

Abstract
Cysteamine induces perforating duodenal ulcers in rats within 24–48 h. This reducing aminothiol generates hydrogen peroxide in the presence of transition metals (e.g., ferric iron), producing oxidative stress, which may contribute to organ-specific tissue damage. Since most intestinal iron absorption takes place in the proximal duodenum, we hypothesized that cysteamine may disrupt regulation of mucosal iron transport, and iron may facilitate cysteamine-induced duodenal ulceration. We show here that cysteamine-induced ulceration was aggravated by pretreatment of rats with Fe3+ or Fe2+ compounds, which elevated iron concentration in the duodenal mucosa. In contrast, feeding rats an iron-deficient diet was associated with a 4.6-fold decrease in ulcer formation, accompanied by a 34% decrease (P < 0.05) in the duodenal mucosal iron concentration. Administration of deferoxamine inhibited ulceration by 65%. We also observed that the antiulcer effect of H2 receptor antagonist cimetidine included a 35% decrease in iron concentration in the duodenal mucosa. Cysteamine-induced duodenal ulcers were also decreased in iron-deficient Belgrade rats (P < 0.05). In normal rats, cysteamine administration increased the iron concentration in the proximal duodenal mucosa by 33% in the preulcerogenic stage but at the same time decreased serum iron (P < 0.05). Cysteamine also enhanced activation of mucosal iron regulatory protein 1 and increased the expression of divalent metal transporter 1 mRNA and protein. Transferrin receptor 1 protein expression was also increased, although mucosal ferroportin and ferritin remained almost unchanged. These results indicate an expansion of the intracellular labile iron pool in the duodenal mucosa, increasing its susceptibility to oxidative stress, and suggest a role for iron in the pathogenesis of organ-specific tissue injury such as duodenal ulcers.
duodenal ulcer disease affects 5–10% of the US population at least once in their lives, and this form of “peptic ulcer” is two to four times more prevalent than gastric ulcer (13, 56). Although a strong association has been established between duodenal ulcers and Helicobacter pylori infection, the molecular mechanisms of ulcer development and healing remain poorly understood. Animal models of duodenal ulcer disease have provided valuable insights into the early, preulcerogenic events and subsequent cellular and biochemical changes that take place in the pathogenesis of duodenal ulceration. Most of this research has been based on ulcer induction using cysteamine (HS-CH2-CH2-NH2, β-mercaptoethylamine), which produces perforating ulcers in the rat proximal duodenum within 24–48 h (54, 57, 60).
Cysteamine is a low-molecular-weight aminothiol that is broadly distributed in organisms ranging from Drosophila to humans (43). Cysteamine is a natural product of coenzyme A metabolism (45). Cysteamine is generated when pantetheinase (which is encoded by the Vanin-1 gene), hydrolyzes pantetheine, thereby recycling pantothenate (vitamin B5) and releasing cysteamine (16). A lack of detectable free cysteamine in the small intestinal mucosa in Vanin-1-null mice appears to provide resistance to oxidative stress (48). These animals are protected against nonsteroidal anti-inflammatory drug- and irradiation-induced intestinal mucosal injury (5, 42).
The molecular mechanisms whereby cysteamine induces duodenal ulceration are incompletely understood. Previous studies from our laboratory demonstrated that cysteamine reduced somatostatin bioavailability and markedly elevated serum gastrin levels, with an associated increase in gastric acid secretion (41, 61, 62), significantly decreased neutralization of acid in the proximal duodenum (2), decreased dopamine levels in glandular stomach and duodenum (59), and inhibited gastric emptying and motility (40). It is also known that certain transcription factors (e.g., early growth response factor 1, hypoxia-inducible factor 1α) and their target genes play a key role in the pathogenesis of cysteamine-induced duodenal ulcers (35, 36), but the complete mechanism of cysteamine-induced duodenal ulceration is still not clear.
In vitro studies have demonstrated that the cytotoxic effect of cysteamine depends primarily on the generation of H2O2 in the presence of transition metals (Mn) such as Fe3+ (6, 11, 63). The thiol-derived H2O2 reacts with the reducing transition metals to produce hydroxyl radicals via the Fenton reaction. These radicals are likely the final mediators of cysteamine cytotoxicity (28, 33), as evidenced by the fact that the effect of cysteamine is diminished by catalase (29). Our previous studies demonstrated that catalase inhibited cysteamine-induced duodenal ulcer in rats (34). Cysteamine also promotes focal intracellular accumulations of iron, which may exacerbate its cytotoxic effects (20, 53, 68). It is not clear, however, whether such iron accumulations have a role in the pathogenesis of cysteamine-induced tissue damage although iron is inherently toxic as a result of its propensity for promoting oxidation through the Fenton reaction (27).
To protect cells from the potentially damaging effects of iron excess, eukaryotes have developed unique mechanisms for the regulation of iron uptake and transport (14). In mammalian cells, iron homeostasis is mediated through binding of iron regulatory protein 1 and 2 (IRP1 and 2) to RNA stem-loop structures called iron-responsive elements (IREs) in transcripts encoding proteins involved in iron metabolism (39, 51). In iron-depleted cells, these proteins bind to IREs in the 3′-untranslated region of transferrin receptor 1 (TfR1) mRNA and in the 5′-untranslated region of ferritin mRNA, thus stabilizing the TfR1 message and inhibiting ferritin synthesis (4, 30, 38, 47). It is noteworthy that cysteamine-induced ulceration in rats occurs mainly in the proximal duodenum, within 10 mm of the pylorus, at the same site where most intestinal iron absorption takes place. The epithelial cell layer of the proximal duodenum is specialized for the uptake and transport of iron from the intestinal lumen to the systemic circulation (17, 69). This raises the question whether the availability of dietary iron within the enterocytes of the proximal duodenum during iron absorption may play a role in ulcerogenesis induced by chemicals such as cysteamine. Iron uptake at the brush border is mediated by divalent metal transporter 1 (DMT1, also known as Nramp2 or DCT1), which transports Fe2+ and other divalent metals (25). DMT1 is encoded by the Slc11a2 gene, which is mutated in the mk mouse strain and in Belgrade rats (19, 50). Mutations in these rodents disrupt DMT1-mediated iron uptake, and animals homozygous for the mutated gene suffer from iron deficiency anemia as a result of impaired intestinal iron absorption (19). The major DMT1 mRNA isoform expressed in the intestine contains an IRE in the 3′-untranslated region and is negatively regulated by iron through inactivation of IRP1 (24). In response to a reduction in dietary iron, upregulation of DMT1 (IRE) takes place in the microvilli of the proximal duodenum (10, 55). Iron transport across the basolateral membrane of the enterocyte to the systemic circulation is dependent on another IRE-containing transcript, ferroportin, although its regulation is more complex (15, 23). IRPs are also subject to regulation by additional, iron-independent signals such as nitric oxide, hypoxia, and oxidative stress, including H2O2 generation (9, 18, 32, 44, 66).
We hypothesized that 1) the duodenal ulcerogen cysteamine may increase iron availability within the duodenal mucosa through enhancement of IRE binding by IRPs, thereby stabilizing DMT1 and TfR transcripts and 2) pharmacological and dietary modulation of the duodenal mucosal iron level might affect the severity of cysteamine-induced duodenal ulcers, essentially implicating iron in the pathogenesis of duodenal ulceration and providing the first mechanistic explanation for the localization of tissue injury after systemic administration of duodenal ulcerogens such as cysteamine.
MATERIALS AND METHODS
Animal experiments.
This study was approved by the Subcommittee for Animal Studies and the Research and Development Committee of VA Long Beach Healthcare System. Female Sprague-Dawley rats (180–210 g) were maintained on standard laboratory rat chow (Harlan Teklad Rodent Diet, 8604, 150–250 ppm Fe; Harlan Teklad, Madison, WI). Rats had unlimited access to food and water. Duodenal ulcers were induced by intragastric administration of cysteamine-HCl (Sigma-Aldrich, St. Louis, MO). Randomized groups of rats (n = 7) were given either saline or cysteamine-HCl in saline (25 mg/100 g body wt) by gavage once and euthanized 0.5, 1, or 2 h later. In other groups, cysteamine was given twice or three times at 4-h intervals and rats were euthanized 6, 12, or 24–48 h after the first dose of cysteamine.
Other groups of rats were pretreated 15 min before each dose of cysteamine with either FeSO4 (20 mg Fe/100 g) or FeCI3 (20 mg/100 g) by gavage or were given ferric gluconate (1 or 5 mg/100 g) intravenously under isoflurane anesthesia or deferoxamine (30 mg/100 g) subcutaneously 30 min before cysteamine administration and euthanized 48 h after the first dose of cysteamine. Additional groups of rats were placed on a low-iron (2–8 ppm Fe) diet (TD 80396, Harlan Teklad) for 4 wk (chronic experiments), and then animals were treated with cysteamine-HCl (25 mg/100 g by gavage × 3 doses within 8 h). The next groups of rats were pretreated with cimetidine (20 mg/100 g, by gavage) 3 and 0.5 h before cysteamine, and animals were euthanized 3 h or 48 h after the first cysteamine dose. We also compared duodenal ulceration after cysteamine administration in groups of homozygous (b/b) and heterozygous (+/b) Belgrade rats (12 mo old) and Sprague-Dawley control rats of similar age. Belgrade rats were purchased from Drs. L. and M. Garrick, State University of New York, Buffalo, NY.
At autopsy 48 h after first cysteamine administration, rat blood specimens were obtained from the abdominal aorta with a 19-gauge needle, placed in BD Vacutainer tubes (BD Biosciences, Franklin, NJ), and centrifuged at 1,300 g for 10 min to separate serum. Mucosal scrapings of proximal duodenum (2.5 cm), stomach, and jejunum (3 cm) and samples of other organs (liver, kidney) were collected and frozen in liquid nitrogen and kept at −80°C until required. RNA extraction and purification were performed using a total RNA isolation kit (Clontech Laboratory, Palo Alto, CA). For protein extraction, tissue specimens were homogenized in lysis buffer (2 M NaCl, 10 mM Tris·HCl, pH 7.4) with protein inhibitors (1 mM PMSF, 1 μg/ml leupeptin, and 1 μg/ml aprotinin; Sigma), sonicated, and centrifuged for 30 min at 16,000 g. Protein concentration was measured by the Bradford assay (Bio-Rad Laboratories, Hercules, CA). Iron concentration was measured in the tissue from different organs, which were homogenized in lysis buffer and centrifuged for 30 min at 16,000 g. Serum iron and gastric, duodenal, jejunal, ileum, colonic, kidney, and hepatic iron concentrations were measured spectrophotometrically with a Roche/Hitachi 917 analyzer using FerroZine (Roche Diagnostics, Indianapolis, IN), and the results of iron concentration in tissue were expressed as μg/g wet weight. In vitro experiments with gastric homogenates and the addition of increased concentration of cysteamine confirmed that cysteamine did not interfere with ascorbate/ferrozine in this colorimetric method. Tissue ferritin measurements were performed on a Bayer Centaur analyzer by immunochemiluminescence (Bayer Diagnostics, Tarrytown, NY).
Gross and histological evaluation of duodenal ulcers.
The duodenal ulcer crater dimensions were measured in millimeters, and the ulcer areas were calculated using the ellipsoid formula. The opened stomachs with 2.5 cm duodenum were fixed in 10% formalin. Sections were embedded in paraffin and stained with hematoxylin and eosin.
Determination of IRP binding activity by EMSA.
32P -labeled RNA containing an IRE was prepared by in vitro transcription of a linearized duodenal ferritin H cDNA clone (gift of Dr. E. Leibold). Transcription was carried out in a reaction mixture containing 1.5 mM each of ATP, GTP, and CTP and 50 μCi [32P]UTP with T7 RNA polymerase. The [32P]UTP-labeled probe was purified by urea gel electrophoresis. Aliquots of cytoplasmic extracts containing 20 μg protein (prepared as described above) were added to a reaction mixture containing 10 mM HEPES, 3 mM MgCl2, 40 mM KCl, 5% glycerol, and 1 × 105 cycles/min 32P-RNA probe, without or with the addition of 2% β-mercaptoethanol to fully activate IRP1 (31). After 10 min of incubation at room temperature, RNase T1 (1 U/reaction) and heparin (5 mg/ml) were added to the reaction mix, and the incubation was continued for 15 min to degrade unbound probe and displace nonspecific protein-RNA interaction. Protein-RNA complexes were resolved by 6% nondenaturing PAGE and exposed on X-ray film for 10–24 h.
RNase protection assay.
Assays were performed on total RNA using riboprobes corresponding to the cDNA sequences for DMT1 (IRE) (GenBank accession no. nt 1413–1628, AF029757) and GAPDH (nt 536–691, AF106860). Riboprobe synthesis was performed using the Riboprobe Combination System SP6/T7 polymerase kit (Promega, Madison, WI) according to the manufacturer's instructions. Each transcription contained 17.5 μM cold UTP and 50 μCi [32P]UTP (3,000 Ci/mmol; NEN Life Science Products, Boston, MA). After synthesis, the template was digested with 1 U RQ RNase-free DNase 1 (Promega), and the riboprobe was purified by urea gel electrophoresis. The RNA samples (20 μg) were hybridized overnight with 105 cycles/min of each probe at 45°C in 50 μl of hybridization buffer (40 mM PIPES, pH 6.4, 400 mM NaCl, 1 mM EDTA, and 80% formamide). After hybridization, the unprotected RNA was digested by the addition of 350 μl of RNase digestion buffer (10 mM Tris·HCl, pH 7.5, 300 mM NaCl, and 5 mM EDTA) containing 80 μg/ml of RNase A and 80 U/ml RNase T1 and incubated at 37°C for 1 h. The reaction was stopped by the addition of 10 μl of 20% SDS and 2.5 μl of 10 mg/ml of proteinase K and incubation at 37°C for 15 min. The reactions were extracted with phenol-chloroform-isoamyl alcohol (25:24:1) and precipitated with ethanol. The pellet was resuspended in 5 μl of loading buffer (80% formamide, 15% glycerol, and 0.25% bromophenol blue) and electrophoresed on a 6% polyacrylamide-50% urea gel for 4 h at 200 V. After electrophoresis, the gel was dried and exposed to X-ray film for 2–96 h.
Western blot.
Proteins (50 or 100 μg) were subjected to SDS-PAGE after dissociation by boiling (5 min) in 2× SDS-sample loading buffer (0.125 M Tris·HCl, pH 6.8, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.05% bromophenol blue). TfR1, DMT1, and ferritin were analyzed using 7.5%, 10%, and 15% SDS-PAGE, respectively. After electrophoresis, proteins were transferred to nitrocellulose membranes. The membranes were incubated with a mouse monoclonal antibody to either TfR1 (Zymed Laboratories, San Francisco, CA), rabbit polyclonal antibodies to DMT1 (with IRE), ferroportin (Alpha Diagnostic International, San Antonio, TX), or H- and l-ferritin subunits (Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were then incubated with secondary antibodies (horseradish peroxidase-labeled anti-mouse or goat anti-rabbit IgG), which were visualized using the ECL detection system (Amersham Biosciences, Pittsburgh, PA). Immunoblot analyses were performed at least five times with each protein to assure the reproducibility of results. The density of protein bands on Western blots was assessed by digital imaging (Eagle EYE 11; Stratagene, La Jolla, CA).
Statistical analysis.
The statistical significance of changes was determined by the Mann-Whitney U-test. Differences resulting in a P value <0.05 were considered to be statistically significant.
RESULTS
Effect of an iron-deficient diet or pretreatment of rats with deferoxamine, ferric or ferrous iron, or ferric gluconate on cysteamine-induced duodenal ulcers.
Cysteamine-induced duodenal ulcers were markedly decreased in size (threefold) in rats pretreated with deferoxamine compared with control rats, whereas Fe2+ (as FeSO4) or Fe3+ (as FeCl3) aggravated duodenal ulceration by 2.2- and 1.7-fold, respectively (Fig. 1, A and B). Pretreatment of anesthetized animals with ferric gluconate in a dose of 5 mg/100 g body wt, but not 1 mg/100 g, significantly increased (37%) cysteamine-induced duodenal ulceration; the anesthesia alone had no effect on ulcer size (Fig. 1C). Duodenal ulceration in iron-deficient animals treated with cysteamine was reduced 4.6-fold compared with controls (Fig. 1B).
Fig. 1.
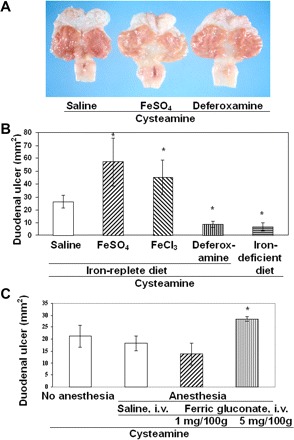
Stimulation or inhibition of cysteamine-induced duodenal ulcerogenesis in rats with increased (Fe+3, Fe+2) or decreased iron levels. A: gross appearance of duodenal ulcers 48 h after cysteamine administration. Animals were pretreated with saline (n = 7), FeSO4 (n = 10), or deferoxamine (n = 10) 15 min before each dose of cysteamine. B: duodenal ulcer size was scored 48 h after cysteamine in animals pretreated with saline, FeCl3, FeSO4, or deferoxamine or fed an iron-deficient diet (chronic experiments, n = 5). The duodenal ulcer crater dimensions were measured in millimeters, and the ulcer areas were calculated using the ellipsoid formula. C: aggravation of duodenal ulcers by ferric gluconate. Duodenal ulcers were compared without and with anesthesia by isoflurane inhalation. Saline and ferric gluconate injections were given intravenously (i.v.) 30 min before cysteamine administration. Results are expressed as means ± SE (*P < 0.01, n = 5).
Effect of an iron-deficient diet or pretreatment of rats with ferric gluconate on iron concentration in duodenal mucosa and serum.
Rats fed a low-iron diet had a 34% decrease in the serum iron concentration compared with control rats maintained on an iron-replete diet, and this was accompanied by a similar decrease in the iron concentration in the duodenal mucosa (P < 0.05; Fig. 2, A and B). The decrease in duodenal iron concentration correlated with the diminished cysteamine-induced duodenal ulcers in iron-deficient animals. The low-iron diet had no effect on gastric iron concentration (Fig. 2C ). Body weight of animals fed low-iron diet was similar to control animals fed the iron-replete diet. At 48 h after cysteamine administration, at a time when ulcers had formed, in both iron-deficient animals and in animals fed a normal diet, cysteamine produced a decrease in the serum iron and the duodenal mucosal iron concentration.
Fig. 2.
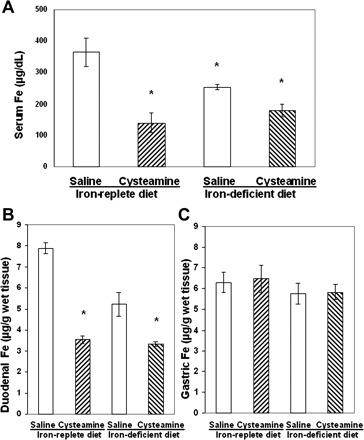
Comparison of the iron concentrations in serum and duodenal mucosa of rats fed with either an iron-replete diet or a low-iron diet for 4 wk before cysteamine treatment. A: serum iron concentration 48 h after saline or cysteamine administration in rats fed with iron-replete or iron-deficient diets. B: decreased iron concentration in 2.5 cm of ulcerated duodenal mucosa 48 h after cysteamine in rats fed iron-replete or iron-deficient diets compared with saline treated animals (*P < 0.05, n = 5 for each group). C: iron concentration in gastric mucosa did not change in animals fed a low-iron diet or an iron-replete diet.
Pretreatment of rats with ferric gluconate (5 mg/100 g), which aggravated cysteamine-induced duodenal ulcers, almost doubled the serum iron concentration and increased (by 63%) iron concentration in the duodenal mucosa 6 h after cysteamine administration (Fig. 3, A and B). Ferric gluconate in dose of 1 mg/100 g, which did not change cysteamine-induced duodenal ulcers, did not affect serum and duodenal iron concentration.
Fig. 3.
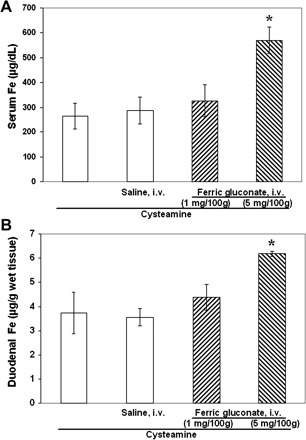
Treatment of anesthetized rats with ferric gluconate (5 mg/100 g) 0.5 h before cysteamine increased serum iron concentration (A) and iron concentration in the proximal duodenum (B) vs. animals treated with cysteamine only, without anesthesia (first column) and anesthetized rats pretreated with saline (i.v.) or ferric gluconate (1 mg/100 g); *P < 0.05, n = 5 for each group.
The antiulcer drug cimetidine, a histamine H2 receptor antagonist, decreased iron concentration in the duodenal mucosa.
Previous studies from our laboratory and others demonstrated that cysteamine increased the gastric acid secretion in rats and that cimetidine inhibited cysteamine-induced gastric acid secretion and decreased cysteamine-induced duodenal ulcers (21, 58, 64). Now we again pretreated animals with cimetidine (20 mg/100 g) at 3 and 0.5 h before cysteamine and demonstrated a 4.5-fold decrease in duodenal ulcers (Fig. 4A). Cimetidine alone or together with cysteamine decreased the serum iron concentration by 48% 3 h after cysteamine administration (Fig. 4B). At the same time, cimetidine also diminished iron concentration by 35% (P < 0.05) in the proximal duodenal mucosa of rats treated with cysteamine (Fig. 4C). These measurements were performed 3 h after cysteamine administration on the basis of our previous studies of the time-course of the inhibitory effect of cimetidine on gastric acid secretion (58).
Fig. 4.
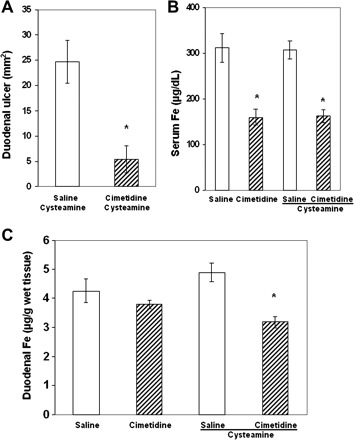
Effect of the histamine H2 receptor antagonist cimetidine on cysteamine-induced duodenal ulcers. A: cimetidine inhibited cysteamine-induced duodenal ulcers. The rats were pretreated with cimetidine 3 and 0.5 h before cysteamine and euthanized 3 h or 48 h after the first cysteamine dose. B: cimetidine decreased serum iron concentration alone or with cysteamine- vs. only saline-treated animals (3 h after cysteamine administration). C: cimetidine given to rats before cysteamine decreased iron concentration in the proximal duodenum vs. saline- or cysteamine-treated rats (3 h after cysteamine administration). Results are expressed as means ± SE (*P < 0.01, n = 5 for each group).
Iron concentrations in the proximal duodenum, jejunum, ileum, and colon.
Our results demonstrated a correlation of cysteamine-induced ulceration in the proximal duodenum with iron concentration in the proximal duodenum; iron pretreatment of rats increased both iron concentration in proximal duodenum and ulcer formation; the iron-deficient diet decreased both duodenal iron and ulcers; and finally cimetidine decreased duodenal iron concentration and protected duodenal mucosa from ulcerative effects of cysteamine. Since most iron absorption takes place in the proximal duodenum and this function gradually decreases from duodenum to colon (70), we determined iron concentration in different parts of small and large intestine. Our results demonstrated that the proximal duodenum has the highest iron concentration compared with jejunum, ileum, and colon, i.e., the concentration gradually decreased from duodenum (4.3 ± 0.4 μg/g wet tissue), jejunum (3.0 ± 0.2 μg/g wet tissue), ileum (1.8 ± 0.2 μg/g wet tissue), to colon (1.3 ± 0.1 μg/g wet tissue).
Early effect of cysteamine on iron concentration in serum and duodenal, gastric, and jejunal mucosa, as well as liver and kidney in the preulcerogenic stage of duodenal ulceration.
Cysteamine administration increased the iron concentration of the proximal duodenal mucosa by 25–33% during the preulcerogenic stage. A statistically significant increase was observed at time points from 0.5 to 12 h after treatment (Fig. 5A), followed by a decrease in the iron content below baseline by 24 h, at which time ulcer formation had occurred. Over the same time course, the iron concentration in the serum decreased during the first 0.5–6 h after cysteamine administration, followed by a return to baseline at 12 h and a subsequent marked decrease by the time of ulcer formation at 24 h (Fig. 5B). There was also a trend toward a slightly higher iron concentration in the liver 0.5 to 12 h after treatment, but the difference was not significant compared with saline-treated control animals. The iron concentrations in the stomach and jejunum initially were much lower than in the duodenum, and cysteamine administration did not induce any significant change in the iron concentration in stomach, jejunum, or kidney at any time point (Fig. 5C).
Fig. 5.
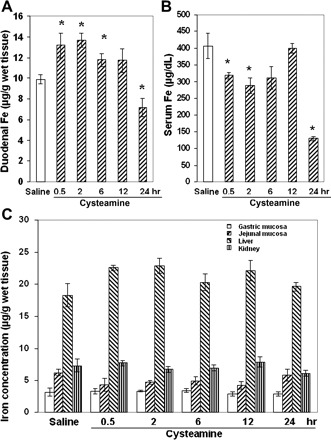
Effect of cysteamine on iron concentration in different organs of rats within the preulcerogenic stage of duodenal ulcer development. A: temporal increase of iron concentration in the proximal duodenum within the first 12 h after cysteamine compared with saline administration. B: iron concentration in rat serum within the first 24 h after cysteamine administration. C: iron concentrations in gastric, jejunal, liver, and kidney tissue of rats treated with cysteamine or saline (*P < 0.05, n = 5 for each group).
IRP1 activity in the duodenal mucosa in the preulcerogenic stage of duodenal ulceration following cysteamine administration.
Previously we demonstrated that cysteamine (β-mercaptoethylamine) as a reducing agent may decrease the redox potential in the proximal duodenum in early stages of duodenal ulceration but not in gastric mucosa (34). It is known that reducing agents (e.g., β-mercaptoethanol) increase the functional activity of IRP1 (31). This raises the possibility that changes in redox potential in the duodenal mucosa may affect IRP-IRE binding by reduction of sulfhydryl groups and disassembly of the 4Fe-4S cluster of IRP1. To examine whether cysteamine may affect IRP1 activity, we assessed IRE binding by IRP1 using EMSA (Fig. 6A). There was a marked increase in IRP1-IRE binding in cysteamine-treated rats during the preulcerogenic stage at 6 h (after two doses of cysteamine), and this was further enhanced at 12 h (after three doses of cysteamine), reaching a level at least 10-fold higher than in saline-treated rats. By 12 h, IRP1 was essentially completely activated compared with the amount of fully activated IRP1, demonstrated by the addition of β-mercaptoethanol. In addition, the total amount of IRP1 visualized in the presence of β-mercaptoethanol was also increased at 6 h, and a further increase was observed at 12 h, indicating increased IRP1 expression (Fig. 6A).
Fig. 6.
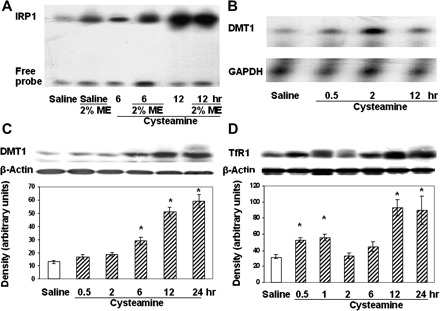
Effect of the duodenal ulcerogen cysteamine on iron regulatory protein 1 (IRP1) activity and divalent metal transporter 1 (DMT1) and transferrin receptor 1 (TfR1) expression in rat duodenal mucosa during the early stages of duodenal ulcer development. A: IRP1-iron-responsive element (IRE) binding activity in the rat duodenal mucosa 6 and 12 h after treatment with cysteamine or saline. 2-Mercaptoethanol (ME, 2%) was added to protein samples to maximize IRE binding activity. The experiments were performed at least 5 times, and representative results are shown. B: RNase protection assay showing DMT1 and GAPDH mRNA expression in the duodenal mucosa 0.5, 2, and 12 h after treatment with cysteamine. C: DMT1 protein expression in rat duodenal mucosa was detected by Western blot at various times after cysteamine treatment. Samples from saline-treated rats were used as controls. D: TfR1 was detected by Western blot in rats treated with saline or cysteamine for various periods of time. The experiments were performed at least 4 times, and representative results are shown.
DMT1 and TfR1 expression in the duodenal mucosa after cysteamine administration.
It is known that IRP1 binds with high affinity to the DMT1 (IRE form), and DMT1 mRNA is regulated through stabilization by the IRE/IRP system, in analogy to TfR mRNA. Thus we next determined whether cysteamine-induced IRP1 activity may affect DMT1 mRNA and protein expression.
The effect of cysteamine on the expression of DMT1 (IRE form) mRNA was assessed by RNase protection assay. DMT1 mRNA expression increased at 0.5, 2, and 12 h after administration, with the greatest enhancement occurring 2 h after cysteamine treatment (Fig. 6B). Although cysteamine did not alter duodenal mucosal DMT1 protein expression at 2 h after the first dose, the DMT1 protein level began to increase at 6 h (after two doses of cysteamine) and reached a maximum of 3.5–4.0-fold at 12 and 24 h after cysteamine treatment (Fig. 6C). Cysteamine treatment of rats induced a time- and dose-dependent increase in TfR1 expression at 6 h (after two doses of cysteamine). At 12 h (after three doses of cysteamine) and at 24 h after the first dose of cysteamine, TfR1 expression was enhanced more than 50% compared with saline-treated animals (Fig. 6D).
Effect of cysteamine on expression of ferroportin and ferritin in the duodenal mucosa.
Ferroportin protein expression in duodenal mucosa did not change significantly after treatment of rats with cysteamine. Slight increases in ferroportin expression were seen only at 6 and 12 h after cysteamine treatment (Fig. 7A). The mean ferritin concentration in the duodenal mucosa was slightly reduced during the early preulcerogenic stages (up to 2 h) and increased during later stages, but these changes were not statistically significant (Fig. 7B). A similar pattern of ferritin expression was observed in Western blots using antibodies to ferritin H- and L-subunits (Fig. 7C), which demonstrated relatively stable levels of H-subunit throughout the time course of cysteamine-induced duodenal ulceration. (No L-ferritin subunit expression was detectable under these conditions.)
Fig. 7.
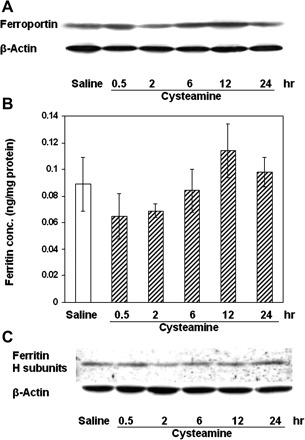
Ferroportin and ferritin protein expression in the duodenal mucosa of rats treated with cysteamine. A: ferroportin was detected by Western blot. B: cysteamine did not induce significant changes in ferritin concentration. C: ferritin (heavy subunit) was also detected by Western blot in rat duodenal mucosa after cysteamine treatment in the same group of rats as used in B. The experiments were performed at least 4 times, and representative results are shown.
Cysteamine-induced duodenal ulcers in heterozygous (+/b) and homozygous (b/b) Belgrade rats.
As shown in Fig. 8, A and B, the mean size of cysteamine-induced duodenal ulcers was similar in heterozygous (+/b) Belgrade rats and Sprague-Dawley rats but was significantly diminished in homozygous (b/b) Belgrade rats. Histology also demonstrated that, when duodenal lesions in (b/b) Belgrade rats were present, they were mostly erosions or superficial ulcers, with limited mucosal necrosis and minimal acute inflammation. Surprisingly, cysteamine also induced ulceration in the gastric antrum of both heterozygous (+/b) and homozygous (b/b) Belgrade rats, and the ulcers were more frequent and severe than in Sprague-Dawley rats (data not shown).
Fig. 8.
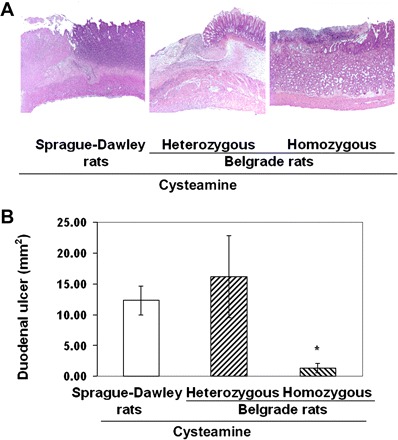
Cysteamine-induced duodenal ulcers in Sprague-Dawley rats and in heterozygous (+/b) and homozygous (b/b) Belgrade rats. A: light microscopy of duodenal ulcers in rats treated with cysteamine (25 mg/100 g) in Sprague-Dawley rats (deep ulcer, left), heterozygous (deep ulcer, middle), and homozygous Belgrade rats (small ulcer, right) (hematoxylin and eosin staining). B: duodenal ulcer crater dimensions in the same groups of rats were measured in millimeters, and the ulcer areas were calculated using the ellipsoid formula. Size of duodenal ulcers is shown as mm2 (means ± SE, *P < 0.01).
DISCUSSION
This study demonstrates several new aspects of the mechanism of cysteamine-induced duodenal ulceration that implicate iron and possibly dysregulation of iron homeostasis in this process. Namely, iron depletion induced by feeding an iron-deficient diet or by deferoxamine administration markedly inhibited cysteamine-induced duodenal ulcers in association with a significant decrease in iron concentration in the proximal duodenal mucosa, whereas iron loading by pretreatment of rats with ferric gluconate or ferric or ferrous iron stimulated ulcerogenesis, which was associated with increased duodenal iron concentration. We demonstrated for the first time that histamine H2 receptor antagonist cimetidine decreased iron concentration in the duodenal mucosa and confirmed its ability to inhibit cysteamine-induced duodenal ulcers. Furthermore, cysteamine increased iron concentration in the duodenal mucosa in preulcerogenic stage but decreased serum iron concentration, without significant changes in iron concentration in the liver, stomach, jejunum, and kidney. The proximal duodenal iron concentration was highest compared with other organs of gastrointestinal tract; these findings suggest a mechanism whereby iron may mediate cysteamine-induced duodenal ulcer development and explain the organ specificity for duodenal ulceration. Administration of cysteamine, an aminothiol with a reducing function, led to increased mucosal IRP1 activation, TfR1 expression, and expression of DMT1 (IRE form) mRNA and proteins. These increases in expression of iron transport proteins may be responsible for the elevated mucosal iron concentration observed in association with cysteamine treatment. In contrast, there was no significant effect on the expression of mucosal ferroportin and ferritin. The combination of increased mucosal iron and almost unchanged ferroportin and ferritin expression under these conditions may favor expansion of the intracellular labile iron pool, possibly leading to an associated increase in susceptibility to oxidative stress (Fig. 9).
Fig. 9.
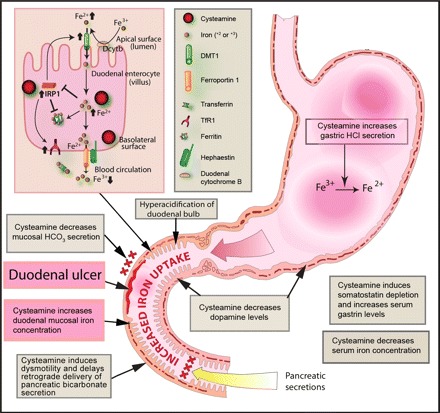
Proposed role of iron in the pathogenesis of cysteamine-induced ulceration in the proximal duodenum of rats. Cysteamine may promote dysregulation of iron absorption by simultaneously increasing iron uptake by duodenal mucosal enterocytes and inhibiting transfer of mucosal iron to the systemic circulation. Cysteamine may enhance iron availability for uptake by duodenal enterocytes through 1) direct reduction of Fe3+ to Fe2+, 2) increased gastric acid secretion following elevation of gastrin levels and inhibition of somatostatin, 3) decreased bicarbonate secretion, and 4) delayed gastric emptying. Within duodenal enterocytes (illustrated here by a composite drawing representing both villus enterocytes and crypt cells expressing TfR1), the reducing agent cysteamine activates IRP1, which normally would be inactivated by increased intracellular iron. Activation of IRP1 in turn would increase DMT1 (IRE) expression, which otherwise would be decreased in the presence of increased intracellular iron, and the increased DMT1 would lead to increased iron transport across the apical membrane, thus further expanding the labile iron pool. IRP1 activation also leads to translational inhibition of ferritin mRNA, which typically would otherwise be increased, and stabilization of TfR1 mRNA, which normally should be decreased, in the presence of increased intracellular iron. TfR1 may take up circulating transferrin-bound iron across the basolateral membrane of crypt cells, thereby also expanding the labile iron pool. Under the conditions of these experiments, however, ferritin protein levels were not affected, suggesting possible competing effects of increased IRP1 activation by cysteamine and increased labile iron pool levels. Although ferroportin 1 mRNA contains IRE or IRE-like moieties, suggesting the possibility of posttranscriptional regulation, the functionality of ferroportin IREs is unclear, and cysteamine administration did not change ferroportin levels. However, iron must be oxidized to Fe3+ for transfer from the enterocyte to the systemic circulation, and the reducing effect of cysteamine may compete with this step, thereby decreasing iron transport across the basolateral membrane and favoring increased intracellular iron levels. Taken together, these processes may expand the labile iron pool and thereby potentiate oxidative stress, which in turn may trigger duodenal ulceration.
Iron is an important catalyst of oxidation-reduction reactions in the cell and is essential for life. However, excess iron is toxic as a result of iron-catalyzed generation of reactive oxygen species that induce damage to cell membranes, proteins, and DNA (28). Divalent iron (ferrous, Fe2+) initiates the Fenton reaction (Fe2+ + H2O2 –> Fe3+ + *OH + OH−) and is frequently used to experimentally induce oxidative damage, including lipid peroxidation. An increase in the size of the intracellular labile iron pool in endothelial cells is associated with increased intracellular oxidative stress induced by H2O2 (65). Our recent studies demonstrated that pretreatment of rats with catalase significantly inhibited cysteamine-induced duodenal ulcers (34). Previous studies have also demonstrated that increased iron accumulation in tissues can lead to organ damage and dysfunction. Iron-related erosive injury is associated with iron deposition in the mucosa of the upper gastrointestinal tract (26). Dogs with elevated duodenal iron concentration in association with experimentally induced exocrine pancreatic insufficiency also develop duodenal mucosal injury (1). In the present study, duodenal mucosal damage was preceded by an increase in the iron concentration in the duodenal mucosa during the preulcerogenic stage of cysteamine-induced mucosal injury, suggesting a role for iron in tissue-specific ulcerogenesis.
The amount of iron entering the body is controlled at the point of absorption in the proximal small intestine, and this process plays a critical role in body iron homeostasis (3). Iron uptake by the proximal 4–6 cm of the duodenum was several fold higher than by the proximal jejunum, stomach, and ileum (7, 22, 49, 69). In mice, iron concentration in the duodenum was two times greater than in stomach, jejunum, ileum, or colon (37). In the present study, we found that iron concentration in the proximal duodenal mucosa of normal rats was higher than in the jejunum, ileum, and colonic mucosa, which appears to be in parallel with the iron absorptive function in these organs. Iron absorption is affected by a wide range of systemic and local intestinal factors. Gastric acid secretion plays a major role in this process (52) by reducing ferric iron to the more readily absorbed ferrous form, and low hydrochloric acid secretion can lead to decreased absorption of ferric iron (12). Previous work from our laboratory showed that cysteamine increases gastric acid secretion (21, 62). In addition, the reducing action of cysteamine may promote the availability of ferrous iron for absorption by the duodenum. Both these properties may act to enhance iron uptake and contribute to the increased iron concentration in the duodenal mucosa that we observed after cysteamine administration. Thus we hypothesized that cysteamine administration might increase iron accumulation in the proximal duodenum, thereby promoting oxidative stress and duodenal mucosal damage that leads to ulceration.
Iron uptake by the duodenal mucosa is regulated through the coordinated expression of iron transport and storage proteins such as DMT1, ferroportin, and ferritin. The increased mucosal iron concentration observed in cysteamine-treated rats would be expected to inhibit IRE/IRP interaction, decrease DMT1 (IRE form) and TfR1 expression, and increase ferritin synthesis. Instead, although cysteamine increased the uptake of iron in duodenal mucosa, DMT1 (IRE form) and TfR1 expression actually increased, and ferroportin and ferritin protein levels did not change significantly. The increased expression of DMT1 (IRE form) and TfR1 can be explained by the observed cysteamine-induced increase in IRP1 activation, which would stabilize DMT1 and TfR1 mRNAs, leading to increased iron uptake by the duodenal mucosa. The interaction of IRP1 with IRE-containing mRNA depends on the assembly/disassembly of a [4Fe-4S] cluster, which makes IRP1 a theoretical target for redox regulation (8). Cysteamine might have the ability to enhance IRP1/IRE binding by modifying the 4Fe-4S cluster of IRP1 directly or through either induction of ischemia or H2O2 production (44, 46, 66). Thus cysteamine increases iron concentration in the duodenal mucosa and causes dysregulated expression of the iron transport proteins DMT1 and TfR1; this in turn could lead to a further increase in iron uptake by the proximal duodenum, thereby inducing oxidative stress and tissue damage. Oxidative damage in the small intestine, as evidenced by lipid peroxidation and a rise in the antioxidant capacity of small intestinal secretions, recently has been demonstrated in humans after perfusion of the duodenum with ferrous gluconate (67).
In summary, our results suggest a new role for iron in cysteamine-induced duodenal ulceration. Cysteamine seems to increase the availability of iron for absorption through induction of gastric acid secretion, which promotes reduction of dietary ferric iron to the ferrous form. In the present study, cysteamine administration also markedly increased the RNA-binding activity of mucosal IRP1. This may be attributable to disassembly of the 4Fe-4S cluster of IRP1 in a manner similar to the action of the structural analog of cysteamine, the sulfhydryl-reducing agent β-mercaptoethanol, which has been shown to activate IRP1 in vitro. The cysteamine-induced increase in expression of TfR1 and DMT1 may contribute to elevated iron uptake by the proximal duodenum. Finally, cysteamine may inhibit oxidation of mucosal iron, maintaining it in the ferrous state, thereby decreasing transfer of Fe+3 across the basolateral membrane and further increasing the iron concentration in the duodenal epithelium. Thus this study demonstrates for the first time a possible role for iron in the mechanism of experimental duodenal ulceration. We also demonstrated for the first time that the histamine H2 receptor antagonist cimetidine decreased iron concentration in the proximal duodenum, which was associated with its antiulcer property. Our results demonstrate that pharmacological and dietary modulation of the duodenal mucosal iron level affects the severity of cysteamine-induced duodenal ulcers, essentially implicating iron in the pathogenesis of duodenal ulceration and providing the first mechanistic explanation for the localization of selective tissue injury after systemic administration of duodenal ulcerogens such as cysteamine.
GRANTS
The present study was supported by funds from the Department of Veterans Affairs, Medical Research Service Merit Review, ChemoPathology ResourCenter, and Cancer Center Support Grant P30-CA-62203 awarded to the University of California, Irvine, CA; Dr. Anderson is the recipient of a Senior Research Fellowship from the National Health and Medical Research Council of Australia.
Acknowledgments
The authors appreciate valuable discussion with Dr. Michael Garrick, Department of Biochemistry, SUNY, Buffalo, NY. We thank Kevin Nguyen, B.S. and Hiroko Matsumoto for technical assistance.
REFERENCES
- 1.Adamama-Moraitou K, Rallis T, Papasteriadis A, Roubies N, Kaldrimidou H. Iron, zinc, and copper concentration in serum, various organs, and hair of dogs with experimentally induced exocrine pancreatic insufficiency. Dig Dis Sci 46: 1444–1457, 2001 [DOI] [PubMed] [Google Scholar]
- 2.Adler RS, Gallagher GT, Szabo S. Duodenal ulcerogens cysteamine and propionitrile decrease duodenal neutralization of acid in the rat. Dig Dis Sci 288: 716–723, 1983 [DOI] [PubMed] [Google Scholar]
- 3.Anderson GJ, Vulpe CD. Regulation of intestinal iron transport. In: Molecular and Cellular Iron Transport, edited by D. Templeton New York: Marcel Dekker, 2001, pp. 559–596.
- 4.Aziz N, Munro HN. Iron regulates ferritin mRNA translation through a segment of its 5′ untranslated region. Proc Natl Acad Sci USA 84: 8478–8482, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berruyer C, Martin FM, Castellano R, Macone A, Malergue F, Garrido-Urbani S, Millet V, Imbert J, Dupre S, Pitari G, Naquet P, Galland F. Vanin-1−/− mice exhibit a glutathione-mediated tissue resistance to oxidative stress. Mol Cell Biol 24: 7214–7224, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biaglow JE, Issels RW, Gerweck LE, Varnes ME, Jacobson B, Mitchell JB, Russo A. Factors influencing the oxidation of cysteamine and other thiols: implications for hyperthermic sensitization and radiation protection. Radiat Res 100: 298–312, 1984 [PubMed] [Google Scholar]
- 7.Bougle D, Vaghefi-Vaezzadeh N, Roland N, Bouvard G, Arhan P, Bureau F, Neuville D, Maubois JL. Influence of short-chain fatty acids on iron absorption by proximal colon. Scand J Gastroenterol 37: 1008–1011, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Bouton C, Hirling H, Drapier JC. Redox modulation of iron regulatory proteins by peroxynitrite. J Biol Chem 272: 19969–19975, 1997 [DOI] [PubMed] [Google Scholar]
- 9.Caltagirone A, Weiss G, Pantopoulos K. Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J Biol Chem 276: 19738–19745, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Canonne-Hergaux F, Gruenheid S, Ponka P, Gros P. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood 93: 4406–4417, 1999 [PubMed] [Google Scholar]
- 11.Capozzi G, Modena G. Oxidation of thiols. In: The Chemistry of the Thiol Group, edited by S. Patai. London, UK: John Wiley & Sons, 1974, pp. 785–840.
- 12.Champagne ET. Low gastric hydrochloric acid secretion and mineral bioavailability. Adv Exp Med Biol 249: 173–184, 1989 [DOI] [PubMed] [Google Scholar]
- 13.Chan FK, Leung WK. Peptic-ulcer disease. Lancet 360: 933–941, 2002 [DOI] [PubMed] [Google Scholar]
- 14.De Silva DM, Askwith CC, Kaplan J. Molecular mechanisms of iron uptake in eukaryotes. Physiol Rev 76: 31–47, 1996 [DOI] [PubMed] [Google Scholar]
- 15.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403: 776–781, 2000 [DOI] [PubMed] [Google Scholar]
- 16.Dupre S, Graziani MT, Rosei MA, Fabi A, Del Grosso E. The enzymatic breakdown of pantethine to pantothenic acid and cysteamine. Eur J Biochem 16: 571–578, 1970 [DOI] [PubMed] [Google Scholar]
- 17.Eichholz A, Howell KE. Binding studies as an approach to the study of intestinal transport. Gastroenterology 62: 647–667, 1972 [PubMed] [Google Scholar]
- 18.Fillebeen C, Pantopoulos K. Redox control of iron regulatory proteins. Redox Rep 7: 15–22, 2002 [DOI] [PubMed] [Google Scholar]
- 19.Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci USA 95: 1148–1153, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frankel D, Schipper HM. Cysteamine pretreatment of the astroglial substratum (mitochondrial iron sequestration) enhances PC12 cell vulnerability to oxidative injury. Exp Neurol 160: 376–385, 1999 [DOI] [PubMed] [Google Scholar]
- 21.Gallagher GT, Szabo S. Direct measurement of duodenal acid-pepsin exposure at site of ulceration in rats. Am J Physiol Gastrointest Liver Physiol 246: G660–G665, 1984 [DOI] [PubMed] [Google Scholar]
- 22.Galy B, Ferring D, Minana B, Bell O, Janser HG, Muckenthaler M, Schumann K, Hentze MW. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 106: 2580–2589, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Ganz T, Nemeth E. Iron imports. IV. Hepcidin and regulation of body iron metabolism. Am J Physiol Gastrointest Liver Physiol 290: G199–G203, 2006 [DOI] [PubMed] [Google Scholar]
- 24.Gruenheid S, Canonne-Hergaux F, Gauthier S, Hackam DJ, Gristein S, Gros P. The iron transport protein NRAMP2 is an integral membrane glycoprotein that colocalizes with transferring in recycling endosomes. J Exp Med 189: 831–841, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388: 482–488, 1997 [DOI] [PubMed] [Google Scholar]
- 26.Haig A, Driman DK. Iron-induced mucosal injury to the upper gastrointestinal tract. Histopathology 48: 808–812, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Halliwell B, Gutteridge JMC. Free radicals, aging and diseases. In: Free Radicals in Biology and Medicine, edited by Halliwell B and Gutteridge JMC. Oxford, UK: Clarendon Press, 1989, pp. 416–508.
- 28.Halliwell B, Gutteridge JMC. The role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol 186: 1–85, 1990 [DOI] [PubMed] [Google Scholar]
- 29.Held KD, Biaglow JE. Mechanisms for the oxygen radical-mediated toxicity of various thiol-containing compounds in cultured mammalian cells. Radiat Res 139: 15–23, 1994 [PubMed] [Google Scholar]
- 30.Hentze MW, Caughman SW, Rouault TA, Barriocanal JG, Dancis A, Harford JB, Klausner RD. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 238: 1570–1573, 1987 [DOI] [PubMed] [Google Scholar]
- 31.Hentze MW, Rouault TA, Harford JB, Klausner RD. Oxidation-reduction and the molecular mechanism of a regulatory RNA-protein interaction. Science 244: 357–359, 1989 [DOI] [PubMed] [Google Scholar]
- 32.Jaffrey SR, Cohen NA, Rouault TA, Klausner RD, Snyder SH. The iron-responsive element binding protein: a target for synaptic actions of nitric oxide. Proc Natl Acad Sci USA 91: 12994–12998, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeitner TM, Lawrence DA. Mechanisms for the cytotoxicity of cysteamine. Toxicol Sci 63: 57–64, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Khomenko T, Deng XM, Jadus MR, Szabo S. Effect of cysteamine on redox-sensitive thiol- containing proteins in the duodenal mucosa. Biochem Biophys Res Commun 309: 910–916, 2003 [DOI] [PubMed] [Google Scholar]
- 35.Khomenko T, Deng X, Sandor Z, Tarnawski AS, Szabo S. Cysteamine alters redox state, HIF-1alpha transcriptional interactions and reduces duodenal mucosal oxygenation: novel insight into the mechanisms of duodenal ulceration. Biochem Biophys Res Commun 317: 121–127, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Khomenko T, Szabo S, Deng XM, Jadus MR, Ishikawa H, Osapay K, Sandor ZS, Chen L. Suppression of early growth response factor-1 with egr-1 antisense oligodeoxynucleotide aggravates experimental duodenal ulcers. Am J Physiol Gastrointest Liver Physiol 290: G1211–G1218, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Kim DW, Kim KY, Choi BS, Youn P, Ryu DY, Klaassen CD, Park JD. Regulation of metal transporters by dietary iron, and the relationship between body iron levels and cadmium uptake. Arch Toxicol 81: 327–334, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Koeller DM, Casey JL, Hentze MW, Gerhardt EM, Chan LN, Klausner RD, Harford JB. A cytosolic protein binds to structural elements within the iron regulatory region of the transferrin receptor mRNA. Proc Natl Acad Sci USA 86: 3574–3578, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leibold EA, Munro HN. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5′ untranslated region of ferritin heavy- and light-subunit mRNAs. Proc Natl Acad Sci USA 85: 2171–2175, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lichtenberger LM, Szabo S, Reynolds ES. Gastric emptying in the rat is inhibited by the duodenal ulcerogens, cysteamine and propionitrile. Gastroenterology 73: 1072–1076, 1977 [PubMed] [Google Scholar]
- 41.Lichtenberger LM, Szabo S, Trier JS. Duodenal ulcerogens, cysteamine and propionitrile, stimulate serum gastrin levels in the rat. Gastroenterology 73: 1305–1308, 1977 [PubMed] [Google Scholar]
- 42.Martin F, Penet MF, Malergue F, Lepidi H, Dessein A, Galland F, de Reggi M, Naquet P, Gharib B. Vanin-1(−/−) mice show decreased NSAID- and Schistosoma-induced intestinal inflammation associated with higher glutathione stores. J Clin Invest 113: 591–597, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller SL, Schlesinger G. Prebiotic syntheses of vitamin coenzymes. I. Cysteamine and 2-mercaptoethanesulfonic acid (coenzyme M). J Mol Evol 36: 302–307, 1993 [DOI] [PubMed] [Google Scholar]
- 44.Mueller S, Pantopoulos K, Hubner CA, Stremmel W, Hentze MW. IRP1 activation by extracellular oxidative stress in the perfused rat liver. J Biol Chem 276: 23192–23196, 2001 [DOI] [PubMed] [Google Scholar]
- 45.Novelli GD, Kaplan NO, Lipmann F. The liberation of pantothenic acid from coenzyme A. J Biol Chem 177: 97–107, 1949 [PubMed] [Google Scholar]
- 46.Pantopoulos K, Hentze MW. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proc Natl Acad Sci USA 95: 10559–10563, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann NY Acad Sci 1012: 1–13, 2004 [DOI] [PubMed] [Google Scholar]
- 48.Pitari G, Malergue F, Martin F, Philippe JM, Massucci MT, Chabret C, Maras B, Dupre S, Naquet P, Galland F. Pantetheinase activity of membrane-bound Vanin-1: lack of free cysteamine in tissues of Vanin-1 deficient mice. FEBS Lett 483: 149–154, 2000 [DOI] [PubMed] [Google Scholar]
- 49.Raja KB, Bjarnason I, Simpson RJ, Peters TJ. In vitro measurement and adaptive response of Fe3+ uptake by mouse intestine. Cell Biochem Funct 5: 69–76, 1987 [DOI] [PubMed] [Google Scholar]
- 50.Russell ES, Nash DJ, Bernstein SE, Kent EL, McFarland EC, Matthews SM, Norwood MS. Characterization and genetic studies of microcytic anemia in house mouse. Blood 35: 838–850, 1970 [PubMed] [Google Scholar]
- 51.Samaniego F, Chin J, Iwai K, Rouault TA, Klausner RD. Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. Structure, function, and post-translational regulation. J Biol Chem 269: 30904–30910, 1994 [PubMed] [Google Scholar]
- 52.Schade SG, Cohen RJ, Conrad MD. Effect of hydrochloric acid secretion on iron absorption. N Engl J Med 279: 672–674, 1968 [DOI] [PubMed] [Google Scholar]
- 53.Schipper HM. Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox Res 1: 57–70, 1999 [DOI] [PubMed] [Google Scholar]
- 54.Selye H, Szabo S. Experimental model for production of perforating duodenal ulcers by cysteamine in rats. Nature 224: 458–459, 1973 [DOI] [PubMed] [Google Scholar]
- 55.Simovich M, Hainsworth LN, Fields PA, Umbreit JN, Conrad ME. Localization of the iron transport proteins Mobilferrin and DMT-1 in the duodenum: the surprising role of mucin. Am J Hematol 74: 32–45, 2003 [DOI] [PubMed] [Google Scholar]
- 56.Sonnenberg A, Everhart JE. Health impact of peptic ulcer in the United States. Am J Gastroenterol 92: 614–20, 1997 [PubMed] [Google Scholar]
- 57.Szabo S. Animal model of human disease. Duodenal ulcer disease animal model: cysteamine-induced acute and chronic ulcer in the rat. Am J Pathol 93: 273–276, 1978 [PMC free article] [PubMed] [Google Scholar]
- 58.Szabo S, Haith LR Jr, Reynolds ES. Pathogenesis of duodenal ulceration produced by cysteamine or propionitrile: influence of vagotomy, sympathectomy, histamine depletion, H-2 receptor antagonists and hormones. Dig Dis Sci 24: 471–477, 1979 [DOI] [PubMed] [Google Scholar]
- 59.Szabo S, Horner HC, Maull H, Schnoor J, Chiueh CC, Palkovits M. Biochemical changes in tissue catecholamines and serotonin in duodenal ulceration caused by cysteamine or propionitrile in the rat. J Pharmacol Exp Ther 2403: 871–878, 1987 [PubMed] [Google Scholar]
- 60.Szabo S, Pihan G. Development and significance of cysteamine and propionitrile models of duodenal ulcer. Chronobiol Int 4: 31–42, 1987 [DOI] [PubMed] [Google Scholar]
- 61.Szabo S, Reichlin S. Somatostatin in rat tissues is depleted by cysteamine administration. Endocrinology 109: 2255–2257, 1981 [DOI] [PubMed] [Google Scholar]
- 62.Szabo S, Reynolds ES, Lictenberger LM, Haith LR Jr, Dzau VJ. Pathogenesis of duodenal ulcer. Gastric hyperacidity caused by propionitrile and cysteamine in rats. Res Commun Chem Pathol Pharmacol 16: 311–323, 1977 [PubMed] [Google Scholar]
- 63.Tahsildar HI, Biaglow JE, Kligerman MM, Varnes ME. Factors influencing the oxidation of the radio protector WR-1065. Radiat Res 113: 243–251, 1998 [PubMed] [Google Scholar]
- 64.Tanaka H, Takeuchi K, Okabe S, Murakami M. Pathogenesis of the earliest cell damage induced by mepirizole and epithelial cysteamine in the rat duodenum. Jpn J Pharmacol 51: 509–519, 1989 [DOI] [PubMed] [Google Scholar]
- 65.Thomas SR, Schulz E, Keaney JF Jr. Hydrogen peroxide restrains endothelium-derived nitric oxide bioactivity—role for iron-dependent oxidative stress. Free Radic Biol Med 41: 681–688, 2006 [DOI] [PubMed] [Google Scholar]
- 66.Toth I, Yuan L, Rogers JT, Boyce H, Bridges KR. Hypoxia alters iron-regulatory protein-1 binding capacity and modulates cellular iron homeostasis in human hepatoma and erythroleukemia cells. J Biol Chem 274: 4467–4473, 1999 [DOI] [PubMed] [Google Scholar]
- 67.Troost FJ, Brummer RJ, Haenen GR, Bast A, van Haaften RI, Evelo CT, Saris WH. Gene expression in human small intestinal mucosa in vivo is mediated by iron-induced oxidative stress. Physiol Genomics 25: 242–249, 2006 [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Manganaro F, Schipper HM. A cellular stress model for the sequestration of redox-active glial iron in the aging and degenerating nervous system. J Neurochem 64: 1868–1877, 1995 [DOI] [PubMed] [Google Scholar]
- 69.Wheby MS, Crosby WH. The gastrointestinal tract and iron absorption. Blood 22: 416–422, 1963 [PubMed] [Google Scholar]
- 70.Wheby MS, Jones LG, Crosby WH. Studies on iron absorption. Intestinal regulatory mechanisms. J Clin Invest 43: 1433–1442, 1964 [DOI] [PMC free article] [PubMed] [Google Scholar]