

Abstract
Intestinal iron absorption involves proteins located in the brush border membrane (BBM), cytoplasm, and basolateral membrane (BLM) of duodenal enterocytes. Ferroportin 1 (FPN1) and hephaestin (Heph) are necessary for transport of iron out of enterocytes, but it is not known whether these two proteins interact during iron absorption. We first examined colocalization of the proteins by cotransfection of HEK293 cells with pDsRed-FPN1 with pEmGFP-Heph or with the COOH-terminal truncated pEmGFP-HephΔ43 or -HephΔ685 and found that FPN1 and Heph with or without the COOH terminus colocalized. In rat duodenal enterocytes, within 1 h of iron feeding prominent migration of FPN1 from the apical subterminal zone to the basal subnuclear zone of the BLM occurred and increased to at least 4 h after feeding. Heph exhibited a similar though less prominent migration after iron ingestion. Analysis using rat duodenal epithelial cell sheets demonstrated that 1) by velocity sedimentation ultracentrifugation, FPN1 and Heph occupied vesicles of different sizes prior to iron feeding and migrated to similar fractions 1 h after iron feeding; 2) by blue native/SDS-PAGE, FPN1, and Heph interacted to form two complexes, one containing dimeric FPN1 and intact Heph and the other consisting of monomeric FPN1 and a Heph fragment; and 3) by immunoprecipitation, anti-Heph or anti-FPN1 antiserum coimmunoprecipitated FPN1 and Heph. Thus the data indicate that FPN1 and Heph migrate and interact during iron feeding and suggest that dimeric FPN1 is associated with intact Heph.
Keywords: DsRed-FPN1, EmGFP-Heph, FPN1 and Heph migration, blue native/SDS-PAGE, coimmunoprecipitation
iron is an essential nutrient absorbed through the duodenal epithelium, and iron homeostasis is achieved by regulation of the absorptive process (20). In the current concept of intestinal iron absorption, dietary iron as Fe3+ is reduced to Fe2+ by the duodenal cytochrome b reductase (Dcytb) (17, 27). The reduced iron is then absorbed through the divalent metal transporter (DMT1) on the brush border membrane (BBM) of duodenal enterocytes (17, 27). The Fe2+ then traverses the enterocyte cytoplasm to the basolateral membrane (BLM) where Fe2+ is transported by ferroportin (FPN1) (1, 9, 27) and converted to Fe3+ by the multicopper ferroxidase hephaestin (Heph) (38) before release to transferrin in the systemic circulation. Since Fe2+ is highly reactive and potentially toxic, the molecular mechanisms for handling the absorbed iron as it transverses the enterocyte from BBM to the BLM are important to elucidate. The detailed molecular relationships of the two membrane transporters, DMT1 and FPN1, and the accessory proteins necessary for iron transport remain largely unknown. The Fe2+ transported into the enterocyte may bind to cytosolic chaperons and iron binding proteins, including iron regulatory proteins (IRPs) that regulate translation of ferritin with the subsequent compartmentalization and storage of iron (2). Alternatively, the Fe3+ reduced to Fe2+ by Dcytb may be taken up by DMT1-containing vesicles that undergo transcytosis and interact with vesicles derived from the BLM (24, 41). Support for this hypothesis is the observation that the internalization of DMT1 occurred in Caco-2 cells after iron treatment and in rat duodenum epithelium with iron ingestion (24, 41).
Iron absorption is, to a large measure, regulated by hepcidin, the iron stores regulator synthesized in the liver. Hepcidin downregulates FPN1 expression both by suppressing FPN1 mRNA synthesis (13, 31, 40) and by promoting FPN1 protein internalization and degradation (30). As a consequence, hepcidin inhibits iron absorption into the circulation. Moreover, the tissue-specific inactivation of FPN1 in mouse intestine causes intestinal accumulation of iron, indicating that FPN1 is essential for iron export from the enterocyte (10). That Heph is also essential for iron export is deduced from the phenotype of the sla mouse, which harbors a mutation in Heph that results in a truncated protein causing enterocytic iron accumulation (38). Since both FPN1 and Heph have a subcellular localization in the enterocyte cytoplasm as well as the BLM (1, 28), it is possible that these two proteins cooperate during iron transport out of the enterocyte. The colocalization of these two proteins has been shown in Caco-2 cells (10), but whether these two proteins physically interact especially in vivo in the intestine is unclear. Furthermore, whether the cellular localization of FPN1 and Heph changes with iron absorption as does DMT1 is also unknown. In this report we examine the physical interactions of these two proteins in several systems. In HEK293 cells, following cotransfections with fluorescently tagged FPN1 and Heph, the two proteins could be seen colocalized subcellularly. In rat duodenal epithelial cells following iron ingestion, FPN1 and Heph were observed by immunofluorescence histochemistry to translocate from the apical to the basal portion of the BLM, although the effect was greater with FPN1. By subjecting rat duodenal epithelial cell lysates to ultracentrifugation velocity sedimentation, we could demonstrate that FPN1 and Heph had dissimilar rates of sedimentation prior to iron ingestion, but after iron ingestion the rates were similar, suggesting that during iron transport the two proteins entered similar compartments. Finally, physical evidence for actual protein-protein interaction during the transport of iron across enterocytes was obtained by the use of blue native (BN)/SDS-PAGE and coimmunoprecipitation of FPN1 and Heph. A preliminary report of these studies has been presented in abstract form in Gastroenterology (39).
MATERIALS AND METHODS
Animals.
Sprague-Dawley rats from Charles River Breeding Laboratories (Wilmington, MA) were bred and raised in our animal quarters with a 12-h light-dark cycle. Six-week-old rats fed Harlan Teklad 22/5 rodent chow were fasted overnight with 5% glucose saline solution ad libitum and were fed a piece of chow (250 mg) containing FeSO4 at a dose of 50 μg iron/g body wt as described previously (41). The chow was always consumed in less than 5 min. The effects of iron feeding via chow are normal physiological effects, given that in an earlier study with intraluminal infusion of FeSO4 similar effects of iron were observed on the internalization of DMT1 (41). Animals were killed at 0, 1, 2, and 4 h after the feeding of the iron-supplemented chow, and the duodenum was removed from the pylorus to the ligament of Treitz. The proximal 1 cm of duodenum 1 cm distal from the pylorus was used for immunofluorescent staining studies and the mucosal scrape of the remainder of the duodenum was used for various biochemical analyses. The animal protocol for these experiments has been approved by the Committee of Animal Care and Use, Louisiana State University Health Sciences Center, Shreveport, LA.
Preparation of antibodies against FPN1 and Heph.
To produce antibodies against rat FPN1, the COOH-terminal 15 amino acids, i.e., CEKVTDESQPNTSVV, were deduced from the nucleotide sequences from GenBank accession number AF39478. For generation of antibody for rat Heph, the COOH-terminal 17 amino acids, i.e., CRRSILDDSFKLLSLKQ, were deduced from GenBank accession number AF246120. The peptides were synthesized, conjugated with keyhole limpet hemocyanin, and used as antigens for rabbit antibody production as described previously (41).
Generation of fluorescent labeled FPN1 and Heph fusion genes.
The pcDsRed (monomeric) vector (Clontech, Mountain View, CA) and the pcDNA-EmGFP/TOPO vector (Invitrogen, Carlsbad, CA) were used to generate various NH2-terminal fusion proteins to preserve the putative PDZ motif of FPN1 and the PDZ domain of intact Heph (41). For positive controls, HEK293 cells were cotransfected with pDsRed + pEmGFP and pDsRed-FPN1 + pEmGFP-FPN1. For the negative control, HEK293 cells were cotransfected with pDsRed-FPN1 + pEmGFP-ferritin heavy chain (HFt), since HFt has been shown to be expressed at high levels independent of iron metabolism (6). The reading frames of FPN1 and Heph were amplified by primer pairs synthesized according to AF39478 and AF246120, respectively. FPN1 was amplified with the primer pairs of sense strand 5′-GAAGAATTCCATGACCAAGTCAAGAGATCAGA-3′ and antisense strand 5′-aagaattctacacaacagatgtattaggc-3′. FPN1 amplicons were digested with EcoR1 and inserted into EcoR1-digested DsRed (monomeric) vectors (Clontech). The entire reading frame of Heph was amplified with primer pairs of sense strand 5′-ATGAAGGCAGGCCATCTTCTC-3′ and antisense strand 5′-CAGCTTGAGAGACAGAAGCTTGA-3′. To exclude the putative membrane anchor that might interfere with interactions of Heph with FPN1, the nucleotides that encode the COOH-terminal 43 amino acids of Heph were deleted by using the antisense strand 5′-TCATATGGCAATCAAAGCAGA-3′ to amplify the HephΔ43 amplicons. To explore whether one putative Cu-ferroxidase site in Heph was sufficient for interaction with FPN1, the COOH-terminal 685 amino acids were deleted by amplification with antisense strand 5′-CTAGAAGACCACCTGGATGGTGT-3′ to amplify HephΔ685 amplicons. For rat HFt, the coding sequences [without iron response element (IRE)] were amplified by primer pairs of sense strand 5′-ATGACCACCGCGTCTCCCTC-3′ and antisense 5′-TTAGCTCTCATCACCGTGTCCC-3′. All amplicons were inserted into pcDNA-EmGFP/TOPO or pDsRed (monomeric) vectors according to the user manual with the sequences subsequently determined to ensure that all of the insertions were correct and in frame.
Cell culture and transfection.
HEK293 cells from ATCC were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml of streptomycin, and 1% l-glutamine. Cells were seeded at 1 × 106 cells per 60-mm plate 16 h before transfection. To verify antiserum specificity, HEK293 cells were used to express fusion gene proteins 72 h after transfection with pDsRed, pEmGFP, pDsRed-FPN1, or pEmGFP-Heph. For examination of colocalization of fusion proteins, HEK 293 cells were cotransfected with combinations of the various DsRed and EmGFP fusion gene constructs (i.e., pDsRed + pEmGFP, pDsRed-FPN1 + pEmGFP-FPN1, pDsRed-FPN1 + pEmGFP-HFt, pDsRed-FPN1 + pEmGFP-Heph, pDsRed-FPN1 + pEmGFP-HephΔ43, or pDsRed-FPN1 + pEmGFP-HephΔ685) by Lipofectamine according to the manufacturer's suggested protocol (Invitrogen). At 48 h after transfection, cells were rinsed with PBS, counterstained with 4,6-diamidino-2-phenylindole, and examined with a Zeiss AxioObserver microscope with Apotome device. Colocalization of fusion proteins was determined from the number of pixels for red, green, and red + green (yellow) along the entire z-section of at least 10 cells by using the Zeiss AxioVision 4.6.3 SP1 colocalization software. Because the levels of expression varied among the different fusion constructs, the pixels for DsRed-FPN1 were set at 100% in relation to the EmGFP-tagged genes so that the expression of EmGFP-FPN1, -HFt, -Heph, -HephΔ43, and -HephΔ685 could be presented as percent colocalization with DsRed-FPN1.
Western blots and immunoprecipitation.
Lysates were prepared from either HEK293 cells or duodenal mucosal scrapes by washing the cells in PBS followed by lysis in RIPA buffer (0.15 mM NaCl, 10 mM NaPO4, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mM sodium orthovanadate; pH 7.4) containing 1% Triton X-100. Aliquots of the total cell extract containing 15 μg of protein were subjected to SDS-PAGE under reducing conditions and transferred to nitrocellulose membranes. Molecular weight standards (Santa Cruz Biotechnology, Santa Cruz, CA) were run in parallel. Membranes were blocked with 5% nonfat powdered milk in a solution of Tris-buffered salt with Tween 20 (20 mM Tris, pH 7.6, 150 mM NaCl, and 0.05% Tween 20) at room temperature for 2 h, incubated with anti-DsRed, anti-GFP (Clontech for both), anti-FPN1 or anti-Heph overnight, washed and incubated with horseradish peroxidase-linked goat anti-rabbit antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. Immunoreactive proteins were detected by ECL Western blotting system (Amersham, Arlington Heights, IL) and were quantitated by transmittance densitometry using volume integration with ImageQuant application software.
For coimmunoprecipitation determinations, duodenal mucosa scrapes were homogenized in ice-cold RIPA buffer as described above and centrifuged at 10,000 g for 5 min, and aliquots of the supernatant were incubated with 20 μl of anti-FPN1 or anti-Heph on a rotator for 1–2 h. The antigen-antibody complexes were absorbed by 40 μl of agarose-protein A suspension for 1 h incubation, washed five times with RIPA buffer by centrifugation at 10,000 g. The agarose pellet was suspended in electrophoresis sample buffer, boiled for 3 min, and subjected to SDS-PAGE under reducing conditions. Portions of the gels were stained with Coomassie blue for visualization of proteins whereas other portions were transferred to nitrocellulose for Western blot analysis with the indicated antiserum.
Indirect immunofluorescent staining of FPN1 and Heph.
To observe subcellular changes in FPN1 and Heph localization, the duodenal segment was cryostat sectioned and the sections were stained with anti-FPN1 (1:250) or anti-Heph (1:300) generated as described above and then with fluorescein-conjugated goat anti-rabbit IgG as described previously (41). The stained sections were examined via a Zeiss Z1 AxioObserver Inverted microscope with an ApoTome device. To quantitate the migration of FPN1 and Heph from one subcellular area to another, the cells were arbitrarily divided into four compartments of equal length along the apical-basal axis of the cell such that zone 1 was the most apical and zone 4 was the most basal compartments. The number of pixels in each zone was then determined by using the AxioVision software in four to six sections of well-orientated villus tops for each of three rats for each time point with each section containing four to six cells.
Ultracentrifugation velocity sedimentation.
Duodenal epithelial cell sheets were isolated by infusing 30 mM EDTA in Hanks’ solution into the left ventricle of the heart as described by Bjerknes and Cheng (4). The resulting cell sheets were homogenized in homogenization buffer (250 mM sucrose, 10 mM Tris·HCl, pH 7.4, 1 mM EDTA, 1 mM PMSF, 1 mM benzamidine) with N2 decompression by use of a cell disruption bomb (Parr Instrument, Moline, IL) at 1,000 psi, the intact cells and nuclei were removed by centrifugation for 5 min at 1,000 g at 4°C, and the supernatant was then layered onto a 30-ml 5–25% sucrose gradient over a 50% sucrose pad and centrifuged for 60 min at 40,000 g in a Beckman SW28 rotor. One-milliliter gradient fractions were collected from the bottom to the top of the gradient, and the proteins in each fraction were precipitated with 10% trichloroacetic acid in the presence of 50 μg/ml bovine insulin and subjected to separation by SDS-PAGE with subsequent Western blot analysis with anti-FPN1 or anti-Heph antiserum to detect the distribution profiles of both proteins. Anti-DMT1 (41) and anti-NHE1 (sodium/hydrogen exchanger 1) (Santa Cruz Biotechnology) were used to demonstrate the sedimentation profiles of DMT1 as BBM protein and NHE1 as BLM protein. NHE1 is a constitutively expressed protein that does not change subcellular localization during sodium absorption (42).
BN/SDS-PAGE.
The BN-PAGE technique was used to detect FPN1 and Heph interactions according to the described methods (36). Duodenal epithelial sheets were suspended in BN buffer (20 mM Bis-Tris, 500 mM ε-aminocaproic acid, 20 mM NaCl, 2 mM EDTA 2, 10% glycerol, and protease inhibitors as described earlier) at pH 7.0. Cells suspended in BN buffer were disrupted with a cell disruption bomb at 1,000 psi. Intact cells and nuclei were removed by centrifugation at 350 g for 5 min with the supernatant then centrifuged at 15,000 g, and the resulting pellet extracted with BN buffer containing 2% Triton X-100, gently mixed every 15 min on ice for 1 h, and centrifuged at 20,000 g for 10 min to collect supernatant for analysis by 5–15% BN-PAGE. Duplicate lanes of the resulting BN-PAGE were cut out, equilibrated for 10 min at 70°C and 20 min at room temperature in 2 × SDS Laemmli loading buffer under reducing conditions, and subjected to a second-dimension separation by 5–15% SDS-PAGE. The resulting gels were subjected to Western blots to detect FPN1 and Heph as described earlier.
Statistical analysis.
Student's t-test was used to determine changes in FPN1, Heph, HFt, and ferritin light chain (LFt) after iron ingestion. Two-way ANOVA was computed from the experimental data related to changes in FPN1 and Heph localization in enterocytes from 0 to 4 h after iron ingestion. When the F value obtained from two-way ANOVA was significant, Bonferroni's test was applied to test for differences among groups. P < 0.05 was considered to be significant.
RESULTS
Characterization of the anti-FPN1 and Heph antisera.
The specificities of the rabbit antisera generated against the COOH-terminal peptides of FPN1 and Heph were demonstrated in HEK293 cells transfected with pDsRed, pDsRed-FPN1, pEmGFP, or pEmGFP-Heph (Fig. 1). In cells transfected with pDsRed (Fig. 1A, lanes 1 and 3) or pDsRed-FPN1 (Fig. 1A, lanes 2 and 4), anti-DsRed antibodies detected proteins only of the appropriate apparent molecular mass of DsRed in lane 1 (30 kDa) and of the DsRed-FPN1 fusion protein in lane 2 (110 kDa), whereas the anti-FPN1 antiserum detected no proteins in lane 3 and the fusion protein of similar molecular mass to that detected by anti-DsRed (110 kDa) in lane 4. Likewise, the anti-EmGFP antibody detected either free EmGFP in lane 5 or the fusion protein of EmGFP and Heph of appropriate molecular mass, 180 kDa, in lane 6 (Fig. 1B), whereas the anti-Heph antiserum detected no proteins in lane 7 and the presumed EmGFP-Heph 180 kDa fusion protein in lane 8 (Fig. 1B). Even under reducing conditions oligomers of the DsRed-FPN1 fusion protein were always detected (Fig. 1A, lane 4). In addition, in parallel Western blot analyses the inclusion of 2 μg each of the COOH-terminal peptides of FPN1 and Heph blocked the antibody detection of DsRed-FPN1 and EmGFP-Heph (not shown).
Fig. 1.
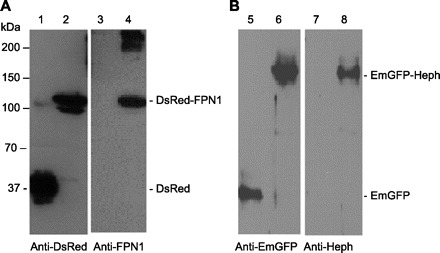
Analysis of specificity of anti-ferroportin 1 (FPN1) and hephaestin (Heph) antisera. HEK293 cells were transfected with pDsRed, pDsRed-FPN1, pEmGFP, or pEmGFP-Heph and cell lysates were subjected to SDS-PAGE under reducing conditions with subsequent Western blot analysis as described in materials and methods. A: lysates from HEK293 cells transfected with pDsRed (lanes 1 and 3) and pDsRed-FPN1 (lanes 2 and 4) were separated by SDS-PAGE and blotted with anti-DsRed (lanes 1 and 2) or anti-FPN1 (lanes 3 and 4). B: lysates from HEK cells transfected with pEmGFP (lanes 5 and 7) or pEmGFP-Heph (lanes 6 and 8) were separated by SDS-PAGE and blotted with anti-EmGFP (lanes 5 and 6) or anti-Heph (lanes 7 and 8).
The antibody specificity was also examined by Western blot analysis of mucosal scrapes from the proximal duodenum (PD, the first 4 cm from the pylorus), the distal duodenum (DD, the distal 4 cm from the pylorus to the ligament of Treitz), the jejunum (J), the ileum (I), and the colon (C) (Fig. 2). In both the PD and DD the anti-FPN1 antiserum detected proteins of approximate molecular mass 70 and 56 kDa as well as two other presumed degradation products with variable amounts of these presumed degradation products in different animals (Fig. 2A). The two 70- and 56-kDa proteins were present at approximately similar amounts in the PD, DD, and J with lesser amounts in the I and C. All of the bands detected by anti-FPN1 were blocked by the addition of 2 μg of the antigen peptide (data not shown). The Western blot analyses with the anti-Heph antiserum showed two bands with approximate molecular masses of 155 and 130 kDa, respectively, representing presumably glycosylated and incompletely glycosylated Heph (32) (Fig. 2B). Interestingly, the 155-kDa Heph band was present with similar intensity along the entire intestine. The minor 80- and 70-kDa bands detected with anti-Heph antiserum appeared to be degradation products (Fig. 2B) since the inclusion of the COOH-terminal peptide of Heph blocked all bands recognized by anti-Heph antiserum (data not shown).
Fig. 2.
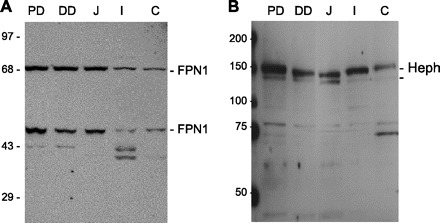
Distribution of FPN1 and Heph along the rat intestine. Rat intestines were longitudinally divided into proximal duodenum (PD, from the pylorus to the first 4 cm of the intestine), distal duodenum (DD, the segment between the distal 4 cm from the pylorus to the ligament of Treitz), jejunum (J, the proximal half of the remainder of the small intestine), ileum (I, the distal half of the small intestine), and colon (C). Mucosal scrapes from these regions were washed with ice-cold PBS, homogenized in RIPA buffer, and subjected to SDS-PAGE under reducing conditions, and Western blot analyses were performed with the antiserum as described in materials and methods either with anti-FPN1 antiserum (A) or anti-Heph antiserum (B). The data presented is a representative example of a Western blot analysis from 2 animals.
Colocalization of pDsRed-FPN1 and pEmGFP-Heph.
FPN1 and Heph have recently been demonstrated to be colocalized in the BLM of Caco-2 cells, a cell line often used as a model system for the study of duodenal transport (19). To substantiate that Heph and FPN1 indeed colocalize and to examine the importance of the Heph COOH terminus for colocalization, a series of constructs encoding fluorescent protein pairs including pDsRed with pEmGFP, pDsRed-FPN1 with pEmGFP-FPN1, pDsRed-FPN1 with pEmGFP-HFt, pDsRed-FPN1 with pEmGFP-Heph, pDsRed-FPN1 with pEmGFP-HephΔ43, and pDsRed-FPN1 with pEmGFP-HephΔ685, were cotransfected into HEK293 cells. The percents of colocalization were calculated as the percent of DsRed-FPN1 in cotransfected cells at 48 h after transfection (Fig. 3, A and B). Transfection of pDsRed with pEmGFP served as a positive control because both fluorescent proteins should occupy the same locations, respectively (Fig. 3Aa). Indeed, the merged images demonstrate a relatively homogeneous yellow (white arrows) pattern representing colocalization of DsRed and EmGFP throughout much of the transfected cells, and the percent colocalization was measured as ∼95% (not shown). An additional positive control of HEK293 cells transfected with pDsRed-FPN1 and pEmGFP-FPN1 showed DsRed-FPN1 and EmGFP-FPN1 in the cytoplasm and cell membranes of cells with 78% colocalization (Fig. 3Ab and Fig. 3B). In contrast was the negative control of cotransfection of pDsRed-FPN1 and pEmGFP-HFt. A priori there is little reason to expect that ferritin should interact with FPN1 (Fig. 3Ac), and colocalization was limited to ∼15% (Fig. 3B). When pDsRed-FPN1 and pEmGFP-Heph were cotransfected into HEK293 cells, colocalization was observed for DsRed-FPN1 and EmGFP-Heph (Fig. 3Ad). The distribution of the fluorescently labeled FPN1 and Heph showed 75% colocalization (Fig. 3B). The same colocalization was observed for the cotransfection pair pDsRed-FPN1 and pEmGFP-HephΔ43 (Fig. 3Ae) and pDsRed-FPN1 and pEmGFP-HephΔ685 (Fig. 3Af) with also ∼75% colocalization (Fig. 3B) observed for both pairs.
Fig. 3.
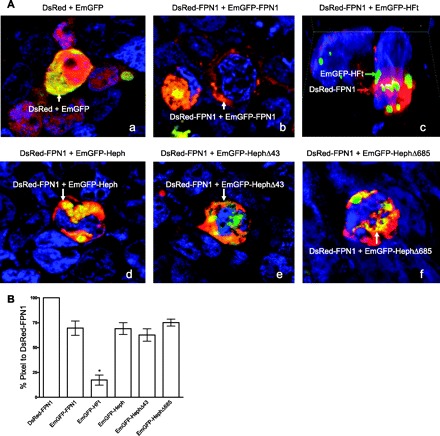
Subcellular localization of DsRed with EmGFP and DsRed-FPN1 with EmGFP-FPN1, EmGFP-ferritin heavy chain (HFt), EmGFP-Heph, EmGFP-HephΔ43, or EmGFP-HephΔ685 48 h after transfection in HEK293 cells. A: HEK293 cells were cotransfected with the vectors pDsRed and pEmGFP (a), pDsRed-FPN1 and pEmGFP-FPN1 (b), pDsRed-FPN1 and pEmGFP-HFt (c), pDsRed-FPN1 and pEmGFP-Heph (d), pDsRed-FPN1 and pEmGFP-HephΔ43 (e), and pDsRed-FPN1 and pEmGFP-HephΔ685 (f). B: the transfected cells were examined with a Zeiss AxioObserver Fluorescence microscope 48 h later after staining the nuclei with 4,6-diamidino-2-phenylindole as described in materials and methods. The colocalization of the red and green fusion proteins after cotransfections with the various combinations of vectors as described in A was determined by summing the pixels along the z-axis for the entire transfected cell and determining the percentage of colocalization using the Zeiss AxioVision 4.6.3 SP1 colocalization software as described in materials and methods. The colocalization is presented as the percentage of pixels of the EmGFP-fusion proteins colocalized with the pixels of DsRed-FPN1. Shown are means ± SE for at least 8 cells for each of the 3 transfection studies. *P < 0.05 vs. other columns. Not included in this analysis is the cotransfection of the vectors alone (Aa), which was always 95%.
Expression of FPN1 and Heph in rat duodenal epithelium.
Prior to examining whether FPN1 and Heph are colocalized in the rat duodenum during iron absorption, we determined FPN1 and Heph protein levels by Western blots. The duodenum expressed constant levels of the 70-kDa FPN1 and 150-kDa Heph bands for 4 h after iron feeding (Fig. 4). The smaller 56-kDa FPN1 band was also expressed at similar levels for up to 4 h (data not shown). The constant levels of FPN1 and Heph after iron absorption confirm previous reports by others (12, 14). As a positive control, HFt and LFt were measured by immunoprecipitation of ferritin from the lysates with anti-rat ferritin antiserum followed by subsequent analysis of HFt and LFt by SDS-PAGE. Both HFt and LFt protein levels increased steadily for the 4 h after iron feeding, demonstrating that the ingested iron was absorbed and elevated ferritin synthesis in the enterocytes (Fig. 4, A and B).
Fig. 4.
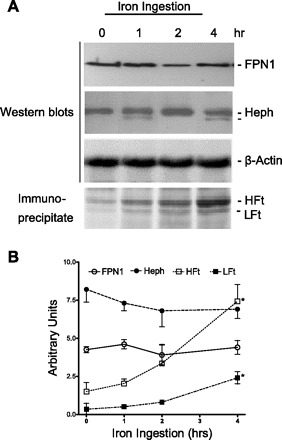
Effect of iron ingestion on duodenal expression of FPN1, Heph, and HFt and ferritin light chain (LFt). A: rat duodenal mucosal scrapes were obtained at the indicated times after iron ingestion, and RIPA extracts were prepared, subjected to SDS-PAGE, and then analyzed by Western blot with anti-FPN1, anti-Heph or anti-β-actin. For analysis of ferritin levels in the RIPA extracts, ferritin was immunoprecipitated with anti-rat ferritin antiserum and subjected to SDS-PAGE and subsequent to Coomassie blue staining showing HFt and LFt. Shown is a representative example of 3 experiments. B: proteins detected as in A were quantitated by gel scanning and the ratio was determined in arbitrary units of FPN1, Heph, HFt, or LFt to β-actin. Shown are means ± SE of 3 experiments. *P < 0.05 vs. the next earlier time point.
The subcellular localization of FPN1 and Heph in rat duodenal epithelium is altered by iron feeding.
Indirect immunofluorescent staining showed that FPN1 was expressed mainly in the BLM as can be seen by examining the epithelial cells in cross section (Fig. 5 Aa). Heph was also localized in the BLM, although with relatively less definition than FPN1, and in supranuclear and juxtanuclear nuclear zones of the enterocytes (Fig. 5Ab). These patterns, commonly known as “chicken-wire” appearance, are consistent with a BLM localization, and the subcellular locations for FPN1 and Heph are in agreement with earlier reports (1, 11, 28). Although FPN1 and Heph protein levels did not change after iron feeding, the subcellular localization of both proteins in longitudinal sections of the epithelium, especially FPN1, was altered by iron feeding (Fig. 5B). After overnight starvation for iron, FPN1 was expressed in the cytoplasmic microsomal compartments under the terminal web and in the BLM of the enterocytes (Fig. 5Ba). At 1 h after iron ingestion, FPN1 migrated to the lower BLM with marked depletion of the subterminal web region (Fig. 5Bb). At 2 and 4 h, FPN1 showed continued migration to the most basal regions of the BLM of the enterocytes (Fig. 5B, c and d). To quantify the changes in intracellular localization of FPN1, cells were arbitrarily divided into equal quarters along the apical-basal axis as indicated in Fig. 5B. The number of pixels in each quarter was summed for at least 10 well-orientated strips of 6–10 upper villus cells (Fig. 5C) with statistically significant redistribution of FPN1 seen at 1 h and continuing for 4 h after iron injection. These changes support the migration of FPN1 from the apical to the basal portions of the enterocytes, Heph also showed migration from the cytoplasmic, subterminal web to the BLM and the more basal portion of the cells (Fig. 5D, a–d). The shifts in localization of Heph were also quantified (Fig. 5F) and, although less marked than for FPN1, the changes were statistically significant and also support the migration of Heph to the basal portion of the cells.
Fig. 5.

Subcellular localization of FPN1 and Heph by immunofluorescent staining after iron ingestion. The cross section of enterocytes at upper nuclear zone showed “chicken-wire” pattern of FPN1 (Aa) and Heph (Ab), although Heph was less confined. FPN1 (B, a–d) and Heph (D, a–d) were detected in rat duodenal upper villus cells at 0, 1, 2, and 4 h after iron ingestion in an longitudinal section through the enterocytes as described in materials and methods. The fluorescent intensity of FPN1 in B and Heph in D in different zones was quantified (C and E) as described in materials and methods by arbitrarily dividing cells along the apical-basal axis into 4 compartments of equal length as shown in B and D and then summing the pixels in each compartment along the z-axis in 4–6 sections each containing 4–6 cells for 3 rats for each time point. Shown are means ± SE for the 3 rats for each time point. Statistical analysis was by 2-way ANOVA. *P < 0.05 vs. 0 h, and §P < 0.05 vs. 2 h.
Ultracentrifugation velocity sedimentation demonstrates that after iron feeding FPN1 and Heph occupy similarly sized vesicles.
Because of the similar changes in distribution of FPN1 and Heph observed after iron feeding, it is possible that both proteins came to occupy similar compartments within the enterocyte. A velocity sedimentation study of subcellular compartments in duodenal enterocytes before and after iron feeding showed that after iron starvation overnight ∼80% of FPN1 was located in the most rapidly sedimenting fractions 1–3 and ∼20% in the most slowly sedimenting and presumably the smallest, microsomal vesicles, fractions 11–13 (Fig. 6, lane 1). One hour after iron feeding, the FPN1 profile changed with the disappearance of FPN1 in the slowest fractions and appearance in fractions 5–7 (Fig. 6, lane 2). In contrast, Heph prior to iron feeding was predominantly distributed in the slower sedimenting fractions 11–13 (Fig. 6, lane 3) and after iron feeding shifted to the more rapidly sedimenting fractions 5–7, coinciding with the distribution of FPN1 (Fig. 6, lanes 1 vs. 3 and 2 vs. 4).
Fig. 6.
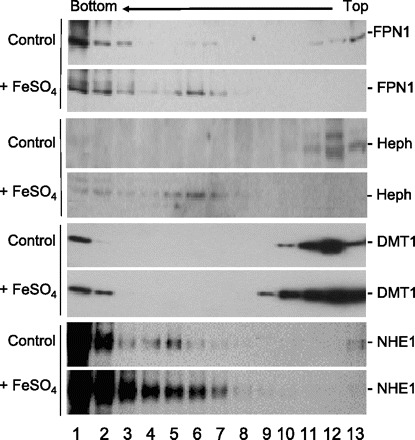
Ultracentrifugation velocity sedimentation profiles of FPN1, Heph, divalent metal transporter (DMT1), and sodium/hydrogen exchanger 1 (NHE1) prior to and after iron ingestion. The postnuclear fractions of the rat duodenal epithelium at 0 and 1 h after iron ingestion were loaded onto a sucrose gradient and centrifuged at 40,000 g for 60 min as described in materials and methods. Fractions were collected from the bottom to the top of the gradient and subjected to SDS-PAGE and Western blot with antibodies for FPN1, Heph, DMT1, or NHE1.
As a control we examined the velocity sedimentation profile of DMT1 (Fig. 6, lanes 5 and 6). We previously reported that under iron-starved conditions DMT1 is located in the BBM and with iron feeding DMT1 is internalized (41). Although the distribution of DMT1 and FPN1 was similar prior to iron feeding (Fig. 6, lanes 1 vs. 5), there was little change in the distribution of DMT1 with iron feeding with the exception of an increase in the slightly more rapidly sedimenting fractions 9–10 and a prominent increase in the rapidly sedimenting fraction 2 (Fig. 6, lanes 2 vs. 6). The increase of DMT1 in these fractions after iron feeding might be related to the specific increase of DMT1 expression observed at 1.5 h after iron ingestion (41). As an additional control, we examined the change in distribution of NHE1, which is known to be expressed on the BLM and whose cellular localization does not change with sodium absorption (42). Before or after iron feeding, there were no changes in NHE1 distribution in the bottom fractions 1–7 (Fig. 6, lanes 7 and 8).
FPN1 and Heph complexes can be detected by BN/SDS-PAGE.
BN/SDS-PAGE is a two-dimensional electrophoretic separation technique that in the first direction allows for protein-protein interactions to be maintained with charge imparted to proteins by binding of Coomassie blue. With a second-dimension separation via SDS-PAGE, proteins that maintained their interactions in the first dimension will appear in the same lane but at their respective apparent molecular masses after the second electrophoresis. When the duodenal subcellular fractions consisting of membrane and microsomes were analyzed by BN-PAGE, it was apparent that FPN1 and Heph were in two complexes (Fig. 7, black arrows and arrowheads). In the more rapidly migrating complex in the first dimension, FPN1 was present as dimers (140 kDa) (black arrow) and Heph was present as a monomer of ∼155 kDa (black arrow). The FPN1 dimer and Heph monomer migrated as a complex. Additionally, FPN1 fragments with molecular masses of ∼56 and 35 kDa (white arrows) (Fig. 7, top) and Heph fragments of ∼56, 48, and 25 kDa (white arrows) (Fig. 7, bottom) were present with migrations similar, but not identical, to the FPN1 dimer or Heph monomer, respectively. Presumably the presence of FPN1 dimers reflects the limited ability of SDS and DTT to gain access to the very hydrophobic FPN1 protein in the gel from the first dimension. In the more slowly migrating complex, FPN1 was present as a monomer (Fig. 7, top, black arrowhead) and Heph was present as a fragment of apparent molecular mass 75 kDa (Fig. 7, bottom, black arrowhead).
Fig. 7.
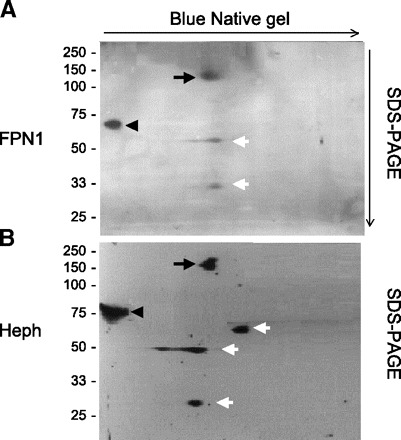
Two-dimensional gel electrophoretic analysis of FPN1 and Heph in rat duodenal epithelium. Two-dimensional gel electrophoresis was applied to a 15,000 g pellet extract obtained from duodenal epithelial cell sheets as described in materials and methods with separation in the first dimension by blue native (BN)-PAGE and in the second dimension by SDS-PAGE as also described in materials and methods. Following the SDS-PAGE the fractionated proteins were transferred to nitrocellulose membrane for Western blot analysis with anti-FPN1 (A) and anti-Heph (B) antisera, respectively. Shown is a representative example from 4 different 2-dimensional electrophoretic analyses each performed on isolated epithelial cells from a separate rat. Black arrows and arrowheads indicate the 2 complexes containing FPN1 and Heph, respectively. White arrows indicate FPN1 and Heph fragments.
Coimmunoprecipitation of FPN1 and Heph.
The interaction of FPN1 and Heph in the intestine was further shown by coimmunoprecipitation. Duodenal epithelial extracts obtained at 0, 1, and 3 h after iron feeding were immunoprecipitated with either anti-Heph or anti-FPN1 antiserum, the precipitates separated by SDS-PAGE, transferred to a nitrocellulose membrane, stained by Ponceau S, and then analyzed by Western blot with the other antisera. With the immunoprecipitate obtained with anti-Heph, a protein of ∼155 kDa was observed in the Ponceau S-stained membrane, consistent with Heph, whereas Western blot analysis of the same membrane with anti-FPN1 demonstrated bands of ∼130 and 70 kDa (Fig. 8, A and B). The 55-kDa IgG heavy chains were present as expected with both analyses. Similarly, with the immunoprecipitate obtained with anti-FPN1, a band of ∼130 kDa could be demonstrated by protein staining (Fig. 8B, left), whereas Western blot analysis with anti-Heph revealed a protein of ∼155-kDa band (Fig. 8B, right).
Fig. 8.
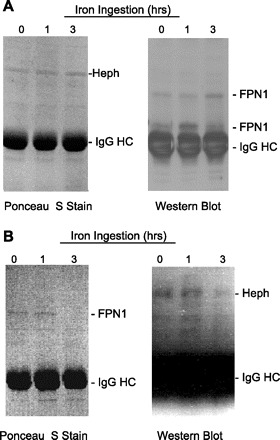
Rat duodenal epithelial FPN1 and Heph coimmunoprecipitate. Rat duodenal epithelium obtained at 0, 1, and 3 h after iron ingestion were homogenized in RIPA buffer containing 1% Triton X-100 and protease inhibitors and the 10,000 g supernatants immunoprecipitated either with anti-Heph or anti-FPN1 antiserum and absorbed to protein A-agarose beads, the immune complexes were subjected to SDS-PAGE, and the proteins were transferred to nitrocellulose membranes as described in materials and methods. The membranes were stained by Ponceau S (left) and then analyzed by Western blot analysis (right) with the immunoprecipitate from anti-FPN1 being analyzed with anti-Heph antisera (A) and the immunoprecipitate from anti-Heph being analyzed with anti-FPN1 (B). HC, heavy chain.
DISCUSSION
The molecular mechanisms involved in the transport of iron across the duodenal epithelium have been clarified in recent years with the description of the multiple proteins involved. Likewise, how iron transport is regulated has been elucidated with the detailed descriptions of hepcidin, the iron stores regulator, and FPN1 (13, 20, 30, 40). Still to be determined is how the iron transporters DMT1 and FPN1 and accessory proteins such as Dcytb and Heph in the duodenum in situ act to absorb iron from the lumen and release iron into the systemic circulation. In the present study, we examined the interaction of FPN1 and Heph, two proteins well described as essential for iron transport across the BLM, during the process of iron absorption.
The present study was initiated in part by our prior observation that with iron feeding DMT1 is internalized (24, 41), undergoing transcytosis and interacting with vesicles derived from the BLM (25). Because of the essential requirement of FPN1 and Heph to release of iron from the enterocyte, we initiated the present studies to see whether with iron feeding either protein exhibited translocation and whether at some point after iron feeding the two proteins were colocalized. Indeed, following iron feeding FPN1 exhibited dramatic changes in subcellular localization by translocating from the terminal web zone prior to iron feeding to the subnuclear region of BLM after iron feeding, consistent with our earlier observation (39). The migration of FPN1 observed after iron feeding is different from that induced by hepcidin, as observed in HEK293 cells where hepcidin induces the internalization of membrane bound FPN1 with subsequent digestion by lysosomes (30). The iron-induced migration of FPN1 is unlikely to be the result of hepcidin production since hepcidin was not significantly generated in the liver within 4 h of iron ingestion (40) and iron feeding did not result in decreased FPN1 expression.
We also observed that Heph exhibited translocation in the enterocytes similar to that observed with FPN1, although a greater amount of Heph remained in the apical cytoplasm compared with FPN1. The patterns as seen in Fig. 5 of the statistically significant changes in the location of FPN1 and Heph with time following iron feeding (i.e., decreasing FPN1 and Heph in zones 1 and 2, the rise and fall in zone 3, and the continued increase with in zone 4) strongly support the migration of these two proteins from the apical to the basal portions of the enterocytes. Several observations support that a portion of Heph appears to be associated, i.e., colocalized, with FPN1: First, following cotransfection of DsRed-FPN1 and EmGFP-Heph, colocalization could be seen by fluorescent microscopy. The extent of colocalization was not affected by elimination of the COOH terminus, i.e., with pEmGFP-HephΔ43, which contains a putative transmembrane domain, nor by the elimination of three of the four putative ferroxidase domains, i.e., with pEmGFP-HephΔ685. Clearly, the pattern of distribution of FPN1 and Heph differed from the immunofluorescent tags alone and the extent of colocalization of FPN1 and Heph was far greater than seen for FPN1 and HFt. Second, the velocity sedimentation profiles of FPN1 and Heph became similar after iron feeding. This observation only supports that these proteins translocate to vesicles of similar size. It is of interest that DMT1, which also translocates following iron feeding, exhibited smaller changes in sedimentation, suggesting that DMT1 was internalized into different vesicles than those containing FPN1 and Heph. Third, the two-dimensional separation of proteins by BN/SDS-PAGE strongly suggests that Heph and FPN1 interact in the duodenal membrane, a conclusion supported by the fourth observation of the coimmunoprecipitation of FPN1 and Heph.
BN/SDS-PAGE has been used to identify various protein-protein interactions in mitochondria, in plasma membranes, and in signaling pathways (3, 34, 36). In the present study, this technique allowed us to examine the interactions of FPN1 and Heph. We observed that Heph was present in two complexes: a complex that migrated faster in the first dimension and in which FPN1 was present as a dimer and a more slowly migrating complex in which FPN1 was present as a monomer. The finding of a dimeric form of FPN1 confirms recent data using cross-linking showing that FPN1 is present as a dimers or multimers (7). Presumably FPN1 dimers persist in the second dimension because of insufficient equilibration of reducing agent and SDS during preparation of the first-dimension gels for the second-dimension electrophoresis. The apparent molecular mass of ∼70 kDa for the FPN1 detected in the more slowly migrating complex also confirms that FPN1 can exist as monomers (15). Although the functional activity of the FPN1-Heph complexes cannot be determined from these studies, the presence of a dimeric form of FPN1 offers a possible explanation as to how heterozygotes for FPN1 mutations can result in iron overload (8, 23).
Copper deficiency compromises the function of Heph as a ferroxidase (5), shortens the half life of Heph (32, 33), and subsequently reduces iron transport across the BLM of enterocytes. It is not known, however, whether under conditions of copper deficiency the interactions and migration of Heph and FPN1 would be altered. It is also unclear why the majority of Heph did not migrate after iron feeding. Perhaps if a larger amount of iron was fed to the rats, a greater percentage of the Heph would translocate. Alternatively, Heph might have additional functions that remain to be elucidated.
The intracellular migration of FPN1 and Heph may serve as a protective mechanism by which the migration to the subnuclear BLM would reduce the accessibility of absorbed iron to FPN1 and Heph complexes. As a result, the accumulation of intracellular iron would reduce the binding of IRPs to the IREs of HFt and LFt mRNAs, subsequently increasing HFt and LFt mRNA translation, resulting in increased ferritin as demonstrated in our studies (Fig. 4). Thus the originally proposed function of ferritin as the mucosal block for iron absorption could be explained by our observation that with iron ingestion there is intracellular migration of FPN1 and Heph and as well as increase in ferritin synthesis to prevent further iron absorption. In addition, the effects of ingested iron on Dcytb and DMT1 expression and internalization may also serve as components of the mucosal block of iron absorption (12, 18, 35).
In our present studies, we observed that iron ingestion increases ferritin, but not FPN1 protein levels. Although iron absorption was not measured directly, the increase of ferritin synthesis demonstrates that iron uptake had occurred. The observed increase of ferritin is in agreement with an earlier report that, in a time course similar to our studies following ingestion of 59Fe, the ingested iron could be found in the enterocytes and in the liver (22). Since FPN1, as well as ferritin, contain an IRE in the 5′-untranslated region, iron ingestion should induce the IRP/IRE response to increase translation of both proteins. The different responses of FPN1 and ferritin indicate that, in addition to the IRE, other factors such as the position of IRE and sequences upstream or downstream of the translational initiation site also play an important role for translational regulation (16, 26, 29).
Heph, unlike its analog ceruloplasmin, contains a predicted COOH-terminal transmembrane sequence with a PDZ domain (37). The PDZ domain may be important for trafficking Heph to the BLM by binding to the COOH-terminal PDZ motif in FPN1 (28). The mutated Heph in sla mice retains the PDZ domain in the COOH terminus but remains in the cytoplasm (21), suggesting that the PDZ domain is either not functional or inaccessible to the PDZ motif of FPN1. The present data further show that DsRed-FPN1 colocalized equally with EmGFP-Heph with or without the COOH-terminal PDZ domain, suggesting that the sequences present in pEmGFP-HephΔ685 may also play a role for the interaction of Heph and FPN1. Since the targeted movement of Heph and FPN1 along the BLM appears to occur in response to iron signaling, the search for molecules responsible for the iron signaling for FPN1-Heph migration in enterocytes is in progress.
GRANTS
This study is supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Research Grants DK-065101 (K. Y. Yeh) and DK-43785 (J. Glass) and by the Feist-Weiller Cancer Center, Louisiana University Health Sciences Center, Shreveport, LA.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 275: 19906–19912, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Arosio P, Levi S. Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med 33: 457–463, 2002 [DOI] [PubMed] [Google Scholar]
- 3.Babusiak M, Man P, Petrak J, Vyoral D. Native proteomic analysis of protein complexes in murine intestinal brush border membranes. Proteomics 7: 121–129, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Bjerknes M, Cheng H. Methods for the isolation of intact epithelium from the mouse intestine. Anat Rec 199: 565–574, 1981 [DOI] [PubMed] [Google Scholar]
- 5.Chen H, Huang G, Su T, Gao H, Attieh ZK, McKie AT, Anderson GJ, Vulpe CD. Decreased hephaestin activity in the intestine of copper-deficient mice causes systemic iron deficiency. J Nutr 136: 1236–1241, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Corsi B, Perrone F, Bourgeois M, Beaumont C, Panzeri MC, Cozzi A, Sangregorio R, Santambrogio P, Albertini A, Arosio P, Levi S. Transient overexpression of human H- and L-ferritin chains in COS cells. Biochem J 330: 315–320, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Domenico I, Ward DM, Musci G, Kaplan J. Evidence for the multimeric structure of ferroportin. Blood 109: 2205–2209, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devalia V, Carter K, Walker AP, Perkins SJ, Worwood M, May A, Dooley JS. Autosomal dominant reticuloendothelial iron overload associated with a 3-base pair deletion in the ferroportin 1 gene (SLC11A3). Blood 100: 695–697, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403: 776–781, 2000 [DOI] [PubMed] [Google Scholar]
- 10.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 1: 191–200, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Frazer DM, Vulpe CD, McKie AT, Wilkins SJ, Trinder D, Cleghorn GJ, Anderson GJ. Cloning and gastrointestinal expression of rat hephaestin: relationship to other iron transport proteins. Am J Physiol Gastrointest Liver Physiol 281: G931–G939, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Frazer DM, Wilkins SJ, Becker EM, Murphy TL, Vulpe CD, McKie AT, Anderson GJ. A rapid decrease in the expression of DMT1 and Dcytb but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut 52: 340–346, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganz T, Nemeth E. Iron imports. IV. Hepcidin and regulation of body iron metabolism. Am J Physiol Gastrointest Liver Physiol 290: G199–G203, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Gleeson F, Ryan E, Barrett S, Russell J, Kelleher B, Crowe J. Duodenal Dcytb and hephaestin mRNA expression are not significantly modulated by variations in body iron homeostasis. Blood Cells Mol Dis 35: 303–308, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Goncalves AS, Muzeau F, Blaybel R, Hetet G, Driss F, Delaby C, Canonne-Hergaux F, Beaumont C. Wild-type and mutant ferroportins do not form oligomers in transfected cells. Biochem J 396: 265–275, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goossen B, Caughman SW, Harford JB, Klausner RD, Hentze MW. Translational repression by a complex between the iron-responsive element of ferritin mRNA and its specific cytoplasmic binding protein is position-dependent in vivo. EMBO J 9: 4127–4133, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388: 482–488, 1997 [DOI] [PubMed] [Google Scholar]
- 18.Hahn PF, Bale WF, Ross JF, Balfour WM, Whipple GH. Radioactive iron absorption by gastro-intestinal tract: influence of anemia, anoxia, and antecedent feeding distribution in growing dogs. J Exp Med 78: 169–188, 1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han O, Kim EY. Colocalization of ferroportin-1 with hephaestin on the basolateral membrane of human intestinal absorptive cells. J Cell Biochem 101: 1000–1010, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 117: 285–297, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Kuo YM, Su T, Chen H, Attieh Z, Syed BA, McKie AT, Anderson GJ, Gitschier J, Vulpe CD. Mislocalisation of hephaestin, a multicopper ferroxidase involved in basolateral intestinal iron transport, in the sex linked anaemia mouse. Gut 53: 201–206, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linder MC, Dunn V, Isaacs E, Jones D, Lim S, Van Volkom M, Munro HN. Ferritin and intestinal iron absorption: pancreatic enzymes and free iron. Am J Physiol 228: 196–204, 1975 [DOI] [PubMed] [Google Scholar]
- 23.Liu XB, Yang F, Haile DJ. Functional consequences of ferroportin 1 mutations. Blood Cells Mol Dis 35: 33–46, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Ma Y, Specian RD, Yeh KY, Yeh M, Rodriguez-Paris J, Glass J. The transcytosis of divalent metal transporter 1 and apo-transferrin during iron uptake in intestinal epithelium. Am J Physiol Gastrointest Liver Physiol 283: G965–G974, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Ma Y, Yeh M, Yeh KY, Glass J. Iron imports. V. Transport of iron through the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol 290: G417–G422, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Mascotti DP, Rup D, Thach RE. Regulation of iron metabolism: translational effects mediated by iron, heme, and cytokines. Annu Rev Nutr 15: 239–261, 1995 [DOI] [PubMed] [Google Scholar]
- 27.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291: 1755–1759, 2001 [DOI] [PubMed] [Google Scholar]
- 28.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 5: 299–309, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Mok H, Jelinek J, Pai S, Cattanach BM, Prchal JT, Youssoufian H, Schumacher A. Disruption of ferroportin 1 regulation causes dynamic alterations in iron homeostasis and erythropoiesis in polycythaemia mice. Development 131: 1859–1868, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: 2090–2093, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 98: 8780–8785, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nittis T, Gitlin JD. Role of copper in the proteosome-mediated degradation of the multicopper oxidase hephaestin. J Biol Chem 279: 25696–25702, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Reeves PG, Demars LC, Johnson WT, Lukaski HC. Dietary copper deficiency reduces iron absorption and duodenal enterocyte hephaestin protein in male and female rats. J Nutr 135: 92–98, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Schagger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem 217: 220–230, 1994 [DOI] [PubMed] [Google Scholar]
- 35.Stewart WB, Yuile CL, Claiborne HA, Snowman RT, Whipple GH. Radioiron absorption in anemic dogs; fluctuations in the mucosal block and evidence for a gradient of absorption in the gastrointestinal tract. J Exp Med 92: 375–382, 1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swamy M, Siegers GM, Minguet S, Wollscheid B, Schamel WW. Blue native polyacrylamide gel electrophoresis (BN-PAGE) for the identification and analysis of multiprotein complexes. Sci STKE 2006: pl4, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Syed BA, Beaumont NJ, Patel A, Naylor CE, Bayele HK, Joannou CL, Rowe PS, Evans RW, Srai SK. Analysis of the human hephaestin gene and protein: comparative modelling of the N-terminus ecto-domain based upon ceruloplasmin. Protein Eng 15: 205–214, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 21: 195–199, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Yeh KY, Yeh M, Glass J. Dietary iron induces ferroportin 1 clustering and migration into the basolateral membrane of the rat intestinal epithelium (Abstract). Gastroenterology 120: A678, 2001 [Google Scholar]
- 40.Yeh KY, Yeh M, Glass J. Hepcidin regulation of ferroportin 1 expression in the liver and intestine of the rat. Am J Physiol Gastrointest Liver Physiol 286: G385–G394, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Yeh KY, Yeh M, Watkins JA, Rodriguez-Paris J, Glass J. Dietary iron induces rapid changes in rat intestinal divalent metal transporter expression. Am J Physiol Gastrointest Liver Physiol 279: G1070–G1079, 2000 [DOI] [PubMed] [Google Scholar]
- 42.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol 67: 411–443, 2005 [DOI] [PubMed] [Google Scholar]