

Abstract
Metastatic prostate cancer (PCa) is initially treated with androgen ablation therapy, which causes regression of androgen-dependent tumors. However, these tumors eventually relapse resulting in recurrent castration-resistant prostate cancer (CRPC). Currently, there is no effective therapy for CRPC and the molecular mechanisms that lead to the development of CRPC are not well understood. Here, we evaluated the hypothesis that combined inhibition of Hedgehog (Hh) and Androgen receptor (AR) signaling will synergistically attenuate the growth of CRPC in vitro and in vivo. Androgen deprivation induced full-length AR protein levels in CRPC cells, but decreased its nuclear localization and transcriptional activity. On the other hand, androgen deprivation also increased a truncated form of AR (lacking ligand binding domain) that possessed transcriptional activity in CRPC cells. Androgen deprivation also promoted the expression of Hedgehog signaling components in castration-resistant prostate cancer cells, xenograft tumors, and the prostate glands of castrated mice. Importantly, while inhibition of either Hh or AR signaling alone was only moderately effective in blocking CRPC cell growth, combination of an Hh pathway inhibitor and a non-competitive AR inhibitor synergistically suppressed the growth of CRPC cells in vitro and in vivo. Finally, non-competitive inhibition of AR, but not competitive inhibition, was effective at limiting the activity of truncated AR leading to the inhibition of CRPC.
Implication Statement
Combined therapy using Hh inhibitors and a non-competitive AR inhibitor may limit CRPC growth.
Keywords: Castration-resistant prostate cancer (CRPC), Hedgehog, Androgen receptor, Smo inhibitor, Pyrvinium pamoate
Introduction
Prostate cancer (PCa) is the second most common cause of death from cancer in men of all ages and is the leading cancer diagnosed, approximately 1 in 6 American men will be diagnosed with prostate cancer during his lifetime (1). Estimated new cases and deaths from prostate cancer in the United States for 2012 include 241,740 new cases and 28,710 deaths (National Cancer Institute Facts and Figure, 2012, www.cancer.gov/cancertopic/types/prostate). Initial treatment for PCa involves androgen ablation therapy which causes regression of androgen-dependent tumors, however patients eventually fail this therapy and die of recurrent castration-resistant prostate cancer (CRPC) (2). Yet, the molecular mechanisms that promote the development of CRPC are not fully understood. Currently there is no effective therapy for CRPC and the median survival after the development of CRPC is 20–24 months (3;4). It is known that prostate tumors can select for genetic and epigenetic changes that confer growth advantage like any other cancer cells (5). Similar processes are believed to be responsible for the development of CRPC (6;7). For example, prostate tumors that recur after androgen ablation therapy have been shown to have amplified AR gene, resulting in increased AR expression (8;9) and hypersensitivity to low levels of circulating androgens. Certain growth factors such as epidermal growth factor (EGF) have been shown to activate AR in the absence of androgen (10). Overexpression of anti-apoptotic protein Bcl-2 is seen in CRPC (11). All of the above mechanisms lead to the development of CRPC under androgen depleted condition.
AR protein is organized into an N-terminal domain that comprises of the transactivation function, DNA binding domain (DBD) and a hinge region connected to the C-terminal domain or ligand binding domain (LBD) (12). AR is a member of a larger family of nuclear receptors that is activated by the binding of androgenic hormones testosterone or dihydrotestosterone (13;14). AR upon ligand binding in the cytosol translocates to the nucleus and acts as a DNA binding transcription factor where it interacts with other factors upon binding to DNA and controls the transcription of androgen-regulated genes, thereby stimulating development and maintenance of male sexual phenotype (13;15–17). Prostate cancer is androgen dependent (18). Because AR activity is dependent on ligand binding, its activity can be blocked by competitive inhibitors such as bicalutamide. On the other hand, Dehm and Tindall demonstrated the expression of truncated AR (lacking LBD) in CRPC cells that were insensitive to bicalutamide treatment(19). Hence, a non-competitive inhibitor of AR is required to inhibit truncated AR activity in CRPC cells. Here we report the use of pyrvinium pamoate, which was shown to be a non-competitive inhibition of AR activity (20), to inhibit AR activity in CRPC cells.
Recent studies have demonstrated that hedgehog (Hh) signaling and other developmentally important signaling pathways are upregulated in cancer stem-like cells leading to their proliferation, invasion, metastasis and development of resistance to drug-induced apoptosis (21;22). Two studies showed that Hh signaling activity is increased in CRPC cells as compared to androgen dependent prostate cancer (ADPC) cells (23;24) suggesting that Hh signaling might contribute to the development and maintenance of CRPC. Mammalian Hh ligands are secreted lipid-modified proteins including sonic (Shh), Indian (Ihh), and Desert (Dhh) hedgehog. Shh is most ubiquitously expressed in mammals (25). They bind their cell surface receptor called Patched. Internalization of Patched after ligand binding leads to derepression and activation of Smoothened (Smo) signaling mediator, which induces activation and nuclear translocation of the Gli family members of transcription factors (21). Many of Gli target gene products are known to promote cell proliferation, self-renewal, cell survival, and epithelial-mesenchymal transition, which are traits of embryonic and adult stem cells as well as carcinoma cells. Aberrant Hh signaling in various carcinomas including prostate cancer have led to the development of several Smo inhibitors, which are currently in clinical trials with some promising outcome (26;27).
Given the important roles played by truncated AR and Hh signaling pathway in CRPC, in the current study, we further characterized their expression and function in various in vitro and in vivo models and tested our hypothesis that the combination of inhibition of both Hh and AR pathway may generate a synergistic blockade of CRPC progression. Our results show that a combined inhibition of Hh and AR signaling pathways using Smo inhibitors and a non-competitive AR inhibitor may be a novel effective therapeutic strategy to inhibit CRPC development and progression.
Materials and Methods
Chemicals
Smo inhibitor LDE225 was provided by Novartis through a material transfer agreement, Other Smo inhibitors Cyclopamine (LC Laboratories, MA) and GDC0449 (LC Laboratories, MA) were purchased. The non-competitive inhibitor of AR (pyrvinium pamoate) and the competitive inhibitor of AR (biclutamide [Casodex]) were purchased from Sigma, MO.
Cell Cultures
LNCaP and 22Rv-1 cells were originally from the American Type Culture Collection (ATCC, VA). The culture was maintained in McCoy’s 5A medium supplemented with pyruvate, L-serine, L-arginine, L-glutamine, 100X nonessential amino acids for MEM, MEM amino acids without L-glutamine, MEM vitamins, penicillin, streptomycin, gentamycin, sodium bicarbonate and 10% fetal bovine serum (FBS). Androgen independent LNCaP and 22Rv-1 cells, which were called LNCaP AI and 22Rv-1 AI, were cultured in the above mentioned McCoy’s 5A medium with 10% charcoal-stripped FBS instead of regular 10% FBS. Benign prostatic hyperplasia (BPH) epithelial cell line BPH-1 was obtained from ATCC and maintained in RPMI-1640 medium supplemented with 10% FBS, penicillin and streptomycin.
Generation of GFP- and luciferase-expressing cell lines
22Rv-1 AI cells were stably transfected with the enhanced green fluorescent protein (GFP) in the retroviral vector, pLXSN (Clontech Laboratories, Inc. CA), to aid in the identification of these cells in animal tissues with green fluorescence imaging. For bioluminescence imaging, the cells were transduced with pLV411G effLuc-flag-IRES-hrGFP (Luc-GFP), a lentiviral expression vector, kindly provided by Dr. Brian Rabinovich at MD Anderson Cancer Center.
Transfections
For transient transfection assays, 5 × 105 cells/well were seeded into six-well plates, incubated for 24h, then transfected with 1μg luciferase reporter plasmid (PSA-Luc or Gli-Luc) and 0.2μg of β-galactosidase expression plasmid using Lipofectamine 2000 (Invitrogen, NY) in a serum-free medium following the manufacturer’s protocol. For stable transfection assay, Human AR cDNA was cloned in the pSDM101 lentiviral vector containing a GFP expression cassette. The empty pSDM101 and AR-pSDM101 vectors were transfected into HEK293 packaging cells, which helps in the packaging of the virus. Viral supernatant were harvested for infection into target cells. Almost all the cells expressed GFP indicating stable transfection of cells.
Animal experiments
Four to five-week-old male athymic nude mice (Harlan Sprague-Dawley, Inc. IN) were used for this study. Animals were maintained under the care and supervision of the Laboratory Animal Research facility at the University of Texas Health Science Center, San Antonio, Texas. The animal protocol was approved and monitored by the Institutional Animal Care and Use Committee.
Orthotopic and Subcutaneous injections
Briefly, 22Rv-1 AI/Luc-GFP cells were harvested from subconfluent, exponentially growing cultures. The cells were inoculated into the dorso-lateral prostate or dorsal lower back of anesthetized male nude mice for orthotopic or subcutaneous injections respectively, with a 27-gauge needle attached to a 1-ml syringe. Each mouse was injected with 1×105 cells in 0.01 ml of PBS for orthotopic injection or 0.05 ml of RPMI medium containing cells mixed with 0.05 ml of Matrigel for subcutaneous injections. Development of orthotopic tumors was monitored at regular intervals with whole body bioluminescence imaging for the detection of Luc-expressing tumor cells as described below. For subcutaneous tumors, the tumor sizes were measured with a caliper in two dimensions. Tumor volumes (V) were calculated with the equation V = (L × W2) × 0.5, where L is length and W is width.
Bioluminescence imaging analysis
Bioluminescence imaging analysis was used to monitor orthotopic tumor growth in mice for ranking and grouping the mice according to their tumor burden for different drug treatments. Mice were anesthetized and D-luciferin (Xenogen, Alameda, CA) was injected i.p at 75 mg/kg in PBS. Xenogen IVIS-Spectrum Imaging system was used to acquire bioluminescence images at 10 min after injection. Acquisition time was set at 60 seconds at the beginning and reduced later on in accordance with signal strength to avoid saturation. Analysis was performed using LivingImage software (Xenogen) by measurement of photon flux (measured in photons/s/cm2/steradian) with a region of interest (ROI) drawn around the bioluminescence signal to be measured. Orthotopic tumor burden was based on photon flux in the ROI around the major bioluminescence signal in the prostate.
Cell proliferation assay
Cells were plated in a 96-well plate at 1,000 cells/well in triplicates. After treatment with LDE225, GDC0449, Cyclopamine, Casodex, and PP individually or in combination for 5 days, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma, MO) solution (2mg/ml) was added and incubated for 2 hours in a tissue incubator. The blue colored formazon product was dissolved in DMSO and quantified at 595 nm wavelength on a Biotek plate reader.
Western Blot Analysis
Cells were harvested and lysed in Laemmli buffer with a cocktail of protease inhibitors. The total protein concentrations were quantified by the BCA protein assay (Thermo Scientific, Rockford, IL). An equal amount of total protein was resolved by SDS-PAGE, transferred to a nitrocellulose membrane under constant voltage and blocked with 5% non-fat dried milk in TBST (10mM Tris pH 7.5, 150mM NaCl and 0.05% Tween-20) followed by washing with TBST. Primary antibodies and secondary antibodies were diluted in TBST and applied with a washing step in between. Proteins were detected using the Amersham ECL Western blotting detection kit (GE Healthcare, Piscataway, NJ, USA). Antibody to AR was from Santa Cruz, CA, to Actin from Sigma, MO and to GAPDH from CalBioChem, MA.
Soft Agar Colony Formation Assay
Cells in 1 ml of 0.4% low melting agarose (Invitrogen, NY) with culture medium were plated at 3,000 cells/well on top of existing 0.8% agarose in 6-well plates. The wells were covered with 1 ml of culture medium containing various treatments and incubated at 37 °C in a 5% CO2 incubator for the indicated number of days. Visualized colonies with diameter >30μm were counted after staining with p-iodonitrotetrazolium violet (Sigma, MO) overnight.
Real-time PCR
Real-time PCR was performed using Brilliant SYBR Green QPCR Master Mix (Stratagene, La Jolla, CA) in the ABI 7900HT (Foster City, CA) according to the manufacturer’s instruction. The relative transcript level for each transcript was calculated according to the equation 2−ctX/2−ctN, where X is the target gene and N is β-actin.
Immunocytochemical Assay of Cell Lines
LNCaP AD and AI cell lines, grown as monolayers in eight-well chamber slides (Nunc International, Rochester, NY), were fixed by incubation for 20 min at RT with 2% formaldehyde in PHEM buffer (PIPES, HEPES, EGTA, MgSO4 pH 7.0). Fixed cells were permeabilized in 0.1% Triton-X 100 in PHEM buffer for 1h. Cells were then incubated for l h at RT with 1% BSA in PHEM buffer to block nonspecific binding sites. After blocking, cells were incubated with Rabbit α-AR (Androgen receptor) primary antibody (Santa Cruz, CA) overnight at 4°C. Following primary antibody incubation, the cells were washed in 1X PHEM buffer and incubated with anti-rabbit secondary antibody conjugated with Alexafluor 568 fluorochrome for 1 hr at room temperature. Control incubations included a primary antibody alone and secondary antibody alone. The slides were mounted with vectashield mounting media (Vector, CA) and viewed under an Olympus Fluoview FV1000 confocal fluorescence microscope.
Small interfering RNA (siRNA)
LNCaP AD cells were transfected with AR siRNA using Lipofectamine 2000 (Invitrogen, NY). AR was silenced using siRNA pool SMARTpool, siAR #1 purchased from Dharmacon (Catalogue number M003400-00, Waltham, MA).
In vitro luciferase assay
Cells were seeded in triplicates in a 12-well plate at a density of 1.8×105 cells/well. When cultures were about 80% confluent, they were co-transfected with 0.2 μg of β-galactosidase expression plasmid and 1μg of PSA or Gli responsive promoter-luciferase construct using Lipofectamine 2000 (Invitrogen, NY) in a serum-free medium following the manufacturer’s protocol. After 5 h, the medium was replaced with the serum containing medium. After overnight incubation, the cells were lysed in a buffer (100 mM K2HPO4, 1 mM DTT, and 1% Triton X-100) and the luciferase activity in the cell lysate was measured as previously described (28). Luciferase activity was normalized for transfection efficiency with β-galactosidase activity.
Statistical analysis
All statistical analyses were performed using the two-tailed Student’s t-tests or One-way analysis of variance followed by Tukey-Kramer post-hoc test. Only values with a P-value < 0.05 was considered as statistically significant. Error bars represent mean ± SEM.
Results
Androgen deprivation increases androgen receptor expression but not androgen receptor activity
To determine the role of androgen receptor (AR) in prostate cancer (PCa) progression under androgen deprivation condition, we assessed the levels of AR expression under in vitro and in vivo conditions in androgen dependent and androgen independent condition. Human PCa cell lines LNCaP and 22Rv-1 were cultured in McCoy’s 5A medium containing 10% regular FBS and designated as androgen-dependent (AD) as the medium contains about 0.1 nM testosterone from FBS. The two cell lines were adapted to grow in McCoy’s 5A medium containing 10% charcoal-stripped FBS for more than 6 months and designated as androgen-independent (AI) as the medium contains less than 3.5 pM testosterone. RNA isolation from AD and AI cells followed by quantitative PCR analysis showed an increase in AR expression in AI cells relative to AD cells (Fig. 1A). Also there was a relative increase in AR protein level in AI cells compared to AD cells as assessed by Western blotting (Fig. 1B). To further confirm these results in an in vivo model, we generated CWR22 human prostate cancer xenograft tumors subcutaneously in athymic male nude mice that were implanted with slow-releasing testosterone pellet for 90 days. After the tumors reached an approximate 800 mm3, the mice were castrated and the testosterone pellet removed to provide an androgen independent condition for the xenograft tumors. Mice 2 and 5 were euthanized and tumors isolated after their tumors reached 800 mm3 in the AD condition, whereas mice 12 and 14 were castrated and testosterone pellet removed after their tumors reached 800 mm3 and these mice were maintained in the AI condition for 14 days before euthanization and isolation of their tumors. RNA isolated from mice 2, 5, 12 and 14 showed a relative increase in AR expression in tumors from mice 12 and 14 (AI condition) compared to tumors from mice 2 and 5 (AD condition) (Fig. 1C). Also there was a relative increase in the level of AR expression in mouse prostate gland after castration compared to mouse prostate gland before castration (Fig. 1D). We also found that the expression of AR was relatively increased in PCa tissues from patients treated with hormone deprivation therapy as compared to PCa tissues from patients without hormone deprivation therapy through an analysis of gene microarray data in Oncomine database from a published study (29) (Fig. 1E).
Figure 1. Androgen deprivation increases AR expression both in vitro and in vivo.
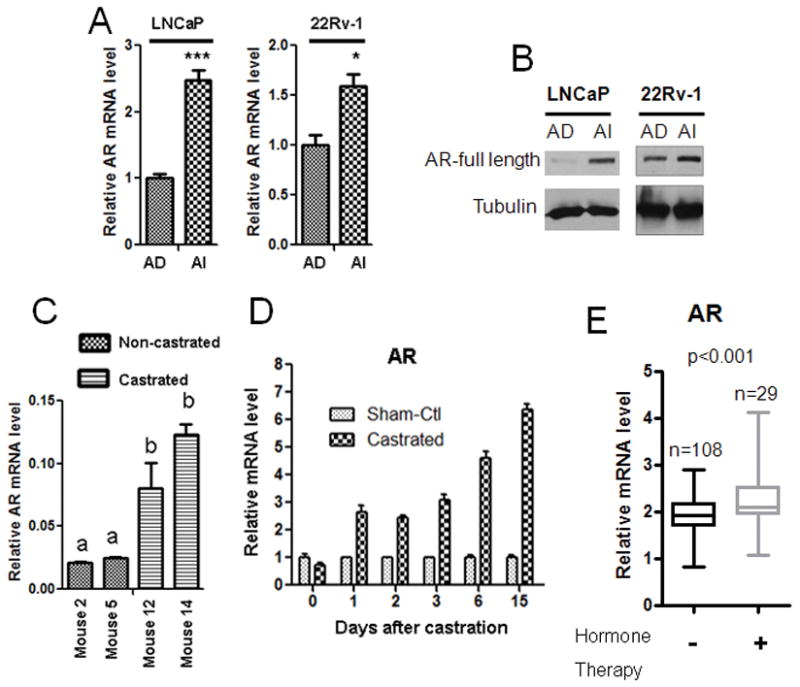
A. Androgen independent (AI) LNCaP and 22Rv-1 cells show higher expression of AR mRNA relative to Androgen dependent (AD) cells. B. LNCaP and 22Rv-1 AI cells show higher expression of AR protein levels relative to their respective AD cells. C. Castrated mice (mouse 12 and 14) show an increase in AR expression in the CWR22 xenografts relative to those of the non-castrated mice (mouse 2 and 5). D. AR mRNA levels was relatively increased in wild-type mouse prostate after castration. Each bar is presented as mean ± SEM from triplicate measurements. *Indicates statistically significant difference between the two groups with a student’s t-test at P<0.05, ***P<0.0001. The bars bearing different letters are significantly different (p<0.05) by ANOVA. E. Hormone deprivation therapy significantly increased mRNA levels of AR. The data were derived from patient samples without or with hormone deprivation therapy (data provided by Dr. Taylor, prostate oncomine database [citation 29]). The bars and box represent 10–90 or 25–75 percentile of the data, respectively, from each group.
We next investigated whether relatively increased AR expression in AI condition compared to AD condition correlates with its activity. Surprisingly, we found a relative decrease in AR activity under AI condition in comparison to AD condition (Fig 2). We assessed for the AR activity by a PSA promoter luciferase reporter assay, in which an AR-responsive promoter sequence of PSA gene with a firefly luciferase (PSA-Luc) reporter plasmid was co-transfected with a β-gal expression plasmid into LNCaP and 22Rv-1 (AD and AI) cells. The relative Luc unit (RLU) is the Luc activity normalized by β-gal activity in the same cell lysate. There was a relative decrease in PSA expression in AI cells as compared to AD cells (Fig. 2A). Also the relative PSA expression was lower in AI cells in comparison to AD cells as assessed by quantitative real time PCR (Fig. 2B). To confirm these results in an in vivo model, we used the previously described CWR22 xenograft mouse model and found that upon castration the PSA mRNA level was relatively lower in comparison to non-castrated or androgen dependent tumors (Fig. 2C). Furthermore, immunocytochemical staining showed a more uniform nuclear localization of AR in AD cells than in AI cells (Fig. 2D). Consistently, immunohistochemical staining also showed that castration led to an increased cytoplasmic localization of AR castrated mouse prostate tissue when compared to the prostate gland tissueof non-castrated mice (Fig. 2E). These data clearly demonstrate that although AR expression is increased under AI condition, the activity of AR is relatively lowered in the AI condition in both in vitro and in vivo studies due to its reduced nuclear localization. Our results demonstrating a decrease in AR activity with increase in AR levels are consistent with a recently published report (30), in which active AR was shown to negatively regulate AR expression.
Figure 2. Androgen deprivation decreases activity and nuclear localization of AR.
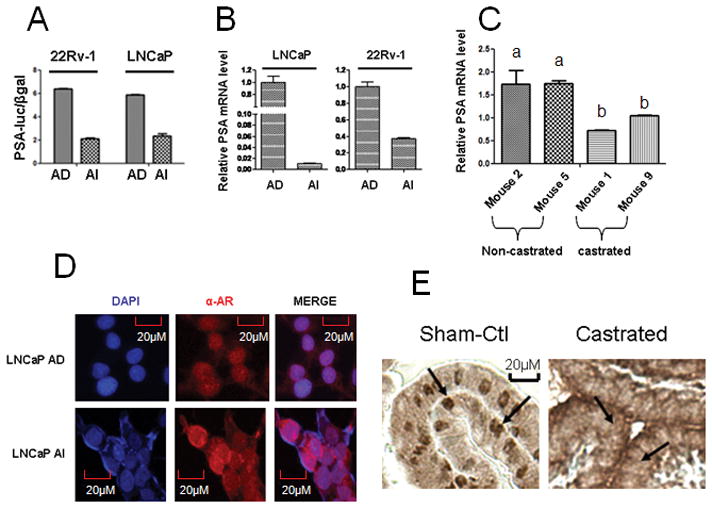
A. AD cells show an increase in AR activity relative to AI cells as assessed by an AR-responsive PSA promoter-luciferase (PSA-Luc) reporter plasmid co-transfected with a β-gal plasmid into the AD and AI cells, the relative Luc unit (RLU) is the Luc activity normalized by β-gal activity in the same cell lysate. B. AD cells show an increase in PSA mRNA expression relative to AI cells. C. Castrated mice (mouse 1 and 9) show a decrease in PSA mRNA expression level relative to non-castrated mice (mouse 2 and 5). Each bar is presented as mean ± SEM from triplicate measurements, and bars bearing different letters are significantly different (p<0.05) by ANOVA. D. DAPI (Blue) stains the nucleus, α-AR (Red - alexafluor 568 fluroescent dye) stains AR protein. Immunocytochemical staining for AR shows an increased AR localization in the cytosol of AI cells compared to AD cells. E. Immunohistochemistry staining for AR shows a decreased AR localization in the nucleus of wild-type mouse prostate cells after castration (arrows point to the nucleus of prostate epithelial cells).
Increased Truncated AR (AR ΔLBD) expression in AI cells
Earlier reports have demonstrated the presence of COOH-terminally truncated AR protein isoform lacking the AR ligand binding domain (LBD) and denoted as AR 3LBD in 22Rv-1 prostate cancer cells (31). Later studies demonstrated the presence of AR ΔLBD in benign and cancerous prostate tissue samples by Western blotting (32). Together, these earlier observations suggest the presence of AR isoform lacking LBD and could be responsible for hormone therapy resistance. Here we confirmed that the level of 75–80kDa AR ΔLBD expression was higher in LNCaP and 22Rv-1 AI cells as compared to their respective AD cells by Western blotting (Fig. 3A). To determine whether the AR ΔLBD is functional in AI cells, we treated 22Rv-1 AD and AI cells with non-competitive AR antagonist, pyrvinium pamoate (PP), which does not interact with AR LBD (20) or a competitive AR antagonist, Casodex, which competes with androgen for interacting with AR LBD (33) and assessed for the transcriptional activity of AR. 22Rv-1 AI cells showed a decrease in AR activity as assessed by MMTV-Luc assay when treated with PP but were insensitive to Casodex treatment (Fig. 3B). However both PP and Casodex inhibited AR activity in 22Rv-1 AD cells (Fig. 3B). Furthermore, we also utilized MDA-Kb2 cell, a breast cancer cell line stably transfected with MMTV-Luc reporter, for stable transfection with AR ΔLBD using a lentiviral vector. We confirmed the expression of AR ΔLBD in stably transfected MDA-Kb2 cells by Western blotting (Fig. 3C). Interestingly, overexpression of AR ΔLBD increased level of the endogenous full length AR, which was also observed in AR ΔLBD-transfected LNCaP AD cells (Fig. S1) indicating that overexpression of AR ΔLBD somehow increased the stability of the full length AR. To determine the effect of PP and casodex on the transcriptional activity of AR ΔLBD in MDA-Kb2 cells, we treated the control and AR ΔLBD-expressing MDA-Kb2 cells with PP or casodex. Our results indicated that PP was much more effective than Casodex in inhibiting the MMTV-driven luciferase activity in AR ΔLBD-expressing cell than in the control cell (Fig. 3D). Together, these results suggest that PP inhibits the activity of both full length AR and AR ΔLBD whereas Casodex was effective only on full length AR and did not inhibit the AR ΔLBD activity in 22Rv-1 AI cells and the transfected AR ΔLBD in MDA-Kb2 cells. Our results highlight the importance of AR ΔLBD in the development of resistance to the treatment with competitive AR antagonists.
Figure 3. PP inhibits full length and truncated AR activity.
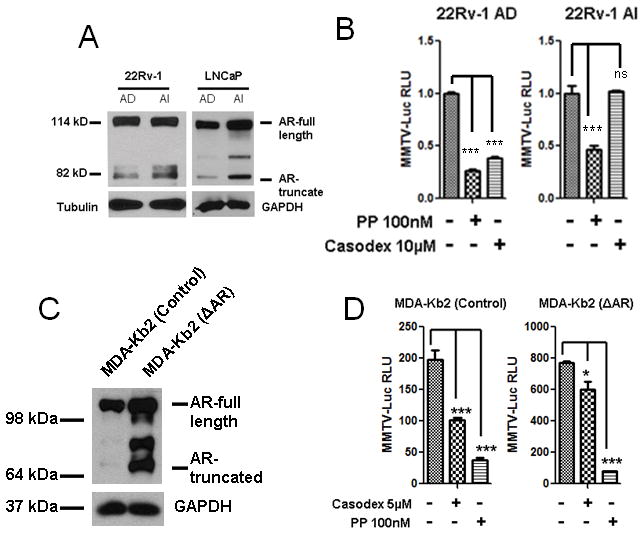
A. Truncated AR (AR ΔLBD) expression is higher in LNCaP and 22Rv-1 AI cells as compared to AD cells as assessed by western blotting. B. PP inhibited endogenous AR activity in both 22Rv-1 AD and AI cells, Casodex inhibited AR activity only in AD cells. C. AR ΔLBD was stably transfected and overexpressed in MDA-Kb2 cells (Human breast cancer cell line). D. MDA-Kb2 cells were stably transfected with the androgen responsive MMTV promoter luciferase gene, and these cells report the activity of androgen receptor. Over-expression of AR ΔLBD in these MDA-Kb2 cells increased luciferase activity as compared to the control cells. PP treatment showed significant inhibition of the over-expressed AR ΔLBD in these MDA-Kb2 cells whereas casodex was less effective in inhibiting the over-expressed AR ΔLBD activity. Each bar is presented as mean ± SEM from triplicate measurements. *Indicates statistically significant difference between the two groups with a student’s t-test at P<0.05, ***P<0.0001, ns represents non-significant.
Activation of Hedgehog (Hh) pathway in CRPC cells and prostate of castrated mice
Recent studies have demonstrated that Hh signaling activity is significantly increased in CRPC cells compared to ADPC cells (23;24). Here we assessed for the expression of Hh signaling components both in vitro and in vivo and also quantified the Hh activity in AD versus AI cells (Fig. 4). Quantitative RT-PCR assays showed a relative increase in Hh pathway components, Gli2, Smo and Shh in LNCaP and 22Rv-1 AI cells when compared to their AD cells (Fig. 4A). We assessed for Gli transcriptional activity using a Gli luciferase assay in which a Gli-responsive promoter luciferase (Gli-Luc) reporter plasmid was co-transfected with a β-gal expression plasmid into LNCaP and 22Rv-1 AD and AI cells. The normalized luciferase activitymeasured in all four cell lines showed a relative increase in Gli activity in AI cells as compared to AD cells (Fig. 4B) suggesting an increased Hh signaling activity in AI cells in comparison to AD cells. To confirm these in vitro results in an in vivo system, we assessed the expression of Hh pathway components, Patched (Ptch), Gli2, and Smo using quantitative PCR from RNA isolated from CWR22 xenograft tumors in castrated and non-castrated mice. Our results showed an increase in Ptch, Gli2 and Smo expression incastrated CWR22 xenograft tumors compared to non-castrated tumors (Fig. 4C). We also observed a relative increase in the expression of Hh pathway components, Ptch, Gli1 and Gli2 mRNA isolated from mouse prostate after castration as compared to prostate from non-castrated mice (Fig. 4D). Furthermore, the expression of Hh pathway components was increased in PCa tissues from patients treated with hormone deprivation therapy through an analysis of gene microarray data in Oncomine from a recent study (29) (Fig. 4E). Together our results demonstrate that Hh pathway components and signaling activity are increased upon androgen deprivation in both in vitro and in vivo conditions suggesting that in addition to truncation mutation of AR, Hh pathway activation may be another mechanism supporting PCa survival and growth under androgen deprivation condition.
Figure 4. Androgen deprivation increases Hedgehog signaling components both in vitro and in vivo.

A. AI cells show an increased mRNA expression of Hedgehog signaling components relative to AD cells. B. A Gli-responsive promoter-luciferase (Gli-Luc) reporter plasmid was co-transfected with a β-gal expression plasmid into the depicted cells, the relative Luc unit (RLU) is the Luc activity normalized by β-gal activity in the same cell lysate. AI cells exhibit an increased Hh signaling activity compared to AD cells. C. Castrated mice (mouse 12 and 14) show an increased mRNA levels for Hedgehog components relative to non-castrated mice (mouse 2 and 5). D. Increased mRNA expression of Hh components are observed in wild type mice prostate after castration. All data are mean±SEM from three independent measurements. Bars bearing different letters are significantly different (p<0.05) by ANOVA. E. Hormone deprivation therapy significantly increased mRNA levels of Shh, Smo, and Gli1. The data were derived from 153 and 20 patient samples without or with hormone deprivation therapy (including neoadjanant hormone therapy) respectively (data provided by Taylor and co-workers (Citation 29 to Oncomine). The bars and box represent 10–90 or 25–75 percentile of the data, respectively, from each group. The mean of each data set is shown as “+”. The P values were obtained with two-tailed Student’s t-tests. The wider distribution of the data of the hormone therapy group is likely due to the small sample size and the variation of hormone therapy duration, which was not provided in the report.
Hh signaling is negatively regulated by androgen dependent AR activity
The fact that Hh signaling activity was up-regulated under androgen deprivation condition indicated that Hh signaling activity is suppressed by AR activity. This was demonstrated in a published study by Sirab et al (34). We took a different approach and showed that overexpression of AR in the untransformed AR negative prostate epithelial cell line BPH-1 (Fig. 5A) resulted in a decrease in expression of Hh pathway components, Gli1, Gli2 and Smo (Fig. 5B). On the other hand, AR siRNA mediated silencing of AR in LNCaP AD cells (Fig. 5C) increased the expression of Hh pathway components, Gli1, Gli2 and Smo (Fig. 5D) and Gli transcriptional activity as assessed by Gli-responsive promoter luciferase assay (Fig. 5E). Together, these results suggest that androgen mediated AR activity suppresses Hh signaling.
Figure 5. AR negatively regulates Hh signaling.
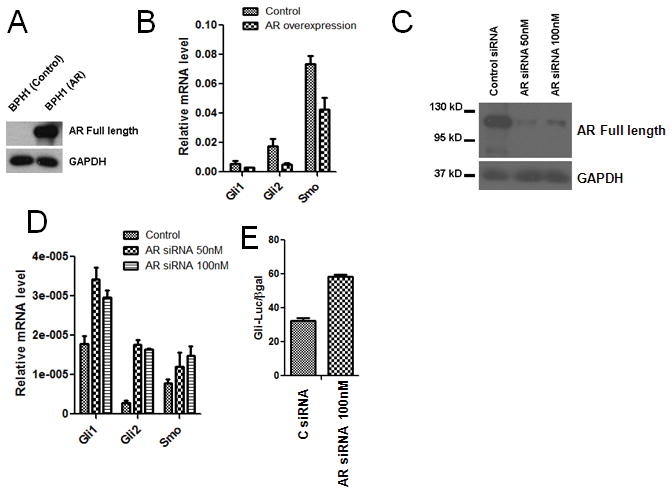
A. Overexpression of AR in BPH1 cells (AR null cells) assessed by Western blot. B. AR overexpression downregulates Hh signaling components Gli1, Gli2 and Smo in BPH1 cells. C. siRNA mediated knockdown of endogenous AR in LNCaP AD cells assessed by Western blot. D. AR knockdown LNCaP AD cells shows increased expression of Hh signaling components Gli1, Gli2 and Smo. E. AR knockdown LNCaP AD cells shows increased Gli-Luciferase activity. Each bar is presented as mean ± SEM from triplicate measurements.
Combined inhibition of Hh and AR signaling was synergistic in inhibiting the growth of PCa cells in vitro
Our results suggest that both Hh and AR signaling are involved in the maintenance of CRPC cell viability and proliferation. Because increased truncated AR expression in AI condition facilitates AR signaling in AI condition, we hypothesized that inhibition of both Hh and AR pathways may synergistically inhibit the growth of CRPC cells. We used Smo inhibitors Cyclopamine and LDE225 in combination with the AR inhibitor PP in a time-course study to assess the inhibition of growth of LNCaP AI cells (Fig. 6A). Our results showed that inhibiting with the Smo inhibitor or AR inhibitor alone was only marginally effective or almost had no inhibitory effects, but the combined inhibition was statistically significant in inhibiting the growth of LNCaP AI cells. Another Smo inhibitor GDC0449 was tested alone and in combination with PP on LNCaP AI cells. Similar results were seen with the combination being very effective in inhibiting the AI cell growth compared to individual treatments (Fig. 6B). To confirm the inhibitory effects on AI cells, we also tested a different AI cell line, 22Rv-1 cells with LDE225 and PP (Fig. 6C). Similar result with a combination being more effective in inhibiting AI cell proliferation was observed. We also tested LDE225 and PP individually and in combination for their effect on anchorage-independent growth of LNCaP AI cells in soft agar and found that LDE225 and PP individually inhibited the anchorage-independent growth of AI cells moderately, whereas the combination was significantly more effective (Fig. 6D). However, we did not see the synergistic inhibition of AI cell growth with Casodex + Cyclopamine and Casodex + LDE225 (Fig. S2), suggesting a significant role of the truncated AR in supporting the growth of the AI cells. We used 5–10 μM of LDE225 and cyclopamine in our MTT assays because this concentration range was found to dose-dependently inhibit Gli transcription activity in the AI cells as shown in the Fig. S3 for LDE225 and Fig. S4 for cyclopamine, yet they showed marginal effect on cell growth (Fig. 6). Similarly, PP concentration of 50 nM or below was used in our MTT assays as it could inhibit AR transcriptional activity in AI cells (Fig. S5), but with limited growth inhibitory activity as shown in Fig. 6. The choice of these drug concentrations led to the demonstration of the synergistic growth inhibition in the AI cells when they were combined.
Figure 6. Combined inhibition of Hh and AR signaling is synergistic in inhibiting the growth of PCa cells in vitro.
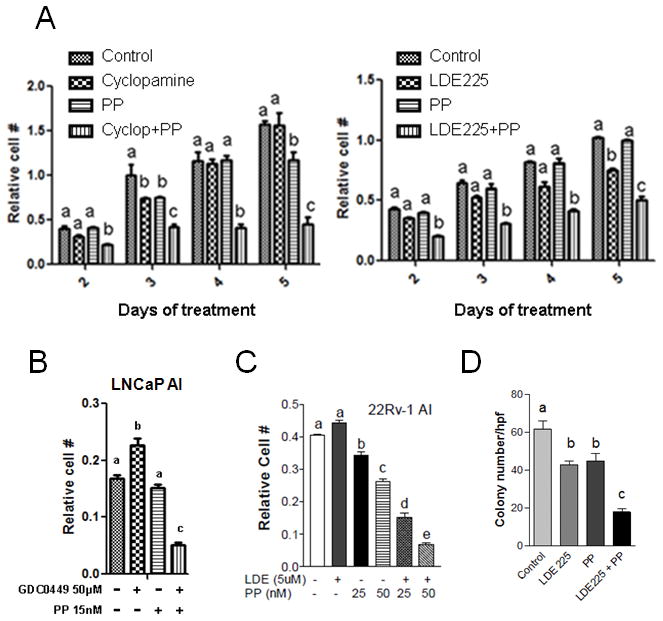
A. Cyclopamine (10μM) or LDE225 (5μM) [Hh inhibitors] in combination with 20nM Pyrvinium Pamoate (PP) (AR inhibitor) synergistically inhibits AI prostate cancer cell proliferation effectively as compared to individual treatments. B. GDC0449 (Hh inhibitor) and PP in combination is effective in preventing AI prostate cancer cell proliferation. C. LDE225 (Hh inhibitor) and PP synergistically inhibit the growth of 22Rv-1 AI cells. D. LDE225 (5μM) and PP (5nM) in combination effectively inhibited anchorage independent growth of LNCaP AI cells. Data is presented as mean of colony number from 6 high power fields in 3 wells for each group. All data are mean±SEM from three independent measurements, bars bearing different letters are significantly different (p<0.05) by ANOVA.
Combined inhibition of Hh and AR signaling was effective in inhibiting CRPC tumor growth in mice
To determine if a combinatorial approach can further enhance the efficacy of inhibiting CRPC-mediated prostate tumor progression in vivo, 22Rv-1 AI cells were orthotopically inoculated into athymic male nude mice. Upon detection of tumors with whole mouse bioluminescence imaging, they were castrated and divided into 4 groups (8 mice in each group) with similar tumor burden and treated everyday for 24 dayswith placebo, cyclopamine at 5 mg/kg/day, PP at 0.2 mg/kg/day, or cyclopamine+PP through intraperitoneal injection. After 24 days of treatment, the mice were euthanized and prostate isolated for measuring tumor weight from each group. Interestingly, the tumor incidence was reduced from 87.5% in the control group to 50% in the cyclopamine group, 55.6% in the PP group, and 33.3% in the combination group. Furthermore, prostate tumors isolated from mice treated with Cyclopamine+PP showed a statistically significant decrease in tumor weight in comparison to the control group (Fig. 7A). To accurately track the growth of tumors as a function of cyclopamine and/or PP treatment, we also inoculated the 22Rv-1 AI cells subcutaneously so that we could measure the tumor volume with a caliper at a regular interval. Subcutaneous tumors started to arise in 2 weeks. The mice were ranked according to their tumor burden and divided into four groups with matched tumor burden. All the mice were castrated following regrouping. Four days post castration, vehicle, cyclopamine at 5 mg/kg/day, PP 0.2 at mg/kg/day, orCyclopamine + PP was given daily through intraperitoneal injection for 11 days and the tumor volume was measured intermittently for each group (Fig. 7B). Here we present the tumor volume data in 2 ways, Figures 7B and 7C. Figure 7B shows tumor growth curve in each mouse with two tumors. We found that the treatment, especially the combination treatment, was effective in inhibiting tumor growth when the tumor volume was equal to or less than 50 mm3 at the start of the treatment. As such, Figure 7C was prepared to show the growth of the tumors in two mice with the lowest tumor burden at the start of the treatment in each group to illustrate the efficacy of each treatment. It is clear that PP alone showed no inhibitory effect while its combination with cyclopamine basically stopped tumor growth. These in vivo results are consistent with our in vitro data (Fig. 6) showing the combined inhibition of Hh and AR signaling using cyclopamine and PP was more effective in inhibiting the growth of AI prostate cancer cells than each of the agent alone.
Figure 7. Combined inhibition of Hh and AR signaling is effective in inhibiting CRPC tumor growth in mice.
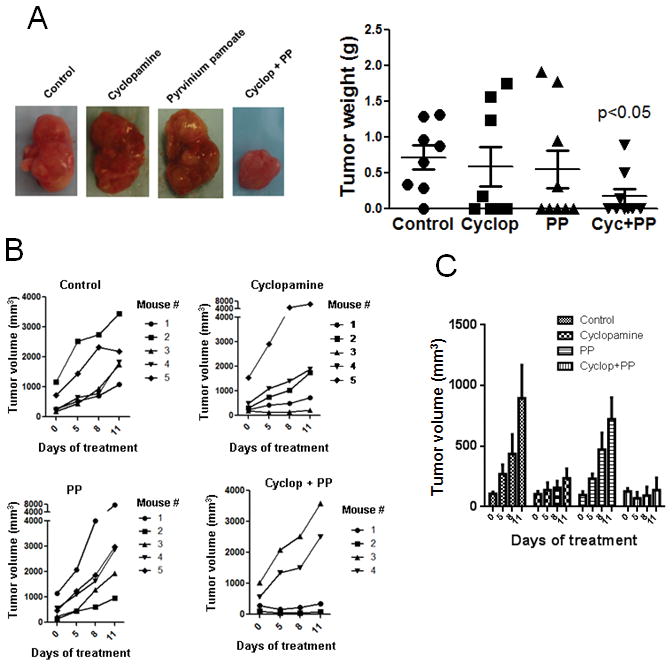
A. 22Rv-1 AI cells were orthotopically inoculated into castrated athymic male nude mice and the mice were treated with cyclopamine at 5 mg/kg/day, PP at 0.2 mg/kg/day, or cyclopamine+PP through intraperitoneal injection for 24 days. Cyclopamine (Hh inhibitor) and PP (AR inhibitor) in combination was effective in inhibiting orthotopically inoculated 22Rv-1 AI (Prostate cancer cell) cell proliferation in mouse prostate as compared to individual treatments. B. 22Rv-1 AI prostate cancer cells were inoculated subcutaneously on the right lower back of male mice and the mice were treated with cyclopamine and PP at a dose of 5mg/kg/day and 0.2 mg/kg/day respectively for 11 days. Caliper measurements of tumors showed a steady increase in tumor volume in control and individual treatment groups, however the cyclopamine and PP combined treatment did not show a gradual increase in the tumor volume in 2 male mice (note: 2 male mice in cyclopamine and PP treatment group did not respond to the drugs as the initial tumor volume before treatment start date was already too big and hence were not responsive to the treatments). C. Each bar is presented as mean ± SEM from 4 subcutaneous tumors from two mice bearing the smallest tumors from each group at the beginning of the treatment. The combination treatment group did not show much increase in tumor volume in a time-course treatment with the Smo and AR inhibitors.
Discussion
Prostate cancer at its early stage is dependent on androgen for growth and survival. Despite initial response rates of 80–90% of early stage tumor regression after androgen ablation therapy, nearly all men develop androgen-independent prostate cancer after 18–36 months (35). Majority of prostate cancer related deaths are due to the development of CRPC and metastasis to various organs. The molecular mechanisms that lead to the development of CRPC are not fully understood and currently there is no effective therapy for it. Although they are refractory to hormone deprivation, CRPC cells are known to express AR and rely on AR activity for survival and proliferation (36). AR activity is maintained under hormone deprivation condition via various mechanisms in CRPC, which include amplification of the AR gene (37;38), missense mutation in AR gene at T877A making it responsive to anti-androgens (39), and ligand-independent activation of AR by various growth factors such as insulin-like growth factor-1 (IGF-1) (10). In our studies, we observed an increase of AR level in CRPC cells and prostate gland of castrated mice when compared to ADPC cells or non-castrated mice. Interestingly, we observed a decreased transcriptional activity of AR in CRPC cells, presumably due to low androgen in the environment and cytoplasmic localization of inactive AR. Our result that a relative increase in AR level in CRPC cells compared to ADPC cells is consistent with a previous finding that increased androgens represses AR expression (30). On the other hand, we observed an increased expression of truncated AR proteins (AR ΔLBD) in both LNCaP and 22Rv-1 AI cells. Previously the AR ΔLBD proteins were shown to be present due to increased proteolytic deletion of the LBD under AI condition in LNCaP cells (40), whereas they are due to insertion mutation of AR gene and differential RNA splicing of AR transcript in 22Rv-1 cells (41). Previous studies have demonstrated increased expression levels of AR ΔLBD and its ability to confer resistance to anti-androgens in androgen refractory PCa cells (42). As expected, AR ΔLBD was found to be insensitive to bicalutamide (Casodex), a competitive AR inhibitor, in our study. However, its transcriptional activity was found to be inhibited by pyrvinium pamoate (PP), a non-competitive AR inhibitor, in CRPC cells suggesting that it is functionally active and may contribute to the development of CRPC as previously suggested (42).
Another mechanism that contributes to the development of CRPC was the activation of Hh signaling pathway upon hormone deprivation. Previous studies have shown that Hh signaling pathway is activated during prostate tumorigenesis and progression (43;44). More recent studies showed that androgen deprivation upregulated the expression of Hedgehog signaling components and consequently Hh signaling activity in prostate cancer cell lines and circulating prostate tumor cells (23;24). Our study not only confirmed the published findings in the prostate cancer cells, but also demonstrated upregulation of Hh pathway components in vivo in the human CWR22 prostate cancer xenograft tumors upon surgical androgen deprivation. Our study also confirmed a previous report demonstrating increased Hh components in the prostate gland of castrated mice (45). Consistent with these findings, genetic approaches with ectopic expression or knockdown of AR with siRNA also showed that AR level is inversely related with Hh pathway components and Gli transcriptional activity suggesting a direct suppression of Hh signaling activity by AR. As a result, androgen deprivation led to an enhanced autocrine Hh signaling activity, which can be dose-dependently inhibited by the treatment with Smo inhibitor, LDE225 in our study (Supplementary Fig. 3), or genetic knockdown of Smo (46).
These findings suggest that the increased Hh signaling and AR ΔLBD expression might contribute to the development and maintenance of CRPC. Thus, to inhibit the development and proliferation of CRPCcells, we used Hh and AR inhibitors independently and in combination both under in vitro and in vivo conditions. We demonstrated that the non-competitive AR inhibitor PP was effective in inhibiting the AR activity in 22Rv-1 AI cells whereas the competitive AR inhibitor casodex was ineffective even though Casodex showed inhibition of AR activity similar to PP in androgen dependent 22Rv-1 cells. Hence a non-competitive AR inhibitor is effective in inhibiting AR activity under AI condition. Significantly, while various Smo inhibitors showed little or no effect on the growth of CRPC cells in micromolar concentrations, they produced synergistic inhibition of CRPC cell growth in combination with suboptimal concentrations of PP. Furthermore, the combination of PP with cyclopamine was found more effective than either agent alone in inhibiting the growth of CRPC tumors in vivo.
In summary, our study demonstrated activation of Hh signaling in both in vitro and in vivo models upon hormone ablation. It also indicated for the first time that the growth of CRPC cells and early stage androgen independent prostate tumors could be effectively inhibited by a Smo inhibitor in combination with a non-competitive AR inhibitor. This concept remains to be validated in clinical trials.
Supplementary Material
Acknowledgments
This work was supported in part by RP120290-IIRA and RP101491 Cancer Research Training Program grants, R01CA079683 grant from NIH, and the Cancer Therapy and Research Center at the University of Texas Health Science Center at San Antonio (UTHSCSA) through the NCI Cancer Center Support Grant 2 P30 CA054174-17. We would like to acknowledge Dr. Donald Tindall at Department of Urology, Mayo Clinic College of Medicine, Rochester, MN for providing us with the AR 3LBD expressing plasmid construct. UTHSCSA’s Optical Imaging Core was used for the optical images presented in this study.
Footnotes
Conflict of interest:
The authors declare no conflict of interest.
Reference List
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Isaacs JT, Furuya Y, Berges R. The role of androgen in the regulation of programmed cell death/apoptosis in normal and malignant prostatic tissue. Semin Cancer Biol. 1994 Oct;5(5):391–400. [PubMed] [Google Scholar]
- 3.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004 Oct 7;351(15):1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 4.Shamash J, Dancey G, Barlow C, Wilson P, Ansell W, Oliver RT. Chlorambucil and lomustine (CL56) in absolute hormone refractory prostate cancer: reinduction of endocrine sensitivity an unexpected finding. Br J Cancer. 2005 Jan 17;92(1):36–40. doi: 10.1038/sj.bjc.6602263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruijter E, van de KC, Miller G, Ruiter D, Debruyne F, Schalken J. Molecular genetics and epidemiology of prostate carcinoma. Endocr Rev. 1999 Feb;20(1):22–45. doi: 10.1210/edrv.20.1.0356. [DOI] [PubMed] [Google Scholar]
- 6.Hyytinen ER, Thalmann GN, Zhau HE, Karhu R, Kallioniemi OP, Chung LW, et al. Genetic changes associated with the acquisition of androgen-independent growth, tumorigenicity and metastatic potential in a prostate cancer model. Br J Cancer. 1997;75(2):190–5. doi: 10.1038/bjc.1997.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pilat MJ, Kamradt JM, Pienta KJ. Hormone resistance in prostate cancer. Cancer Metastasis Rev. 1998;17(4):373–81. doi: 10.1023/a:1006166511344. [DOI] [PubMed] [Google Scholar]
- 8.Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997 Jan 15;57(2):314–9. [PubMed] [Google Scholar]
- 9.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995 Apr;9(4):401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 10.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994 Oct 15;54(20):5474–8. [PubMed] [Google Scholar]
- 11.Colombel M, Symmans F, Gil S, O’Toole KM, Chopin D, Benson M, et al. Detection of the apoptosis-suppressing oncoprotein bc1-2 in hormone-refractory human prostate cancers. Am J Pathol. 1993 Aug;143(2):390–400. [PMC free article] [PubMed] [Google Scholar]
- 12.Lamb DJ, Weigel NL, Marcelli M. Androgen receptors and their biology. Vitam Horm. 2001;62:199–230. doi: 10.1016/s0083-6729(01)62005-3. [DOI] [PubMed] [Google Scholar]
- 13.Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science. 1988 Apr 15;240(4850):327–30. doi: 10.1126/science.3353727. [DOI] [PubMed] [Google Scholar]
- 14.Roy AK, Lavrovsky Y, Song CS, Chen S, Jung MH, Velu NK, et al. Regulation of androgen action. Vitam Horm. 1999;55:309–52. doi: 10.1016/s0083-6729(08)60938-3. [DOI] [PubMed] [Google Scholar]
- 15.Tan JA, Joseph DR, Quarmby VE, Lubahn DB, Sar M, French FS, et al. The rat androgen receptor: primary structure, autoregulation of its messenger ribonucleic acid, and immunocytochemical localization of the receptor protein. Mol Endocrinol. 1988 Dec;2(12):1276–85. doi: 10.1210/mend-2-12-1276. [DOI] [PubMed] [Google Scholar]
- 16.Tan J, Marschke KB, Ho KC, Perry ST, Wilson EM, French FS. Response elements of the androgen-regulated C3 gene. J Biol Chem. 1992 Apr 15;267(11):7958. [PubMed] [Google Scholar]
- 17.Ho KC, Marschke KB, Tan J, Power SG, Wilson EM, French FS. A complex response element in intron 1 of the androgen-regulated 20-kDa protein gene displays cell type-dependent androgen receptor specificity. J Biol Chem. 1993 Dec 25;268(36):27226–35. [PubMed] [Google Scholar]
- 18.Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol. 2002 Feb;167(2 Pt 2):948–51. [PubMed] [Google Scholar]
- 19.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011 Oct;18(5):R183–R196. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones JO, Bolton EC, Huang Y, Feau C, Guy RK, Yamamoto KR, et al. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc Natl Acad Sci U S A. 2009 Apr 28;106(17):7233–8. doi: 10.1073/pnas.0807282106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009 Sep;9(7):873–86. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- 22.Medina V, Calvo MB, az-Prado S, Espada J. Hedgehog signalling as a target in cancer stem cells. Clin Transl Oncol. 2009 Apr;11(4):199–207. doi: 10.1007/s12094-009-0341-y. [DOI] [PubMed] [Google Scholar]
- 23.Chen M, Tanner M, Levine AC, Levina E, Ohouo P, Buttyan R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle. 2009 Jan 1;8(1):149–57. doi: 10.4161/cc.8.1.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw G, Price AM, Ktori E, Bisson I, Purkis PE, McFaul S, et al. Hedgehog signalling in androgen independent prostate cancer. Eur Urol. 2008 Dec;54(6):1333–43. doi: 10.1016/j.eururo.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 25.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008 Sep 15;22(18):2454–72. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 26.Low JA, de Sauvage FJ. Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol. 2010 Dec 20;28(36):5321–6. doi: 10.1200/JCO.2010.27.9943. [DOI] [PubMed] [Google Scholar]
- 27.Merchant AA, Matsui W. Targeting Hedgehog--a cancer stem cell pathway. Clin Cancer Res. 2010 Jun 15;16(12):3130–40. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM, et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006 Jul 1;66(13):6714–21. doi: 10.1158/0008-5472.CAN-05-3565. [DOI] [PubMed] [Google Scholar]
- 29.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010 Jul 13;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011 Oct 18;20(4):457–71. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF, et al. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002 Nov 15;62(22):6606–14. [PubMed] [Google Scholar]
- 32.Libertini SJ, Tepper CG, Rodriguez V, Asmuth DM, Kung HJ, Mudryj M. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res. 2007 Oct 1;67(19):9001–5. doi: 10.1158/0008-5472.CAN-07-1072. [DOI] [PubMed] [Google Scholar]
- 33.Furr BJ, Tucker H. The preclinical development of bicalutamide: pharmacodynamics and mechanism of action. Urology. 1996 Jan;47(1A Suppl):13–25. doi: 10.1016/s0090-4295(96)80003-3. [DOI] [PubMed] [Google Scholar]
- 34.Sirab N, Terry S, Giton F, Caradec J, Chimingqi M, Moutereau S, et al. Androgens regulate Hedgehog signalling and proliferation in androgen-dependent prostate cells. Int J Cancer. 2012 Sep 15;131(6):1297–306. doi: 10.1002/ijc.27384. [DOI] [PubMed] [Google Scholar]
- 35.Lamont KR, Tindall DJ. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Mol Endocrinol. 2011 Jun;25(6):897–907. doi: 10.1210/me.2010-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crawford ED, Petrylak D. Castration-resistant prostate cancer: descriptive yet pejorative? J Clin Oncol. 2010 Aug 10;28(23):e408. doi: 10.1200/JCO.2010.28.7664. [DOI] [PubMed] [Google Scholar]
- 37.Brown RS, Edwards J, Dogan A, Payne H, Harland SJ, Bartlett JM, et al. Amplification of the androgen receptor gene in bone metastases from hormone-refractory prostate cancer. J Pathol. 2002 Oct;198(2):237–44. doi: 10.1002/path.1206. [DOI] [PubMed] [Google Scholar]
- 38.Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 2003 Aug 4;89(3):552–6. doi: 10.1038/sj.bjc.6601127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaddipati JP, McLeod DG, Heidenberg HB, Sesterhenn IA, Finger MJ, Moul JW, et al. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res. 1994 Jun 1;54(11):2861–4. [PubMed] [Google Scholar]
- 40.Harada N, Inoue K, Yamaji R, Nakano Y, Inui H. Androgen deprivation causes truncation of the C-terminal region of androgen receptor in human prostate cancer LNCaP cells. Cancer Sci. 2012 Jun;103(6):1022–7. doi: 10.1111/j.1349-7006.2012.02250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marcias G, Erdmann E, Lapouge G, Siebert C, Barthelemy P, Duclos B, et al. Identification of novel truncated androgen receptor (AR) mutants including unreported pre-mRNA splicing variants in the 22Rv1 hormone-refractory prostate cancer (PCa) cell line. Hum Mutat. 2010 Jan;31(1):74–80. doi: 10.1002/humu.21138. [DOI] [PubMed] [Google Scholar]
- 42.Dehm SM, Tindall DJ. Ligand-independent androgen receptor activity is activation function-2-independent and resistant to antiandrogens in androgen refractory prostate cancer cells. J Biol Chem. 2006 Sep 22;281(38):27882–93. doi: 10.1074/jbc.M605002200. [DOI] [PubMed] [Google Scholar]
- 43.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004 Oct 7;431(7009):707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 44.Sheng T, Li C, Zhang X, Chi S, He N, Chen K, et al. Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer. 2004 Oct 13;3:29. doi: 10.1186/1476-4598-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang XD, Wang BE, Soriano R, Zha J, Zhang Z, Modrusan Z, et al. Expression profiling of the mouse prostate after castration and hormone replacement: implication of H-cadherin in prostate tumorigenesis. Differentiation. 2007 Mar;75(3):219–34. doi: 10.1111/j.1432-0436.2006.00135.x. [DOI] [PubMed] [Google Scholar]
- 46.Chen M, Feuerstein MA, Levina E, Baghel PS, Carkner RD, Tanner MJ, et al. Hedgehog/Gli supports androgen signaling in androgen deprived and androgen independent prostate cancer cells. Mol Cancer. 2010;9:89. doi: 10.1186/1476-4598-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.