

Summary
Fibronectin and its major receptor, integrin α5β1 are required for embryogenesis. These mutants have similar phenotypes, although, defects in integrin α5-deficient mice are milder. In this paper, we examined heart development in those mutants, in which the heart is formed, and discovered that both fibronectin and integrin α5 were required for cardiac morphogenesis, and in particular, for the formation of the cardiac outflow tract. We found that Isl1+ precursors are specified and migrate into the heart in fibronectin- or integrin α5- mutant embryos, however, the hearts in these mutants are of aberrant shape, and the cardiac outflow tracts are short and malformed. We show that these defects are likely due to the requirement for cell adhesion to fibronectin for proliferation of myocardial progenitors and for Fgf8 signaling in the pharyngeal region.
Keywords: fibronectin, integrin a5, heart, cardiac, outflow tract, morphogenesis
Introduction
Cardiovascular morphogenesis is a complex process that requires extensive communication and interactions between progenitors and cells derived from all germ layers (Rentschler et al., 2010). Cellular components of the three germ layers and the neural crest participate in these interactions, and signaling by growth factors from and to specific tissues in proximity with cardiac progenitors is important (Astrof, 2013). However, how various signaling pathways are orchestrated and integrated during cardiac development is not well understood.
Gene knock out studies have shown that extracellular matrix (ECM) proteins play essential roles in organ morphogenesis in vivo (Aszodi et al., 2006; Wickstrom et al., 2011). For example, laminins containing γ1 or β1 chains are essential for the generation of endodermal cell polarity and formation of the Reichert’s membrane, and their absence leads to peri-implantation lethality (Miner et al., 2004; Miner and Yurchenco, 2004); Fibronectin (FN), fibulin-1, collagen V are required for vascular development (Aszodi et al., 2006). In addition, ECM proteins have long been known to modulate growth factor signaling and influence cell fate decisions through interactions with their cellular receptors such as integrins (Hynes, 2009). For example, mesenchymal stem cells adopt different cell fates depending on the stiffness of the ECM substrate (Engler et al., 2006). Endothelial cells plated on fibronectin (FN) proliferate, while those plated on laminin 111, exit from cell cycle and form endothelial tubes/vascular channels (Giancotti and Tarone, 2003). However, despite exciting new progress in understanding the roles of ECM proteins in vivo (Lindsay and Dietz, 2011), molecular mechanisms underlying functions of the majority of the ECM components in vivo are not well-understood.
FN is a large ECM glycoprotein encoded by a single gene in mice and humans. FN is unique to vertebrates (Hynes, 2012). It has been hypothesized that FN evolved with vertebrate features such as closed, endothelial-lined vasculature, the multi-potent neural crest and bones and is important for the development and function of these vertebrate innovations (Hynes, 2012). Indeed, global FN-null mouse embryos die by mid-gestation with severe defects in vascular and neural crest development, and conditional ablation of FN protein from the blood plasma leads incomplete blood clotting and aberrant wound healing following brain ischemia (George et al., 1997; Mittal et al., 2010; Ni et al., 2003; Sakai et al., 2001).
In order to understand molecular roles of FN in vertebrate morphogenesis, one first needs to understand the features of embryonic development dependent on FN’s function. In our previous work, we reported that FN is required for the morphogenesis of the mouse node and establishment of the left-right asymmetry at the early stages of mouse development (Pulina et al., 2011). We found that FN is also important for the maintenance of the embryonic left-right body plan by regulating expression of Lefty1 at the midline and maintaining integrity of the notochord. FN is also required for the proper development of the heart and early embryonic and extraembryonic blood vessels (George et al., 1997; George et al., 1993; Georges-Labouesse et al., 1996). In addition, prior experiments indicated that FN is important for formation of a single cardiac tube from bilateral cardiac progenitors both in zebrafish and in mice (George et al., 1997; Trinh and Stainier, 2004).
We demonstrated that at later stages of development, in those mouse embryos, in which heart tube is formed, FN is required for proliferation and survival of the cardiac neural crest (Mittal et al., 2010). However, particular aspects of cardiac development affected by the absence of FN in these mice have not been reported. Experiments in this paper were designed to analyze cardiac morphogenesis in FN-null mutants, in which a single cardiac tube has formed. We show that FN is required for morphogenesis of the cardiac outflow tract. Furthermore, our studies suggest that this effect is due to the requirement for cell adhesion to FN for proliferation of pre-cardiac mesoderm and for Fgf8 signaling, and that integrin α5β1 is a major mediator of FN signaling during cardiac development.
Materials and methods
Embryos
FN-null and control embryos were obtained from matings of FN-het mice (George et al., 1993). Similarly, integrin α5-null embryos were obtained from matings of integrin α5-het mice (Yang et al., 1993). For in situ hybridization, embryos were fixed in 4% paraformaldehyde at 4°C overnight, dehydrated and stored in 100% methanol at −20°C. Plasmids encoding probes to detect ANF, Wnt11, and BMP4 were obtained from Drs. Benoit Bruneau, Sylvia Evans and Brigid Hogan, respectively. Cited1 mouse cDNA was from Open Biosystems, and Cyclin D2 fragment was obtained by PCR using mouse genomic DNA and primers 5′-gatatgcggccgcGGTCTTTTAGAAGTGAGAGG-3′ and 5′-gaattcgtcgacCCGTCTATGGTCTCCATCTGCC-3′, and the 666-bp DNA fragment was subcloned into pBS-KS II vector using Not I and Sal I restriction sites. DIG-labeled probes were synthesized and in situ hybridization was performed according to standard methods (Henrique et al., 1995).
Scanning electron microscopy
Embryos were collected at E9.5 and fixed in 0.25% glutaraldehyde in PBS for at least 48 h at 4 °C, dehydrated in graded series of ethanol and stored in 100% ethanol at −20 °C until use. Embryos were critical-point dried and mounted using a sewing needle onto a double-sided adhesive tape attached to a metal stub. Embryos were then coated with gold/palladium in a DentonVacuum Desk IV sputter coater and photographed using Zeiss Field Emission Scanning Electron Microscope Supra 25.
Fate mapping studies Isl1Cre/+
mice were mated with either FN-het or integrin α5-het mice. FN+/−; Isl1Cre/+ males were mated with FN+/−; R26R/R26R females to obtain FN−/ −; R26R; Isl1Cre/+ embryos and controls. Alternatively, integrin α5+/−; Isl1Cre/+ males were mated with integrin α5+/−; R26R/R26R females. Embryos were collected at E9.5, fixed and stained using bluo-gal (Sigma) to detect LacZ according to standard protocols. R26R reporter mice were obtained from the Jackson labs, cat #3504. This strain exhibits weak LacZ activity following Cre expression in embryos.
Immunostaining and measurements
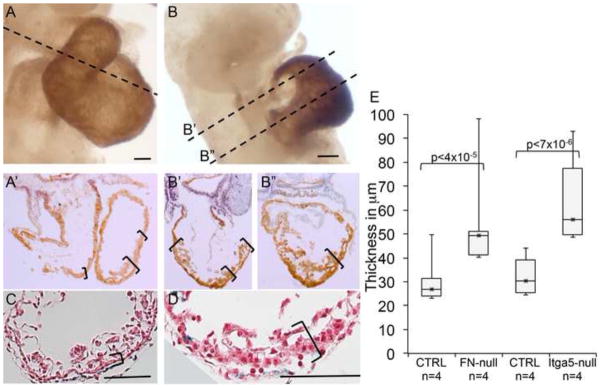
Embryos collected at E8.75–E9.5 were fixed in 4% PFA overnight at 4°C, and washed in PBS. MF20 antibody (Iowa developmental hybridoma bank) was used for staining at 1:100 dilution. Primary antibody was detected using anti-mouse secondary antibody conjugated to horseradish peroxidase, and detected using DAB (Vector labs). Stained embryos were dehydrated, embedded into paraffin, sectioned, boiled in citric acid buffer and stained using anti-Isl1 antibody 39.4D5, obtained from the Iowa Developmental Hybridoma Bank. Staining using anti-pHH3 antibody and anti-cleaved caspase 3 antibody was done as described (Mittal et al., 2010). Proliferation was quantified by counting splanchnic mesodermal cells (based on histological of DAPI-labeled nuclei) and pHH3+ cells among them. Serial sections from 4 control and 5 mutant embryos isolated at E8.75 were used. At this stage, control and mutant embryos analyzed looked morphologically similar (Mittal et al., 2010). Thickness of the myocardium in control and FN-null embryos was measured using serial sections from 4 control and 4 mutant E8.75 embryos. Ventral wall of the left ventricle was measured in all cases. Similarly, the thickness of ventricular walls in controls (n=4) and integrin α5-null embryos (n=4) isolated at E9.0 was analyzed. Similar looking control and integrin α5-null embryos were chosen for these analyses (Mittal et al., 2010). Box plots were generated using Excel. The error bars span the data from the minimum to the maximum numbers. The box contains the data points within the second and third quartiles. The line through the box marks the median.
Gene set enrichment analysis
Gene expression analyses in this paper were reported in (Astrof et al., 2007). In brief, we generated labeled mRNA from 8 FN-null mutants (4 embryos isolated from C57BL/6J strain and 4 embryos from 129S4 strain) and 7 control embryos isolated at the 6th somite stage and derived from C57BL/6J (n=4) and 129S4 genetic background. The data reported here represents the analysis of all FN-null vs all wild-type embryos, and represents changes in gene expression between mutants and controls independent of genetic background. In order to find pathways and biological processes enriched in our data set, we used DAVID Bioinformatics Resources 6.7 (http://david.abcc.ncifcrf.gov/) and the list of 609 genes, which were statistically significantly upregulated more than 1.7 fold in FN-null mutants compared with controls, and had non-redundant DAVID IDs (See supplemental file FN_upregulaed.xls).
Results
Early studies indicated that FN-null mutants die before mid-gestation due to pronounced cardiovascular defects, the severity of which is dependent on the genetic background (Astrof et al., 2007; George et al., 1997). In 129S4 genetic background, FN is required for the coalescence of the bilateral cardiac primordia to form a single heart tube, such that FN-null embryos in this strain exhibit cardia bifida. However, in C57BL/6J genetic background, FN-null embryos contain a single heart tube (Astrof et al., 2007; George et al., 1997). This difference in phenotype between the two strains was mapped to a one-mega base interval on mouse chromosome 4 (Astrof et al., 2007).
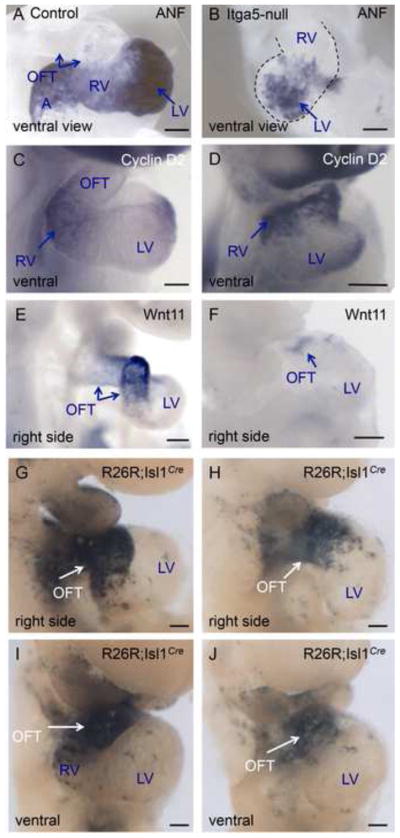
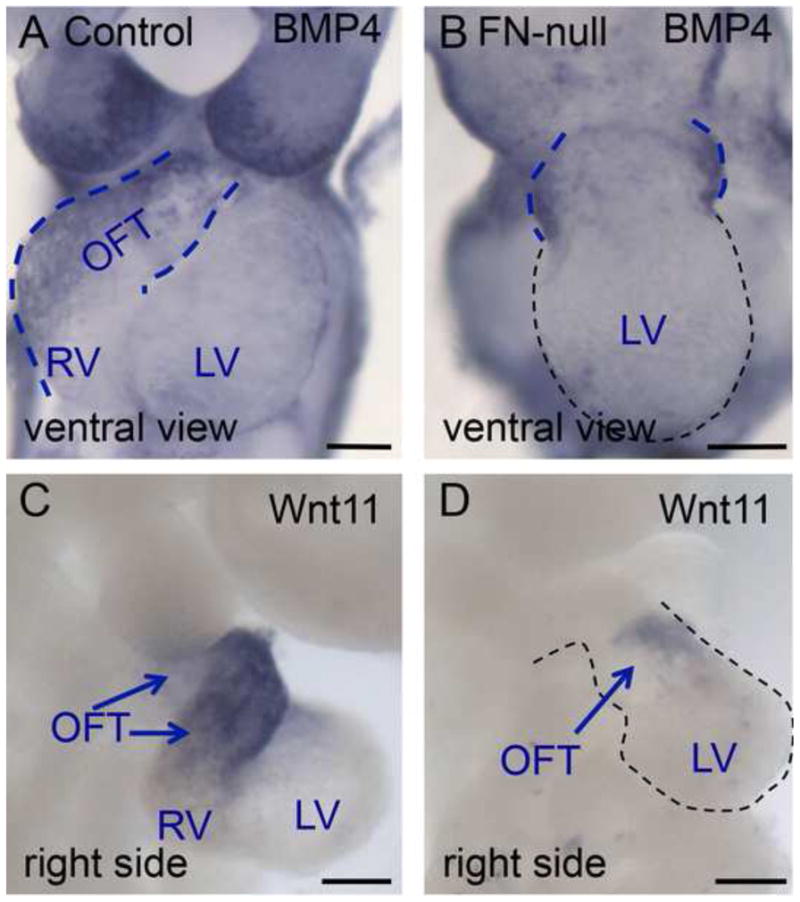
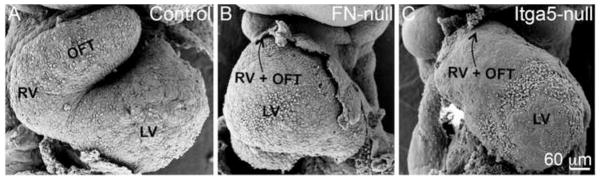
However, even though the beating heart formed in FN-null mutants in C57BL/6J background, we noticed that it was not normal (Fig. 1). Scanning electron microscopy (SEM) showed that FN-null mutants contain a linear heart tube lacking distinctive left and right ventricles, and the stereotypical outflow tract (Fig. 1B). In order to investigate cardiac development in FN-null embryos, we performed in situ hybridization (ISH) to study gene expression patterns of cardiac-expressed genes. At E9.5, atrial natriuretic factor (ANF) is mainly expressed in the left ventricle, atria and to a lesser extent in the right ventricle (Fig. 2A, C). ISH staining of FN-null embryos showed that this pattern is also preserved in the mutants (Fig. 2B, D). Cited1 is expressed in the left ventricle in control E9.5 embryos (Fig. 2E). And while we don’t see a morphologically-distinct left ventricle in FN-null embryos, we observed a localized expression of Cited1 mRNA in the ventral part of the linear heart tube in FN-null hearts, indicating that left-ventricular identity is contained within the lower part of the linear heart tube in these mutants (Fig. 2F). Investigation of BMP4 and Wnt11 mRNA expression demonstrated that segments containing the cardiac outflow tract and the right ventricle identities are present but are much smaller in FN-null mutants compared with controls (Fig. 3).
Figure 1. FN and integrin α5 are required for cardiac morphogenesis.
Scanning electron micrographs of control (A), FN-null (B) and integrin α5-null (C) embryos isolated at E9.5. All views are ventral. LV-left ventricle, RV-right ventricle, OFT – outflow tract. In B and C, arrow points toward the connection of the heart with the systemic circulation. Note dysmorphic and short RV and OFT in the mutants. Magnification is the same in all panels.
Figure 2. FN is not required for cardiac chamber specification.
A–D. Expression of ANF mRNA in control (A, C) and FN-null (B, D) hearts. ANF is expressed in the atria, the left ventricle and to a limited extent, in the right ventricle. The expression patterns of ANF in control and FN-null embryos are comparable. Long arrow in A points to the proximal OFT, and short arrow – to the distal. E. Cited 1 mRNA is confined to the left ventricle in control embryos. Inset in E shows the right side view. F. Expression of Cited 1 mRNA in FN-null heart is limited to the lower region of the straight heart tube, the presumptive left ventricle. E9.5 embryos are shown. A- atria, other abbreviations are as in Fig. 1. Scale bars in all figures are 100 μm unless otherwise stated.
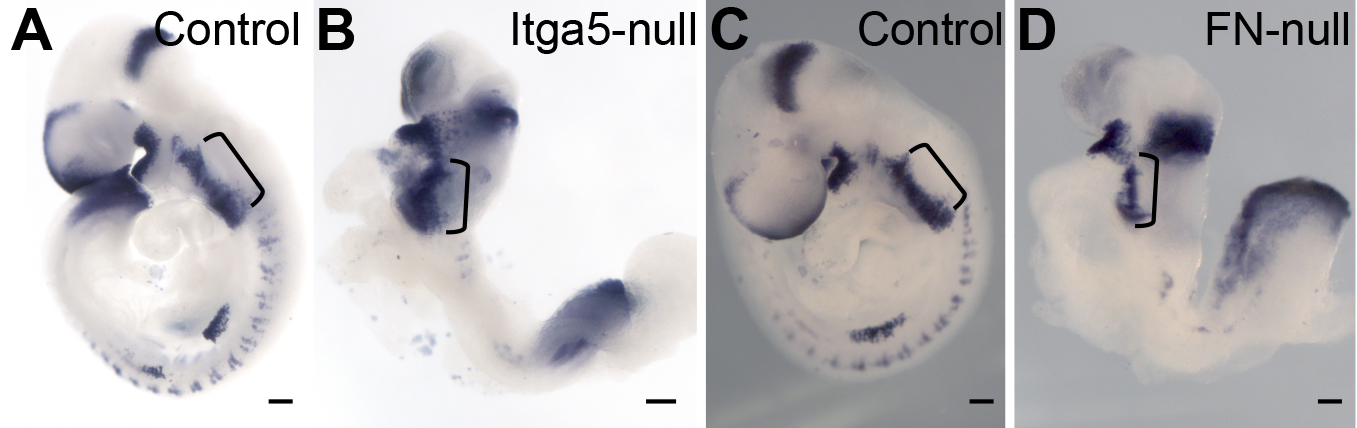
Figure 3. The outflow tract and the right ventricle are specified in the absence of FN.
BMP4 mRNA is expressed in the outflow tract of control embryo, blue dotted line (A). In FN-null, BMP4 expression is limited to the short OFT connecting the LV to the embryo, blue dotted line. C–D. Expression of Wnt11 mRNA. Wnt11 is expressed in the OFT of control embryo, arrows (C) and in the short OFT of FN-null. All embryos were collected at E9.5.
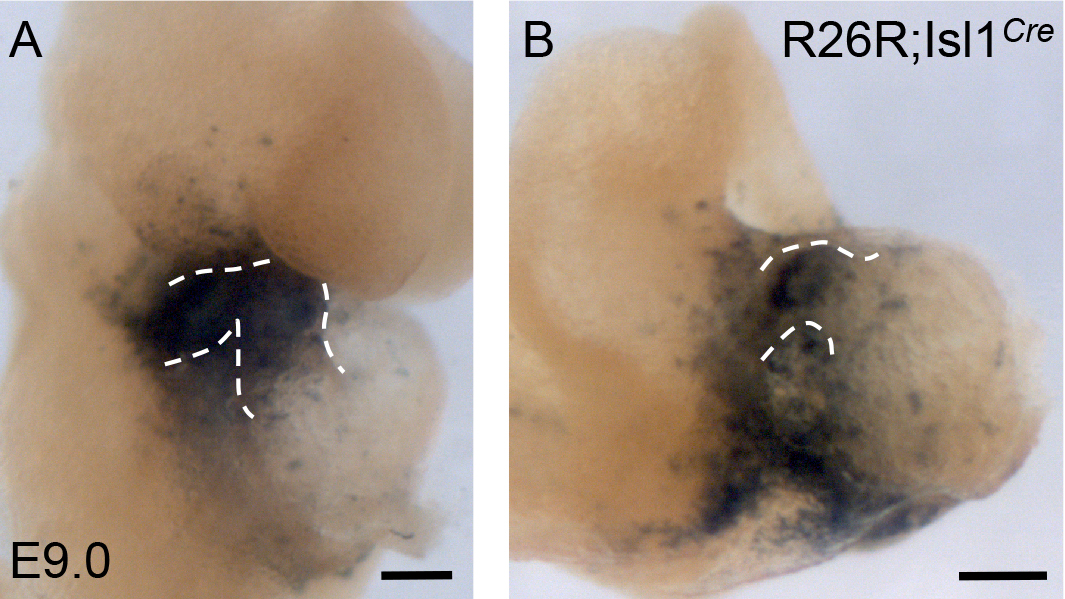
Shortened outflow tract and right ventricle have been observed in Isl1-null, FoxH1-null and Fgf8-hypomorphic mutants (Cai et al., 2003; Meyers et al., 1998; von Both et al., 2004). These mutants contain hypoplastic right ventricles and short outflow tracts, and similar phenotype of FN-null mutants suggest that FN is important for the development of these structures. Cell adhesion to FN in vitro permits cellular migration, therefore, it was possible that the short outflow tracts in FN-null mutants resulted from defective migration of cardiac progenitors into the cardiac outflow tract. To examine this possibility, we checked the location of Isl1+ cells and their descendants in FN-null mutants, using Isl1Cre knock in strain of mice and R26R reporter mice bred into FN-null background. The use of a weak R26R reporter variant (Jackson lab, cat #3504) allowed us to follow the fate of the cardiac progenitors comprising the anterior/second heart field, while progenitors of the first heart field, contributing the to left ventricle remained mostly unlabeled. Similar to controls, mutant Isl1+ cells and their descendants where already present in the heart’s outflow tracts at E9.0 (Supplemental Fig. 1). At e9.5, Isl1+ cells and their descendants populated the hearts of controls and mutants, however, the outflow portion of the mutant hearts (the segment connecting the left ventricle to the arterial circulation) was abnormally short (Fig. 4). Histological sectioning of control and FN-null embryos showed that Isl1+ cells and their descendants are present at the right embryonic positions within pharyngeal arches and the outflow tract, and that there is no aberrant accumulation of Isl1-labeled cells in the mutants (Fig. 4E, F). We also did not observe ectopic Isl1+ cells located outside their expected positions in the embryos. Taken together, these studies indicate that the migration of Is1+ cells into the heart occurs in the expected spatio-temporal manner and does not depend on FN. These observations are consistent with previous studies demonstrating that FN is not required for cell migration in vivo (George et al., 1997; Georges-Labouesse et al., 1996).
Figure 4. FN is required for the morphogenesis of the cardiac outflow tract and the right ventricle.
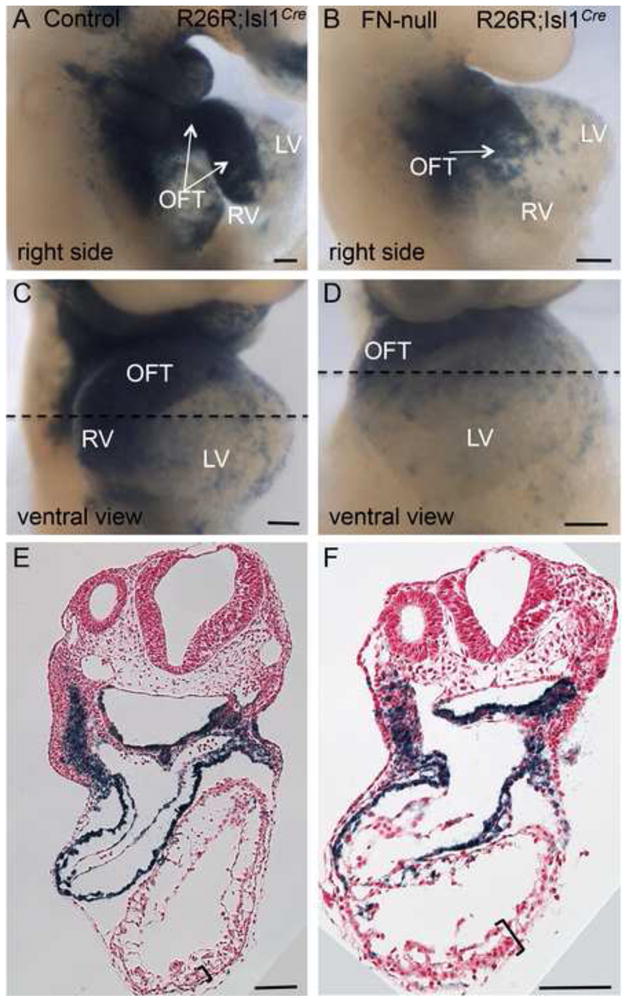
Fate mapping of Isl1+ cells and their descendants in control (A, C, E) and FN-null (B, D, F) embryos isolated at E9.5. Note the presence of blue cells (β-gal+) in the outflow tracts of the mutants. Dotted line is the plane of sections shown in E–F. Regions outlined by brackets in E–F show H&E stained sections of thickened myocardial wall in FN-null embryos, they are expanded in Fig. 6.
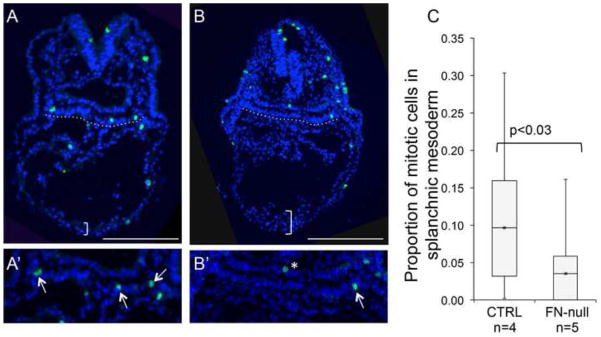
Short outflow tracts could be due to defective proliferation or survival of myocardial cells or their precursors. Aberrant cell survival is unlikely to contribute to the observed defect in the outflow tract because we found only 10 cleaved caspase 3-positive cells (6 in the myocardium and 4 in the splanchnic and pharyngeal mesoderm) when we examined 59 serial sections from 5 FN-null embryos isolated at E8.75. Moreover, our previous studies showed no increased apoptosis in Isl1+ mesodermal cells in FN-null embryos (Mittal et al., 2010). However, we detected nearly three-fold decrease in cell proliferation within the splanchnic mesoderm of our mutants (n=5) compared with controls (n=4) at E8.75 (Fig. 5). These observations suggest that short outflow tracts in FN-null embryos result from defective proliferation of splanchnic mesodermal cells, known to harbor cardiomyocyte precursors.
Figure 5. FN is required for proliferation of cells in splanchnic mesoderm.
A–B. E8.75 control (A) and mutant (B) embryos were stained with anti-pHH3 antibody (green) and DAPI (blue). Representative sections are shown. Splanchnic mesoderm is underlined by the dotted lines and shown enlarged in A′–B′. C. Quantification of the fraction of pHH3+ labeled cells in splanchnic mesoderm. 4–5 sections from each embryo were analyzed, total of 413 control and 537 mutant nuclei were counted per genotype.
Even though the outflow tracts and the right ventricles were small in our mutants, we noticed that overall, hearts in FN-null embryos were disproportionately large compared to the overall embryo size (Astrof et al., 2007). Indeed measuring the thickness of left ventricles at their apex/thickest part, we found aberrant thickening of ventricular walls in FN-null mutants (n=4) compared with controls (n=4) (Fig. 6). We hypothesized that the thickened myocardial wall was due to the presence of additional layers of cardiomyocytes, and stained control and mutant embryos using the MF20 antibody recognizing heavy chain of myosin II. Our experiments indicate the presence of ectopic MF20+ cardiomyocytes in mutants, and show that FN and integrin α5 are not required for cardiomyocyte differentiation (Fig. 6 and data not shown) consistent with previous observations (George et al., 1997; Georges-Labouesse et al., 1996). Moreover, mRNA expression profiling studies comparing FN-null embryos and controls consisting of wild-type and heterozygous embryos isolated at E8.0 (6 somites) demonstrated a high, statistically significant enrichment in genes with a role in cardiomyocyte function and hypertrophic cardiomyopathy among the upregulated genes in FN-null embryos (Table 1). We did not observe increased myocardial proliferation in FN-null embryos, suggesting that ectopic cardiomyocytes could have resulted from aberrant differentiation of cardiac precursors in FN-null embryos, or aberrant allocation of cardiac precursors into the first heart field vs the SHF. Taken together, these studies demonstrate a requisite role of FN in proliferation of the splanchnic mesodermal cells, descendants of which form the cardiac outflow tract, and even though distinct cardiac parts could be identified due to marker expression and fate mapping, we found that FN is required for the overall morphogenesis of the proper cardiac shape.
Figure 6. FN is required for normal ventricular morphogenesis.

A–B. Expression of myosin heavy chain is visualized using MF20 antibody. Dotted lines show planes of sections in A′, B′ and B″ below. Blue nuclei show expression of Isl1 protein. Note thickened myocardial wall in the hearts of FN-null embryos (B′, B″, D) compared with controls (A′, C,). E. Quantification of myocardial thickness in serial sections from control and mutant embryos. Ventral ventricular walls in 4–6 sections of serialy-sectioned embryos were measured. In this figure, H&E panels C and D are magnified views of regions indicated by the brackets in Fig. 4E–F.
Table 1.
Biological processes enriched among the genes upregulated more than 1.7 fold in FN-null embryos (n=8) compared with controls (n=7). P is the value of probability adjusted for multiple testing using the FDR (false discovery rate) correction. The last column lists genes in each GO category upregulated in FN-null mutants.
| GO Category | Fold enrichment | P (FDR) | Genes |
|---|---|---|---|
| Myofibril | 14 | 5.22E-11 | ACTA1, MYL2, CRYAB, MYBPC3, LDB3, ACTN2, MYH7, MYH6, TNNI3, TTN, TRIM63, CSRP3, PGM5, ITGB1BP2, RYR2, SLC4A1, MYOM |
| Contractile fiber | 13.6 | 1.06E-10 | ACTA1, MYL2, CRYAB, MYBPC3, LDB3, ACTN2, MYH7, MYH6, TNNI3, TTN, TRIM63, CSRP3, PGM5, ITGB1BP2, RYR2, SLC4A1, MYOM1 |
| Sarcomere | 15 | 1.25E-10 | ACTA1, CRYAB, MYBPC3, LDB3, ACTN2, MYH7, MYH6, TNNI3, TTN, TRIM63, CSRP3, PGM5, ITGB1BP2, RYR2, SLC4A1, MYOM1 |
| Z disk | 16.5 | 8.58E-06 | PGM5, CRYAB, ITGB1BP2, LDB3, RYR2, ACTN2, SLC4A1, TTN, TRIM63, CSRP3 |
| I band | 14 | 3.19E-05 | PGM5, CRYAB, ITGB1BP2, LDB3, RYR2, ACTN2, SLC4A1, TTN, TRIM63, CSRP3 |
| Myosin complex | 12 | 1.14E-04 | MYL4, MYL2, DYNLL2, MYL3, MYBPC3, MYH7, MYH6, MYOM1, TTN, MYL9 |
| Actin cytoskeleton | 5.5 | 6.32E-04 | MYL4, ACTA1, MYL2, MYL3, MYBPC3, ACTN2, MYH7, MYH6, TTN, MYL9, ANXA2, ARPC1B, ARPC3, DYNLL2, MYOM1 |
| Hypertrophic cardiomyopathy | 7 | 0.009 | SLC8A1, MYL2, MYL3, MYBPC3, RYR2, MYH7, MYH6, TNNI3, TTN, CACNA2D2 |
In order to examine potential downstream mediators of FN signal in the outflow tract development, we tested the role of integrin α5β1 in this process. Integrins are heterodimers of one alpha and one beta chains, and constitute a major class of cellular receptors that bind ECM proteins and mediate their intracellular signaling. α5β1 integrin heterodimer is a major cellular receptor for FN during early embryogenesis, and genetic ablation of integrin α5 gives rise to similar, albeit milder, morphogenetic defects, leading to embryonic lethality by mid gestation as seen with FN-null mutants (Yang et al., 1999; Yang et al., 1993).
Heart development in integrin α5-null mouse mutants has not been investigated in detail, although, our prior studies demonstrated aberrant cardiac looping in α5-null mice and α5-mutant zebrafish due to the requirement of integrin α5 in the establishment of the left-right embryonic body plan (Pulina et al., 2011). In order to investigate the role of integrin α5 in cardiac development, we examined the hearts of α5-null mice using SEM and ISH (Figs. 1 and 7). These analyses demonstrated that similar to FN, integrin α5 was required for morphogenesis of the heart’s proper shape. ISH experiments demonstrated chamber-specific gene expression was preserved in integrin α5-null hearts, however, hearts were misshapen and portions of the heart corresponding to the outflow tract and the right ventricle were greatly reduced (Fig. 7A–F).
Figure 7. Integrin α5 is not required for specification of cardiac chambers.
Control embryos-A, C, E, G, I; integrin α5-nulls – B, D, F, H, J. A–F. In situ hybridization. G–J. Fate mapping of Isl1+ cells. Abbreviations are as in Fig. 1. Scale bars are 100 μm.
We next investigated the fate of the Isl1+ cardiac progenitors in integrin α5-null mutants, and found that Isl1+ cells and their descendants migrate into the heart, and form a short stalk connecting the left ventricle with the arterial circulation (Fig. 7G–J). Histological sectioning indicated increased thickness of the myocardial wall due to ectopic presence of MF20+ cardiomyocytes (Fig. 6E and data not shown). Taken together these studies indicate the indispensible role of integrin α5β1 in cardiac morphogenesis. The similarity in the phenotypes of FN-null and integrin α5-null mutants, and prior literature indicating the role of integrin α5 in transducing FN signaling (George and Hynes, 1994; Giancotti and Tarone, 2003; Yang et al., 1999; Yang et al., 1993), suggest that integrin α5β1 mediates FN signaling during cardiac morphogenesis.
Cell adhesion to FN mediated by integrin α5β1 has long been known to regulate signaling by receptor tyrosine kinases, including Fgf receptors (Miyamoto et al., 1996). In addition, FN binds all known Fgf ligands (Martino and Hubbell, 2010) and syndecan-4 (Saoncella et al., 1999), a co-receptor required for Fgf and FN-integrin mediated signaling. Thus we hypothesized that FN modulates outflow tract development by regulating Fgf signaling. Fgf8 is the main Fgf isoform regulating early cardiac development and severe Fgf8 hypomorphs exhibit morphological defects similar to those observed in FN-null and integrin α5-null mutants (Frank et al., 2002; Meyers et al., 1998; Park et al., 2006; Park et al., 2008). Therefore, we assayed Fgf8 mRNA expression levels in our mutants. Our ISH experiments indicated comparable expression of Fgf8 mRNA in FN-null, integrin α5-null and control embryos (Supplemental Fig. 2), indicating that neither FN nor integrin α5 are required to regulate Fgf8 expression. Therefore, we tested the possibility that FN and integrin α5 modulate Fgf8 signaling by assaying expression of genes belonging to Fgf8 syn-expression group that were previously shown to be regulated by Fgf8 signaling in early embryos (Furthauer et al., 2002; Niehrs and Meinhardt, 2002). Our experiments indicate that transcriptional targets downstream of Fgf8 signaling are downregulated in FN-null and integrin α5-null mutants compared with controls (Fig. 8A–D).
Figure 8. Fibronectin and integrin α5 regulate Fgf8 signaling in vivo and in vitro.
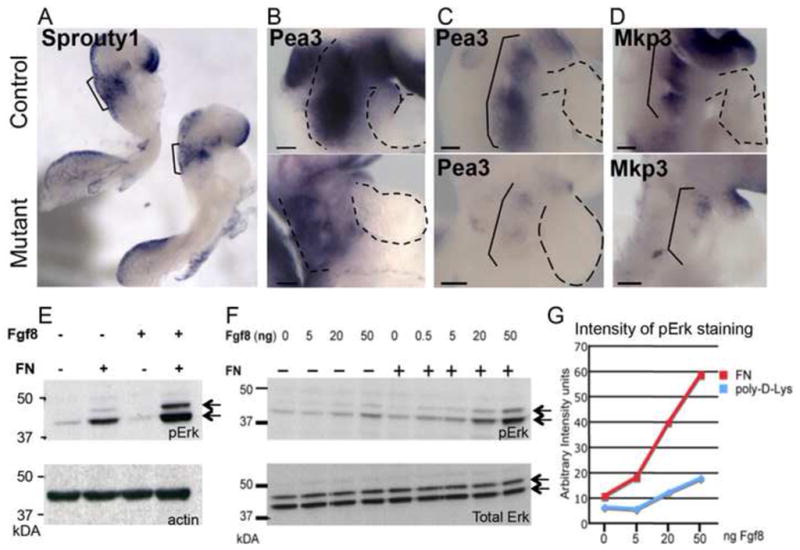
mRNA expression of downstream signaling reporters of Fgf8 signaling in E8.75 embryos. A–B. Control (top) and FN-null mutants (bottom). C–D. Control (top) and integrin α5-null mutants (bottom). E. Cell adhesion to FN but not to poly-d-Lys potentiates Fgf8 activation of Erk (double arrows). F. Cell adhesion to FN mediates Fgf8 signaling in a dose-dependent manner and does not lead to increases in Erk protein levels (bottom gel). Activation of Erk in response to cell adhesion to FN and increased doses of Fgf8 is quantified in G.
In order to determine whether cell adhesion to FN was required to mediate Fgf8 signaling, we used a well-established in vitro assay to test the requirement for cell adhesion to FN in potentiating Fgf8 signaling. Prior experiments indicated that binding of integrin α5β1 to FN is required for the phosphorylation and activation of Erk following stimulation of cells with Fgf2 (Miyamoto et al., 1996). Therefore, we tested whether cell adhesion to FN co-operates with Fgf8 signaling in activating Erk. Our experiments demonstrated that cell adhesion to FN leads to increased levels of phosphorylated forms of Erk and that this response is dose-dependent (Fig. 8E–G). In contrast, in the absence of cell adhesion to FN, addition of Fgf8 leads only to minimal activation of Erk (Fig. 9E–G). These results are consistent with prior literature demonstrating that integrin α5β1-mediated cell adhesion to FN potentiates growth factor signaling regulated by receptor tyrosine kinases (Giancotti and Tarone, 2003). However, our experiments are the first demonstration of the requirement for FN in growth factor signaling in vivo. Prior studies demonstrated that decreased Fgf8 expression or signaling in vivo leads to decreased proliferation of cardiac progenitors, giving rise to defective development of the cardiac outflow tract (Frank et al., 2002; Hutson et al., 2010; Hutson et al., 2006; Meyers et al., 1998; Park et al., 2006; Park et al., 2008). Taken together, our studies suggest that the short outflow tracts in FN mutants are due defective Fgf8 signaling and attenuated proliferation of myocardial progenitors, and that FN and integrin α5β1 regulate morphogenesis of the arterial pole of the heart by potentiating Fgf8 signaling.
Discussion
FN-null and integrin α5-null mutants developing within the C57BL6/J genetic background survive up to E10.5 (Astrof et al., 2007). Even though these mutants are smaller and phenotypically different compared with controls, gene expression studies indicate that they are not underdeveloped, as progenitors and descendants of all examined embryonic lineages are specified in similar spatio-temporal patterns in mutants and controls (George et al., 1997; Georges-Labouesse et al., 1996; Mittal et al., 2010). While all examined embryonic lineages are specified in these mutants, morphogenesis of organs and structures such as notochord, somites and blood vessels is defective (George et al., 1997; Georges-Labouesse et al., 1996). Taken together, these studies indicated that defects observed in FN-null and integrin α5-null embryos are not due to developmental delay but due to defective morphogenesis of correctly specified precursors into appropriate three-dimensional structures.
Initial studies describing the phenotypes of FN-null and integrin α5-null mutants were performed before the discovery of the second heart field and genetic networks regulating cardiac morphogenesis. While dissecting FN-null embryos for a different project, we noticed that hearts in these mutants look similar to the hearts of Isl1-null mutants that were just published at that time. This suggested that FN regulates morphogenesis of the SHF derivatives, the outflow tract and the right ventricle. Our ISH and fate mapping studies support this observation.
Our previous studies demonstrated that FN and integrin α5 are required for the establishment of the left-right embryonic body plan, explaining the reason for aberrant cardiac looping in FN-null and α5-null mutants (Pulina et al., 2011). In this paper, we demonstrate that FN is required for morphogenesis of the cardiac outflow tract and the right ventricle, as both structures are very small in the mutants, and that this is likely due to defective proliferation of the cardiac progenitors in the splanchnic mesoderm. Intriguingly, ventricular walls of FN-null and integrin α5-null mutants are thicker and more muscular compared with controls, and our gene expression profiling data indicated upregulation of many genes involved in cardiomyocyte structure and function, and a highly statistically significant enrichment of these genes among the upregulated genes overall in FN-mutant embryos (Table 1). This upregulation of cardiomyocyte structural genes was already evident at E8.0 (~6 somite stage), a very early stage in heart development. Even though the myocardial wall was thicker, we did not note increase in cardiomyocyte proliferation. This suggests that FN may modulate the balance between proliferation and differentiation of cardiac progenitors.
FN is expressed by myocardial progenitors in the splanchnic mesoderm (Mittal et al., 2010) and facilitates their proliferation (this study). In the absence of FN (in our FN-null mutants), cardiac progenitors may interact with other ECM glycoproteins, (e.g. laminins) and proceed to differentiate. This possible scenario is similar to endothelial cells, which proliferate when plated on FN but exit from cell cycle and differentiate when plated on laminin (Wary et al., 1996). Depending on the timing of this regulation, the absence of FN may lead to increased differentiation of Isl1+ cells providing ectopic cardiomyocytes for the left ventricle, and depleting the number of Isl1+ cells allocated for the outflow tract and the right ventricle. Decreased Fgf8 expression and signaling is already known to cause precautious differentiation of cardiac precursors into cardiomyocytes (Hutson et al., 2010; Tirosh-Finkel et al., 2010); therefore, another possible cause of the short outflow tract and the right ventricle, and ectopic cardiomyocyte differentiation could be the imbalance in Fgf8 signaling observed in FN and integrin α5 mutants.
FN signals to cells by interacting with cellular receptors, and integrin α5β1 is one of the major FN receptors in vivo (Yang et al., 1999; Yang et al., 1993). In vitro, multiple studies demonstrated that FN binds α5β1 and stimulates α5β1 signaling, activating pathways regulating cell proliferation, survival, migration, remodeling of actin cytoskeleton and others (Giancotti and Tarone, 2003; Schwartz and Assoian, 2001). In vivo, a major role for integrin α5β1 as a FN receptor is supported by gene knock out studies, which showed that the phenotype of integrin α5-null embryos is similar to that of FN-nulls (Mittal et al., 2010; Pulina et al., 2011; Takahashi et al., 2007; Yang et al., 1999; Yang et al., 1993). Binding of FN to integrin α5β1 expressed on cellular surfaces leads to formation of FN fibrils, and this process is defective in integrin α5-null embryos (Pulina et al., 2011), indicating that in early embryogenesis, FN binding and signaling to cells is mainly dependent on integrin α5β1. Cardiac abnormalities seen with integrin α5-null mutants are similar to those observed in FN-null mutants. Both mutants exhibit defects in Fgf8 signaling, while Fgf8 mRNA expression levels are not significantly altered. We also showed that cell adhesion to FN is necessary to mediate Fgf8 signaling in vitro. Taken together, these studies support a model in which cell adhesion of cardiac precursor cells to FN mediated by integrin α5β1 and probably additional FN-binding integrins such as those containing integrin αv chain, regulate Fgf8 signaling and proliferation of pre-cardiac mesoderm, and consequently, the morphogenesis of the cardiac outflow tract. While detailed mechanisms remain to be investigated, our studies unequivocally show that both FN and integrin α5 are required for cardiac morphogenesis. Future studies using these and conditional mutants will be instrumental to determining cellular requirements for FN, integrin α5β1 and molecular mechanisms downstream of their interactions in heart development.
Supplementary Material
{kind=link}
{kind=link}
Highlights.
Fibronectin (FN)-null and integrin α5-null mutants have dysmorphic hearts
Mutants have small outflow tracts and right ventricles but thickened left ventricles
Specification & migration of Isl1+ cardiac precursors is independent of FN or Itga5
FN mediates proliferation of cardiac progenitors in splanchnic mesoderm
FN and Itga5 facilitate Fgf8 signaling in vivo
Acknowledgments
Funding. This work was supported by NIH NHLBI R01 HL103920, W.W. Smith and American Heart Association Innovative Research Grant 12IRG9130012 to S.A.
We thank Jeff Lee for advice on sample preparation for scanning electron microscopy and Michael Snitkovsky for generating the box plots. Nina Lampen at the Memorial Sloan-Kettering Cancer Center for assistance with scanning EM. Dr. Sylvia Evans for the Isl1Cre mice, and Drs. Benoit Bruneau, Sylvia Evans and Brigid Hogan for plasmids to prepare mRNA probes, and Dr. Anne Moon for advice and reading the manuscript. The MF20 and Isl1 monoclonal antibodies developed by Drs. Fischman and Jessell, respectively, were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA 52242. This work was supported by the grants from the NIH NHLBI HL103920, AHA SDG 0835556D, AHA IRG 12IRG9130012, and W.W. Smith Charitable Trust to S.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Astrof S. Interactions between neural crest-derived cells and extracellular microenvironment during cardiovascular development. In: Desimone DW, Mecham RP, editors. Extracellular Matrix in Development. Springer Verlag; Berlin: 2013. pp. 105–131. [Google Scholar]
- Astrof S, Kirby A, Lindblad-Toh K, Daly M, Hynes RO. Heart development in fibronectin-null mice is governed by a genetic modifier on chromosome four. Mech Dev. 2007;124:551–558. doi: 10.1016/j.mod.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Aszodi A, Legate KR, Nakchbandi I, Fassler R. What mouse mutants teach us about extracellular matrix function. Annual review of cell and developmental biology. 2006;22:591–621. doi: 10.1146/annurev.cellbio.22.010305.104258. [DOI] [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furthauer M, Lin W, Ang SL, Thisse B, Thisse C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat Cell Biol. 2002;4:170–174. doi: 10.1038/ncb750. [DOI] [PubMed] [Google Scholar]
- George EL, Baldwin HS, Hynes RO. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood. 1997;90:3073–3081. [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- George EL, Hynes RO. Gene targeting and generation of mutant mice for studies of cell-extracellular matrix interactions. Methods Enzymol. 1994;245:386–420. doi: 10.1016/0076-6879(94)45021-8. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse EN, George EL, Rayburn H, Hynes RO. Mesodermal development in mouse embryos mutant for fibronectin. Dev Dyn. 1996;207:145–156. doi: 10.1002/(SICI)1097-0177(199610)207:2<145::AID-AJA3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Tarone G. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu Rev Cell Dev Biol. 2003;19:173–206. doi: 10.1146/annurev.cellbio.19.031103.133334. [DOI] [PubMed] [Google Scholar]
- Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish-Horowicz D. Expression of a Delta homologue in prospective neurons in the chick. Nature. 1995;375:787–790. doi: 10.1038/375787a0. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Zeng XL, Kim AJ, Antoon E, Harward S, Kirby ML. Arterial pole progenitors interpret opposing FGF/BMP signals to proliferate or differentiate. Development. 2010;137:3001–3011. doi: 10.1242/dev.051565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson MR, Zhang P, Stadt HA, Sato AK, Li YX, Burch J, Creazzo TL, Kirby ML. Cardiac arterial pole alignment is sensitive to FGF8 signaling in the pharynx. Developmental biology. 2006;295:486–497. doi: 10.1016/j.ydbio.2006.02.052. [DOI] [PubMed] [Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Evolution: The evolution of metazoan extracellular matrix. J Cell Biol. 2012;196:671–679. doi: 10.1083/jcb.201109041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–316. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino MM, Hubbell JA. The 12th–14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010;24:4711–4721. doi: 10.1096/fj.09-151282. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nature genetics. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Miner JH, Li C, Mudd JL, Go G, Sutherland AE. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development. 2004;131:2247–2256. doi: 10.1242/dev.01112. [DOI] [PubMed] [Google Scholar]
- Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annual review of cell and developmental biology. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- Mittal A, Pulina M, Hou SY, Astrof S. Fibronectin and integrin alpha 5 play essential roles in the development of the cardiac neural crest. Mech Dev. 2010;127:472–484. doi: 10.1016/j.mod.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Gutkind JS, Yamada KM. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J Cell Biol. 1996;135:1633–1642. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Yuen PS, Papalia JM, Trevithick JE, Sakai T, Fassler R, Hynes RO, Wagner DD. Plasma fibronectin promotes thrombus growth and stability in injured arterioles. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2415–2419. doi: 10.1073/pnas.2628067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C, Meinhardt H. Modular feedback. Nature. 2002;417:35–36. doi: 10.1038/417035a. [DOI] [PubMed] [Google Scholar]
- Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–2433. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Watanabe Y, Smyth G, Miyagawa-Tomita S, Meyers E, Klingensmith J, Camenisch T, Buckingham M, Moon AM. An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development. 2008;135:3599–3610. doi: 10.1242/dev.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulina MV, Hou SY, Mittal A, Julich D, Whittaker CA, Holley SA, Hynes RO, Astrof S. Essential roles of fibronectin in the development of the left-right embryonic body plan. Dev Biol. 2011;354:208–220. doi: 10.1016/j.ydbio.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentschler S, Jain R, Epstein JA. Tissue-tissue interactions during morphogenesis of the outflow tract. Pediatr Cardiol. 2010;31:408–413. doi: 10.1007/s00246-009-9611-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Johnson KJ, Murozono M, Sakai K, Magnuson MA, Wieloch T, Cronberg T, Isshiki A, Erickson HP, Fassler R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- Saoncella S, Echtermeyer F, Denhez F, Nowlen JK, Mosher DF, Robinson SD, Hynes RO, Goetinck PF. Syndecan-4 signals cooperatively with integrins in a Rho-dependent manner in the assembly of focal adhesions and actin stress fibers. Proc Natl Acad Sci U S A. 1999;96:2805–2810. doi: 10.1073/pnas.96.6.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Assoian RK. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci. 2001;114:2553–2560. doi: 10.1242/jcs.114.14.2553. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Leiss M, Moser M, Ohashi T, Kitao T, Heckmann D, Pfeifer A, Kessler H, Takagi J, Erickson HP, Fassler R. The RGD motif in fibronectin is essential for development but dispensable for fibril assembly. J Cell Biol. 2007;178:167–178. doi: 10.1083/jcb.200703021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh-Finkel L, Zeisel A, Brodt-Ivenshitz M, Shamai A, Yao Z, Seger R, Domany E, Tzahor E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development. 2010;137:2989–3000. doi: 10.1242/dev.051649. [DOI] [PubMed] [Google Scholar]
- Trinh LA, Stainier DY. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev Cell. 2004;6:371–382. doi: 10.1016/s1534-5807(04)00063-2. [DOI] [PubMed] [Google Scholar]
- von Both I, Silvestri C, Erdemir T, Lickert H, Walls JR, Henkelman RM, Rossant J, Harvey RP, Attisano L, Wrana JL. Foxh1 is essential for development of the anterior heart field. Dev Cell. 2004;7:331–345. doi: 10.1016/j.devcel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Wickstrom SA, Radovanac K, Fassler R. Cold Spring Harbor perspectives in biology. 2011. Genetic analyses of integrin signaling; p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JT, Bader BL, Kreidberg JA, Ullman-Cullere M, Trevithick JE, Hynes RO. Overlapping and independent functions of fibronectin receptor integrins in early mesodermal development. Dev Biol. 1999;215:264–277. doi: 10.1006/dbio.1999.9451. [DOI] [PubMed] [Google Scholar]
- Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development. 1993;119:1093–1105. doi: 10.1242/dev.119.4.1093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.