKIN | 6q25 | Proteosomal degradation | 67/251 | 27 | 32 |
| THSBS2 | 6q27 | Cell adhesion | 42/80 | 52 | 30 |
| sFRP4 | 7p14 | Wnt signaling | 55/261 | 21 | 35 |
| MDR1 | 7q21 | Transmembrane transporter | 36/80 | 45 | 30 |
| sFRP1 | 8p12 | Wnt signaling | 99/261 | 38 | 35 |
| P15 | 9p21 | Cell cycle control | 17/45, 20/46, 17/34, 19/80,73/251 | 32 (23–50) | 16–18, 30,32 |
| GNA14 | 9q21 | G protein | 27/44 | 59 | 33 |
| DBC1 | 9q32 | Inhibitor of Sirt1 | 29/170 | 17 | 26 |
| c-ABL | 9q34 | Kinase | 6/80 | 8 | 30 |
| DAPK | 9q34 | Apoptosis | 33 | 13 | 32 |
| PTEN | 10q23 | Phosphatase | 50 | 20 | 32 |
| sFRP5 | 10q24 | Wnt signaling | 73 | 28 | 35 |
| BNIP3 | 10q26 | Apoptosis | 5/34 | 15 | 27 |
| Calcitonin | 11p15 | Calcium metabolism | 6/7, 13/14, 44/47, 45/105 | 62 (42–93) | 12–15 |
| Dkk3 | 11p15 | Wnt signaling | 60/183 | 33 | 35 |
| P57 | 11p15 | Cell cycle control | 31/63, 45/251 | 30 (18–50) | 30,32 |
| SLC2A14 | 12p13 | Glucose transport | 12/16 | 75 | 34 |
| WIF1 | 12q14 | Wnt signaling | 78 | 30 | 35 |
| APAF-1 | 12q23 | Apoptosis | 85 | 34 | 32 |
| DDX51 | 12q24 | 8/16 | 50 | 34 | |
| HDPR1 | 14q23 | Wnt signaling | 68/261 | 26 | 35 |
| THSBS1 | 15q15 | Cell adhesion | 16/80 | 20 | 30 |
| NOPE | 15q22 | 13/16 | 81 | 34 | |
| SALL1 | 16q12 | Zn finger protein | 41/41 | 100 | 33 |
| E-cadherin | 16q22 | Cell adhesion | 18/33, 92/251 | 39 (37–54) | 21, 30 |
| CDH-13 | 16q24 | Cell adhesion | 87/251 | 35 | 32 |
| DCC | 18q21 | Putative tumor suppressor gene | 14/16 | 87 | 34 |
| MY05B | 18q21 | Protein traffick | 36/36 | 100 | 33 |
| ZNF382 | 19q13 | Zn finger protein | 24/46 | 52 | 33 |
| NES-1 | 19q13 | Serine protease | 143/251 | 57 | 32 |
| Hck | 20q11 | Kinase | 9/44 | 20 | 25 |
| TMS-1 | 20q13 | 22/251 | 9 | 32 | |
| MN1 | 22q12 | Involved in meningioma | 45/53 | 85 | 33 |
In general, gene specific studies are limited by selection bias. Genome-wide analysis allows unmasking of unanticipated genes and molecular pathways. Several efforts have been made to create an unbiased methylation profile in ALL33,34. Kuang et al used the Methylated CpG Island Amplification (MCA) technique coupled with an established promoter array33: in excess of 400 genes were uncoverd as potential methylation targets in ALL33. Using bisulfite pyrosequencing, 26 of these genes were validated in leukemia cell lines and 15 (Table 1) in primary ALL samples. Genes were evenly distributed through all autosomes (Figure 1) and could be clustered in specific molecular pathways encompassing multiple functions33. There was overrepresentation of Wnt related genes and kinases such as the Ephrin family of ligands and receptors. Independent studies have already shown the importance of Wnt signaling epigenetic alterations in ALL35. The inactivation of Ephrin (a large family of kinases) mediated signaling also seems common in ALL (Kuang et al in preparation)36 and may be related to altered Akt signaling36. In a parallel study, Taylor et al34 also reported results of a large scale methylation analysis in ALL using a different type of array. Using this technique, this group identified 262 differentially methylated genes in ALL and validated 10 of them in a small cohort of patients (Table 1). Of interest, there was little overlap between the results of the 2 array experiments33,34, with only 9 genes concordant between both studies. These differences are probably related to the CpG islands covered by each array platform and the results are likely complementary. Recently, Taylor et al also have shown the feasibility of performing “deep-sequencing” methylation studies in acute leukemia37.
Figure 1. Chromosomal distribution of potential methylation targets uncovered by MCA/Agilent array33.

The figures on top of each chromosome indicate the number of the chromosome and the number of genes (an percentage) located in each individual chromosome.
Analysis of aberrant DNA methylatyion as a tool to predict prognosis in ALL
Methylation of multiple genes/pathways is common in ALL. Because aberrant DNA methylation can suppress tumor suppressor genes, it is possible that the methylation/suppression of these genes may confer distinct clinical pathological characteristics to these patients, including worse prognosis. One of the first studies to demonstrate such an effect, an analysis of 3 genes (p15, p73 and p57)38, is illustrative, in that none of these genes when analyzed individually showed clear prognostic value. For instance, p57 was first found to be methylated in leukemia39. Subsequent studies showed the gene to be methylated in close to 50% of adult patients and to be correlated with methylation of p73 and p15, but not with any other significant patient characteristic. p73 is a p53 homologue that is upstream of p5740; these three genes have a role in cell cycle progression. Patients who showed methylation of more than 1 gene of this triad had a median survival of 52 weeks (the equivalent of Ph + ALL in the preimatinib era) that was significantly worse than that of patients with methylation of only 0 or 1 gene of this triad (Figure 2). The inference from these data was that the cell cycle control check point controlled by p73/p57/p15 had evolved requiring redundant systems (ie p15 and p57)and therefore the need of complete epigenetic suppression to have an effect on survival. Indeed, in a limited set of patients, there was clear cell cycle dysregulation in patients with methylation of this pathway38. Because it is presumed that methylation results in silencing, the same group of investigators analyzed the prognostic value of protein expression (the reverse of methylation) of the p73/p15/p57 triad in ALL41 using a tissue microarray platform constructed with the bone marrow samples that had been used for methylation analysis38. Methylation of these genes inversely correlated with protein expression, and those patients with evidence of protein expression had a significant better outcome by multivariate analysis. These results have several implications: 1) that methylation and expression studies can be complementary and 2) that the identification of methylation marks may result in the development of widely available clinical assays.
Figure 2. Translational implications of using analysis of DNA methylation in adult Ph negative ALL.
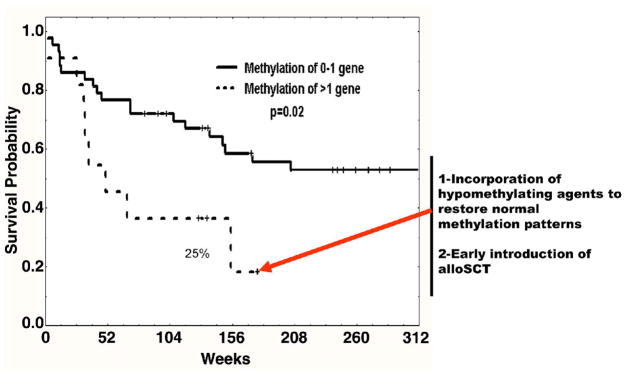
The impact on survival of methylation of a triad of genes composed of p73/p15 and p57 was evaluated in a cohort of adult patients with Ph negative ALL treated homogenously with hyper-CVAD therapy38. Patients with methylation of 2 or 3 genes had a significantly worse prognosis compared with those with methylation of 0 or 1 genes38. This data was confirmed at the protein level41. This information could be used to incorporate hypomethylating agents or to consider the introduction of allogeneic stem cell transplantation.
Others have also shown that the methylation of multiple genes and pathways is associated with worse outcome in ALL32 and that the larger the number of genes methylated, the worse the outcome32,33. Although in general results of most groups have been concordant or complementary, there has been some controversy surrounding the p21 gene. In the original study, p21 methylation was shown to be a strong prognostic marker in ALL42. Subsequent investigations failed to find this association43, a finding confirmed by several other groups44. [unclear syntax: what is being confirmed, the original or the second study?] The most likely reason for this discrepancy is probably use in the original study of a non-bisulfite assay, prone to false positives and in some cases difficult to interpret.
Are there methylation differences between pediatric and adult ALL?
An obvious question was whether differences in prognosis known to differentiate adult from pediatric ALL could be related, in part, to DNA methylation: quantitatively (number of genes methylated) or qualitatively (differences in methylation patterns). The first report45 indicated that there were no obvious differences in terms of the frequency of methylation observed in children and adult ALL. However, there were several limitations to this study, as the number of patients with pediatric ALL studied was small and insufficient genotypic subsets of patients were studied. However, aberrant methylation of multiple genes is common in pediatric ALL45,46: the Spanish group also demonstrated that the high frequency of methylation in the younger group of patients and a potential correlation between methylation and prognosis32. These data were against expectations, founded on the concept that methylation increases with aging47 and therefore older patients should have a higher frequency of aberrant methylation. Prognostic differences between children and adults possibly are not related to quantitative methylation but to the inactivation of specific pathways. One example may be the p73/p15/p57 pathway38,48: epigenetic inactivation was observed in close to 25% of adult patients but was extremely rare in the younger patients48, despite that individual methylation of any of these three genes was not significantly different from the adult subgroup48. Although these results need to be confirmed in other larger cohort of patients, they suggest that prognostic differences between age groups could be related in part to differences in methylation patterns. Methylation of p16, a rare event in primary ALL30,49, has been shown to be present in pediatric cases with MLL alterations50. The same phenomenon has also been the cases for the FHIT gene51,52. Specific patterns and genetic associations may have a role in the prognosis of pediatric patients with ALL.
Can the analysis of DNA methylation be used as a marker of minimal residual disease?
Another question is whether methylation patterns at the time of relapse are stable in ALL (Figure 3); the answer would have important implications. If stable, it could be proposed that these methylation alterations are a key molecular component of the malignant clone, and the detection of residual levels of methylation might usefully indicate presence of residual leukemia in patients in morphological remission (Figure 3A). Methylation profiles of 5 genes (ER, MDR1, p73, p15 and p16) was determined in a group of patients before therapy and at the time of morphological relapse53. Overall, methylation patterns were stable in about 70% of patients. Genes such as p73 (92%), ER (88%) and MDR1 (72%) were stable at the time of relapse, whereas p15 was only concordant in around 60% of cases. Of interest, there was an increase in p16 methylation accompanied by gain of p15 methylation (Figure 3B), suggesting that gain of methylation may have a role in relapse/resistance mechanisms in ALL. Overall, this study indicated that methylation patterns are stable in a large fraction of patients with ALL, and therefore it might be possible to design minimal residual disease assays using detection of aberrant DNA methylation in ALL. Whether the methylation changes observed in the 30% of patients at relapse was the result of the emergence of a new clone or epigenetic alterations in the original clone remains uncertain but of with potential implications for our understanding of relapse dynamics and patterns of resistance.
Figure 3. Analysis of DNA methylation as a tool to detect minimal residual disease.

A. A model of methylation dynamics in ALL. Pretreatment bone marrow of patients with ALL should be populated by two or three clones: a dominant malignant clone (red), residual normal hematopoiesis (black) and potential a subdominant malignant clone (green). With intensive chemotherapy, most patients achieve complete remission and the marrow is now dominated by the normal clone. But because a large fraction of adult patients will relapse with conventional therapy it is possible that residual molecular levels of both the dominant and possibly the subdominant clone are present during remission. At the time of relapse approximately 70% of patients relapse with a methylation pattern similar to that of the initial presentation and 30% with a different profile53. B. Gene specific dynamics of 5 genes at the time of relapse53. Stable indicates that the methylation status of the gene does not change from initial presentation to the time of relapse. C. The data presented in A and B allows the hypothesis that detection of residual levels of methylation at the time of remission could predict worse outcomes. This has been shown for the p73 gene54.
Based on these data, detection of residual methylation as a predict or of relapse in ALL has been sought (Yang et al submitted)54, DNA was extracted from 199 patients with Ph negative ALL at the time of morphological remission (around day 14 to 21 after standard hyperCVAD based chemotherapy55). Three genes were analyzed: p15, p73 and p57, using a real-time bisulfite PCR assay especially developed for this analysis. Residual methylation of p73 was detected in 10% of patients, p15 in 17%, and p57 in 4%. In all, 25% of the patients had evidence of residual methylation. The presence of residual p73 methylation was associated with a significantly worse outcome (HR 2.68, p=−.003) (Figure 3C). Although these results are exploratory due to the limited number of genes analyzed, they show the feasibility of methylation based assays to detect residual leukemia, which could complement other flow or molecular assays. A more systematic analysis of genes in ALL may provide a useful tool to predict outcome in patients in remission with ALL. These results also allow the consideration of incorporating hypomethylating agents as consolidation/maintenance strategies in ALL.
Incorporating hypomethylating-based therapy in ALL
At the present time, two drugs with hypomethylating activity are approved in the US for patients with myelodysplastic syndrome7,8. These agents also have activity in AML56. In vitro data using cellular systems has indicated that the selective reactivation of a gene specifically inactivated in ALL (in this case p57) results in ALL cell death, only in cells in which the gene is epigenetically silenced and not in leukemia cells in which the gene is not methylated57. These results indicate that reactivation of epigenetically silenced genes can have an anti-leukemia effect, perhaps selective, and they reinforces consideration ofhypomethylating therapy in ALL, either as single agent or in combination with other standard forms of therapy exploiting possible synergistic effects58. There is only limited experience with the hypomethylating agents in ALL. In one trial59, relative high doses of 5-azacitidine (150 mg/m2 as a continous infusion daily x 5) were combined with cytarabine in patients that had previously failed cytarabine. The hypothesis was that treatment with 5-azacitidine could induce expression of deoxycytidine kinase. Two complete responses (CR) were observed in 17 patients treated. In another study60, decitabine was combined with either amsacrine or idarubicin in patients with acute leukemia. There was a CR rate of 36% (23 out 63) with a median disease free survival of 8 months. An ongoing trial is evaluating the role of decitabine in patients with advanced relapsed/refractory ALL61. In this study two phase 1 trials are conducted in parallel. In the first phase, patients are treated with a 5-day schedule of decitabine every other week. In the second phase, patients who had received decitabine in the first phase but did not respond or relapsed to single agent decitabine can then participate in a phase 1 study of decitabine and hyper-CVAD62 combination. The reasons for the more frequent schedule of decitabine in ALL were twofold: first, data from in vitro modeling indicating the activity of decitabine in ALL cell lines especially when using chronic exposure63, and, second, the rapid proliferative nature of the leukemia clone in patients with relapse disease. To date, doses up to 100 mg/m2 IV daily x 5 every other week have been safely administered, without excess toxicity and with evidence of clinical activity in patients with multiple relapse/refractory leukemia.
Summary and future research
The study of epigenetic alterations in ALL is transitioning from a relative obscure field of research to the potential development of new biomarkers and therapeutic alternatives for patients with this disease. Here, we have summarized data that multiple tumor suppressor genes and molecular pathways are inactivated in ALL. This information potentially can be utilized to predict response to therapy, detect patients at risk that are in morphological relapse, and to target the incorporation of hypomethylating agents in ALL. Large scale validation studies and trials are needed to confirm these early data and to allow its into clinical practice. The analysis of specific histone modification patterns, and the role of histone deacetylase inhibitors9 in combination with conventional chemotherapeutic agents58 or hypomethylating agents64,65, as well as the role of microRNAs4, should be studied in conjunction in ALL.
Acknowledgments
This work was supported by the Leukemia and Lymphoma Society of America, a Physician-Scientist Award from MD Anderson Cancer Center and NIH grants CA 100067, CA105771 and CA126457 (all to GGM) and by a Leukemia SPORE (P50 CA100632) Career Development Award (S-Q K)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 2.Rice JC, Allis CD. Code of silence. Nature. 2001;414:258–261. doi: 10.1038/35104721. [DOI] [PubMed] [Google Scholar]
- 3.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 4.Fabbri M, Garzon R, Andreeff M, Kantarjian HM, Garcia-Manero G, Calin GA. MicroRNAs and noncoding RNAs in hematological malignancies: molecular, clinical and therapeutic implications. Leukemia. 2008;22:1095–1105. doi: 10.1038/leu.2008.30. [DOI] [PubMed] [Google Scholar]
- 5.Benetti R, Gonzalo S, Jaco I, et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15:268–279. doi: 10.1038/nsmb.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 8.Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Manero G, Issa JP. Histone deacetylase inhibitors: a review of their clinical status as antineoplastic agents. Cancer Invest. 2005;23:635–642. doi: 10.1080/07357900500283119. [DOI] [PubMed] [Google Scholar]
- 10.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong HL, Byun HM, Kwan JM, et al. Rapid and quantitative method of allele-specific DNA methylation analysis. Biotechniques. 2006;41:734–739. doi: 10.2144/000112305. [DOI] [PubMed] [Google Scholar]
- 12.Ritter M, de Kant E, Huhn D, Neubauer A. Detection of DNA methylation in the calcitonin gene in human leukemias using differential polymerase chain reaction. Leukemia. 1995;9:915–921. [PubMed] [Google Scholar]
- 13.Leegwater PA, Lambooy LH, De Abreu RA, Bokkerink JP, van den Heuvel LP. DNA methylation patterns in the calcitonin gene region at first diagnosis and at relapse of acute lymphoblastic leukemia (ALL) Leukemia. 1997;11:971–978. doi: 10.1038/sj.leu.2400688. [DOI] [PubMed] [Google Scholar]
- 14.Thomas X, Teillon MH, Belhabri A, et al. Hypermethylation of calcitonin gene in adult acute leukemia at diagnosis and during complete remission. Hematol Cell Ther. 1999;41:19–26. doi: 10.1007/s00282-999-0019-5. [DOI] [PubMed] [Google Scholar]
- 15.Roman J, Castillejo JA, Jimenez A, et al. Hypermethylation of the calcitonin gene in acute lymphoblastic leukaemia is associated with unfavourable clinical outcome. Br J Haematol. 2001;113:329–338. doi: 10.1046/j.1365-2141.2001.02764.x. [DOI] [PubMed] [Google Scholar]
- 16.Batova A, Diccianni MB, Yu JC, et al. Frequent and selective methylation of p15 and deletion of both p15 and p16 in T-cell acute lymphoblastic leukemia. Cancer Res. 1997;57:832–836. [PubMed] [Google Scholar]
- 17.Iravani M, Dhat R, Price CM. Methylation of the multi tumor suppressor gene-2 (MTS2, CDKN1, p15INK4B) in childhood acute lymphoblastic leukemia. Oncogene. 1997;15:2609–2614. doi: 10.1038/sj.onc.1201428. [DOI] [PubMed] [Google Scholar]
- 18.Wong IH, Ng MH, Huang DP, Lee JC. Aberrant p15 promoter methylation in adult and childhood acute leukemias of nearly all morphologic subtypes: potential prognostic implications. Blood. 2000;95:1942–1949. [PubMed] [Google Scholar]
- 19.Corn PG, Kuerbitz SJ, van Noesel MM, et al. Transcriptional silencing of the p73 gene in acute lymphoblastic leukemia and Burkitt’s lymphoma is associated with 5′ CpG island methylation. Cancer Res. 1999;59:3352–3356. [PubMed] [Google Scholar]
- 20.Kawano S, Miller CW, Gombart AF, et al. Loss of p73 gene expression in leukemias/lymphomas due to hypermethylation. Blood. 1999;94:1113–1120. [PubMed] [Google Scholar]
- 21.Corn PG, Smith BD, Ruckdeschel ES, Douglas D, Baylin SB, Herman JG. E-cadherin expression is silenced by 5′ CpG island methylation in acute leukemia. Clin Cancer Res. 2000;6:4243–4248. [PubMed] [Google Scholar]
- 22.Issa JP, Zehnbauer BA, Civin CI, et al. The estrogen receptor CpG island is methylated in most hematopoietic neoplasms. Cancer Res. 1996;56:973–977. [PubMed] [Google Scholar]
- 23.Roman-Gomez J, Jimenez-Velasco A, Agirre X, et al. Transcriptional silencing of the Dickkopfs-3 (Dkk-3) gene by CpG hypermethylation in acute lymphoblastic leukaemia. Br J Cancer. 2004;91:707–713. doi: 10.1038/sj.bjc.6602008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jimenez-Velasco A, Roman-Gomez J, Agirre X, et al. Downregulation of the large tumor suppressor 2 (LATS2/KPM) gene is associated with poor prognosis in acute lymphoblastic leukemia. Leukemia. 2005;19:2347–2350. doi: 10.1038/sj.leu.2403974. [DOI] [PubMed] [Google Scholar]
- 25.Hoshino K, Quintas-Cardama A, Yang H, Sanchez-Gonzalez B, Garcia-Manero G. Aberrant DNA methylation of the Src kinase Hck, but not of Lyn, in Philadelphia chromosome negative acute lymphocytic leukemia. Leukemia. 2007;21:906–911. doi: 10.1038/sj.leu.2404615. [DOI] [PubMed] [Google Scholar]
- 26.San Jose-Eneriz E, Agirre X, Roman-Gomez J, et al. Downregulation of DBC1 expression in acute lymphoblastic leukaemia is mediated by aberrant methylation of its promoter. Br J Haematol. 2006;134:137–144. doi: 10.1111/j.1365-2141.2006.06131.x. [DOI] [PubMed] [Google Scholar]
- 27.Murai M, Toyota M, Satoh A, et al. Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br J Cancer. 2005;92:1165–1172. doi: 10.1038/sj.bjc.6602422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toyota M, Kopecky KJ, Toyota MO, Jair KW, Willman CL, Issa JP. Methylation profiling in acute myeloid leukemia. Blood. 2001;97:2823–2829. doi: 10.1182/blood.v97.9.2823. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Manero G, Daniel J, Smith TL, et al. DNA Methylation of Multiple Promoter-associated CpG Islands in Adult Acute Lymphocytic Leukemia. Clin Cancer Res. 2002;8:2217–2224. [PubMed] [Google Scholar]
- 31.Shteper PJ, Siegfried Z, Asimakopoulos FA, et al. ABL1 methylation in Ph-positive ALL is exclusively associated with the P210 form of BCR-ABL. Leukemia. 2001;15:575–582. doi: 10.1038/sj.leu.2402026. [DOI] [PubMed] [Google Scholar]
- 32.Roman-Gomez J, Jimenez-Velasco A, Castillejo JA, et al. Promoter hypermethylation of cancer-related genes: a strong independent prognostic factor in acute lymphoblastic leukemia. Blood. 2004;104:2492–2498. doi: 10.1182/blood-2004-03-0954. [DOI] [PubMed] [Google Scholar]
- 33.Kuang S-K, Tong W-T, Yang H, et al. Genome-Wide Identification of Aberrant Promoter Associated CpG island Methylation in Adult Acute Lymphocytic Leukemia Leukemia. doi: 10.1038/leu.2008.130. in press. [DOI] [PubMed] [Google Scholar]
- 34.Taylor KH, Pena-Hernandez KE, Davis JW, et al. Large-scale CpG methylation analysis identifies novel candidate genes and reveals methylation hotspots in acute lymphoblastic leukemia. Cancer Res. 2007;67:2617–2625. doi: 10.1158/0008-5472.CAN-06-3993. [DOI] [PubMed] [Google Scholar]
- 35.Roman-Gomez J, Cordeu L, Agirre X, et al. Epigenetic regulation of Wnt-signaling pathway in acute lymphoblastic leukemia. Blood. 2007;109:3462–3469. doi: 10.1182/blood-2006-09-047043. [DOI] [PubMed] [Google Scholar]
- 36.Kuang S-Q, Fang Z-H, Lopez G, Tong W-G, Yang H, Garcia-Manero G. Eph Receptor Tyrosine Kinases and Ephrin Ligands Are Epigenetically Inactivated in Acute Lymphoblastic Leukemia and Are Potential New Tumor Suppressor Genes in Human Leukemia. Blood. 2007;110:2128. [Google Scholar]
- 37.Taylor KH, Kramer RS, Davis JW, et al. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res. 2007;67:8511–8518. doi: 10.1158/0008-5472.CAN-07-1016. [DOI] [PubMed] [Google Scholar]
- 38.Shen L, Toyota M, Kondo Y, et al. Aberrant DNA methylation of p57KIP2 identifies a cell-cycle regulatory pathway with prognostic impact in adult acute lymphocytic leukemia. Blood. 2003;101:4131–4136. doi: 10.1182/blood-2002-08-2466. [DOI] [PubMed] [Google Scholar]
- 39.Kikuchi T, Toyota M, Itoh F, et al. Inactivation of p57KIP2 by regional promoter hypermethylation and histone deacetylation in human tumors. Oncogene. 2002;21:2741–2749. doi: 10.1038/sj.onc.1205376. [DOI] [PubMed] [Google Scholar]
- 40.Blint E, Phillips AC, Kozlov S, Stewart CL, Vousden KH. Induction of p57(KIP2) expression by p73beta. Proc Natl Acad Sci U S A. 2002;99:3529–3534. doi: 10.1073/pnas.062491899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bueso-Ramos C, Xu Y, McDonnell TJ, et al. Protein expression of a triad of frequently methylated genes, p73, p57Kip2, and p15, has prognostic value in adult acute lymphocytic leukemia independently of its methylation status. J Clin Oncol. 2005;23:3932–3939. doi: 10.1200/JCO.2005.02.998. [DOI] [PubMed] [Google Scholar]
- 42.Roman-Gomez J, Castillejo JA, Jimenez A, et al. 5′ CpG island hypermethylation is associated with transcriptional silencing of the p21(CIP1/WAF1/SDI1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood. 2002;99:2291–2296. doi: 10.1182/blood.v99.7.2291. [DOI] [PubMed] [Google Scholar]
- 43.Shen L, Kondo Y, Issa JP, Garcia-Manero G. Lack of p21(CIP1) DNA methylation in acute lymphocytic leukemia. Blood. 2002;100:3432–3433. doi: 10.1182/blood-2002-07-1990. author reply 3433–3434. [DOI] [PubMed] [Google Scholar]
- 44.Ying J, Srivastava G, Gao Z, et al. Promoter hypermethylation of the cyclin-dependent kinase inhibitor (CDKI) gene p21WAF1/CIP1/SDI1 is rare in various lymphomas and carcinomas. Blood. 2004;103:743–746. doi: 10.1182/blood-2003-09-3193. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Manero G, Jeha S, Daniel J, et al. Aberrant DNA methylation in pediatric patients with acute lymphocytic leukemia. Cancer. 2003;97:695–702. doi: 10.1002/cncr.11090. [DOI] [PubMed] [Google Scholar]
- 46.Gutierrez MI, Siraj AK, Bhargava M, et al. Concurrent methylation of multiple genes in childhood ALL: Correlation with phenotype and molecular subgroup. Leukemia. 2003;17:1845–1850. doi: 10.1038/sj.leu.2403060. [DOI] [PubMed] [Google Scholar]
- 47.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 48.Canalli AA, Yang H, Jeha S, et al. Aberrant DNA methylation of a cell cycle regulatory pathway composed of P73, P15 and P57KIP2 is a rare event in children with acute lymphocytic leukemia. Leuk Res. 2005;29:881–885. doi: 10.1016/j.leukres.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 49.Herman JG, Civin CI, Issa JP, Collector MI, Sharkis SJ, Baylin SB. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res. 1997;57:837–841. [PubMed] [Google Scholar]
- 50.Nakamura M, Sugita K, Inukai T, et al. p16/MTS1/INK4A gene is frequently inactivated by hypermethylation in childhood acute lymphoblastic leukemia with 11q23 translocation. Leukemia. 1999;13:884–890. doi: 10.1038/sj.leu.2401437. [DOI] [PubMed] [Google Scholar]
- 51.Stam RW, den Boer ML, Passier MM, et al. Silencing of the tumor suppressor gene FHIT is highly characteristic for MLL gene rearranged infant acute lymphoblastic leukemia. Leukemia. 2006;20:264–271. doi: 10.1038/sj.leu.2404074. [DOI] [PubMed] [Google Scholar]
- 52.Zheng S, Ma X, Zhang L, et al. Hypermethylation of the 5′ CpG island of the FHIT gene is associated with hyperdiploid and translocation-negative subtypes of pediatric leukemia. Cancer Res. 2004;64:2000–2006. doi: 10.1158/0008-5472.can-03-2387. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Manero G, Bueso-Ramos C, Daniel J, Williamson J, Kantarjian HM, Issa JP. DNA methylation patterns at relapse in adult acute lymphocytic leukemia. Clin Cancer Res. 2002;8:1897–1903. [PubMed] [Google Scholar]
- 54.Garcia-Manero G, Bueso-Ramos C, Xiao L, et al. Detection of Residual p73 DNA Methylation Predicts for Shorter Disease Free and Overall Survival in Patients (pts) with Philadelphia (Ph) Chromosome Negative Acute Lymphocytic Leukemia (ALL) in Remission. Blood. 2006;108:abstract 2333. [Google Scholar]
- 55.Kantarjian HM, O’Brien S, Smith TL, et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J Clin Oncol. 2000;18:547–561. doi: 10.1200/JCO.2000.18.3.547. [DOI] [PubMed] [Google Scholar]
- 56.Silverman LR, McKenzie DR, Peterson BL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006;24:3895–3903. doi: 10.1200/JCO.2005.05.4346. [DOI] [PubMed] [Google Scholar]
- 57.Kuang SQ, Ling X, Sanchez-Gonzalez B, Yang H, Andreeff M, Garcia-Manero G. Differential tumor suppressor properties and transforming growth factor-beta responsiveness of p57KIP2 in leukemia cells with aberrant p57KIP2 promoter DNA methylation. Oncogene. 2007;26:1439–1448. doi: 10.1038/sj.onc.1209907. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez-Gonzalez B, Yang H, Bueso-Ramos C, et al. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood. 2006;108:1174–1182. doi: 10.1182/blood-2005-09-008086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Avramis VI, Mecum RA, Nyce J, Steele DA, Holcenberg JS. Pharmacodynamic and DNA methylation studies of high-dose 1-beta-D-arabinofuranosyl cytosine before and after in vivo 5-azacytidine treatment in pediatric patients with refractory acute lymphocytic leukemia. Cancer Chemother Pharmacol. 1989;24:203–210. doi: 10.1007/BF00257619. [DOI] [PubMed] [Google Scholar]
- 60.Willemze R, Suciu S, Archimbaud E, et al. A randomized phase II study on the effects of 5-Aza-2′-deoxycytidine combined with either amsacrine or idarubicin in patients with relapsed acute leukemia: an EORTC Leukemia Cooperative Group phase II study (06893) Leukemia. 1997;11 (Suppl 1):S24–27. [PubMed] [Google Scholar]
- 61.Garcia-Manero G, Thomas D, Rytting M, et al. Phase I Study of 5-aza-2′-Deoxycitidine, Alone or in Combination with Hyper-CVAD, in Relapsed or Refractory Acute Lymphocytic Leukemia (ALL) Blood. 2007;110:abstract 2826. [Google Scholar]
- 62.Garcia-Manero G, Kantarjian HM. The hyper-CVAD regimen in adult acute lymphocytic leukemia. Hematol Oncol Clin North Am. 2000;14:1381–1396. x–xi. doi: 10.1016/s0889-8588(05)70192-1. [DOI] [PubMed] [Google Scholar]
- 63.Yang H, Hoshino K, Sanchez-Gonzalez B, Kantarjian H, Garcia-Manero G. Antileukemia activity of the combination of 5-aza-2′-deoxycytidine with valproic acid. Leuk Res. 2005;29:739–748. doi: 10.1016/j.leukres.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 64.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soriano AO, Yang H, Faderl S, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]