

Abstract
The transduction function for ADP stimulation of mitochondrial ATP synthesis in skeletal muscle was reconstructed in vivo and in silico to investigate the magnitude and origin of mitochondrial sensitivity to cytoplasmic ADP concentration changes. Dynamic in vivo measurements of human leg muscle phosphocreatine (PCr) content during metabolic recovery from contractions were performed by 31P-NMR spectroscopy. The cytoplasmic ADP concentration ([ADP]) and rate of oxidative ATP synthesis (Jp) at each time point were calculated from creatine kinase equilibrium and the derivative of a monoexponential fit to the PCr recovery data, respectively. Reconstructed [ADP]-Jp relations for individual muscles containing more than 100 data points were kinetically characterized by nonlinear curve fitting yielding an apparent kinetic order and ADP affinity of 1.9 ± 0.2 and 0.022 ± 0.003 mM, respectively (means ± SD; n = 6). Next, in silico [ADP]-Jp relations for skeletal muscle were generated using a computational model of muscle oxidative ATP metabolism whereby model parameters corresponding to mitochondrial enzymes were randomly changed by 50–150% to determine control of mitochondrial ADP sensitivity. The multiparametric sensitivity analysis showed that mitochondrial ADP ultrasensitivity is an emergent property of the integrated mitochondrial enzyme network controlled primarily by kinetic properties of the adenine nucleotide translocator.
Keywords: mitochondria, nuclear magnetic resonance, mathematical modeling, regulation, adenosine 5′-diphosphate
the metabolic regulation underlying energy balance in mammalian cells has long been the subject of investigation, in particular regulation of mitochondrial ATP synthesis (1). At first, a relatively straightforward picture emerged from studies in isolated mitochondria; a feedback control loop involving transduction of changes in the extramitochondrial concentrations of the ATP hydrolysis products ADP and Pi to the intramitochondrial ATP synthetic network during cellular work explained energy balance (10, 13). 31P-NMR spectroscopy (11) and computational modeling (28) later showed that mitochondrial sensing of concentration changes in ADP alone sufficed to explain energy balance in skeletal muscle. However, in cardiac muscle, near-constant phosphocreatine (PCr), and thereby ADP, concentrations were measured during work jumps (3). This observation led to the proposition of a second, if not alternative, mitochondrial metabolic control mechanism in excitable cells such as cardiac muscle, i.e., a feedforward control loop involving direct or indirect transduction of intracellular calcium concentration changes to the mitochondrial ATP synthetic network (1). More recently, yet another alternative respiratory control mechanism of particular relevance to energy balance in the heart has been identified (2). It involves a mix of feedforward and feedback kinetic effects of Pi on multiple reactions in the oxidative ADP phosphorylation pathway (7).
What has been relatively lacking for each of the postulated respiratory control signals, in particular calcium, is a thorough kinetic characterization of the corresponding mitochondrial transduction function f([X],Jp), where [X] is the extramitochondrial concentration of signal molecule X and Jp equals the rate of mitochondrial ATP synthesis. The need for explicit knowledge of these transduction functions in respiratory control model validation has recently become all the more pressing due to a surge in availability of computational models of mitochondrial oxidative metabolism that require proper validation criteria (4, 15, 31, 33, 42, 46). Of these wanting kinetic functions, the transduction function f([ADP],Jp) has been investigated most thoroughly (10, 13, 17, 23, 24, 29). Characterized initially as “approximately hyperbolic” with a “Km” in the range of 20–30 μM (10, 13), it was later reported that this function is in fact sigmoidal with an apparent kinetic order of at least two (24). Two independent studies have since, albeit indirectly, confirmed the non-Michaelis-Menten (MM) nature of the ADP stimulation of mitochondrial ATP synthesis (14, 45).
According to Koshland et al.'s (27) classification of biological sensory systems, a sigmoidal ADP transduction function with a Hill coefficient (nH) >1 would indicate that mitochondria are ultrasensitive to variations in extramitochondrial ADP concentration changes. Normal sensitivity was defined as the sensitivity corresponding to a hyperbolic input-output function such as in MM kinetics (i.e., nH = 1) (27). Importantly, mitochondrial ultrasensitivity, but not normal sensitivity to ADP, was shown to explain energy balance by feedback respiratory control for a range of mammalian cell types, including skeletal and cardiac muscle (24). However, the hypothesis of mitochondrial ADP ultrasensitivity has generally not been embraced. Quantitative formalisms based on MM kinetics of ADP stimulation of mitochondrial ATP synthesis have continued to be used in the field of muscle energetics to evaluate mitochondrial function on the basis of 31P-NMR spectroscopy measurements (34). A possible explanation may be the lack of any verified mechanistic basis for second-order kinetic behavior of ADP stimulation of mitochondria. Non-MM kinetics of ATP/ADP exchange across the inner membrane catalyzed by the adenine nucleotide translocator (ANT) has been proposed but not confirmed as the origin of mitochondrial ADP ultrasensitivity (24).
Here, the magnitude and control of mitochondrial ADP sensitivity are investigated further. We collected multiple high-time resolution 31P-NMR spectroscopy data sets on PCr concentration dynamics in human muscle during recovery from exercise to reconstruct the in vivo mitochondrial ADP transduction function with a high number of data points for accurate kinetic characterization. The result confirmed second-order kinetics of ADP stimulation of mitochondrial respiration. Next, we investigated the control of mitochondrial ADP sensitivity by conducting a network analysis of a computational model of mitochondrial oxidative ADP phosphorylation in muscle (46). Hereto, a multiparametric sensitivity analysis (MPSA) was performed, which involved generation of multiple random sets of parameter values for all mitochondrial enzymes in the model followed by reconstruction and characterization of the mitochondrial ADP transduction function for each set. The results indicated that mitochondrial ADP ultrasensitivity is an emergent property of the integrated mitochondrial metabolic network determined primarily by the kinetic properties of the ANT.
MATERIALS AND METHODS
Reconstruction and Analysis of the In Vivo Mitochondrial ADP Transduction Function in Muscle
Subjects.
Six healthy normally active subjects (4 males, 2 females; mean age ± SD 31 ± 12 yr) participated in the study. The nature and the risks of the experimental procedures were explained to the subjects, and all gave their written informed consent to participate in the study, which was approved by the local Medical Ethics Committee of the Máxima Medical Center, Veldhoven, The Netherlands.
31P-magnetic resonance spectroscopy.
31P-magnetic resonance spectroscopy (MRS) was performed at 1.5 Tesla (Gyroscan S15/ACS; Philips Medical Systems, Best, The Netherlands), as described previously (41). Briefly, after localized shimming, 31P signals were collected using a 6-cm-diameter surface coil placed over the m. vastus lateralis (spectral width, 2,000 Hz; no. of data points, 1,024). From the dimension of the coil and the size and geometry of a typical upper leg, it was estimated that the majority of the signal in the unlocalized 31P-MRS measurements originated from the m. vastus lateralis, with minimal contaminations from the adjacent m. rectus femoris and underlying m. vastus intermedius. Spectra were acquired during a rest-exercise-recovery protocol, with a repetition time of 3 s and 2 scans yielding a time resolution of 6 s. The first 20 spectra (2 min) were measured at rest, after which the subjects started the exercise. Exercise consisted of dynamic, single-leg extensions every 1.5 s in the supine position using a home-built MR compatible ergometer (41). The initial workload varied per subject and ranged between 7.5 and 12.5 W. This level was maintained for the first minute, and the workload was then increased by 5 W each minute. Subjects performed exercises of different durations, and eight to 12 data sets were collected per subject during four to eight different sessions, with ≥15 min rest between different protocols within one session. The position of the 31P surface coil was marked on the leg during the first session, and the coil was placed at the same location in subsequent sessions.
31P-MRS data analysis.
Spectra were fitted in the time domain by using a nonlinear least squares algorithm (43) in the jMRUI software package (32), as described previously (41). PCr, Pi, and ATP signals were fitted to Lorentzian line shapes. Absolute concentrations of the phosphorylated metabolites were calculated after correction for partial saturation and assuming that [ATP] is 8.2 mM at rest (39). Intracellular pH was calculated from the chemical shift difference between the Pi and PCr resonances (38). All data sets had an end-exercise pH of ≥6.7. The free cytosolic ADP concentration ([ADP]) was calculated from pH and [PCr] using a creatine kinase equilibrium constant (Keq) of 1.66 × 109 M−1 (30) and assuming that 15% of the total creatine is unphosphorylated at rest (8), using the following equation (Eq. 1):
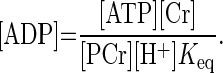 |
(1) |
The molar free energy of cytosolic ATP hydrolysis was calculated according to Eq. 2,
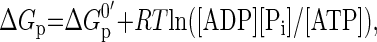 |
(2) |
where ΔGp0′ is −31.8 kJ/mol at 37°C (21).
The PCr recovery time course, PCr(t), was fitted to a monoexponential function (Eq. 3):
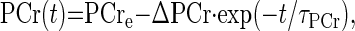 |
(3) |
where PCre is the PCr level after recovery, ΔPCr is the difference between the PCr levels after recovery and at the end of exercise, and τPCr is the time constant for PCr resynthesis. The PCr resynthesis rate at time t [VPCr(t)] was calculated from the derivative of the fitted PCr recovery time course (Eq. 3). During recovery from exercise, PCr is resynthesized purely as a consequence of oxidative ATP synthesis (35, 37, 38). Because the creatine kinase reaction is much faster than oxidative ATP production (45), VPCr(t) reflects mitochondrial oxidative phosphorylation flux. Covariations of VPCr(t) with thermodynamic (ΔGp) and kinetic ([ADP]) adenine nucleotide concentration functions were analyzed by nonlinear curve fitting of a Hill function of the form (Eq. 4):
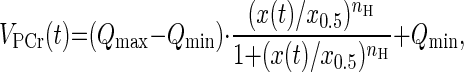 |
(4) |
with Qmax and Qmin the maximal and minimal net ADP phosphorylation fluxes1 , x0.5 the ΔGp or [ADP] value at half-maximal VPCr, and nH the Hill coefficient (24). This analysis was performed for each subject separately, using the pooled data from all the exercise protocols of that subject (8–12 data sets/subject). Only data points with VPCr(t) > 0.02 mM/s were included in the analysis. All nonlinear curve fitting was performed using Matlab (version 7.3; Mathworks, Natick, MA).
Statistical analysis.
All data are expressed as means ± SD. Statistical analyses were performed using the SPSS 15.0 software package (SPSS, Chicago, IL). Because of the small number of subjects, the nonparametric Wilcoxon signed-rank test was used for paired comparisons of the data. The level of significance was set at P < 0.05.
Simulation and Analysis of In Silico Mitochondrial ADP Transduction Functions in Muscle
The computational model of skeletal muscle oxidative ATP metabolism featuring a detailed biophysical model of mitochondrial oxidative ADP phosphorylation by Wu et al. (46) was used as platform for all in silico investigations of the origin of mitochondrial ADP ultrasensitivity. First, an analysis of the sensitivity of the macroscopic parameters Qmax, K50, and nH of in silico-reconstructed [ADP]-Jp relations for muscle toward 19 mitochondrial parameters in the Wu et al. (46) model was performed (Table 1). This particular set consisted of 15 mitochondrial parameters of which the value had previously been estimated by model fitting (denoted by Ref. 5c in Table 2 of Ref. 46) and four mitochondrial parameters (kO2, Km,ADP, θ, and β, respectively) of which the value had been taken from a computational model (denoted by Ref. 26 in Table 2 of Ref. 46). The design of this MPSA was based on methods described elsewhere (47), including statistical analysis (20). Next, the MPSA results were used to investigate whether any set of model parameter values that would give rise in silico to mitochondrial ADP ultrasensitivity existed. Hereto, the model was fitted to an in vivo [ADP]-Jp data set from an individual muscle, whereby only model parameters with a significant Kolmogorov-Smirnov (K-S) test score in the MPSA (see below) were varied.
Table 1.
Overview of the mitochondrial model parameters tested in the multiparametric sensitivity analysis of the Wu et al. (46) model of oxidative ATP metabolism in muscle
| Parameter No. | Model Parameter | Description |
|---|---|---|
| 1 | R | Dehydrogenase parameter |
| 2 | kPi,1 | Dehydrogenase parameter |
| 3 | kPi,2 | Dehydrogenase parameter |
| 4 | XDH | Dehydrogenase parameter |
| 5 | XC1 | Complex I activity |
| 6 | XC3 | Complex III activity |
| 7 | kPi,3 | Complex III parameter |
| 8 | kPi,4 | Complex III parameter |
| 9 | XC4 | Complex IV activity |
| 10 | kO2 | Complex IV parameter |
| 11 | XF1 | F0F1-ATPase activity |
| 12 | XANT | ANT activity |
| 13 | Km,ADP | Michaelis constant ANT |
| 14 | θ | ANT parameter |
| 15 | kPiHt | H+/Pi− cotransporter parameter |
| 16 | XPiHt | H+/Pi− cotransport activity |
| 17 | XKH | K+/H+ antiporter activity |
| 18 | XHle | H+ leak activity |
| 19 | β | Mitochondrial matrix buffering capacity |
ANT, adenine nucleotide translocator.
Table 2.
Results of the unconstrained curve fitting of Eq. 4 to the (ΔGp, VPCr) data for the 6 subjects
| Qmax, mM/s | Qmin, mM/s | ΔGp,0.5, kJ/mol | nH | |
|---|---|---|---|---|
| Subject | ||||
| 1 | 0.81 | −0.04 | −58.1 | 24.9 |
| 2 | 0.76 | −0.02 | −58.9 | 25.9 |
| 3 | 0.69 | −0.09 | −58.8 | 21.8 |
| 4 | 0.70 | −0.04 | −57.9 | 23.5 |
| 5 | 0.86 | −0.03 | −57.8 | 23.8 |
| 6 | 0.65 | −0.03 | −57.9 | 22.7 |
| Means ± SD | 0.74±0.08 | −0.04±0.02 | −58.2±0.5 | 23.8±1.5 |
Qmax, maximal net phosphocreatine (PCr) resynthesis flux; Qmin, minimal net PCr resynthesis flux; ΔGp,0.5, molar free energy of cytosolic ATP hydrolysis at half-maximal VPCr; nH, Hill coefficient.
MPSA
Generation of random sets of parameter values.
Multiple random samples were taken from a 50–150% range of the default value for each of the 19 kinetic parameters of the mitochondrial enzymes in the model (Table 1) plus five dummy parameters yielding multiple random sets of parameter values for simulation and analysis of in silico [ADP]-Jp relations for muscle. The introduction of dummy parameters in this analysis (but not in the model) was necessary to dissect the contribution of sensitive vs. insensitive model parameters in a particular set to any changes in Qmax, K50, and nH of the corresponding in silico [ADP]-Jp relation. An optimized Monte Carlo sampling scheme was used to sample from the multidimensional distribution while guaranteeing that individual parameter ranges were evenly covered. In this case, the parameters were sampled from a logarithmic uniform distribution within the defined range. The vectors containing the parameter samples were combined to obtain N parameter combinations. It was verified that the number of Monte Carlo runs, N, was sufficiently large to guarantee a good representation of all possible parameter value combinations (see below). The sampling and combination process was done with Latin hypercube sampling, whereby each parameter range was divided into N equally probable intervals from which only one sample was drawn. These samples were permuted and stored in a vector (with N samples for each parameter). Subsequently, these permuted vectors of all p parameters were combined in an N-by-p matrix. After the initial combination process, the minimum distance between the sample points was maximized. The Latin hypercube was generated using the function lhsdesign of the Statistics Toolbox from Matlab. The number of Monte Carlo runs (N) was 5,000.
Simulations.
The model was simulated for each set of parameter values and characterized with respect to the particular Qmax, K50, and nH of the corresponding in silico [ADP]-Jp relation by fitting of a three-parameter Hill function (Eq. 4 with Qmin set at 0; in this case, x0.5 = [ADP]0.5 = K50). This particular Hill function rather than Eq. 4 was used for computational ease on the grounds that Qmin was very small compared with Qmax and not significantly different from 0 (see results and Table 2). To quantify the similarity of each of the simulations with respect to the reference simulation, the sum of squared differences was calculated as criterion function. Each set of parameter values was then classified as either “unacceptable” or “acceptable” by comparing the value of the criterion function to a certain threshold. Specifically, the set of parameter values was scored unacceptable if the criterion function value was greater than the threshold and acceptable in all other cases. The average value of the criterion function for the total ensemble of simulations was used as threshold value (20).
Sensitivity analysis.
The influence of each parameter on the model output was evaluated statistically using the K-S test (20). Hereto, the distributions of the individual parameter values associated with the unacceptable and the acceptable cases were compared. For each parameter, the cumulative frequency was calculated for all unacceptable and acceptable cases. The sensitivity is evaluated by a measure of the separation of the two cumulative frequency distributions. The K-S test for the ith parameter is represented as Eq. 5,
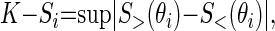 |
(5) |
where S> and S< are the cumulative frequency functions corresponding to unacceptable and acceptable cases, respectively, and θi is the parameter. The value of K-S is determined as the maximum vertical distance between the cumulative frequency distribution curves for n unacceptable and m acceptable cases, n + m = N. A larger K-S score indicates that the model is sensitive to variation in that parameter. The highest K-S score of the dummy parameters was used as threshold for statistical significance; i.e., parameters with a K-S score above the threshold were classified as sensitive. Finally, the K-S scores were summarized in a ranking of the sensitive parameters. This ranking was also used to verify that the number of Monte Carlo runs, N, was sufficiently large to guarantee a good representation of all possible parameter value combinations. For sufficiently large N, the ranking of the sensitive parameters is independent of the exact number of samples (47).
Model Fitting
To test whether any set of model parameter values existed that would give rise in silico to mitochondrial ADP ultrasensitivity, the model was fitted to an in vivo [ADP]-Jp data set from an individual muscle (subject 2) whereby only model parameters with a significant K-S score were adjusted. Model fitting was performed using a nonlinear least-squares optimization method employing the lsqnonlin algorithm in the parameter estimation toolbox in Matlab. Subsequently, in silico [ADP]-Jp covariations were computed for the model with the fitted parameter values and kinetically characterized by curve fitting of Eq. 4.
RESULTS
Reconstruction and Analysis of the In Vivo Mitochondrial ADP Transduction Function in Muscle
Figure 1 shows typical examples of 31P-MR spectra obtained from the vastus lateralis muscle of an individual subject 1) at rest (Fig. 1A), 2) at the end of exercise (Fig. 1B), and 3) at two time points during recovery (Fig. 1, C and D). Figure 2, solid line, shows the corresponding plot of the PCr concentration against recovery time together with the monoexponential fit of the recovery data. The PCr resynthesis rate was calculated from the derivative of the fitted PCr recovery time course for each time point sampled during recovery. For this particular population of six subjects, the maximal PCr resynthesis rate was 0.73 ± 0.05 mM/s. End-exercise ADP concentration (maximal) and molar Gibbs free energy of cytosolic ATP hydrolysis (lowest) were 86 ± 8 μM and −52.8 ± 0.4 kJ/mol, respectively.
Fig. 1.
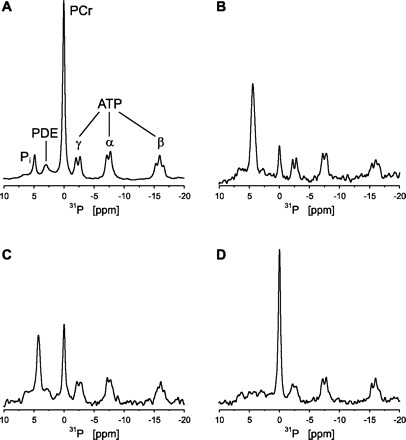
Typical m. vastus lateralis 31P-MR spectra for 1 subject at rest (A; no. of scans = 60), at the end of exercise (B: no. of scans = 2), and at 15 and 117 s of recovery (C and D, respectively; no. of scans = 2). Spectra were processed with 5 Hz line broadening. PDE, phosphodiesters; PCr, phosphocreatine. α, β and γ indicate the 3 phosphate groups of ATP. For this subject the PCr depletion at the end of exercise (B) was 81%, and the corresponding end-exercise pH was 6.72.
Fig. 2.
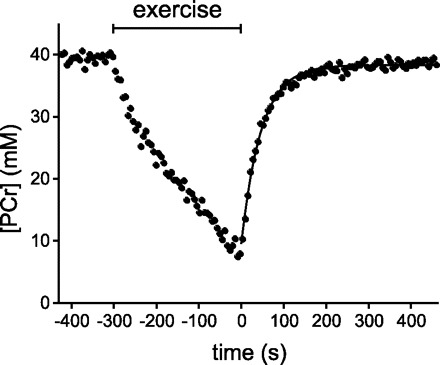
PCr concentration during rest, exercise, and recovery obtained from the corresponding data set shown in Fig. 1 (time resolution 6 s). The recovery of PCr (starting at t = 0) was fitted to a monoexponential function (solid line). The time constant for PCr recovery was 46.8 s.
To estimate the PCr resynthesis rate asymptotes Qmax and Qmin in the muscle cells, we analyzed the thermodynamic flow-force relation of PCr resynthesis (36, 40). This relation was characterized by unconstrained fitting of Eq. 4 to the (ΔGp, VPCr) data (Fig. 3). Almost the full range of sustainable energy balance states was covered by the experimental data points, resulting in accurate estimation of the flux asymptotes Qmax and Qmin (Table 2). The group mean value for ΔGp at half-maximal VPCr was −58.2 ± 0.5 kJ/mol.
Fig. 3.
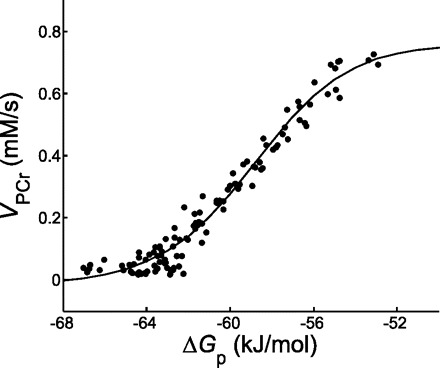
PCr resynthesis rate as a function of the molar free energy of cytosolic ATP hydrolysis (•) for an individual muscle (subject 2). The unconstrained, 4-parameter fit of Eq. 4 to the data is shown by the solid line.
The values for Qmax and Qmin determined from the thermodynamic flow-force relation were next imposed as constraints on the fit of Eq. 4 to the ([ADP], VPCr) data (Fig. 4, solid line) to determine the apparent affinity and kinetic order of mitochondrial ADP sensing. The results of the analysis for each of the subjects studied are summarized in Table 3. Group mean values for [ADP]0.5 and nH were 22 ± 3 μM and 1.9 ± 0.2, respectively. Eq. 4 was also fitted to the ([ADP], VPCr) data without imposing any constraints on the flux asymptotes (Fig. 4, dotted line, and Table 4). The fitted values for Qmax and Qmin were similar to the values obtained from the thermodynamic flow-force relation (Table 2), except for one muscle (subject 3). Fitted estimates of [ADP]0.5 and nH were not different from the values obtained from the constrained fit (Table 3). Last, the PCr resynthesis rate, VPCr(t), was also correlated with the ADP concentration at each measurement point using a hyperbolic function (nH = 1 in Eq. 4) corresponding to the classic MM ADP respiratory control model (12). The result for a single muscle is shown in Fig. 4 (dashed line). The MM ADP control model was incompatible with the experimental data at high flux values, causing overestimation of the maximal net PCr resynthesis flux Qmax [0.98 ± 0.13 vs. 0.74 ± 0.08 mM/s (Table 2), P < 0.05]. In addition, the MM ADP control model predicted a sevenfold higher net mitochondrial ATP hydrolysis rate at low ADP concentrations than the second-order control model [Qmin −0.27 ± 0.04 vs. −0.04 ± 0.02 mM/s (Table 2); P < 0.05].
Fig. 4.

PCr resynthesis rate as a function of ADP concentration (•) for an individual muscle (subject 2). Eq. 4 was fitted to the data: 2-parameter fit with maximal (Qmax) and minimal net ADP phosphorylation fluxes (Qmin) constrained to the values obtained from the thermodynamic flow-force relation (solid line), unconstrained 4-parameter fit (dotted line), and 3-parameter fit with Hill coefficient (nH) constrained to 1 (dashed line).
Table 3.
Results of the curve fitting of Eq. 4 to the ([ADP], VPCr) data for the 6 subjects with Qmax and Qmin constrained to the values obtained from the thermodynamic flow-force relation (Table 2)
| [ADP]0.5, μM | nH | |
|---|---|---|
| Subject | ||
| 1 | 25 | 1.9 |
| 2 | 19 | 2.1 |
| 3 | 18 | 1.7 |
| 4 | 23 | 1.9 |
| 5 | 24 | 1.8 |
| 6 | 22 | 1.7 |
| Means ± SD | 22±3 | 1.9±0.2 |
[ADP]0.5, ADP concentration at half-maximal VPCr.
Table 4.
Results of the unconstrained curve fitting of Eq. 4 to the ([ADP], VPCr) data for the 6 subjects
| Qmax, mM/s | Qmin, mM/s | [ADP]0.5, μM | nH | |
|---|---|---|---|---|
| Subject | ||||
| 1 | 0.78 | −0.03 | 24 | 2.1 |
| 2 | 0.72 | −0.03 | 18 | 2.3 |
| 3 | 0.62 | −0.01 | 18 | 2.3 |
| 4 | 0.65 | −0.05 | 21 | 2.0 |
| 5 | 0.83 | −0.07 | 22 | 1.7 |
| 6 | 0.64 | −0.05 | 21 | 1.7 |
| Means±SD | 0.71±0.09 | −0.04±0.02 | 21±2 | 2.0±0.3 |
Simulation and Analysis of In Silico Mitochondrial ADP Transduction Functions in Muscle
MPSA.
The results of the MPSA are summarized in Fig. 5. The K-S scores with respect to the Qmax, ADP affinity (K50), and nH of the mitochondrial ADP transduction function for each of the 19 mitochondrial kinetic parameters and five dummy parameters (parameters 20–24; Table 1) are shown in Fig. 5, A, B, and C, respectively. The threshold score for significant sensitivity determined by the dummy parameter K-S scores (20) was 0.03–0.04 (Fig. 5, A–C). On this basis, all three macroscopic kinetic parameters of the mitochondrial ADP transduction function were found to exhibit significant sensitivity to the particular kinetic properties of three mitochondrial enzymes in the model, i.e., the lumped tricarboxylic acid dehydrogenase activity (TCA-DH; parameters 1–4), respiratory chain complex III (CIII; parameters 6–8), and the ANT (parameters 12–14) (Fig. 5, A–C). Within this subset, the ANT parameter sensitivity was dominant (Fig. 5, A–C). Specifically, the Qmax of the mitochondrial ADP transduction function exhibited significant sensitivity to parameters 1–4, 7, 12, and 14 corresponding to TCA-DH parameters kPi,1 and kPi,2 and r and XDH, CIII parameter kPi,3, and ANT parameters XANT and θ, respectively (Table 1), whereby the sensitivity to the ANT parameter XANT was dominant (K-S score 0.46 vs. 0.05–0.07 for TCA-DH and CIII parameters, respectively; Fig. 5A). Likewise, the apparent kinetic order of the ADP transduction function (nH) exhibited significant sensitivity only to all TCA-DH parameters (parameters 1–4; Table 1) and all ANT parameters (parameters 12–14; Table 1), whereby the sensitivity to ANT parameter θ was dominant (K-S score 0.63 vs. 0.09–0.13 for all other ANT and TCA-DH parameters, respectively; Fig. 5B). Finally, the overall mitochondrial ADP affinity K50 exhibited significant sensitivity to the TCA-DH parameters kPi,1, r, and XDH (parameters 1, 3, and 4, respectively; Table 1), CIII parameters XC3 and kPi,3 (parameters 6 and 7, respectively), and ANT parameters Km-ANT and θ (parameters 13 and 14, respectively), whereby the sensitivity to θ was again dominant (K-S score 0.47 vs. 0.04–0.07 for all other significant parameters) (Fig. 5C).
Fig. 5.
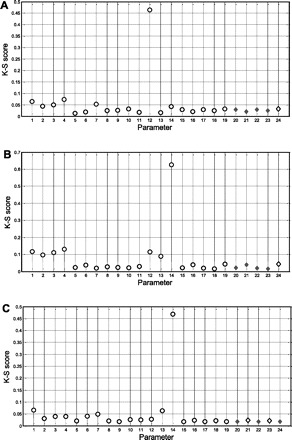
Kolmogorov-Smirnov (K-S) scores of the 19 model parameters (○) and 5 dummy parameters (◊) from the multiparametric sensitivity analysis of the model for each of the 3 macroscopic kinetic parameters of the mitochondrial transduction function: Qmax, (A) nH (B), and K50 (C). Shaded areas indicate the threshold of significance set by the K-S scores of the 5 dummy parameters in each graph.
Model fitting.
The MPSA results were next used to investigate whether any set of model parameter values that would give rise to in silico mitochondrial ADP ultrasensitivity existed. First, the transduction function for the default parameterization of the model (46) was characterized. Figure 6A shows the [ADP]-Jp covariation computed for the default set of model parameter values together with a set of experimental [ADP]-Jp data points obtained in an individual muscle. By scaling XANT and XDH, the in silico Qmax of oxidative phosphorylation was adjusted to 0.8 mM/s, corresponding to the in vivo estimate for this particular muscle (subject 2; Table 2). This required a twofold increase of XANT and a threefold increase of XDH to 0.016 and 0.260 mM/s, respectively. Curve-fitting of Eq. 4 to the simulated data showed that the ADP transduction function for the default parameterization of the model was hyperbolic (nH 1.06) with Qmax and Qmin of 0.80 and −0.02 mM/s, respectively, and a K50 of 0.16 mM. Clearly, the first-order in silico mitochondrial ADP sensitivity for the default model parameterization was incompatible with the measured in vivo response of Jp to [ADP] changes in muscle (Fig. 6A). Figure 6, B and C, shows the computed variations of the mitochondrial membrane potential (Δψm) and redox potential {([NADH]/[NAD])m}, respectively, with [ADP] for the default parameterization. Both potentials were predicted first to rapidly increase at very low [ADP] and subsequently gradually drop toward limit values of 170 mV and 1.3, respectively (Fig. 6, B and C).
Fig. 6.
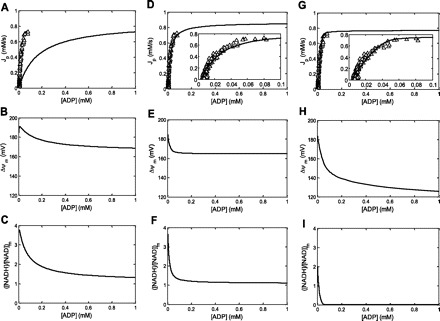
Simulations of the covariation of Jp (in mM/s), ΔΨm (in mV), and ([NADH]/[NAD])m with cyoplasmic ADP concentration (in mM) for 3 different model parameterizations. Open triangles correspond to measured in vivo ADP concentration ([ADP])-Jp covariation in an individual muscle (subject 2). A–C: default model parameterization with XANT and XDH is 0.016 and 0.269 mM/s, respectively. A: the simulated relation was kinetically characterized by unconstrained, 4-parameter curve fitting of Eq. 4 yielding fitted estimates for with Qmax, Qmin, nH, and K50 of 0.80 mM/s, −0.02 mM/s, 1.06, and 0.16 mM, respectively. D–F: fitted model parameterization I; XANT = 0.41 mM/s and θ = 1.0. XDH was kept at 0.269 mM/s. D: the simulated relation was kinetically characterized by unconstrained, 4-parameter curve fitting of Eq. 4 yielding fitted estimates for with Qmax, Qmin, nH, and K50 of 0.82 mM/s, −0.02 mM/s, 1.50, and 0.025 mM, respectively. G–I: fitted model parameterization II; XANT = 0.32 mM/s, XDH = 0.12 mM/s, and r = 3.1. G: the simulated relation was kinetically characterized by unconstrained, 4-parameter curve fitting of Eq. 4 yielding fitted estimates for with Qmax, Qmin, nH, and K50 of 0.81 mM/s, −0.02 mM/s, 2.10, and 0.023 mM, respectively.
Figure 6, D–I, shows the results of the model fitting to the same in vivo [ADP]-Jp data set for two cases. In the first case, only the model parameters with the highest MPSA K-S score (i.e., ANT parameters XANT and θ; Fig. 5, A–C) were adjustable parameters in the fitting yielding XANT = 0.041 mM/s and θ = 1.0 mM/s (vs. 0.016 and 0.35 mM/s, respectively, in the default case). Figure 6D shows the corresponding fit of Eq. 4 to the computed [ADP]-Jp covariation for this particular model parameterization compared with the measured [ADP]-Jp covariation in the individual muscle. The in silico ADP transduction function in this case was sigmoidal with a nH of 1.5 and a K50 of 0.025 mM corresponding to ADP ultrasensitivity (27). The fitted estimates for Qmax and Qmin were 0.82 and −0.02 mM/s, respectively. Figure 6, E and F, shows the computed variations with [ADP] of Δψ and ([NADH]/[NAD])m for this particular model parameterization. Both potentials exhibited a steeper drop over the physiological range of ADP concentration changes in muscle compared with the default model parameterization (but without any initial rise at low [ADP]) followed by a more rapid stabilization at 165 mV and 1.1, respectively, at ADP concentrations >0.2 mM. Figure 6, G–I, shows the results for an alternative model fitting. In this case, all model parameters with a significant K-S score except θ were adjustable parameters in the fitting, yielding XANT = 0.32 mM/s, XDH = 0.12 mM/s, and r = 3.1 (vs. 0.016 mM/s, 0.26 mM/s, and 4.6, respectively, for the default case in Fig. 6A). The ADP transduction function for this model parameterization was likewise sigmoidal with fitted estimates of Qmax, Qmin, and K50 of 0.81 mM/s, −0.02 mM/s, and 0.023 mM, respectively (Fig. 6G). The nH was in this case even higher than in the former case (i.e., 2.1 vs. 1.5, respectively). However, for this particular model parameterization, ([NADH]/[NAD])m collapsed over the physiological range of ADP concentration changes in muscle accompanied by ΔΨ falling <150 mV (Fig. 6, H and I).
DISCUSSION
The present in vivo and in silico investigations of the magnitude and origin of mitochondrial sensitivity to cytoplasmic ADP concentration changes in human skeletal muscle have yielded two main results. First, it was found by 31P-MRS that the in vivo affinity and kinetic order of ADP stimulation of mitochondrial oxidative ADP phosphorylation in human skeletal muscle were 0.022 ± 0.003 mM and 1.9 ± 0.2, respectively. Second, it was found by computational analysis that these kinetic characteristics of mitochondrial ADP sensing and transduction are determined primarily by the kinetic properties of the mitochondrial adenine nucleotide transporter. Below, these results and aspects of the underlying analysis are discussed.
In Vivo Mitochondrial ADP Transduction Function: Analysis
The previous investigation of the mitochondrial ADP transduction function used, among others, in vivo data sets on the covariations of the mitochondrial ATP synthesis rate (Jp) with cytosolic ΔGp and [ADP] in contracting forearm muscle (24). Hereto, muscle PCr, Pi, and ATP concentrations and pH were measured in individual subjects typically at six electrical nerve stimulation frequencies (24). As such, the ΔGp-Jp and [ADP]-Jp relations in individual subjects were only sparsely sampled (7 points in each data set). In the present study, sampling of these relations was highly improved when an alternative approach was taken. First of all, the relationship between mitochondrial ATP synthesis flux and ΔGp and [ADP] was reconstructed from the densely sampled (6-s time resolution) PCr recovery time course following muscle contractions (Figs. 3 and 4). Second, multiple data sets were obtained from each subject, resulting in individual data sets containing 100+ (range: 109–206) data points, including improved sampling of the ΔGp-Jp covariation at very low fluxes (Figs. 3 and 4). As a result, accurate estimation of the mitochondrial flux asymptotes (and therefore nH) was achieved in individual subjects (Tables 2 and 3). In fact, fully unconstrained fitting of a sigmoidal relation to individual ([ADP], Jp) data sets yielded the same results as two-parameter fits with flux asymptotes fixed at values obtained from the thermodynamic flow-force analysis (nH: 2.0 ± 0.3 vs. 1.9 ± 0.2; Tables 3 and 4, respectively). These values were not different from the value obtained previously for skeletal muscle [i.e., 2.1 (24)].
A second methodological difference with the previous investigation of the mitochondrial ADP transduction function regarded the calculation of the mitochondrial ATP synthesis rate (Jp) corresponding to each [ADP] data point. In both cases, the derivative of the exponential fit to the time course of PCr was used to compute the net ATP turnover flux at each time point. However, in the previous study, a subsequent subtraction step was necessary to correct for any nonoxidative ATP synthesis flux evidenced from progressive acidification of the contracting muscle fibers (24). The magnitude of this flux was estimated from the pH time course. In the present study, no such correction was necessary because the muscle was electrically silent during recovery. It has previously been shown that anaerobic ATP synthesis flux is negligible under these conditions (5, 16, 35, 37, 38). Indeed, fitting of a biexponential function to the PCr time course during recovery failed to detect any significant second source of ATP synthesis (data not shown). If anything, only the first PCr concentration time point at 3 s into metabolic recovery may have had a contribution from nonmitochondrial ATP synthesis (19). In that case, the data points in Fig. 4 at the highest ADP concentrations would need to be correlated with lower mitochondrial Jp values. If anything, this would render the fitted transduction function even more, not less, sigmoidal.
In Vivo Mitochondrial ADP Transduction Function: Magnitude of the Apparent Kinetic Order (nH)
The first main result of the present investigation of the magnitude and origin of mitochondrial ADP ultrasensitivity is that the apparent kinetic order of the in vivo mitochondrial transduction function f([ADP],Jp) in skeletal muscle is 1.9 ± 0.02 (range: 1.7–2.1; Table 3). This value is in close agreement with the outcome of a previous investigation of the precise value of this macroscopic kinetic parameter in isolated mitochondria and human forearm muscle (2.4 and 2.1, respectively) (24). As such, the present investigation constitutes the first direct in vivo confirmation of the previously formulated hypothesis that mitochondria are ultrasensitive to extramitochondrial ADP concentration changes (24). Previous confirmations had come only from indirect evidence (14, 45).
This result impacts the field of bioenergetics in two ways. First, after having previously been dismissed as an irrelevant regulatory mechanism in cardiac energetics (3), feedback control of mitochondrial ATP synthesis has recently returned to the center of attention (2). This renewed interest has been spurred by the discovery of multiple stimulatory effects of Pi on the mitochondrial metabolic network involved in oxidative ADP phosphorylation in cardiac and skeletal muscle (7). Indeed, a recent review of contemporary knowledge of cardiac energetics elaborately discusses the impact of this discovery on understanding energy balance in the heart (2). However, no mention was made of the other ATP hydrolysis product, ADP, and its particular role in feedback control of ATP synthesis in the heart (2). Yet it was shown previously that mitochondrial ADP ultrasensitivity may in large part explain the measured covariation of [ADP] and myocardial oxygen consumption (24). Therefore, the present affirmation of the hypothesis of mitochondrial ADP ultrasensitivity suggests that both this particular mechanism as well as the multiple stimulatory roles of Pi contribute to the efficacy of feedback respiratory control in striated muscle energetics. Importantly, the apparent allosteric stimulatory effects of ADP and Pi on mitochondrial ATP synthesis appear to operate independently. This conclusion is based on the finding that the magnitude of mitochondrial ADP ultrasensitivity quantified by nH of the transduction function (27) was sensitive only to a subset of the parameters in the mitochondrial metabolic network model whereby the multiple stimulatory roles of Pi were mathematically implemented (Fig. 5B). Second, the result impacts computational modeling of mammalian cell energetics. Specifically, the affirmation of second order for the mitochondrial ADP transduction function provides a firm and tractable validation criterion for evaluation of past (4, 15, 31, 33, 42, 46) and future computational models of mitochondrial oxidative metabolism.
In Vvo Mitochondrial ADP Sensing and Transduction: Origin of ADP Ultrasensitivity
Koshland identified three distinct generic biochemical mechanisms that may endow a biological network with ultrasensitivity (27). For the particular case of mitochondrial ultrasensitivity to cytoplasmic ADP concentration changes, concrete indications exist for the possible involvement of two of these mechanisms, i.e., the presence of allosteric network elements and multisite network activation (27). Specifically, three alternative biochemical implementations of multisite activation have been demonstrated in mitochondria, i.e., multisite kinetic activation by calcium (1), multisite kinetic activation by Pi (7), and multisite phosphorylation (22). Likewise, in vitro evidence has been obtained for non-MM kinetics of ADP-ATP exchange catalyzed by the ANT (9, 18). The latter mechanism was previously invoked to explain mitochondrial ADP ultrasensitivity in the original communication (24). However, that particular hypothesis for the origin of mitochondrial ADP ultrasensitivity has awaited validation. The present investigation has yielded new evidence that the kinetic properties of ANT indeed principally determine the macroscopic mitochondrial property of ADP sensitivity. This is the second main result of the investigation.
The evidence for this conclusion that was obtained in this study was twofold. First, the MPSA of the computational model of skeletal muscle oxidative ATP metabolism, including a detailed biophysical model of mitochondrial oxidative ADP phosphorylation (46), identified an ANT parameter as the primary determinant of mitochondrial ADP sensitivity with respect to both ADP sensing (K50) and transduction (nH) (Fig. 5, B and C). This particular parameter, θ, is a phenomenological parameter in the rate equation for the ADP-ATP exchange reaction catalyzed by ANT of the model that was analyzed (see Ref. 46 and appendix). The K-S scores for this parameter were one order of magnitude higher than the statistical threshold and five- to 10-fold higher than any other model parameter (Fig. 5, B and C). Second, the results of the model fitting to an in vivo [ADP]-Jp data set from an individual muscle showed that mitochondrial ultrasensitivity to ADP concomitant with homeostasis of the mitochondrial membrane and redox potentials could be obtained in silico if and only if the ANT parameter θ was included in the set of adjustable parameters in the model fitting (Fig. 6).
The default ADP sensitivity of the mitochondrial network in the model was insufficient, as indicated by the hyperbolic nature of the simulated [ADP]-Jp relation for the default model parameterization (Fig. 6A). Changing θ from its default value of 0.35 to the fitted optimum of 1.0 concomitant with a 2.5-fold increase of the Vmax of ANT resulted in an almost fivefold increase in mitochondrial ADP sensitivity compared with the default parameterization of the model (nH 1.5 vs. 1.06 mM and K50 0.025 vs. 0.16 mM, respectively; Fig. 6, A and D). Importantly, these specific values of in silico mitochondrial ADP affinity and transduction closely agreed with the experimentally determined values of these macroscopic mitochondrial kinetic parameters in muscle (Table 3). Furthermore, simulation showed that homeostasis of the mitochondrial membrane and redox potential at high ADP concentrations was superior compared with the default model parameterization (Fig. 6, B and C vs. E and F, respectively). In contrast, ΔΨm and particularly ([NADH]/[NAD])m collapsed at physiological ADP concentrations in muscle for the alternative combination of fitted model parameters (including the Vmax of ANT but not θ) that yielded mitochondrial ADP ultrasensitivity (Fig. 6, H and I, respectively). Specifically, simulation showed that ([NADH]/[NAD])m in this case near-instantaneously collapsed to almost zero when mitochondria were activated by cytoplasmic accumulation of ADP and ΔΨm dropped below the critical value of 150 mV at an only moderately elevated cytoplasmic ADP concentration for muscle of 0.07 mM (Fig. 6, H and I). It has been well established that the reaction catalyzed by mitochondrial F1-ATPase reverses from net ATP synthesis to ATP hydrolysis for ΔΨm values below 150 mV (31).
Finally, it could perhaps be argued that the maximal nH for in silico mitochondrial ADP transduction that was obtained by fitting of θ and XANT falls short of reproducing the in vivo estimate of this macroscopic parameter [1.5 vs. 1.9 ± 0.02 (range: 1.7–2.1), respectively; Table 3]. In that case, there would maximally be need for another twofold increase in sensitivity by some kinetic mechanism that remained unidentified in the present computational studies likely because it was not included in the particular mitochondrial computational model that was analyzed. Since multisite kinetic activation of the mitochondrial network by Pi has been explicitly incorporated in the model (46), another kinetic mechanism must be involved. In that case, some form of kinetic regulation by calcium may appear to be a tenable first hypothesis for several reasons. First of all, the computational mitochondrial model platform for our computational studies did not yet include implementation of calcium balance [nor of any metabolic regulatory effect of calcium (46)]. Second, it has been well established that calcium can modify TCA-DH activity in mitochondria (1). Third, the kinetic parameters of the TCA-DH were found to have significant control, albeit small compared with θ, of the nH of the mitochondrial ADP transduction function (Fig. 5B). However, we were unable to further increase nH above 1.5 by additionally changing any of the TCA-DH parameter values within a 10-fold range (data not shown), indicating that, if anything, none of the known calcium effects on TCA-DH activity (1) would be involved. Any significant contribution of calcium activation of other enzymes in the mitochondrial network [e.g., direct activation of ATP synthase (1) or indirect activation of the redox proton pumps via covalent modification by kinases (22)] also appears unlikely since none of these mitochondrial enzymes had any significant MPSA K-S scores (Fig. 5B). As such, it may well prove difficult to identify any ancillary kinetic mechanisms and their biochemical implementation in the mitochondrial metabolic network that may further increase the macroscopic kinetic order of ADP transduction from 1.5 to 1.9. However, the present study illustrates that computational modeling and network analysis provide powerful tools to conduct such an investigation.
Implications for Kinetic Modeling of ANT
The adjustment of the value of ANT parameter θ from 0.35 to 1.0 that, together with a 2.5-fold increase in Vmax of ATP/ADP exchange, transforms the ADP sensitivity of the mitochondrial metabolic network in the Wu et al. (46) model from normal to ultrasensitivity represents not merely an arbitrary model optimization. Instead, it has a significant mechanistic implication. In the Wu et al. (46) model, θ is a phenomenological partition coefficient that determines the magnitude of the effective ΔΨm components on the intermembrane space and matrix sides of the inner membrane (ΔΨm–i and ΔΨm–x, respectively) with respect to the transport of ADP and ATP across the inner membrane (see appendix). The phenomenological partitioning of ΔΨm into ΔΨm–i = θ × ΔΨm and ΔΨm–x = (1-θ) × ΔΨm was originally introduced in the kinetic modeling of the ATP/ADP exchange reaction catalyzed by the ANT by Korzeniewski and Froncisz (26) and later parameterized by fitting of in vitro mitochondrial adenine nucleotide uptake data yielding θ = 0.35 (25). It has since been used in other computational models of mitochondrial ATP synthesis, including the model analyzed here (44, 46).
The results of the present investigation invalidate this particular kinetic model of mitochondrial ATP/ADP exchange as a suitable component of any computational model that seeks to simulate the behavior of mitochondria in living cells. Specifically, the result of the model fitting that θ should be 1 rather than 0.35 to adequately simulate ATP metabolism in muscle indicated that the partitioning of ΔΨm into any cytoplasmic and matrix component introduced on the basis of in vitro data is not appropriate in vivo and should be omitted. The adjusted ANT rate equation is shown in the appendix (Eq. A3). The mechanistic implication of this result is that the kinetic effect of the mitochondrial membrane potential on the ATP4−/ADP3− exchange rate supported by the ANT can be mathematically accounted for by its effect in the matrix.
APPENDIX
The rate equation for ATP/ADP exchange across the inner mitochondrial membrane catalyzed by the ANT in the model of Wu et al. (46) is
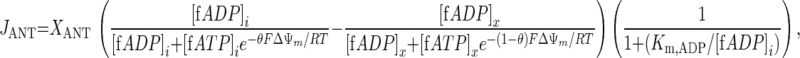 |
(A1) |
where [fX]i and [fX]x are the magnesium-unbound species of ATP and ADP in the mitochondrial intermembrane space and matrix, respectively, and ΔΨm is the mitochondrial membrane potential (46). The kinetic parameters XANT and Km,ADP (parameters 12 and 13 in Table 1, respectively) correspond to the ANT activity (in mol·s−1·l mito−1) and the ANT Michaelis constant (in M), respectively (46). Parameter θ is a phenomenological coefficient introduced by Korzeniewski and Froncisz (26) that determines the magnitude of the effective ΔΨm components on the intermembrane space and matrix sides of the inner membrane (ΔΨm–i and ΔΨm–x, respectively) with respect to the transport of ADP and ATP across the inner membrane according to Eqs. A2a and A2b:
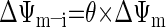 |
(A2a) |
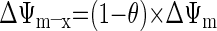 |
(A2b) |
The default value of θ in the Wu model was 0.35 (46) and identical to the value derived previously by Korzeniewski (25) on the basis of model fitting to in vitro data of adenine nucleotide uptake in isolated mitochondria.
For the case θ = 1, Eq. A1 reduces to:
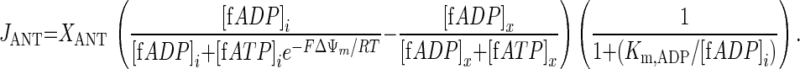 |
(A3) |
GRANTS
This research was funded in part by the National Heart, Lung, and Blood Institute through a subcontract of grant HL-072011.
Acknowledgments
We are grateful to Larry de Graaf for technical assistance with the MR data acquisition and to Fan Wu and Dan Beard for sharing their computational model of muscle ATP metabolism.
Footnotes
The terms Qmax and Qmin rather than Vmax and Vmin are used here to denote the extreme net organelle fluxes. The term x0.5 rather than K50 was chosen since Eqn. [4] was used to analyze both ΔGp-VPCr and [ADP]-VPCr covariations.
REFERENCES
- 1.Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol 34: 1259–1271, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Balaban RS. Domestication of the cardiac mitochondrion for energy conversion. J Mol Cell Cardiol 46: 832–841, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balaban RS, Kantor HL, Katz LA, Briggs RW. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 232: 1121–1123, 1986 [DOI] [PubMed] [Google Scholar]
- 4.Beard DA. A biophysical model of the mitochondrial respiratory system and oxidative phosphorylation. PLoS Comput Biol 1: e36, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blei ML, Conley KE, Kushmerick MJ. Separate measures of ATP utilization and recovery in human skeletal muscle. J Physiol 465: 203–222, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blei ML, Conley KE, Odderson IB, Esselman PC, Kushmerick MJ. Individual variation in contractile cost and recovery in a human skeletal muscle. Proc Natl Acad Sci USA 90: 7396–7400, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bose S, French S, Evans FJ, Joubert F, Balaban RS. Metabolic network control of oxidative phosphorylation: multiple roles of inorganic phosphate. J Biol Chem 278: 39155–39165, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Boska M. ATP production rates as a function of force level in the human gastrocnemius/soleus using 31P MRS. Magn Reson Med 32: 1–10, 1994 [DOI] [PubMed] [Google Scholar]
- 9.Brandolin G, Le Saux A, Trezeguet V, Lauquin GJ, Vignais PV. Chemical, immunological, enzymatic, and genetic approaches to studying the arrangement of the peptide chain of the ADP/ATP carrier in the mitochondrial membrane. J Bioenerg Biomembr 25: 459–472, 1993 [DOI] [PubMed] [Google Scholar]
- 10.Chance B. Reaction of oxygen with the respiratory chain in cells and tissues. J Gen Physiol 49, Suppl: 163–195, 1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chance B, Eleff S, Bank W, Leigh JS Jr, Warnell R. 31P NMR studies of control of mitochondrial function in phosphofructokinase-deficient human skeletal muscle. Proc Natl Acad Sci USA 79: 7714–7718, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chance B, Leigh JS Jr, Clark BJ, Maris J, Kent J, Nioka S, Smith D. Control of oxidative metabolism and oxygen delivery in human skeletal muscle: a steady-state analysis of the work/energy cost transfer function. Proc Natl Acad Sci USA 82: 8384–8388, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem 217: 383–393, 1955 [PubMed] [Google Scholar]
- 14.Cieslar JH, Dobson GP. Free [ADP] and aerobic muscle work follow at least second order kinetics in rat gastrocnemius in vivo. J Biol Chem 275: 6129–6134, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Cortassa S, Aon MA, Marban E, Winslow RL, O'Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J 84: 2734–2755, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowther GJ, Kemper WF, Carey MF, Conley KE. Control of glycolysis in contracting skeletal muscle. II. Turning it off. Am J Physiol Endocrinol Metab 282: E74–E79, 2002 [DOI] [PubMed] [Google Scholar]
- 17.De Martino C, Floridi A, Marcante ML, Malorni W, Scorza Barcellona P, Bellocci M, Silvestrini B. Morphological, histochemical and biochemical studies on germ cell mitochondria of normal rats. Cell Tissue Res 196: 1–22, 1979 [DOI] [PubMed] [Google Scholar]
- 18.Duyckaerts C, Sluse-Goffart CM, Fux JP, Sluse FE, Liebecq C. Kinetic mechanism of the exchanges catalysed by the adenine-nucleotide carrier. Eur J Biochem 106: 1–6, 1980 [DOI] [PubMed] [Google Scholar]
- 19.Forbes SC, Paganini AT, Slade JM, Towse TF, Meyer RA. Phosphocreatine recovery kinetics following low- and high-intensity exercise in human triceps surae and rat posterior hindlimb muscles. Am J Physiol Regul Integr Comp Physiol 296: R161–R170, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groenendaal W, Jeneson JA, Verhoog PJ, van Riel NA, Ten Eikelder HM, Nicolay K, Hilbers PA. Computational modelling identifies the impact of subtle anatomical variations between amphibian and mammalian skeletal muscle on spatiotemporal calcium dynamics. IET Syst Biol 2: 411–422, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Guynn RW, Veech RL. The equilibrium constants of the adenosine triphosphate hydrolysis and the adenosine triphosphate-citrate lyase reactions. J Biol Chem 248: 6966–6972, 1973 [PubMed] [Google Scholar]
- 22.Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, Harris RA, Balaban RS. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry 45: 2524–2536, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobus WE, Moreadith RW, Vandegaer KM. Mitochondrial respiratory control. Evidence against the regulation of respiration by extramitochondrial phosphorylation potentials or by [ATP]/[ADP] ratios. J Biol Chem 257: 2397–2402, 1982 [PubMed] [Google Scholar]
- 24.Jeneson JA, Wiseman RW, Westerhoff HV, Kushmerick MJ. The signal transduction function for oxidative phosphorylation is at least second order in ADP. J Biol Chem 271: 27995–27998, 1996 [DOI] [PubMed] [Google Scholar]
- 25.Korzeniewski B. Simulation of oxidative phosphorylation in hepatocytes. Biophys Chem 58: 215–224, 1996 [DOI] [PubMed] [Google Scholar]
- 26.Korzeniewski B, Froncisz W. An extended dynamic model of oxidative phosphorylation. Biochim Biophys Acta 1060: 210–223, 1991 [DOI] [PubMed] [Google Scholar]
- 27.Koshland DE Jr, Goldbeter A, Stock JB. Amplification and adaptation in regulatory and sensory systems. Science 217: 220–225, 1982 [DOI] [PubMed] [Google Scholar]
- 28.Kushmerick MJ. Energy balance in muscle activity: simulations of ATPase coupled to oxidative phosphorylation and to creatine kinase. Comp Biochem Physiol B Biochem Mol Biol 120: 109–123, 1998 [DOI] [PubMed] [Google Scholar]
- 29.Kushmerick MJ, Meyer RA, Brown TR. Regulation of oxygen consumption in fast- and slow-twitch muscle. Am J Physiol Cell Physiol 263: C598–C606, 1992 [DOI] [PubMed] [Google Scholar]
- 30.Lawson JW, Veech RL. Effects of pH and free Mg2+ on the Keq of the creatine kinase reaction and other phosphate hydrolyses and phosphate transfer reactions. J Biol Chem 254: 6528–6537, 1979 [PubMed] [Google Scholar]
- 31.Magnus G, Keizer J. Minimal model of β-cell mitochondrial Ca2+ handling. Am J Physiol Cell Physiol 273: C717–C733, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Naressi A, Couturier C, Devos JM, Janssen M, Mangeat C, de Beer R, Graveron-Demilly D. Java-based graphical user interface for the MRUI quantitation package. MAGMA 12: 141–152, 2001 [DOI] [PubMed] [Google Scholar]
- 33.Nguyen MH, Dudycha SJ, Jafri MS. Effect of Ca2+ on cardiac mitochondrial energy production is modulated by Na+ and H+ dynamics. Am J Physiol Cell Physiol 292: C2004–C2020, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Prompers JJ, Jeneson JA, Drost MR, Oomens CC, Strijkers GJ, Nicolay K. Dynamic MRS and MRI of skeletal muscle function and biomechanics. NMR Biomed 19: 927–953, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Quistorff B, Johansen L, Sahlin K. Absence of phosphocreatine resynthesis in human calf muscle during ischaemic recovery. Biochem J 291: 681–686, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rottenberg H. The thermodynamic description of enzyme-catalyzed reactions. The linear relation between the reaction rate and the affinity. Biophys J 13: 503–511, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahlin K. Intracellular pH and energy metabolism in skeletal muscle of man. With special reference to exercise. Acta Physiol Scand Suppl 455: 1–56, 1978 [PubMed] [Google Scholar]
- 38.Taylor DJ, Bore PJ, Styles P, Gadian DG, Radda GK. Bioenergetics of intact human muscle. A 31P nuclear magnetic resonance study. Mol Biol Med 1: 77–94, 1983 [PubMed] [Google Scholar]
- 39.Taylor DJ, Styles P, Matthews PM, Arnold DA, Gadian DG, Bore P, Radda GK. Energetics of human muscle: exercise-induced ATP depletion. Magn Reson Med 3: 44–54, 1986 [DOI] [PubMed] [Google Scholar]
- 40.van Dam K, Westerhoff HV. Thermodynamics and Control of Biological Free Energy Transduction. Amsterdam: Elsevier, 1987
- 41.van den Broek NM, De Feyter HM, de Graaf L, Nicolay K, Prompers JJ. Intersubject differences in the effect of acidosis on phosphocreatine recovery kinetics in muscle after exercise are due to differences in proton efflux rates. Am J Physiol Cell Physiol 293: C228–C237, 2007 [DOI] [PubMed] [Google Scholar]
- 42.van Stiphout RG, van Riel NA, Verhoog PJ, Hilbers PA, Nicolay K, Jeneson JA. Computational model of excitable cell indicates ATP free energy dynamics in response to calcium oscillations are undampened by cytosolic ATP buffers. Syst Biol (Stevenage) 153: 405–408, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Vanhamme L, van den Boogaart A, Van Huffel S. Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J Magn Reson 129: 35–43, 1997 [DOI] [PubMed] [Google Scholar]
- 44.Vendelin M, Kongas O, Saks V. Regulation of mitochondrial respiration in heart cells analyzed by reaction-diffusion model of energy transfer. Am J Physiol Cell Physiol 278: C747–C764, 2000 [DOI] [PubMed] [Google Scholar]
- 45.Vicini P, Kushmerick MJ. Cellular energetics analysis by a mathematical model of energy balance: estimation of parameters in human skeletal muscle. Am J Physiol Cell Physiol 279: C213–C224, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Wu F, Jeneson JA, Beard DA. Oxidative ATP synthesis in skeletal muscle is controlled by substrate feedback. Am J Physiol Cell Physiol 292: C115–C124, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Zi Z, Cho KH, Sung MH, Xia X, Zheng J, Sun Z. In silico identification of the key components and steps in IFN-gamma induced JAK-STAT signaling pathway. FEBS Lett 579: 1101–1108, 2005 [DOI] [PubMed] [Google Scholar]