

Abstract
Sepsis is an acute inflammatory condition that can result in multiple organ failure and acute lung injury (ALI). Growth arrest-specific protein 6 (Gas6) is a broad regulator of the innate immune response involved with the NF-κB signaling pathway. We hypothesized that Gas6 could have a protective role in attenuating the severity of ALI and sepsis. Male mice were subjected to sepsis by cecal ligation and puncture (CLP) after which recombinant murine Gas6 (rmGas6; 5 μg/mouse) or normal saline (vehicle) was administered intravenously. Blood and lung tissues were collected at 20 h after CLP for various measurements. Treatment with rmGas6 significantly reduced serum levels of the injury markers AST, ALT and LDH as well as proinflammatory cytokines IL-6 and IL-17, compared to the vehicle group (P<0.05). The parenchyma of the lungs damaged by CLP was attenuated by rmGas6 treatment. Lung mRNA levels of TNF-α, IL-1β, IL-6, IL-17 and MIP-2 were decreased by 60%, 86%, 82%, 93% and 82%, respectively, with rmGas6 treatment as determined by real time RT-PCR (P<0.05). The degradation of IκB-α induced by CLP in the lungs was inhibited by rmGas6 treatment. The number of neutrophils and myeloperoxidase activity in the lungs were significantly reduced in the rmGas6 group. Moreover, rmGas6 reduced the in-vitro migration of differentiated human promyelocytic HL60 cells by 64%. Finally, the 10-day survival rate of mice subjected to CLP was increased from 31% in the vehicle group to 67% in the rmGas6 group (P<0.05). Thus, Gas6 has potential to be developed as a novel therapeutic agent to treat patients with sepsis and acute lung injury.
Keywords: Gas6, acute lung injury, sepsis, inflammation, neutrophil
INTRODUCTION
Sepsis is a major cause of admission to intensive care units (ICU) and a large proportion of septic patients will succumb to multi-organ system injury and failure (1,2). Acute lung injury (ALI) is a common affliction incurred during sepsis, responsible for an estimated 74,500 deaths per year (3). The diagnosis of sepsis and severe sepsis is on the rise and although mortality has decreased, ALI mortality still remains unacceptably high (4). Much research surrounds ALI pathogenesis and potential therapies, focusing on dampening local and systemic hyperinflammation, decreasing interstitial edema and lowering neutrophil accumulation in damaged organs (5-7). However despite these efforts, few therapies have emerged as potential candidates to ameliorate sepsis-induced ALI. The need to explore and understand the response which induces ALI is a financial imperative for hospitals and is invaluable to patient care.
Neutrophils are activated during sepsis which then transmigrate and infiltrate the lung parenchyma contributing to ALI (8). Although intended to protect the body from deleterious events, this mechanism can be injurious to the lung tissue by causing endothelial cell damage and unrestrained inflammation (9). Local inflammation via the release of myeloperoxidase (MPO), tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, IL-6 and IL-17 and other damaging mediators occurs due to the septic response (10). Infiltration by neutrophils is aided by the presence of chemoattractants such as the CXCs, namely KC (CXCL1) and MIP-2 (CXCL2) (11). These chemokines are under the regulation of the transcriptional factor NF-κB, whose translocation to the nucleus (and thus action) is dependent on the cytoplasmic cleavage of the IκB-α subunit (12). The suppression of NF-κB can be regulated by receptor-ligand binding of the TAM family receptors, Tyro3, Axl, and Mer. In contrast, TAM-deficient macrophages exhibit higher NF-κB activity than wild-type cells. In fact, inhibition of the Mer receptor has been shown to heighten inflammatory cytokines and increase organ damage when stimulated with lipopolysaccharide (LPS) (13-16). Additionally, mice with absent intracellular Mer domains exhibit an exaggerated inflammatory response to LPS (17). The TAM/NF-κB regulatory pathway is a protective strategy employed by endogenous ligands during inflammation.
Growth arrest-specific protein 6 (Gas6) is a biological agonist for the TAM receptors. Gas6 is a vitamin K-dependent protein found to be similar to Protein S, though lacking anticoagulation activity and differing in its binding affinity to the TAM receptors (16). Gas6 contains a GLA (γ-carboxyglutamic acid) domain which is able to interact with phosphatidylserine on the surface of apoptotic cells and aid in phagocytosis (18). During inflammation Gas6 is expressed at high levels, existing in the plasma bound to its soluble receptor Axl (sAxl) and soluble Mer (sMer), effectively inactivating Gas6 activity (19-21). Gas6, once bound to TAM receptors, has been shown to have an anti-apoptotic effect as well as pro-survival capability. During TNF-α-mediated cytotoxicity of human umbilical vein endothelial cells, Gas6 is able to protect against apoptosis and prolong survival when growth factors are depleted (22-24). In hepatic ischemia/reperfusion injury, Gas6 is found to be protective from systemic injury as well as potentiating survival of hepatocytes (25). Its actions upon Mer and Axl receptors have been well studied, demonstrating Gas6’s down-regulation of the NF-κB pathway and thusly inhibition of inflammation in vitro. (15,26).
With the function of Gas6 partly elucidated during inflammatory conditions and disorders, we hypothesized that this protein could abate sepsis-induced inflammation and subsequent sequelae. In the present study, we used a mouse model of sepsis induced by cecal ligation and puncture (CLP) to examine the effect of recombinant murine Gas6 (rmGas6) treatment on organ injury and systemic inflammation in sepsis. We then compared the severity of lung damage between vehicle- and rmGas6-treated septic animals. We also assessed the effects of rmGas6 treatment on pulmonary levels of cytokines and activation of NF-κB pathway as well as neutrophil migration in vivo and in vitro. Finally, we conducted a survival study to evaluate the efficacy of rmGas6 in treating animals suffering from sepsis.
MATERIALS AND METHODS
Animal model of sepsis
Male C75BL/6 mice (20-25 g; Taconic, Albany, NY) were allocated to three groups, sham, vehicle, and treatment groups. Mice were anesthetized by isoflurane inhalation. A 1-cm abdominal incision was made to expose the cecum. The cecum was ligated with 4-0 silk suture 0.5-cm away from the base of the ileocecal valve. Double puncture with a 22-gauge needle was preformed to perforate the cecum. A small amount of stool was squeezed from both holes and the cecum was returned to the abdominal cavity. The abdomen was then closed in 2 layers. One milliliter of resuscitative normal saline was given subcutaneously. At 20 h after CLP, the animals were euthanized and blood and tissue samples were collected for various analyses. All experiments were performed in accordance with the guidelines for the use of experimental animals by the National Institute of Health and were approved by the Institutional Animal Care and Use Committee of The Feinstein Institute of Medical Research.
Administration of rmGas6
Immediately after performing CLP, a small incision on the neck was made over the internal jugular vein (IJ) and exposure was gained. Normal saline (vehicle) in 200 μl or rmGas6 (5 μg/mouse; R&D Systems, Minneapolis, MN) was delivered by bolus injection via a PE-10 catheter with the IJ. The proximal and distal ends of the jugular vein were completely ligated and the wound was closed with one interrupted 4-0 nylon suture. The dosage of rmGas6 was based on a study by Llacun et al (25).
Measurement of serum levels of injury markers
Blood samples were centrifuged at 2,000g for 15 min to collect serum and then stored at −80°C for measuring the activity of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and lactate dehydrogenase (LDH) by using assay kits from Pointe Scientific (Canton, Mich).
Measurement of serum cytokines
IL-6 and IL-17 levels in the serum were measured by BD FACSArray Bioanalyzer (BD Bioscience; San Jose, CA) directly from a 96-well plate through multiplexed BD Cytometric Bead Arrays (CBAs), according to the manufacturer’s instruction. Data was processed using FCAP array software. New to the pro-inflammatory cytokine milieu, IL-17 has been shown to augment inflammation and neutrophil migration and activation, prompting our investigation (27).
Histological analysis and immunostaining
Lung tissue was fixed in 10% formalin and then embedded in paraffin. Tissue was sectioned at a thickness of 5 μm, placed onto glass slides and stained with hematoxylin and eosin (H&E). Examination of these tissue sections was evaluated under light microscopy in a blinded manner. Interstitial edema, hemorrhage, and alveolar congestion were assessed and graded. The severity of lung injury was scored from 1 to 10 with 3 being the lowest possible score. Ten high-powered fields per sample were scored and averaged to represent each animal. For neutrophil staining, paraffin-embedded lung tissues were dewaxed in xylene and rehydrated in a graded series of ethanol. The slides were heated in 0.92% citric acid buffer (Vector Laboratories, Burlingame, CA) at 95°C for 30 min. After cooling, the slides were incubated with 2% H2O2/60% methanol and blocked in normal rabbit serum/Tris-buffered saline. The anti-Gr-1 antibody (BioLegend; San Diego, CA) was applied and incubated over night. The detection was carried out as per the instructions provided by an immunohistochemistry kit with NovaRED substrate from Vector Laboratories.
Reverse transcriptase-polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from lung tissues using Trizol (Invitrogen, Carlsbad, CA) and was reverse-transcribed into cDNA using murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA). A PCR reaction was done in 25 μl of final volume containing 0.08 μmol of each forward and reverse primer, cDNA, and 12.5 μl SYBR Green PCR Master Mix (Applied Biosystems). Amplification was conducted in an Applied Biosystems 7300 real-time PCR machine under the temperature of 50°C for 2 min, 95°C for 10 min and 45 cycles of 95°C for 15 seconds and 60°C for 1 min. Mouse β-actin was used for normalization. Expression of mRNA was represented as fold change in comparison to sham tissue levels. The primers used are listed in Table 1.
TABLE 1. A list of primer sequences.
| Gene | GenBank | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|---|
| TNF-α | NM_013693 | AGACCCTCACACTCAGATCATCTTC | TTGCTACGACGTGGGCTACA |
| IL-1β | NM_008361 | CAGGATGAGGACATGAGCACC | CTCTGCAGACTCAAACTCCAC |
| IL-6 | NM_031168 | CCAGAGGAGACTTCACAG | CAGAATTGCCATTGCACAAC |
| IL-17 | NM_008371 | CAGGGAGAGCTTCATCTGTGT | AGGAAGTCCTTGGCCTCACT |
| MIP-2 | NM_009140 | CATCCAGAGCTTGATGGTGA | CTTTGGTTCTTCCGTTGAGG |
| β-Actin | NM_007393 | CGTGAAAAGATGACCCAGATCA | TGGTACGACCAGAGGCATACAG |
Western blotting
Lung tissue was homogenized in lysis buffer (10 mM Tris-HCl pH 7.5, 120 mM NaCl 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) with protease inhibitor (Roche Diagnostics; Indianapolis, IN) by sonication. Total lysate was fractioned on Bis-Tris gels (4-12%) and transferred to PVDF membranes. The membranes were then blocked with 5% milk in 0.2× PBS and then incubated with anti-Iκβ-α or β-actin primary antibodies (Santa Cruz Biotechnologies, Santa Cruz, CA). After washing, the membranes were incubated with a fluorescently-labeled secondary antibody. Scanning the membranes was completed with the Odyssey image system (LI-COR, Lincoln, NE) and the band intensity measured by the Odyssey densitometric software.
Measurement of lung myeloperoxidase (MPO) activity
Lung tissue was homogenized in KPO4 buffer containing 0.5% hexa-decyl-trimethyl-ammonium bromide by sonication. The samples were centrifuged and the supernatant was diluted in the reaction solution containing o-dianisidine hydrochloride and H2O2 in phosphate buffer in a 96-well plate. The rate of change in absorbance for 1 min was measured at 460 nm to calculate MPO activity units per gram of protein.
In vitro neutrophil migration assay
Human promyelocytic HL60 cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in RPMI medium (Invitrogen) containing 10% fetal bovine serum and supplemented with penicillin and streptomycin. HL60 cells were differentiated into neutrophil-like cells by adding DMSO (12.7 μl/ml per million cells) for 5 days. The migration assays were conducted in a modified 24-well (3.0 μm) Boyden chamber (BD Biosciences). The differentiated HL-60 cells were incubated with PBS (vehicle) or rmGas6 (10 ng/ml) for 2 h. In the mean time, cells were labeled with Vybrant DiO (Invirogen). The labeled cells were plated in the upper well and medium containing 1 ng/ml IL-8 (R&D Systems) was placed in the lower well as a chemotactic stimulus. After a 2 h incubation period, the upper surface of the filter was washed and swabbed with cotton to remove nonmigratory cells. Migrated cells were fixed with 10% formalin. Five random microscopic fields per well were counted.
Survival study
In the survival study, mice were subjected to the previously stated CLP and treated immediately after operation with the antibiotic Imipenem (0.5 μg/kg), and resuscitative fluid. At 2 h post-CLP, mice were administered rmGas6 (5 μg/mice) or an equivalent volume of normal saline via the internal jugular vein. The animals were then followed daily for 10 days.
Statistical analysis
Data are expressed as mean ± standard error (SE) and compared via one way analysis of variance (ANOVA) and Student-Newman-Keuls (SNK) test for multiple group comparisons. Student’s t-test is applied only for a pair comparison. Survival rate was determined by the Kaplan-Meier estimator and compared by a log-rank test. Significance was considered if P < 0.05 for all data.
RESULTS
rmGas6 decreases organ injury and systemic inflammation after CLP
Clinical markers of organ injury used to asses the extent and severity of sepsis were measured 20 h after CLP in mice. The levels of the liver enzymes AST and ALT as well as LDH were significantly elevated after CLP (Fig. 1). In rmGas6-treated mice the levels of AST, ALT and LDH were reduced by 38%, 74% and 43%, respectively, compared to the vehicle group (P < 0.05, Fig. 1). Similarly, serum levels of IL-6 and IL-17 were dramatically affected, with vehicle-treated animal levels of 19.3 ± 3.3 ng/ml and 16.8 ± 4.7 pg/ml, respectively, compared to sham levels of undetectable and 2.2 ± 0.3 pg/ml, respectively. In contrast, rmGas6 treatment significantly decreased IL-6 and IL -17 levels by 47% and 78%, respectively, compared to the vehicle group (Fig. 2).
FIG. 1. Effect of rmGas6 on serum levels of organ injury markers in mice after CLP.
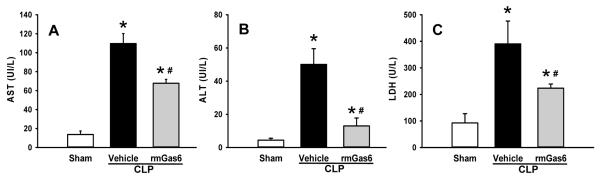
Sham, vehicle, and rmGas6-treated groups underwent CLP and the serum was collected at 20 h for measuring AST (A), ALT (B), and LDH (C). Data presented as means ± SE (n=4-6/group) and compared by one-way ANOVA and SNK method; *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
FIG. 2. Effect of rmGas6 on the serum levels of pro-inflammatory cytokines in mice after CLP.
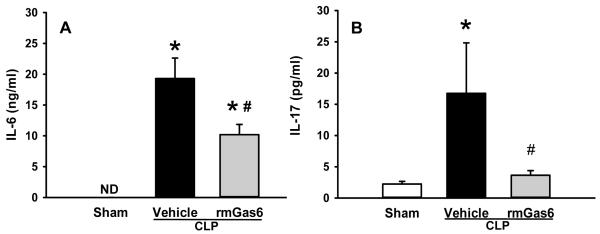
Sham, vehicle, and rmGas6-treated groups underwent CLP and the serum was collected at 20 h post-CLP for measuring the levels of IL-6 (A) and IL-17 (B) by cytometric bead array. Data presented as means ± SE (n=4-6/group) and compared by one-way ANOVA and SNK method; *P < 0.05 vs. sham; #P < 0.05 vs. vehicle. ND, non-detectable.
rmGas6 protects against CLP-induced ALI
To elaborate the effect of rmGas6 on the acute injury of the lungs caused by CLP, lung tissue underwent histological examination using H&E staining and the severity of injury was graded with an established scoring system as described in Materials and Methods. Representative histological architecture of the sham, vehicle, and rmGas6-treated mice are shown in Figure 3A. Evidence of edema, hemorrhage, hyaline-filled alveoli and neutrophil infiltration were enhanced in the vehicle group as compared to sham, while it was attenuated by rmGas6 treatment. As quantified in Figure 3B, rmGas6 treatment reduced the histologically-graded damage of the lungs (P < 0.05).
FIG. 3. Effect of rmGas6 on histological architecture of the lungs in mice after CLP.

Lung tissue was isolated at 20 h post-CLP and subjected to H&E staining. (A) Representative photomicrographs of the tissue sections. Sham tissues show normal architecture, while the vehicle group has enhanced edema, hemorrhage and polymorphonuclear leukocyte infiltration. The structure of rmGas6-treated group is of more resemblance to the sham. Magnification, ×200. (B) Histological scoring of lung injury after CLP was judged by a blinded observer. Data presented as means ± SE (n=4-6/group) and compared by one-way ANOVA and SNK method; *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
Lung cytokine and chemokine production is inhibited by rmGas6 after CLP
Excessive production of pro-inflammatory cytokines and chemokines are major contributing factors for the tissue injury. Lung tissue was harvested 20 h after CLP and subjected to RT-PCR analysis for determining the change of mRNA levels of cytokines and chemokines. The vehicle group demonstrated a fold increase of 47, 76, 73, 3,584 and 1,379 times of TNF-α, IL-1β, IL-6, IL-17 and MIP-2, respectively, compared to sham animals (Fig. 4). In rmGas6 treatment group, there was a 60%, 86%, 82%, 93% and 82% reduction of the respective cytokine or chemokine, compared to the vehicle group (Fig. 4).
FIG. 4. Effect of rmGas6 on the expression of proinflammatory cytokines and chemokines in the lungs after CLP.

Lung tissues from sham, vehicle, and rmGas6-teated groups were collected at 20 h after CLP. The mRNA levels of TNF-α (A), IL-1β (B), IL-6 (C), IL-17 (D) and MIP-2 (E) were performed by RT-PCR analysis and normalized to β-actin. The sham expression level was designated as 1 for comparison. Data presented as means ± SE (n=4-6/group) and compared by one-way ANOVA and SNK method; *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
rmGas6 attenuates IκB-α degradation in the lungs after CLP
Gas6 is a ligand for the TAM receptors, which can counteract the signal for NF-κB activation and subsequently attenuate the expression of downstream target genes of the NF-κB pathway. To elicit the status of NF-κB activation in which Gas6 acts during sepsis, cellular levels of IκB-α, an endogenous inhibitor of NF-κB, were determined by Western blotting. Lung tissues were collected 20 h after CLP in all groups. We observed a 62% decrease of IκB-α levels after CLP, whereas rmGas6-treated animals displayed the levels comparable to the sham (Fig. 5). This result indicates that NF-κB pathway is activated after CLP, while it is inhibited by rmGas6.
FIG. 5. Effect of rmGas6 on IκB-α levels in the lung after CLP.

Lung tissues from sham, vehicle, and rmGas6-teated groups were collected at 20 h after CLP. Total cell lysates were subjected to Western blotting. (A) Representative blots against IκB-α and corresponding β-actin are shown. (B) Blots were scanned and quantified with densitometry. Band intensity of IκB-α was normalized to the corresponding band intensity of β-actin. The level of protein expression in the sham group is designated as 1 for comparison. Data presented as means ± SE (n=3/group) and compared by one-way ANOVA and SNK method; *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
rmGas6 reduces neutrophil infiltration in the lungs after CLP
ALI is mostly caused by the deleterious inflammatory mediators released by neutrophils; this damage is reflected in the number of neutrophils and MPO activity within the lung. Gr-1 (a marker of neutrophils) staining in lung tissues 20 h after CLP demonstrated a statistically lower number of Gr-1 positively stained cells in the rmGas6 treatment group than in the vehicle group (Figs. 6A and B). Furthermore, the MPO activity in the vehicle group was 12 times higher than that in the sham, while rmGas6 reduced this activity by 69% in comparison with the vehicle group (Fig. 6C).
FIG. 6. Effect of rmGas6 on neutrophil transmigration into the lung parenchyma after CLP.

Lung tissues from sham, vehicle, and rmGas6-teated groups were collected at 20 h after CLP. (A) Representative photomicrographs of the tissue sections with immunostaining against Gr-1. Arrows demarcate examples of areas of staining. Magnification, ×400. (B) A graphical representation of Gr-1-positive staining cells averaged over 10 microscopic fields per animal. (C) Lung tissue myeloperoxidase (MPO) activity was determined spectrophotometrically. Data presented as means ± SE (n=4-6/group) and compared by one-way ANOVA and SNK method; *P < 0.05 vs. sham; #P < 0.05 vs. vehicle.
rmGas6 inhibits neutrophil migration in vitro
To evaluate the direct effect of rmGas6 on neutrophil migration, human leukocyte HL60 cells were subjected to a transwell chemotaxis assay with IL-8 as a chemoattractant. In the PBS-treated HL60 cells, a massive number of migrated neutrophils were detected at the bottom of the micropore membrane, while pretreatment with rmGas6 resulted in a 64% reduction of cell migration (Figs. 7A-B). To exclude the possibility that the reduction of cell migration was due to the cytotoxicity of rmGas6, we performed an MTS assay to compare the cell viability between PBS- and rmGas6-treated cells. There was no significant difference between these two groups (data not shown).
FIG. 7. Effect of rmGas6 on migration of human promyelocytic HL60 cells in vitro.
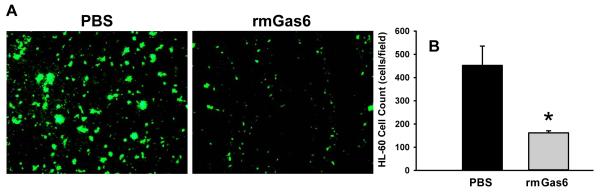
Differentiated HL60 cells were incubated with PBS (vehicle) or rmGas6 (10 ng/ml) as well as Vybrant DiO for 2 h and then subjected to a transwell migration assay with IL-8 as a chemoattractant. (A) A representative image of migrated HL60 cells labeled with Vybrant DiO (green fluorescence) on the bottom of the transwell membrane. Magnification, ×100. (B) Five random microscopic fields per well were counted. Data presented as means ± SE (n=3 independent experiments) and compared by Student’s t-test; *P < 0.05 vs. PBS.
Post-treatment with rmGas6 improves survival after CLP
To assess a long-term beneficial effect of rmGas6’s in our model of CLP-induced sepsis, we followed the vehicle and rmGas6-treated animals for 10 days. To be more clinically relevant, rmGas6 was administered 2 h after CLP in the survival study. As shown in Figure 8, the survival was significantly improved to 67% in rmGas6-treated mice (n=15) in comparison with 31% in vehicle treated mice (n=16; P < 0.05).
FIG. 8. Effect of post-treatment with rmGas6 on the survival of mice after CLP.

Mice underwent CLP and were administered PBS (vehicle) or rmGas6 at 2 h after CLP. Mice were monitored daily for 10 days. The survival rate was analyzed by Kaplan-Meier survival analysis and compared by the log-rank test. *P < 0.05 vs. vehicle.
DISCUSSION
The prevalence of sepsis and subsequent ALI is a constant burden in intensive care units (28). This ailment has plagued patients and clinicians at an increasing rate and new methods of attenuating the damage incurred sepsis are sorely needed. Research into this area of inflammation and immunology is invaluable in understanding new therapies and modalities of treatment. Thus, we have shown a novel function of Gas6 in sepsis-induced acute lung injury. Exaggerated cytokine expression and neutrophil migration has been induced in our model of murine sepsis and with administration of rmGas6 this inflammatory cascade is attenuated as evidenced histologically, biochemically and in survival rate. Surprisingly, inhibition of neutrophil migration into the lung parenchyma is demonstrated and is a novel action of Gas6.
In vitro, Gas6 is known to dampen the inflammatory response by downregulating NF-κB pathway through binding receptors of the TAM family. Using this Gas6-mediated pathway, Sharif et al. showed a decrease of cytokine expression by upregulating Twist, a transcription repressor that negatively regulates NF-κB activity (29). In LPS-stimulated macrophages, Deng et al. found suppression of TNF-α, IL1-β and IL-6 by the Gas6/TAM receptor pathway, implicating the TAM receptors in the regulation of TLR4-induced inflammation (15). This is of utmost importance during the hyperinflammatory state in which most septic patients are likely to present clinically. Dampening this phase of the septic response is beneficial, as immunosupression at this point shows positive results in clinical and animal research (30). In this fashion, we have shown that the early administration of rmGas6 is beneficial in our model of CLP-induced sepsis. There is a limitation of our study with early administering rmGas6, which may not have benefit under clinical scenario. Detailed studies for optimizing the dose and time-course (treatment window) as well as possible toxicity analysis are needed to further develop rmGas6 as a therapeutic agent for treating sepsis.
Another potential mechanism through which Gas6 aids in the acute inflammatory process is by way of phagocytosis and efferocytosis. As previously shown in literature, the interaction between phosphatidylserine, Gas6 and the TAM receptors aids in the engulfment of apoptotic cells (31). Apoptotic immune cell clearance should be enhanced by the presence of Gas6 as it acts as a “bridging molecule” between the target and the phagocyte. Through effective and efficient removal of apoptotic cells, inflammation can be reduced and the deleterious effects of secondary necrosis (leakage of inflammatory agents) can be abated (32). In this way, Gas6 can dampen the response to damage incurred by sepsis. A late time-point measurement as described in this study only indicated that rmGas6 treatment facilitated the septic animals to recover from hyperinflammation stage. A series of time point studies is needed to further address the effect of rmGas6 on the progression of cellular changes during the development of sepsis. Whether rmGas can protect endothelial or epithelial cells from apoptosis induced by sepsis will be another mechanism needs further examination.
In addition to the known properties of Gas6 in inflammatory states, we have demonstrated inhibited neutrophil recruitment into the lungs during sepsis. Neutrophil involvement in the development in ALI has been known to be detrimental to the lung parenchyma (33). Treatment with rmGas6 improved the organ damage seen in untreated ALI and decreased MPO activity as well as tissue mRNA levels of chemoattractants and inflammatory cytokines. This mechanism of avoiding lung injury is well recognized, but is a novel Gas6 function. This action could be due to the down regulation of IL-17 and MIP-2, however, our in-vitro migration assay clearly demonstrates that the inhibition likely occurs by directly regulating neutrophil movement. In this study, we focused on studying the activated neutrophils that were able to transmigrate to the lungs and cause the tissue damage. As such, we did not determine the change of total neutrophil count in the circulation after sepsis. However, rmGas6 did not cause a cytotoxic effect on neutrophils analyzed by MTS cell viability assay. Whether rmGas6 has other activity on lowering the neutrophil population in the circulation, leading to reduction of neutrophil infiltration after sepsis, needs further investigation.
Past studies involving septic patients and animals agree that during sepsis, Gas6 is elevated in the serum. In fact, the disease severity and extent of organ damage can be correlated to the level of Gas6 (20). This, however, proves to be a subtherapeutic level of Gas6 as described in literature. Hurtado et al. described that during sepsis Gas6 is dramatically increased, however it exists bound to soluble Axl rendering Gas6 inactive (19). Also, it has been shown that Gas6 can be inhibited by the extracellular portion of Mer, again depleting the efficacy of endogenous Gas6 (34). Additionally, Sather et al. demonstrated that Gas6’s function is indeed inhibited by this soluble form of the Mer receptor, in that there was inhibition of macrophage engulfment of apoptotic cells (35). Binding affinities differ between the TAM receptors, (Axl>Tyro3>>Mer) (13) suggesting that even when abundant, the biologically expressed levels of Gas6 may not produce a clinically effective depression of inflammation. However, copious free exogenous Gas6 is able to activate Axl and Mer receptors, leading to the downward cascade which aids in phagocytosis and inhibits NF-κB and neutrophil migration. We did not determine the serum Gas6 levels due to its binding to soluble TAM receptors, which may not reflect the true value of its biological activity. Administration of rmGas6 as conducted in this study should compensate Gas6 levels high enough to overcome the interference from soluble TAM receptors by showing the beneficial effects after sepsis. Regarding to the stability and half-life of rmGas6 will be an important issue for the future study.
Highlighting the effectiveness of Gas6, post-treatment in sepsis improved overall 10-day mortality after CLP-induced sepsis. According to our previous experience, lethality after 10 days is rarely occurred in the CLP model. The late mortality (> 28 days) of patients undergoing sepsis is the standard outcome measurement for clinical trials. However, a 10-day survival study in mice may be equivalent to multi-week observation in humans. Therefore, our survival study can be considered as a long-term evaluation of rmGas6 effect. The dampening of the systemic inflammatory response in immunocompetent individuals is an effective method to avoid damage in the immediate hyperinflammatory time point. As expected, mice which received rmGas6 2 hours after CLP-induced sepsis survived this insult and more rapidly exhibited normal behavior (data not shown). The post-treatment model of administration is more clinically applicable, as patients commonly present well into septic sequelae. With Gas6-mediated decrease in injury, we expect that treatment will result in fewer ventilator days and an avoidance of multi-organ dysfunction (and interventions which are required) in those with sepsis-induced ALI.
In summary, the presented data confirms Gas6’s role as an anti-inflammatory agent capable of abrogating sepsis-induced organ dysfunction and neutrophil-induced ALI. Neutrophil migration inhibition is a novel function of Gas6, first shown here. However, there is always a gap to translate the results from the animal studies to human clinical conditions. To identify the effects of rmGas6 on sepsis, we performed a severe CLP model in this study. Although there is a high mortality in septic patients, the severity among patient population is varied. In addition, genetic variation between humans may contribute to different responses even under similar clinical conditions. In contrast, a single mouse strain was used in this study. Therefore, it should be cautious when interpreting the animal data and linked it to humans. Overall, this study indicates that administration of Gas6 early after onset of infection may have the ability to be a therapy that abrogates the development of sepsis.
Acknowledgments
This study was supported by National Institutes of Health grants, HL076179, GM057468, and GM053008 (PW).
Footnotes
Disclosure of Financial Interests and Potential Conflicts of Interest: All authors reported no financial interests or potential conflicts of interest.
This work was awarded The Shock Society New Investigator Award at the 36th Annual Conference on Shock, San Diego, California. June 1-4, 2013.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Gaieski DF, Edwards M, Kallan M, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41:1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 4.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2005;33:319–327. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moine P, McIntyre R, Schwartz MD, Kaneko D, Shenkar R, Le Tulzo Y, Moore EE, Abraham E. NF-kappaB regulatory mechanisms in alveolar macrophages from patients with acute respiratory distress syndrome. Shock. 2000;13:85–91. doi: 10.1097/00024382-200013020-00001. [DOI] [PubMed] [Google Scholar]
- 6.Liu SF, Malik AB. NF-κB activation as a pathologic mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 7.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31(Suppl):S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 8.Aziz M, Matsuda A, Yang WL, Jacob A, Wang P. Milk fat globule-epidermal growth factor-factor 8 attenuates neutrophil infiltration in acute lung injury via modulation of CXCR2. J Immunol. 2012;189:393–402. doi: 10.4049/jimmunol.1200262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7:1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito S, Ley K. Critical role of endothelial cxcr2 in LPS-induced neutrophil migrations into the lung. J Clin Invest. 2006;116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto Y, Gaynor R. IkB kinases: key regulators of NF-kB pathway. Trends Biochem Sci. 2004;2:72–9. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Lemke G, Rothlin C. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–336. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YJ, Han JY, Byun J, Park HJ, Park EM, Chong YH, Cho MS, Kang JL. Inhibiting Mer receptor tyrosine kinase suppresses STAT1, SOCS1/3, and NF-κB activation and enhances inflammatory responses in lipopolysaccharide-induced acute lung injury. J Leukoc Biol. 2012;91:921–932. doi: 10.1189/jlb.0611289. [DOI] [PubMed] [Google Scholar]
- 15.Deng T, Zhang Y, Chen Q, Yan K, Han D. Toll-like receptor-mediated inhibition of Gas6 and ProS expression facilitates inflammatory cytokine production in mouse macrophages. Immunology. 2012;135:40–50. doi: 10.1111/j.1365-2567.2011.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez-Fernandez L, Bellido-Martin L, Garcia de Frutos P. Growth arrest specific gene 6 (Gas6). An outline of its role in haemostasis and inflammation. Thromb Haemost. 2008;100:604–610. doi: 10.1160/th08-04-0253. [DOI] [PubMed] [Google Scholar]
- 17.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jenette JC, Roubey RA, Earp HS, Masushima G, Reap EA. Delayed apaptotic clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118:539–553. doi: 10.1242/jcs.01632. [DOI] [PubMed] [Google Scholar]
- 19.Hurtado B, de Frutos PG. Gas6 in systemic inflammatory disease: with and without infection. Crit Care. 2010;14:1003. doi: 10.1186/cc9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giobt S, Massin F, Carvoisy A, Dupays R, Barraud D, Nace L, Bollaert PE. Growth arrest-specific protein 6 plasma concentrations during septic shock. Crit Care. 2007;11:R8. doi: 10.1186/cc5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekman C, Linder A, Akesson P, Dahlback B. Plasma concentrations of Gas6 (growth arrest specific protein 6) and its soluble tyrosine kinase receptor sAxl in sepsis and systemic inflammatory response syndromes. Crit Care. 2010;14(4):R158. doi: 10.1186/cc9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Donnell K, Harkes IC, Dougherty L, Wicks IP. Expression of receptor tyrosine kinase Axl and its ligand Gas6 in rheumatoid arthritis: evidence for a novel endothelial cell survival pathway. Am J Pathol. 1999;154:1171–1180. doi: 10.1016/S0002-9440(10)65369-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shankar SL, O’Guin K, Kim M, Varnum B, Lemke G, Brosnan CF, Shafit-Zagardo B. Gas6/Axl signaling activates the phosphatidylserine 3-kinase/Akt1 survival pathway to protect oligodendrocytes from tumor necrosis factor alpha-induced apoptosis. J Neurosci. 2006;26:5638–5648. doi: 10.1523/JNEUROSCI.5063-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasanbasic I, Cuerguis J, Varnum B, Blostein MD. Intracellular signaling pathways involved in Gas6-Axl mediated survival of endothelial cells. Am J Physiol Hear Circ Physiol. 2004;287:1207–1213. doi: 10.1152/ajpheart.00020.2004. [DOI] [PubMed] [Google Scholar]
- 25.Llacun L, Barcena C, Bellido-Martin L, Fernandez L, Stefanovic M, Mari M, Garcia-Ruiz C, Fernandez-Checa JC, Garcia de Frutos P, Morales A. Growth arrest-specific protein 6 is heptaoprotective against murine ischemia/rerperfusion injury. Hepatology. 2010;52:1371–1379. doi: 10.1002/hep.23833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Png KJ, Halberg N, Yoshida M, Tavazoie SF. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature. 2011;481:190–194. doi: 10.1038/nature10661. [DOI] [PubMed] [Google Scholar]
- 27.Song X, Qian Y. The activation and regulation of IL-17 receptor mediated signaling. Thorax. 2013;62:175–182. doi: 10.1016/j.cyto.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 28.Engel C, Brunkhorst FM, Bone HG, Brunkhorst R, Gerlach H, Grond S, Gruendling M, Huhle G, Jaschinski U, John S, et al. Epidemiology of sepsis in Germany: results from a national prospective multicenter study. Intensive Care Med. 2007;33:606–618. doi: 10.1007/s00134-006-0517-7. [DOI] [PubMed] [Google Scholar]
- 29.Sharif MN, Sosic D, rothlin CV, Kelly E, Lemke G, Olson EN, Ivashkiv LB. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. 2006;203:1891–1901. doi: 10.1084/jem.20051725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hotchkiss RS, Monneret G, Payen D. Immunosupression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lanct Infect Dis. 2013;13:260–268. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemke G, Burstyn Cohen T. TAM receptors and the clearance of apoptotic cells. Ann NY Acad Sci. 2010;1209:23–29. doi: 10.1111/j.1749-6632.2010.05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miksa M, Wu R, Dong W, Das P, Yang D, Wang P. Dendritic cell-derived exosomes containing milk fat globule epidermal growth factor VIII attenuate proinflammatory responses in sepsis. Shock. 2006;25:586–593. doi: 10.1097/01.shk.0000209533.22941.d0. [DOI] [PubMed] [Google Scholar]
- 33.Uriarte SM, Rane MJ, Merchant ML, Jin S, Lentsch AB, Ward RA, McLeish KR. Inhibition of neutrophil exocytosis amerliorates acute lung injury in rats. Shock. 2013;39:286–292. doi: 10.1097/SHK.0b013e318282c9a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinger JG, Omari KM, Marsden K, Raine CS, Shafit-Zagardo B. Up-regulation of soluble Axl and Mer receptor tyrosine kinases negatively correlates with Gas6 in established multiple sclerosis lesions. Am J Pathol. 2009;175:283–293. doi: 10.2353/ajpath.2009.080807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum B, Henson PM, Graham DK. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109:1026–1033. doi: 10.1182/blood-2006-05-021634. [DOI] [PMC free article] [PubMed] [Google Scholar]