

Abstract
Label-free quantitative strategies are commonly used in shot-gun proteomics to detect differences in protein abundance between biological sample groups. Here, we have employed a combination of two such approaches, spectral counting (SpC) and average MS/MS total ion current (MS2 TIC), for the analysis of rat kidney mitochondria in response to metabolic acidosis. In total, forty nine proteins were observed to be significantly altered in response to metabolic acidosis (p-value < 0.05). Of these, 32 proteins were uniquely observed as significantly different by SpC, 14 by MS2 TIC, and only 3 by both approaches. Western blot analysis was performed on a subset of these proteins to validate the observed abundance differences. This study illustrates the utility and ease of combining these two label-free quantitative approaches to increase the number of detected protein abundance differences in the shot-gun analysis of complex biological samples.
Keywords: Spectral counting, total ion current, label-free, quantitative proteomics, metabolic acidosis, MS/MS TIC, spectral TIC
INTRODUCTION
Mass spectrometry based quantitative profiling enable the observation of protein abundance differences between disease states and have been extensively utilized for the discovery of potential protein biomarkers.1, 2, 3, 4 The ability to detect quantitative differences within complex biological systems is essential to further our understanding of cellular processes and disease progression.5 The analytical approaches employed for relative protein quantitation can be grouped into two major categories, labeling and label-free.6 Examples of labeling approaches include isotope coded affinity tags (ICAT)7, stable isotope labeling by amino acids in cell culture (SILAC)8, tandem mass tags (TMT)9 and isobaric tag for relative and absolute quantitation (iTRAQ).10 While labeling methods provide accurate quantitation, they suffer from dynamic range limitations (Asara 2008). Furthermore, labeling methods require complex sample preparation, are limited in sampling size, have issues with incomplete labeling, are expensive, and involve time consuming data analysis.11 Moreover, cell culture specific methods, such as SILAC, cannot be employed for the analysis of primary tissues which are often important for biomarker investigation in clinical studies.12
Label-free approaches have become widely employed for the detection of relative protein abundance differences due to their ease of use and diverse applications.13 They are ideally suited for large scale discovery experiments with the goal of detecting significant differences between biological states to drive follow up studies. Additionally, label-free methods have been shown to enable relative quantitation over a larger dynamic range as compared to stable isotope labeling approaches.13 One frequently employed label-free approach is spectral counting (SpC) in which relative protein abundance is measured by the number of MS/MS spectra identified for all the peptides assigned to a protein. Liu, et al. demonstrated that this spectral count value strongly correlates with protein abundance in a complex matrix.14 Additionally, SpC analysis has been shown to be highly sensitive for the detection of subtle changes in protein abundance.15 However, Old, et al. illustrated a major limitation of SpC analysis for low abundance proteins (≤ 4 spectral counts) where quantitative values were found to be inaccurate.15 While SpC can suffer from inaccuracy in the quantitative measurement (i.e. fold change) it is extremely reproducible and easy to implement.
Another example of label-free quantitation is the use of ion intensity as a quantitative measure. Most approaches base the quantitative value on the intensity of the parent ion peak which requires high resolution measurements to ensure neighboring peaks are distinguished.16 This strategy is highly accurate but requires high resolution precursor ion measurements. An alternative strategy is instead based on the average or sum of the total ion current of the MS/MS spectra (MS2 TIC) assigned to all peptides for a given protein. This strategy is particularly useful for low resolution experiments (e.g. ion trap instrumentation). Fragment ion intensity measurements have been used in targeted quantitative approaches such as multiple reaction monitoring 17 and non-targeted labeling approaches such as iTRAQ.10 Recently, two studies have successfully used either sum or average MS/MS TIC measurements in shot-gun proteomics experiments to identify phosphotyrosine binding proteins as well as to explore protein differences between total lung homogenates and lung endothelial plasma membrane subfractions.18,19 Asara, et al. demonstrated that MS2 TIC displayed an improved dynamic range for detecting relative protein abundance changes as compared to SILAC.19 MS2 TIC methods have also been shown to provide improved sensitivity (as compared with SpC) for the detection of relative changes in low abundance proteins.20 However, MS2 TIC is not as reproducible as SpC due to inherent variation in sampling across a chromatographic peak over all samples.
One of the primary advantages of a label-free approach is that the data required for the analysis is collected as part of the standard analytical workflow and further experimentation or sample preparation is not required. Thus, both SpC and MS2 TIC analysis can easily be performed within the same experiment. Here, we present a detailed comparison of these two label-free approaches applied to the same dataset. Our results indicate that they are complementary and the use of both approaches increases the value of the experiment by detecting a larger number of proteins found to be significantly changing between biological states. This dual strategy is particularly useful for laboratories working with older or low resolution instrumentation which often do not yield a large number of biologically important candidates to explore in follow up studies.
The analysis presented here was performed in the context of a larger study focused on understanding proteome remodeling in the mitochondria of the proximal convoluted tubule during the renal response to metabolic acidosis.21 Briefly, metabolic acidosis is a common clinical condition that is caused by a decrease in blood pH and bicarbonate concentration.22 Previous studies have established that expression of a few mitochondrial proteins, including key enzymes of glutamine metabolism such as kidney-type glutaminase (KGA) 23, 24 and glutamate dehydrogenase (GDH) 25, are increased during metabolic acidosis.26 Thus, the relative abundance changes observed for KGA and GDH were used as positive controls for this study. The results describe the comparison of mitochondrial proteins that exhibit significant differences in protein abundance between control and acidotic rat kidneys as determined by SpC and MS2 TIC analyses.
MATERIALS AND METHODS
Proteomic sample preparation
The study compared mitochondrial fractions of proximal convoluted tubules isolated from kidneys of control rats (n=3) and 7 d acidotic rats (n=3). Control rats drank tap water and rats that were made acidotic for 7 d were provided with 0.28 M NH4Cl as their sole source of drinking water. Proximal convoluted tubule isolation and mitochondria enrichment procedures were described in detail.21 All of the procedures were approved by the Institutional Animal Care and Use Committee at Colorado State University. Both Bradford 27 and bicinchoninic acid (Pierce) protein assays 28 were performed to determine protein concentrations. Briefly, 50 μg of the mitochondrial proteins underwent acetone precipitation, were resuspended in 8 M urea and 0.2% ProteaseMAX surfactant (Promega), reduced with dithiothreitol, and alkylated with idoacetamide. The proteins underwent tryptic digestion for 3 h at 37°C and then stopped with 0.5% trifluoroacetic acid. Peptides were dried in a speed-vac and purified using a reverse phase C18 TopTip (Glygen). Purified peptides were dried and reconstituted in 50 μl of 0.1% formic acid/3% acetonitrile.
Mass spectrometry
Briefly, 10 μg aliquot of each sample was injected onto a strong cation exchange (SCX) column (ZORBAX BioSCX Series II, 300 μm × 35 mm, 3.5 μm column, Agilent) and peptides were eluted using 20 μl salt bumps of 10, 15, 25, 35, 50, 75, 150, and 500 mM NaCl. The peptides were further fractionated on a reverse phase column (1200 nanoHPLC, Zorbax C18, 5μm, 75 μm ID x 150mm column, Agilent) using a 90 min linear gradient from 25%–55% buffer B (buffer B = 90% acetonitrile and 0.1% formic acid) at a flow rate of 300 nl/min. Peptides were eluted directly into the mass spectrometer (LTQ linear ion trap, Thermo Scientific) and spectra were collected over a m/z range of 200–2000 Da using a dynamic exclusion limit of 3 MS/MS spectra of a given peptide mass for 30 sec and an exclusion duration of 90 sec. A window of 2.0 m/z was used for precursor ion selection and relative collision energy of 35 V for fragmentation. The compound lists from the resulting spectra were produced using Xcalibar 2.2 software (Thermo Scientific) with an intensity threshold of 5,000 and 1 scan/group. Duplicate 2D-LC analysis was performed for each biological sample and sample order was randomized before analysis. The addition of 2D-LC to a relative quantitation workflow has been shown to not effect quantitation and results in an overall increase in peptide identifications.29
Bioinformatics
The tandem mass spectra (MS/MS) were searched against the Uniprot-KB Rattus norvegicus protein sequence database (74,140 sequence entries) which was concatenated with a reverse database using both the Mascot (version 2.3.02, Matrix Science) and SEQUEST (version v.27, rev. 11, Sorcerer, Sage-N Research) database search engines. The following search parameters were used: average mass, peptide mass tolerance of 2.5 Da, fragment ion mass tolerance of 1.0 Da, complete tryptic digestion allowing two missed cleavages, variable modifications of methionine oxidation and lysine acetylation, and a fixed modification of cysteine carbamidomethylation. Peptide identifications from both search engine results were combined in Scaffold 3 (Version 3.6.0, Proteome Software) using protein identification algorithms. Search results from all raw files associated with a biological sample (i.e. all 2D-LC fractions and duplicate injections) were summed within the Scaffold 3 software. Peptide probability thresholds of 90% and protein of 99% were applied with Peptide and Protein Prophet algorithms in Scaffold 3.30 Proteins containing shared peptides were grouped by Scaffold 3, satisfying the laws of parsimony. Using a target decoy approach a peptide false discovery rate (FDR) of 0.2% was determined.31 Proteins that were identified by a minimum of 2 unique peptides in at least 1 biological replicate were considered confidently identified. Using these criteria, 901 total proteins were identified in all the samples.21
Label-free protein quantitation
Box plots were generated to assess data quality between technical and biological replicates. Variance in the biological replicates was assessed by determining the number of proteins identified per sample, the total spectra identified, and the total peptides identified (Figure S1). Correlation of biological and technical replicates was tested using Pearson’s correlation test (Figure S2). The statistical analysis was performed using the R v2.15 statistics package (http://r-project.org) and DanteR v1.0.2 (http://omics.pnl.gov/software/DanteR.php.
MS/MS data to be used for quantitation was normalized between samples in Scaffold 3 by using the sum of the unweighted spectral counts for each sample to determine a sample specific scaling factor that was then applied to all proteins in that sample. The impact of this normalization on both SpC and MS2 TIC values is illustrated in Figure S3. Quantitative analysis using SpC and MS2 TIC were separately performed on the normalized data. The average MS/MS total ion current value was chosen because it has been shown to be less biased by protein length than the sum.19 For SpC analysis, a pseudo value of 1 was added to all normalized values to eliminate zero values. For MS2 TIC a pseudo count of 2,753.8 was added to all normalized values. This number represents the lowest observed MS2 TIC value in the dataset. To test for significant changes in protein abundance between treatments, the Student’s t-test was performed on the normalized spectral count or MS2 TIC values. For a protein to be included in the quantitative analysis the following requirements had to be met; there must have been at least two unique peptides in at least two out of three biological replicates and there must have been a sum of ≥ 10 spectra overall in the biological replicates. Manual validation of MS/MS spectra was performed for protein identifications that met these thresholds and showed significant (p < 0.05) change in abundance between treatments, but only had two unique peptide assignments. Criteria for manual spectral validation included the following: 1) a minimum of at least 3 theoretical y or b ions in consecutive order with intensities greater than 5% of the maximum intensity; 2) an absence of prominent unassigned peaks greater than 5% of the maximum intensity; and 3) indicative residue specific fragmentation, such as intense ions N-terminal to proline and immediately C-terminal to aspartate and glutamate. These conservative thresholds coupled with manual spectral validation ensure that the MS2 TIC values are not largely influenced by signal from chimeric peptides. Fold changes were calculated as the ratio of the mean normalized SpC or MS2 TIC for the three biological replicates for each condition. Data quality was also assessed using the Power Law Global Error Model (PLGEM) as implemented within the Scaffold software (Figure S4) 32
Western blot analyses
Additional mitochondrial fractions isolated from renal proximal convoluted tubules were prepared to enable validation of the results observed by SpC and MS2 TIC analysis. For western blotting, control and acidotic samples containing 10 μg of protein were separated by 8 or 10% SDS-PAGE. Western blot analysis was carried out as described previously.21 Additionally, the rabbit polyclonal heat shock cognate 71 kDa protein (HSP7C) (Abcam) antibody was used to validate the change identified by MS2 TIC.
RESULTS AND DISSCUSION
The experimental workflow beginning with mitochondrial isolation from three control and three acidotic rat kidneys is illustrated in Figure 1. Key to this workflow is the use of two label-free relative quantitative approaches (SpC & MS2 TIC) from the same set of mass spectrometry data. Spectral counts, by definition, are assigned in whole number increments where the total spectral count value for a protein is the sum (total number) of all spectra assigned to peptides from that protein. Conversely, MS2 TIC values are by definition much larger values where the summation of the total ion current for the MS/MS spectra are averaged for the assigned peptides from a given protein. The use of this total ion current value for each tandem mass spectra allows for a more weighted value to be assigned to each spectrum. Both SpC & MS2 TIC values can be easily extracted from any dataset without special or additional data acquisition.
Figure 1.
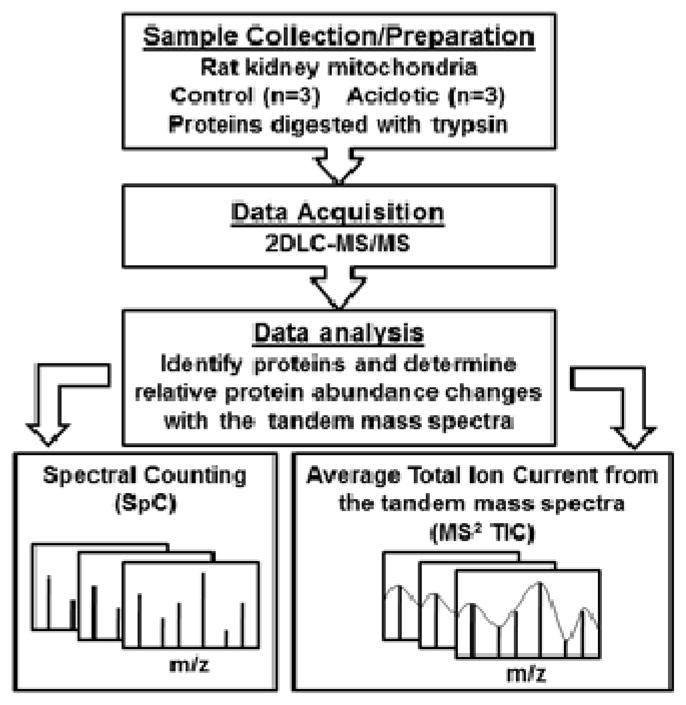
The experimental workflow for proteomic analysis of rat renal mitochondrial proteins. Spectral counting (SpC) and average total ion current from the tandem mass spectra (MS2 TIC) was used to determine changes in protein abundance occurring in response to metabolic acidosis.
Careful design of a label-free quantitative experiment is critical to accurately identify protein changes. 33, 34 One metric of quantitative data quality is that the number of proteins, peptides, and spectra identified in each sample are similar. Variance in the raw data was assessed by calculating the coefficient of variance between samples (CV). The following % CV were calculated for number of proteins identified per sample (% CV for control 4.7 and acidotic 1.4), the total spectra identified (% CV for control 15.7 and acidotic 2.5), and the total peptides identified (% CV for control 20.4 and acidotic 4.1) (Figure S1). The number of identified proteins, peptides, and spectra were within 20% for all the samples, both control and acidotic. Pearson’s correlation calculations reported R2 values ranging from 0.89–1.00 between all the samples (Figure S2). Box plots for both pre and post-normalization of quantitative values from SpC and MS2 TIC showed minimal variation and the effects of normalization (Figure S3). Overall, the data showed acceptable qualitative features for quantitative analysis.
Normalization was performed on both the SpC and MS2 TIC values to account for analytical variations between samples. The Student’s t-test was used to determine statistically significant changes in protein abundance (p-values ≤ 0.05). Each protein with a significant p-value had to meet the additional requirement of a sum of ≥ 10 spectra over the three biological replicates and have a minimum of two manually validated unique peptides in at least two biological replicates. Biological relevance is often assessed by calculating the fold change, although it relies solely on the ratio of the quantitative values, not the variance between samples. Therefore, fold changes were only calculated for those proteins meeting the above requirements and a fold change cut off was not applied.
Of the 901 proteins identified a total of 49 proteins were observed to be significantly altered (p-value < 0.05) in response to metabolic acidosis. Of these, 32 proteins were observed only in the SpC analysis (Table 1), 14 were observed only in the MS2 TIC analysis (Table 2), and 3 were observed by both approaches (Fig. 2). The three common proteins, apolipoprotein A-IV, peroxisomal bifunctional enzyme (EHHADH), and glutamate dehydrogenase (GDH), are denoted in bold and italicized in the tables. Two proteins served as positive controls, KGA 23,24 and GDH 25, both of which are known to increase in abundance during acidosis.26 In this analysis, both KGA and GDH were observed to be significantly increased upon acidosis by SpC. However, only GDH was observed to significantly different by MS2 TIC. Furthermore, the fold change observed for GDH by the MS2 TIC method was actually a slight decrease (0.9). The significant p-value indicates that the MS2 TIC values for GDH had very little variance among biological replicates. GDH is an extremely abundant protein in these samples (mean control MS2 TIC = 2.0×105), thus the lack of an applicable fold change is likely due to saturation of the MS2 TIC signal (Table 2). While not within the significance threshold, KGA was observed to change by MS2 TIC with a fold change of 1.3 (p-value = 0.08).
Table 1.
Proteins observed by SpC with significant (p-value < 0.05) abundance changes In response to metabolic acidosis
| Protein name | Accession # | SpC
|
Mean Normalized SPC Values
|
Mean Normalized MS
2 TIC Values |
|||
|---|---|---|---|---|---|---|---|
| P-value | Fold Change | Control (S.E.) | Acidotic (S.E.) | Control (S.E.) | Acidotic (S.E.) | ||
|
| |||||||
| UDP-glucuronosyltransferase 2B15 | P36511 | 0.00022 | 6.4 | 1.8 (1.0) | 17.0 (0.7) | 1.9E+05 (1.7E+05) | 7.0E+04 (2.2E+04) |
| UDP-glucuronosyltransferase 1–1 (UGT1A1) | Q64550 | 0.00086 | 4.6 | 11.0 (1.8) | 54.1 (4.5) | 6.3E+04 (2.0E+04) | 6.8E+04 (4.6E+03) |
| Peroxisomal multifunctional enzyme type 2 (HSD17B4) | P97852 | 0.0018 | 2.0 | 48.1 (3.6) | 96.3 (6.3) | 1.8E+06 (2.8E+04) | 1.6E+06 (1.6E+04) |
| Dimethylglycine dehydrogenase. mitochondrial | Q63342 | 0.0024 | 1.8 | 40.9 (1.8) | 75.0 (4.7) | 1.4E+05 (1.4E+04) | 1.6E+05 (2.2E+04) |
| Glutamate dehydrogenase 1, mitochondrial (GDH) | P10860 | 0.0035 | 1.7 | 431.5 (45.2) | 715.7 (7.9) | 2.0E+05 (7.3E+03) | 1.7E+05 (3.1E+03) |
| 3-ketoacyl-CoA thiolase B. peroxisomal (ACAA1) | F1LPD6 | 0.0049 | 2.4 | 11.3 (1.1) | 28.4 (2.8) | 4.3E+04 (1.1E+04) | 3.8E+04 (6.3E+03) |
| Enoyl-CoA hydratase domain-containing protein 3 | Q3MIE0 | 0.0061 | 3.6 | 0.4 (0.4) | 3.8 (0.6) | 1.5E+04 (1.5E+04) | 6.4E+04 (2.2E+04) |
| Vitamin D-binding protein | P04276 | 0.0062 | 2.6 | 15.0 (4.4) | 40.2 (1.9) | 1.8E+05 (1.3E+05) | 1.0E+05 (1.4E+04) |
| Calnexin | P35565 | 0.0069 | 0.4 | 35.1 (3.6) | 14.8 (1.5) | 6.1E+04 (1.7E+04) | 7.7E+04 (9.0E+03) |
| Catalase (CAT) | P04762 | 0.0095 | 2.2 | 55.1 (12.0) | 123.1 (8.3) | 1.1E+05 (2.3E+04) | 1.3E+05 (1.6E+04) |
| Fumarylacetoacetate hydrolase domain-containing protein 2 | B2RYW9 | 0.011 | 2.0 | 29.6 (5.9) | 61.1 (3.8) | 1.2E+05 (2.9E+04) | 1.7E+05 (7.8E+03) |
| Ras-related protein Rab-21 | Q8AXT5 | 0.011 | 4.1 | 1.5 (0.8) | 9.5 (1.6) | 3.8E+04 (2.8E+04) | 5.5E+04 (1.1E+04) |
| Epoxide hydrolase 1 | P07687 | 0.013 | 3.1 | 11.1 (3.3) | 36.3 (4.9) | 2.2E+05 (6.1E+04) | 2.2E+05 (3.4E+04) |
| Cytochrome b5 | P00173 | 0.014 | 1.7 | 37.5 (5.8) | 64.7 (2.8) | 2.2E+05 (1.3E+04) | 2.6E+05 (2.2E+04) |
| Peroxisomal bifunctional enzyme (EHHADH) | P07896 | 0.014 | 2.1 | 14.3 (3.5) | 31.5 (2.0) | 8.2E+04 (1.7E+04) | 2.2E+05 (1.7E+04) |
| Uncharacterized protein | D4A0Y1 | 0.014 | 4.0 | 1.2 (0.7) | 7.7 (1.4) | 1.2E+04 (6.8E+03) | 3.0E+04 (2.3E+03) |
| Ectonucleoside triphosphate diphosphohydrolase 5 | Q6P6S9 | 0.018 | 2.1 | 8.0 (0.8) | 18.2 (2.5) | 6.6E+04 (8.3E+03) | 4.4E+04 (4.2E+03) |
| Kidney-type glutaminase (KGA) | P13264 | 0.019 | 3.2 | 17.7 (2.1) | 69.3 (10.7) | 0.2E+04 (3.1E+03) | 1.1E+05 (1.0E+04) |
| 4F2 cell-surface antigen heavy chain | Q794F9 | 0.02 | 0.5 | 105.6 (13.0) | 50.5 (6.8) | 1.3E+05 (2.7E+03) | 1.2E+05 (2.1E+04) |
| Peroxisomal acyl-coenzyme A oxidase 1 | P07872 | 0.021 | 1.9 | 26.2 (6.8) | 51.4 (0.5) | 5.1E+04 (5.1E+03) | 7.1E+04 (1.6E+04) |
| Cytochrome P450 4A2 | P20818 | 0.023 | 1.9 | 91.2 (22.0) | 175.1 (7.7) | 3.8E+04 (3.8E+03) | 4.7E+04 (9.3E+03) |
| Filamin-B | D3ZD13 | 0.023 | 0.6 | 4.2 (0.5) | 2.1 (0.3) | 4.0E+04 (1.2E+04) | 3.6E+04 (1.1E+04) |
| Protein AMBP | Q64240 | 0.024 | 5.0 | 2.0 (0.9) | 13.8 (3.2) | 8.1E+04 (5.7E+04) | 7.2E+04 (1.9E+04) |
| ATP synthase-coupling factor 6. mitochondrial | P21571 | 0.027 | 0.8 | 115.6 (4.4) | 96.9 (3.3) | 2.7E+05 (2.9E+04) | 2.4E+05 (1.3E+04) |
| Enoyl-CoA delta isomerase 1. mitochondrial | P23965 | 0.035 | 1.5 | 76.7 (6.5) | 116.2 (10.8) | 1.0E+05 (2.0E+04) | 1.3E+05 (9.5E+03) |
| Protein FAM151A | Q642A7 | 0.036 | 0.4 | 32.8 (6.3) | 11.7 (2.5) | 5.2E+04 (1.1E+04) | 9.1E+04 (2.2E+04) |
| ATP-binding cassette sub-family D member 3 | P16970 | 0.037 | 1.8 | 29.3 (6.3) | 52.5 (4.0) | 8.1E+04 (2.5E+04) | 1.1E+05 (8.4E+03) |
| Probable D-lactate dehydrogenase. mitochondrial | F1LVD7 | 0.037 | 1.7 | 27.0 (4.0) | 45.7 (4.5) | 3.0E+05 (8.6E+04) | 2.4E+05 (3.1E+04) |
| GrpE protein homolog 1. mitochondrial | P97576 | 0.04 | 1.5 | 10.2 (0.9) | 16.3 (1.8) | 2.3E+05 (4.7E+04) | 2.8E+05 (6.9E+04) |
| Apollpoprotein A-IV | P02651 | 0.041 | 1.8 | 46.3 (10.0) | 83.6 (7.6) | 1.6E+05 (1.5E+03) | 2.0E+05 (1.3E+04) |
| All-trans-13. 14-dihydroretinol saturase. isoform CRA_b | G3V7V6 | 0.043 | 4.9 | 0.7 (0.4) | 7.5 (2.3) | 5.8E+04 (3.5E+04) | 1.1E+05 (7.7E+03) |
| NADPH--cytochrome P450 reductase | P00388 | 0.044 | 1.6 | 13.2 (1.4) | 21.6 (2.6) | 8.5E+04 (1.2E+04) | 8.7E+04 (7.1E+03) |
| Ribonuclease 4 | O55004 | 0.044 | 2.2 | 7.5 (1.4) | 17.7 (3.2) | 1.4E+05 (3.8E+04) | 1.6E+05 (7.5E+03) |
| Electron transfer flavoprotein subunit beta | Q68FU3 | 0.046 | 1.3 | 92.3 (9.3) | 125.5 (6.5) | 1.6E+05 (2.1E+04) | 1.7E+05 (7.2E+03) |
| Lon protease homolog. mitochondrial | Q924S5 | 0.049 | 0.7 | 86.5 (10.1) | 57.5 (2.7) | 8.6E+04 (1.7E+04) | 6.2E+04 (4.2E+03) |
Bold & itlaicized are common to both methods.
Table 2.
Proteins observed by MS2 TIC with significant p-value < 0.05) abundance changes in response to metabolic acidosis
| Protein name | Accession # | MS2 TIC
|
Mean Normalized MS2 TIC Values
|
Mean Normalized SpC Values
|
|||
|---|---|---|---|---|---|---|---|
| P-Value | Fold Change | Control (S.E.) | Acidotic (S.E.) | Control (S.E.) | Acidotic (S.E) | ||
|
| |||||||
| Carbonic anhydrase 6B. mitochondrial (CA6B) | Q88HG8 | 9.9E-06 | 76.6 | 0.0E+00 (0.0E+00) | 2.1E+06 (1.3E+04) | 0.0 (0.0) | 4.7 (1.8) |
| RCG41951. isoform CRA_a | D3ZCZ9 | 0.00073 | 2.2 | 6.2E+04 (6.5E+03) | 1.4E+05 (5.3E+03) | 15.2 (5.1) | 21.3 (1.3) |
| Histone H4 | P62804 | 0.0028 | 0.4 | 4.2E+05 (2.6E+04) | 1.7E+05 (2.9E+04) | 6.6 (22) | 9.5 (1.2) |
| Tropomyosin alpha-3 chain | F1LRP5 | 0.004 | 0.2 | 5.8E+04 (4.0E+03) | 7.5E+03 (7.5E+03) | 17.7 (8.9) | 0.3 (0.3) |
| Saccharopine dehydrogenase-like oxidoreductase | Q6AY30 | 0.0072 | 0.7 | 5.9E+04 (2.0E+03) | 4.0E+04 (3.1E+03) | 25.1 (9.4) | 17.6 (1.4) |
| Uncharacterized protein | E9PT15 | 0.0086 | 1.4 | 2.2E+05 (1.3E+04) | 3.1E+05 (1.2E+04) | 71.9 (7.1) | 92.0 (10.5) |
| Heat shock cognate 71 kDa protein (HSP7C) | P63018 | 0.016 | 0.7 | 2.0E+05 (4.3E+03) | 1.4E+05 (1.2E+04) | 94.3 (25.0) | 73.1 (3.8) |
| EH domain-containing protein 1 | Q641Z6 | 0.017 | 0.8 | 8.2E+04 (3.7E+03) | 6.2E+04 (3.7E+03) | 57.3 (19.8) | 40.4 (2.1) |
| Peroxisomal bifunctional enzyme (EHHADH) | P07898 | 0.019 | 1.7 | 8.2E+04 (1.7E+04) | 2.2E+06 (1.7E+04) | 14.3 (3.6) | 31.6 (2.0) |
| Glutamate dehydrogenase 1, mitochondrial (GDH) | P10860 | 0.022 | 0.9 | 2.0E+05 (7.3E+03) | 1.7E+05 (3.1E+03) | 431.5 (45.2) | 715.7 (7.9) |
| Tubulin beta-5 chain | P69897 | 0.023 | 1.4 | 7.0E+04 (5.8E+03) | 1.0E+05 (5.9E+03) | 106.4 (30.0) | 89.7 (8.5) |
| Apollpoprotein A-IV | P02651 | 0.026 | 1.3 | 1.5E+05 (1.5E+03) | 2.0E+05 (1.3E+04) | 46.3 (10.0) | 83.6 (7.6) |
| Xaa-Pro aminopeptidase 1 | O54975 | 0.028 | 2.1 | 6.2E+04 (1.8E+04) | 1.3E+05 (1.0E+04) | 6.9 (23) | 7.9 (3.8) |
| Glutamyl aminopeptidase | P50123 | 0.035 | 0.6 | 1.4E+05 (1.4E+04) | 8.8E+04 (9.6E+03) | 38.8 (10.4) | 20.8 (8.0) |
| Sulfate anion transporter 1 | P45380 | 0.038 | 2.3 | 7.7E+04 (1.1E+04) | 1.8E+05 (3.1E+04) | 6.8 (0.9) | 3.9 (1.1) |
| NADH dehydrogenase 1 alpha subcomplex. 8 | D4A311 | 0.042 | 0.5 | 2.7E+05 (4.7E+04) | 1.3E+05 (9.1E+03) | 41.9 (10.3) | 49.1 (4.4) |
| Triosephosohate isomerase | P48600 | 0.042 | 0.8 | 1.1E+06 (8.4E+03) | 9.0E+04 (3.3E+03) | 64.7 (9.0) | 32.1 (2.9) |
Bold & itlaicized are common to both methods.
Figure 2.
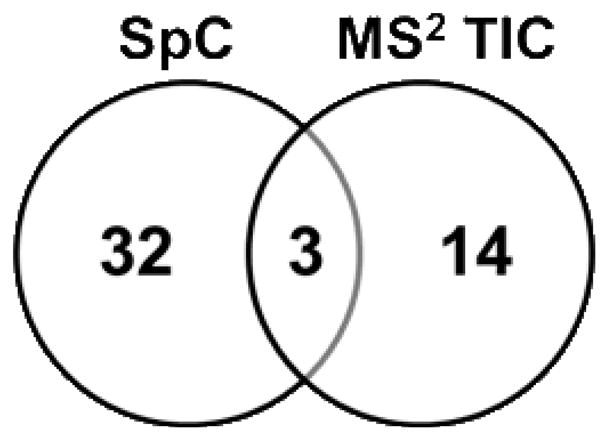
Venn diagram of identified proteins observed to be significantly changing (p-values < 0.05) in response to metabolic acidosis using both spectral counting (SpC) and average MS/MS total ion current (MS2 TIC).
Western blot analysis was performed as an independent method to validate several of the proteins observed to be significantly changing in abundance in response to metabolic acidosis. Western blotting was performed with control (n=3) and acidotic (n=3) rat renal mitochondria samples (Fig. 3). Blots were probed with specific antibodies to nine proteins and normalized by probing with an antibody to a mitochondrial protein that served as a loading control (Fig. 3A). UDP-glucuronosyltransferase 1–1 (UGT1A1) was observed to be changing only by SpC (p-value = 0.0009, fold change = 4.6). However, western blotting of UGT1A1 revealed a fold change of only 1.6. The larger fold change observed by SpC could be an artificial inflation due to the detection of shared peptides from other isoforms, whereas the antibody is specific only to the 1A1 isoform. Peroxisomal multifunctional enzyme type 2 (HSD17B4), 3-ketoacyl-CoA thiolase B (ACAA1), catalase (CAT), and kidney-type glutaminase (KGA) were all detected as significantly changing only by SpC. All four of these proteins yielded statistically equivalent fold changes by western blotting with specific antibodies (Fig. 3B). Two of the proteins uniquely observed by MS2 TIC were also validated by western blotting; carbonic anhydrase 5B (CA5B) and heat shock cognate 71 kDa protein (HSP7C) (Fig 3A). MS2 TIC fold change for CA5B is exaggerated as this protein was not detected in control samples. CA5B narrowly fell outside the significant threshold in the SpC results, (p-value = 0.068). However, the fold change observed by SpC (5.7-fold) agrees well with the western blotting results, (3.7-fold). CA5B had a mean spectral count value of only 4.7 in the acidotic samples and 0 in the control samples which illustrates the limitation of SpC for low abundance proteins. The decrease in HSP7C, identified uniquely by MS2 TIC, was also confirmed by western blotting. Of the three proteins observed by both approaches, two were further evaluated by western blotting: peroxisomal bifunctional enzyme (EHHADH) and glutamate dehydrogenase (GDH). EHHADH did not show a significant fold change by western blotting. This result may be due to the large amount of non-specific binding observed with this antibody. Statistically equivalent fold changes were observed by western blot analysis and SpC for GDH.
Figure 3.
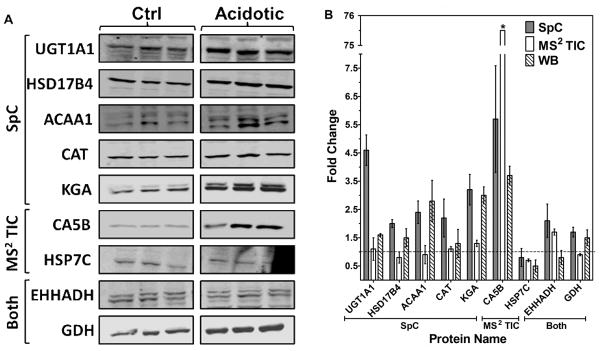
Western blot analysis validating proteins found to be significantly changing in response to metabolic acidosis (p-values < 0.05) by either spectral counting (SpC), average total ion current (MS2 TIC) or both. A: The control (ctrl) and acidotic samples protein bands were imaged and quantified and each blot was normalized to a loading control. B: Fold changes from SpC, MS2 TIC and western blotting (WB). The reported data are the mean ± the S.E. of triplicate biological samples. The * indicates error not shown for CA5B MS2 TIC, ± 4.8.
Previous studies have shown that SpC is not accurate for proteins at low abundance (i.e. ≤ 4 spectral counts) and MS2 TIC is not accurate for proteins with ≤ 5 identified MS/MS spectra.15, 35, 19 To evaluate the trend within our dataset, the mean spectral counts of the triplicate acidotic samples were plotted against the calculated p-values (Fig. 4A). The majority of the data is represented by proteins with < 100 spectral counts. As the spectral counts decrease the p-values tend to be less significant, illustrating the limitation of this method for proteins at low abundance. This is further represented in the inset of figure 4A which shows a clear shift in the distribution of proteins with p-values > 0.05, above which corresponds to proteins with very low SpC values. The range of the mean normalized SpC values for proteins with p-values < 0.05 is broad, ranging from 3.8 to 715.7. Conversely, the MS2 TIC values do not exhibit this trend (Fig. 4B). The scale for the MS2 TIC data is notably larger compared to the SpC data (x-axis of Fig 4A vs 4B). As mentioned above, this is due to the fact that in SpC a value of 1 is assigned for each spectra whereas in MS2 TIC the value for each protein corresponds the average ion signal of all the MS/MS spectra (a number much larger than 1). The difference in scale results in an increase of the quantifiable dynamic range of the MS2 TIC approach.20 The disadvantage of this larger scale is that saturation may occur at very high MS2 TIC values. This is illustrated in Fig 4B as there are no proteins with a p-value < 0.05 at MS2 TIC mean values > 3.2×105. Another notable trend in the data presented in Figure 4 is that the MS2 TIC data is more disperse than the SpC data. This difference highlights the superb reproducibility of SpC analysis. MS2 TIC measurements tend to be less reproducible due to the fact that in each analytical run the peptide will not be sampled at the exact same point in the chromatographic peak and this will be reflected in the MS2 TIC value.
Figure 4.
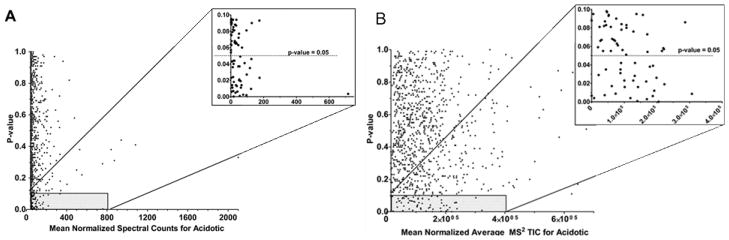
Quantitative value vs. p-value. A: The mean SpC of triplicate acidotic samples (x-axis) vs. p-values B: The mean MS2 TIC of triplicate acidotic samples (x-axis) vs. p-values. Both insets have only the data with p-values < 0.1 (y-axis).
While SpC and MS2 TIC are excellent methods for detecting protein differences, they are known to suffer from inaccuracy. Therefore, in this study, only a significance level threshold (p-value < 0.05) was employed. Often, however, fold changes are desired and are important in assessing biological relevance. To assess the use of fold change values in our results, fold change calculations from SpC or MS2 TIC analyses were compared to the corresponding p-values from each method (Fig. 5). For both approaches a convergence to a fold change of 1 was observed with increasing p-value, this trend is expected and adds confidence to the results. Additionally, the lack of data points present at a fold change of 1 with p-values < 0.05 is also encouraging. The prominent difference in the data for the two approaches is that the MS2 TIC analysis results in much larger fold change values (Fig. 5B is missing the top ten MS2 TIC fold changes ranging from 20–140). The exaggerated fold changes are a result of the larger scale of MS2 TIC values and thus the pseudo count approach is not as effective in dealing with zero values.
Figure 5.
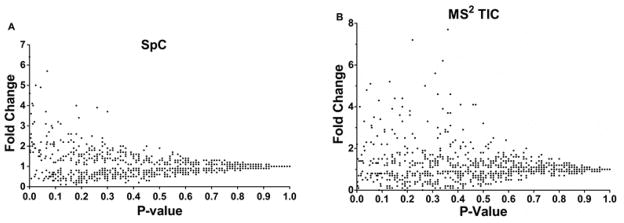
P-value vs. fold change. A: The p-values from SpC analysis (x-axis) and the calculated fold changes (y-axis). B: The p-values from average MS2 TIC (x-axis) and the calculated fold changes (y-axis). Ten values are not plotted here; largest fold change was 137 for MS2 TIC.
CONCLUSIONS
Label-free relative quantitation methods are extremely important in large scale proteomics experiments to further our understanding of disease states and enable biomarker discovery. In this type of study, the goal is to detect significant differences between biological states which can generate hypotheses and drive future research. Samples are often very complex and the proteins of biological interest can be at low abundance within a high background. The ability to maximally mine proteomic data to improve our ability to identify significant differences between disease states is essential. There is no current proteomic profiling strategy that is a capable of identifying the entire proteome or for determining all quantitative changes between disease states. Spectral counting is advantageous in its ease of use and high reproducibility but can fail for proteins at low abundance. Conversely, MS2 TIC measurements can be employed over a larger dynamic range but suffer from irreproducibility and signal saturation for highly abundant proteins. While each of these methods has specific advantages and disadvantages, together they enable a more comprehensive picture of protein abundance differences over a wide dynamic range of protein concentrations. The dual approach is straightforward and requires minimal added effort to implement. Importantly, no additional samples or mass spectrometry analyses are needed to obtain both SpC and MS2 TIC values. Therefore, the added value gained by employing a dual approach cost only the additional time for data analysis. In summary, the results of this study clearly illustrate the benefit of employing a dual label-free relative quantitative approach to increase the value of a shot-gun proteomics dataset through the detection of more biologically relevant targets.
Supplementary Material
Acknowledgments
Funding Sources:
This research was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-37124 and DK-75517 awarded to N. P. Curthoys. All mass spectrometry was performed in the Proteomics and Metabolomics Facility at Colorado State University (http://www.pmf.colostate.edu).
We would like to thank Dr. N. P. Curthoys for his funding support and assistance with review of this manuscript. We would like to also acknowledge Brian Searle from Proteome Software for his thoughtful review of this manuscript.
ABBREVIATIONS
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- TIC
total ion current
Footnotes
Author Contributions
The manuscript was written through contributions of all authors.
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Coombs KM. Quantitative proteomics of complex mixtures. Expert review of proteomics. 2011;8(5):659–77. doi: 10.1586/epr.11.55. [DOI] [PubMed] [Google Scholar]
- 2.Filiou MD, Turck CW, Martins-de-Souza D. Quantitative proteomics for investigating psychiatric disorders. Proteomics. Clinical applications. 2011;5(1–2):38–49. doi: 10.1002/prca.201000060. [DOI] [PubMed] [Google Scholar]
- 3.Liang S, Xu Z, Xu X, Zhao X, Huang C, Wei Y. Quantitative proteomics for cancer biomarker discovery. Comb Chem High Throughput Screen. 2012;15(3):221–31. doi: 10.2174/138620712799218635. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q, Faca V, Hanash S. Mining the plasma proteome for disease applications across seven logs of protein abundance. J Proteome Res. 2011;10(1):46–50. doi: 10.1021/pr101052y. [DOI] [PubMed] [Google Scholar]
- 5.Cravatt BF, Simon GM, Yates JR., III The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450(7172):991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 6.Xie F, Liu T, Qian WJ, Petyuk VA, Smith RD. Liquid chromatography-mass spectrometry-based quantitative proteomics. The Journal of biological chemistry. 2011;286(29):25443–9. doi: 10.1074/jbc.R110.199703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature biotechnology. 1999;17(10):994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 8.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 9.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C. Tandem Mass Tags:_ A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry. 2003;75(8):1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 10.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Panchaud A, Affolter M, Moreillon P, Kussmann M. Experimental and computational approaches to quantitative proteomics: status quo and outlook. Journal of proteomics. 2008;71(1):19–33. doi: 10.1016/j.jprot.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Nikolov M, Schmidt C, Urlaub H. Quantitative mass spectrometry-based proteomics: an overview. Methods in molecular biology (Clifton, NJ) 2012;893:85–100. doi: 10.1007/978-1-61779-885-6_7. [DOI] [PubMed] [Google Scholar]
- 13.Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Analytical and bioanalytical chemistry. 2007;389(4):1017–31. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 14.Liu H, Sadygov RG, Yates JR. 3rd, A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 15.Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4(10):1487–502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Zhu W, Smith JW, Huang CM. Mass spectrometry-based label-free quantitative proteomics. Journal of biomedicine & biotechnology. 2010;2010:840518. doi: 10.1155/2010/840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104(14):5860–5. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat Biotech. 2010;28(1):83–89. doi: 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asara JM, Christofk HR, Freimark LM, Cantley LC. A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. PROTEOMICS. 2008;8(5):994–9. doi: 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
- 20.Wu Q, Zhao Q, Liang Z, Qu Y, Zhang L, Zhang Y. NSI and NSMT: usages of MS/MS fragment ion intensity for sensitive differential proteome detection and accurate protein fold change calculation in relative label-free proteome quantification. The Analyst. 2012;137(13):3146–53. doi: 10.1039/c2an35173k. [DOI] [PubMed] [Google Scholar]
- 21.Freund DM, Prenni JE, Curthoys NP. Response of the Mitochondrial Proteome of Rat Renal Proximal Convoluted Tubules to Chronic Metabolic Acidosis. American Journal of Physiology - Renal Physiology. 2012 doi: 10.1152/ajprenal.00526.2012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halperin ML. Metabolic aspects of metabolic acidosis. Clin Invest Med. 1993;16(4):294–305. [PubMed] [Google Scholar]
- 23.Curthoys NP, Lowry OH. The distribution of glutaminase isoenzymes in the various structures of the nephron in normal, acidotic, and alkalotic rat kidney. J Biol Chem. 1973;248(1):162–8. [PubMed] [Google Scholar]
- 24.Wright PA, Knepper MA. Phosphate-dependent glutaminase activity in rat renal cortical and medullary tubule segments. Am J Physiol. 1990;259(6 Pt 2):F961–70. doi: 10.1152/ajprenal.1990.259.6.F961. [DOI] [PubMed] [Google Scholar]
- 25.Wright PA, Knepper MA. Glutamate dehydrogenase activities in microdissected rat nephron segments: effects of acid-base loading. Am J Physiol. 1990;259(1 Pt 2):F53–9. doi: 10.1152/ajprenal.1990.259.1.F53. [DOI] [PubMed] [Google Scholar]
- 26.Curthoys NP, Taylor L, Hoffert JD, Knepper MA. Proteomic analysis of the adaptive response of rat renal proximal tubules to metabolic acidosis. American Journal of Physiology - Renal Physiology. 2007;292(1):F140–F147. doi: 10.1152/ajprenal.00217.2006. [DOI] [PubMed] [Google Scholar]
- 27.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 28.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 29.Patel NA, Crombie A, Slade SE, Thalassinos K, Hughes C, Connolly JB, Langridge J, Murrell JC, Scrivens JH. Comparison of One- and Two-dimensional Liquid Chromatography Approaches in the Label-free Quantitative Analysis of Methylocella silvestris. J Proteome Res. 2012;11(9):4755–63. doi: 10.1021/pr300253s. [DOI] [PubMed] [Google Scholar]
- 30.(a) Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical chemistry. 2002;74(20):5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]; (b) Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 31.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 32.Pavelka N, Fournier ML, Swanson SK, Pelizzola M, Ricciardi-Castagnoli P, Florens L, Washburn MP. Statistical similarities between transcriptomics and quantitative shotgun proteomics data. Mol Cell Proteomics. 2008;7(4):631–644. doi: 10.1074/mcp.M700240-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Lundgren DH, Hwang SI, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert Rev Proteomics. 2010;7(1):39–53. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- 34.Oberg AL, Vitek O. Statistical design of quantitative mass spectrometry-based proteomic experiments. J Proteome Res. 2009;8(5):2144–56. doi: 10.1021/pr8010099. [DOI] [PubMed] [Google Scholar]
- 35.Wong JW, Sullivan MJ, Cagney G. Computational methods for the comparative quantification of proteins in label-free LCn-MS experiments. Briefings in bioinformatics. 2008;9(2):156–65. doi: 10.1093/bib/bbm046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.