

Abstract
Oligodendrocytes, the myelin-forming cells of the CNS, exquisitely tailor the thickness of individual myelin sheaths to the diameter of their target axons to maximize the speed of action potential propagation, thus ensuring proper neuronal connectivity and function. Following demyelinating injuries to the adult CNS, newly formed oligodendrocytes frequently generate new myelin sheaths. Following episodes of demyelination such as those that occur in patients with multiple sclerosis, however, the matching of myelin thickness to axon diameter fails leaving remyelinated axons with thin myelin sheaths potentially compromising function and leaving axons vulnerable to damage. How oligodendrocytes determine the appropriate thickness of myelin for an axon of defined size during repair is unknown and identifying the signals that regulate myelin thickness has obvious therapeutic implications. Here, we show that sustained activation of extracellular-regulated kinases 1 and 2 (ERK1/2) in oligodendrocyte lineage cells results in accelerated myelin repair after injury, and is sufficient for the generation of thick myelin sheaths around remyelinated axons in the adult mouse spinal cord. Our findings suggest a model where ERK1/2 MAP kinase signaling acts as a myelin thickness rheostat that instructs oligodendrocytes to generate axon-appropriate quantities of myelin.
Introduction
Perturbations in CNS myelination underlie a wide range of neurological diseases including multiple sclerosis (MS), which affects nearly 2.5 million people worldwide (Compston and Coles, 2008). Patients with MS typically exhibit a relapsing/remitting disease course where periods of active demyelination are followed by periods of repair when new myelin sheaths are generated. Importantly, the correlation between axon diameter and myelin sheath thickness attained during development of the nervous system is rarely achieved following remyelination in the adult, making axons with unusually thin myelin sheaths relative to their diameter a pathological hallmark of CNS remyelination (Suzuki et al., 1969; Blakemore, 1974; Ludwin and Maitland, 1984). Thinly myelinated axons likely have perturbed conductivity and may be more vulnerable to insults as new demyelinating activity has been demonstrated in previously remyelinated MS plaques (Prineas et al., 1993). Since axonal loss represents the major pathological correlate of functional deterioration in MS patients (Bjartmar et al., 2003; Lappe-Siefke et al., 2003; Edgar et al., 2004), therapies directed at increasing both the rate of remyelination and myelin thickness may have the greatest impact on axonal preservation and, consequently, limit the severity of patient symptoms. It remains unclear why myelin repair results in thin myelin.
Phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling plays a critical role in the control of myelin thickness during development as downregulation leads to thin myelin sheaths in the mouse (Hu et al., 2006; Narayanan et al., 2009; Tyler et al., 2009), and overactivation results in hypermyelination (Ye et al., 1995; Flores et al., 2008; Goebbels et al., 2010; Harrington et al., 2010). In the setting of demyelinating injuries, however, activation of PI3K signaling does not increase the speed of remyelination and the newly formed myelin sheaths are thinner than the originals (Harrington et al., 2010). We have shown that the ERK MAP kinase pathway is also critical for proper myelination during development in the mouse CNS. Loss of ERK2 leads to delayed forebrain myelination (Fyffe-Maricich et al., 2011), and deletion of Erk1 and Erk2 results in severe hypomyelination (Ishii et al., 2012). Moreover, recent in vitro (Xiao et al., 2012) and in vivo (Ishii et al., 2013) studies point to a role for ERK1/2 signaling in the promotion of developmental CNS myelination; however, it is unclear whether this developmental role for ERK1/2 is conserved in the adult after CNS injury.
In this study, we assessed whether sustained ERK1/2 MAP kinase activation was sufficient for enhanced myelin repair in the adult CNS. We generated mice that express a constitutively active variant of the Erk1/2 upstream kinase Mek1 (Mek1DD; Srinivasan et al., 2009) specifically in 2′-3′ cyclic nucleotide 3′-phosphodiesterase (CNP+) oligodendrocyte progenitor cells (OPCs) and mature oligodendrocytes (OLs; CNP-Cre; Mek1DD). We used a local injection of the glial toxin lysolecithin (l-α-lysophosphatidylcholine, LPC) to induce an area of focal demyelination and examined the rate of remyelination and the thickness of newly generated myelin sheaths in the dorsal spinal cord.
Materials and Methods
Experimental animals.
CNP-Cre/+ mice (Lappe-Siefke et al., 2003) on a mixed 129 C57BL/6 background were interbred with homozygous R26StopFLMEK1DD mice (Srinivasan et al., 2009; The Jackson Laboratory) to generate CNP-Cre; Mek1DD/+ mice, referred to as “mutants” and WT;Mek1DD/+ littermates, referred to as “controls.” Cre-positive mice exhibit constitutively active MAP kinase signal transduction in CNP-positive OPCs and OLs as a result of the expression of MEK1DD, a mutant form of rat MAPKK1 (the ERK1/2 kinase) that has been rendered constitutively active by two serine to aspartic acid substitutions (S218D/S222D) within the catalytic domain. Expression of MEK1DD can be followed due to the addition of an EGFP marker. All mice were kept in micro-isolation in a pathogen-free environment in the Animal Resource Center of Case Western Reserve University, and all procedures were conducted according to approved Institutional Animal Care and Use Committee guidelines.
Demyelination by lysolecithin injection.
All LPC injections were performed on sex-matched pairs of control and mutant adult mice (both males and females) aged 8–10 weeks. Dorsal laminectomy was performed at the T11/T12 level and 1 μl of 1% LPC (Sigma) in a 0.9% sodium chloride solution was microinjected using a sterile beveled microcapillary glass needle into the dorsal column of the spinal cord at a rate of 8 nl/s. To prevent liquid reflux at the completion of the injection, the needle was left in place for 3 min before removal. Importantly, charcoal was used to mark the site of LPC injection so that the area of tissue at the lesion center could be unambiguously identified even after remyelination was complete. Lesion sizes were determined by staining serial sections with luxol fast blue to define the rostrocaudal extent of the lesion along the dorsal spinal cord.
Immunolabeling of frozen sections.
Mice were perfused in 4% paraformaldehyde (PFA)/PBS, postfixed overnight followed by overnight cryoprotection in 20% sucrose/PBS. Coronal cryosections (20 μm) through the thoracic spinal cord were rinsed in PBS, subjected to antigen retrieval in citrate buffer, pH 6.0, washed with PBS, and then blocked and permeabilized with 0.3% Triton X-100 and 4% goat serum in PBS for 30 min. Sections were incubated with primary antibodies at 4°C overnight, then incubated with secondary antibodies at room temperature for 1 h. Sections were counterstained with DAPI for 3 min. Primary antibodies used were anti-myelin basic protein (MBP; 1:500; SMI-99; Covance), anti-CC1 (1:100; anti-APC; Oncogene), anti-Ki67 (1:250; BD PharMingen), anti-Olig2 (1:250; Millipore Bioscience Research Reagents), anti-GFP (1:500; Invitrogen and 1:1000 chicken anti-GFP; Aves Laboratories), and anti-pERK1/2 (1:250; Cell Signaling Technology). Alexa Fluor-conjugated IgG (1:500; Invitrogen) was used for secondary antibodies. Myelin loss was identified using standard luxol fast blue histochemical staining (0.1% solution; Electron Microscopy Sciences) and immunostaining for MBP. Cell counts were done from a minimum of three mice per genotype and data were analyzed using Student's t test.
Immunoblotting.
Immunoblotting was performed as previously described (Fyffe-Maricich et al., 2011). Equal amounts of total protein were loaded from two to three mice per genotype onto SDS-PAGE gels, transferred to PVDF membranes, and probed for pERK1/2 (1:1000; Cell Signaling Technology) and GAPDH (1:10,000; Santa Cruz Biotechnology).
Electron microscopy.
Postnatal day 30 (P30) or adult mice were perfused with 4% PFA and 2% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4 (Electron Microscopy Sciences). Samples were taken from the thoracic spinal cord and postfixed in 1% OsO4. Samples were dehydrated through graded ethanols, stained en bloc with uranyl acetate, and embedded in a Poly/Bed812 resin (Polysciences). Thin (1 μm) sections were stained with toluidine blue and ultrathin (0.1 μm) sections from matching areas of experimental and control tissue blocks were cut and visualized at 3000× or 50,000× magnification using an electron microscope (JEOL1200CX) at 80 kV. Sections were analyzed from three mutant and three control mice. G-ratios were calculated from at least 100 axons per mouse. G-ratios were measured using Adobe Photoshop software and are presented as the axon diameter/ total diameter of the axon plus the myelin sheath. Linear regression was used for indicating the differences between the groups in myelin thickness across the range of axon diameters. Average g-ratio was compared using Student's t test.
Results
Myelin thickness is increased in uninjured juvenile and adult mice after sustained activation of ERK1/2
Double-immunolabeling of spinal cord sections from CNP-Cre;Mek1DD mice with CC1 and GFP showed a near complete overlap (99% ± 0.5), indicating expression of MEK1DD-EGFP in virtually all OLs in the spinal cord during development (P8–P12; Fig. 1B). Upregulation of the activated form of ERK1/2 (pERK1/2) was observed in 71 ± 4.4% of CC1+ OLs (Fig. 1C). Immunoblot analysis of spinal cord lysates from adult mutant and control littermates confirmed a moderate but significant increase (∼1.8-fold) in pERK1/2 expression in CNP-Cre;Mek1DD mice (Fig. 1D). Analysis using electron microscopy (EM) followed by g-ratio calculation (axon diameter/ total outer diameter of myelinated fiber) revealed significantly lower g-ratios in uninjured spinal cords from mice with increased levels of pERK1/2 at P30 (control = 0.758 ± 0.004, CNP-Cre;Mek1DD = 0.697 ± 0.005; p = 5.69 × 10−22) and in the adult (control = 0.767 ± 0.004, CNP-Cre;Mek1DD = 0.647 ± 0.006; p = 7.95 × 10−44; Fig. 2A–F), confirming increased baseline myelin thickness in this mouse model. These data provide support for a recent study that focused on the development of hypermyelination using the same transgenic mouse model (Ishii et al., 2013). Not only were the myelin sheaths unusually thick, but measurement of OL soma diameter revealed significant OL hypertrophy in CNP-Cre;Mek1DD (9.2 ± 0.2 μm) compared with control littermates (7.7 ± 0.1 μm, p = 5.7 × 10−6; Fig. 2G,H), suggesting that myelin thickness may be related to the size of the myelinating cell. Although the major effect of upregulation of pERK1/2 is significantly thicker myelin sheaths during the period of active developmental myelination, it appears to drive continuous myelin formation at a low level in uninjured adult mice. The average g-ratio for CNP-Cre;Mek1DD mice was significantly lower at 4 months of age (0.647 ± 0.006) compared with P30 (0.697 ± 0.005, p = 2.4 × 10−11), indicating increased myelin thickness over time (Fig. 2D,F). The magnitude of this post developmental period increase in myelin thickness was small and may reflect the continuous generation of OLs and myelin in the adult animal as was recently demonstrated (Psachoulia et al., 2009). These data are in contrast to other models that result in elevated CNS myelin thickness, such as the constitutive activation of AKT1, where myelin formation appears to continue at a relatively high level for the lifetime of the mice, eventually resulting in pathological consequences such as axonal compression and degeneration (Flores et al., 2008; Harrington et al., 2010).
Figure 1.
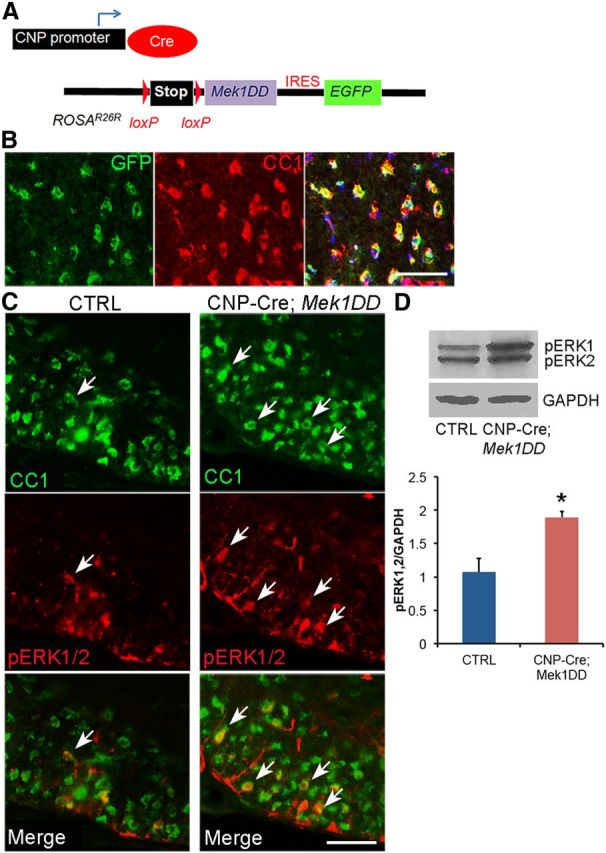
Expression of MEK1DD in CNP+ cells following Cre-mediated recombination leads to an upregulation of pERK1/2 in OLs. A, Schematic representation of the constructs used to create the transgenic mice. B, GFP labels cells that express MEK1DD and CC1 labels mature OLs. DAPI marks cell nuclei in blue. C, Mature CC1+ OLs express high levels of pERK1/2 (white arrows) in the developing spinal cord of CNP-Cre;Mek1DD mice. D, Immunoblotting confirms a significant increase in pERK1/2 expression in the adult CNP-Cre;Mek1DD spinal cord. Error bars indicate SEM. *p < 0.05. Scale bars: 50 μm. CTRL, control.
Figure 2.

Elevated pERK1/2 expression drives OL hypertrophy and the production of thicker myelin sheaths in juvenile and adult mice. A–C, EM analysis and (D) myelin thickness as well as (E and F) g-ratio calculations demonstrate thicker myelin sheaths in CNP-Cre;Mek1DD mouse spinal cords at P30 and P120. Solid lines show linear trend lines. G, H, Toluidine blue staining of 1 μm epon sections reveals thicker myelin sheaths and significantly larger OL cell bodies in the dorsal spinal cord of CNP-Cre;Mek1DD mice at P30 (n = 3 per genotype). Scale bars: (in C) A–C, 2 μm; (in H) G, H, 25 μm. CTRL, control.
Myelin thickness is increased around remyelinated CNS axons in mice with sustained activation of ERK1/2
To determine the effect of sustained ERK1/2 activation on adult myelin repair, a focal area of demyelination was induced in CNP-Cre;Mek1DD and control littermate mice via LPC injection into the midline of the dorsal spinal cord. In this model, demyelination occurs rapidly and the lesioned area is largely devoid of myelin within 2 d. Spontaneous remyelination (characterized by thin myelin sheaths) is apparent after 14 d in WT mice (Jeffery and Blakemore, 1995; Blakemore and Franklin, 2008). Cre-mediated recombination and upregulation of the activated form of ERK1/2 (pERK1/2) in OLs after demyelinating injury was confirmed in the CNP-Cre;Mek1DD mice by immunohistochemistry using triple labeling with antibodies against GFP, CC1, and pERK1/2 (Fig. 3A). Similar to early postnatal development, we saw a near complete overlap of GFP and CC1 demonstrating that nearly all (96 ± 1.2%) OLs expressed MEK1DD-EGFP but that only 66 ± 4.2% of OLs expressed pERK1/2. In contrast, only 11 ± 2.3% of OLs expressed pERK1/2 in the control mice revealing a significant upregulation of OL-specific pERK1/2 expression in CNP-Cre;Mek1DD mice (p = 2.2 × 10−5). Analysis of 1 μm sections and by EM 7 weeks after LPC injection revealed that remyelination was complete in both mutant and control mice; however, the majority of remyelinated axons in CNP-Cre;Mek1DD mice were covered by strikingly thicker myelin compared with remyelinated axons of comparable size in controls (Fig. 3B,F). Average g-ratios were significantly lower in CNP-Cre;Mek1DD mice (control = 0.800 ± 0.007 vs CNP-Cre;Mek1DD = 0.681 ± 0.011, p = 2.4 × 10−15; Fig. 3G) revealing that the upregulation of pERK1/2, specifically in cells of the OL lineage, is sufficient to drive the production of thick myelin sheaths surrounding remyelinated axons. When individual g-ratios were plotted, two populations were evident in the CNP-Cre;Mek1DD mice: one that overlapped with the control samples (population A) and one that did not (population B) (Fig. 3C–E). These results likely reflect the fact that only 66% of mutant OLs were found to express pERK1/2 after demyelination. Comparison of average g-ratios from uninjured control mice versus remyelinated control mice and uninjured CNP-Cre;Mek1DD versus remyelinated CNP-Cre;Mek1DD mice revealed a similar increase in g-ratio as a result of remyelination in both genotypes (Fig. 3G). Importantly, however, when the average g-ratio of axons from CNP-Cre;Mek1DD mice that cluster in population B is considered separately; there is no significant difference between uninjured (0.647 ± 0.006) and remyelinated axons (0.628 ± 0.009, p = 0.08), demonstrating that around a significant number of axons in the mutant mice, myelin thickness returns to baseline levels following remyelination.
Figure 3.

Increased levels of pERK1/2 are sufficient to drive the production of thick myelin sheaths following remyelination. A, Transverse sections of remyelinating spinal cords were immunolabeled for GFP, CC1, and pERK1/2. White arrows mark mature CC1+ OLs that express MEK1DD-EGFP and high levels of pERK1/2 in the CNP-Cre;Mek1DD mice. A significantly smaller number of OLs in the control (CTRL) mice express pERK1/2 at 7 weeks postlesion (7wpl; white arrows). A very small number of CC1+ OLs in the mutant mice do not express MEK1DD (asterisk). White arrowheads identify a MEK1DD-expressing OL that does not show pERK1/2 expression. B, Toluidine blue staining of 1 μm sections from the dorsal spinal cord at 7 wpl demonstrates thin myelin sheaths around remyelinated axons in control littermates (dashed circle). Two populations of OLs were detected in CNP-Cre;Mek1DD mice. OLs with very large cell bodies are surrounded by thick myelin sheaths (solid circle) whereas smaller OLs are surrounded by remyelinated axons with thin myelin sheaths (dashed circle). Black arrowheads indicate the OL cell body. C, Scatter plot analysis of individual g-ratios from mutant mice also demonstrates segregation into two groups: population A (above the dashed line) and population B (below the dashed line). Histogram analysis shows the percentage of axons with specific ranges of g-ratios from (D) control or (E) CNP-Cre;Mek1DD mice. B marks population B and A marks population A. F, Analysis by EM confirms the findings from toluidine blue staining. Blue arrowheads point to axons in mutant and control mice that are covered by myelin sheaths of comparable thickness. G, Comparison of average g-ratios from uninjured adult mice at P120 and mice at 7 wpl. **p < 0.001, *p < 0.01. Error bars indicate SEM. Scale bars: A, 50 μm; B, 25 μm; F, 2 μm.
Increased levels of activated ERK1/2 in OLs leads to accelerated myelin repair
To determine whether increased levels of pERK1/2 led to an accelerated rate of myelin repair, lesioned tissue was analyzed at 7 d postlesion (dpl). The average lesion size for CNP-Cre;Mek1DD was significantly smaller compared with controls (control = 5808 μm ±568 vs mutant = 2880 μm ± 337, p = 0.004), suggesting accelerated remyelination (Fig. 4A,B). Furthermore, a striking increase in MBP expression was noted, especially along the lesion borders in the CNP-Cre;Mek1DD mice (Fig. 4C,D). The early appearance of MBP-positive cells in the lesioned area suggested that elevated pERK1/2 could be promoting the accelerated differentiation of OPCs. To address this possibility the expression of the mature OL marker CC1 was assayed at 7 dpl. No significant difference in the number of CC1+ mature OLs was detected between CNP-Cre;Mek1DD mice (101 ± 4.7) and controls (93 ± 10.7, p = 0.5), demonstrating that the increased expression of MBP was a result of early myelin protein production by individual OLs and not likely due to accelerated OPC differentiation. Analysis of 1 μm sections at 14 dpl showed that the percentage of axons that had undergone remyelination around the lesion border was significantly higher in CNP-Cre;Mek1DD mice (85% ± 0.9) compared with control littermates (54% ± 6.7, p = 0.018), confirming that the early expression of MBP seen at 7 dpl leads to the accelerated production of new myelin sheaths (Fig. 4E,F).
Figure 4.
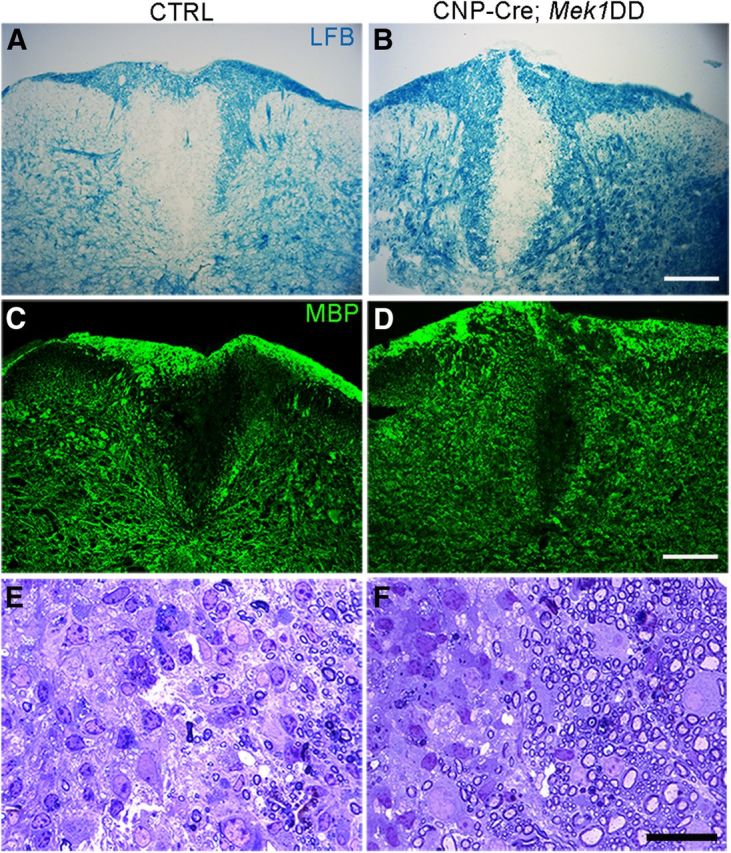
Upregulation of pERK1/2 leads to accelerated remyelination. A, B, Labeling of myelin by luxol fast blue (LFB) from adult CNP-Cre;Mek1DD mice at 7 dpl demonstrates a significantly smaller area of demyelination compared with controls (CTRL; n = 5 control and n = 4 mutants). C, D, Immunostaining reveals increased expression of MBP along the lesion border in CNP-Cre;Mek1DD mice at 7 dpl. E, F, Toluidine blue staining of thin sections shows significantly more remyelinated axons along the lesion border in CNP-Cre;Mek1DD mice at 14 dpl (n = 4 control and n = 3 mutants). Scale bars: A–D, 100 μm; E, F, 25 μm.
CNP-Cre;Mek1DD mice display a similar acute response to demyelination as littermate controls
Smaller lesion sizes in CNP-Cre;Mek1DD mice could be explained if increased baseline myelin thickness rendered the mice less sensitive or resistant to the demyelinating effects of LPC. To address this question we examined the acute response of adult CNP-Cre; Mek1DD mice to LPC-induced demyelination. At 2 dpl, lesion sizes were similar between CNP-Cre; Mek1DD mice (4673 μm ± 897, n = 3) and control littermates (4200 μm ± 738, p = 0.703, n = 4), demonstrating that both mutant and WT mice are equally susceptible to LPC-induced demyelination. Furthermore, both the total number of OLIG2+ OPCs (control = 82 ± 11 vs mutant = 85 ± 5.6, p = 0.843) and the percentage of Ki67+/OLIG2+ proliferating OPCs (control = 36% ± 1.2 vs mutant = 37% ± 2.4, p = 0.964) in the lesioned area were similar between control and CNP-Cre;Mek1DD mice, demonstrating that the upregulation of pERK1/2 does not lead to changes in OPC survival or proliferation.
Discussion
Over 50 years ago, Rushton (1950) proposed that for a given axon diameter, there is a specific myelin thickness that maximizes conduction velocity. Alterations in this ratio affect the speed of nerve impulse transmission and thus the timing of neuronal signals, potentially leading to aberrant circuit connectivity, and/or rendering axons vulnerable to damage. Myelin sheath thickness must therefore be carefully controlled in the CNS. For reasons that remain unknown, demyelinated lesions that have undergone remyelination in both mouse models as well as in patients with MS fail to achieve this optimal ratio and are characterized by the presence of very thin myelin sheaths around axons regardless of their diameter. Here, we provide what is to our knowledge the first demonstration of a signaling pathway critical for the control of CNS myelin thickness in the adult mouse spinal cord after demyelinating injury. Increased expression of the activated form of ERK1/2 (pERK1/2), specifically in CNP+ cells, was sufficient for the generation of thick myelin sheaths surrounding remyelinated axons. Interestingly, however, not all remyelinated axons in the CNP-Cre;Mek1DD mice were covered by thick myelin sheaths (note the large scatter of g-ratios in Fig. 3C). In Figure 2 we demonstrated that hypermyelinated CNP-Cre;Mek1DD mice have OL cell bodies of increased size compared with littermate controls. Following demyelination, the smaller OLs in the CNP-Cre;Mek1DD mice were often surrounded by axons with thin myelin sheaths (Fig. 3B, dashed circle; C, population A on scatter plot), while large OL cell bodies were surrounded by hypermyelinated axons suggesting that increased cell size may contribute to enhanced myelination. The two populations of g-ratios may be explained by the fact that only 66% of CNP-Cre;Mek1DD OLs were found to upregulate pERK1/2 in the dorsal spinal cord following demyelination. The ERK MAP kinase cascade is involved in multiple important cellular processes, and therefore potent negative feedback mechanisms exist to ensure tight regulation of this pathway. It is therefore not surprising that high levels of pERK1/2 are not constantly maintained in 100% of OLs in the mutant mice, especially after demyelinating injury. One complication with this interpretation is that a similar percentage of OLs upregulate pERK1/2 during development (71%), and yet there does not appear to be an obvious separation of g-ratios into two groups. These data suggest that there may be important differences between how the remyelinating OLs that are derived from adult OPCs and the OLs derived from OPCs during development respond to increased ERK MAP kinase signaling. There may be heterogeneity within the OL population that is derived from adult OPCs such that specific downstream targets of ERK1/2 MAP kinase necessary to drive myelin sheath growth could be differentially expressed in particular OL subpopulations following injury.
ERK1/2 are known to integrate and transduce multiple upstream signals, and thus elucidation of the molecular mechanisms by which these proteins promote increased myelin thickness in the setting of demyelinating injury will likely be challenging. One promising candidate for an upstream regulator of this pathway is the epidermal growth factor receptor (EGFR). CNP promoter-driven overexpression of the human EGFR has been shown to result in increased myelin thickness around remyelinated axons after focal demyelination in the corpus callosum (Aguirre et al., 2007). G-ratios in this study were calculated at a much earlier time point in the repair process (21 dpl) compared with what we report here (7 weeks post lesion), raising the possibility that the increased myelin thickness they observed may simply have been a consequence of accelerated myelin repair compared with WT littermates. If myelin thickness in mice that overexpress EGFR remains elevated at later time points once remyelination is complete in WT mice, these data would be consistent with what we report here, suggesting that EGFR may be at least one of the important upstream receptors that signals through the ERK MAP kinase pathway to control myelin thickness during repair.
In addition to thicker myelin sheaths, we observed that the increased activation of ERK1/2 resulted in accelerated myelin repair. CNP-Cre;Mek1DD mice were found to express MBP throughout all but the very center of the lesioned area at 7 dpl, a time point quite early in the repair process in the spinal cord. This early increase in MBP expression resulted in the accelerated remyelination noted at 14 dpl. The increased expression of MBP did not reflect early differentiation of OPCs, however, since the number of CC1+ mature OLs was not increased in the mutant mice at this time point. Together, these data demonstrate that after demyelination in the adult CNS, sustained activation of ERK1/2 in CNP+ OL lineage cells is sufficient to drive early expression of the major myelin protein, MBP, leading to the accelerated production of new myelin sheaths that are much thicker than their WT counterparts, while the number of mature OLs remains unchanged. We have also demonstrated that OPC proliferation and survival appear to be unaffected by the sustained activation of ERK1/2 at 2 dpl, suggesting that the primary role for ERK MAP kinase signaling in this process is to promote myelin growth.
The data presented here, along with our previous studies showing that in the absence of Erk1/2 thin myelin sheaths are generated around axons of all diameters (Ishii et al., 2012), have led us to propose a model where ERK1/2 MAP kinase signaling serves as a rheostat for establishing axonal appropriate thickness of CNS myelin sheaths. According to this model, subtle changes in the intensity of ERK1/2 activation control the precise amount of myelin generated around each axon. During development, the threshold for ERK1/2 activation is likely to be low, ensuring relatively high levels of pERK1/2 to drive appropriate myelin thickness. In the adult following demyelination, we propose that an increased threshold for ERK1/2 activation results in thin myelin sheaths around axons of all sizes. Future studies will address whether perturbations in ERK MAP kinase signaling characterize demyelinated lesions in human tissue.
Our current findings suggest that therapies targeted to upregulate signaling through this pathway may prove useful not only to drive accelerated remyelination, but also to generate thicker myelin sheaths potentially rendering CNS axons less vulnerable to future episodes of demyelination. An important future goal is to determine the nature of the signals that activate ERK1/2 MAP kinase signaling to convey information regarding axon size and to elucidate the downstream targets of ERK1/2 that upregulate myelin-specific genes, thereby controlling myelin thickness in the CNS.
Footnotes
This work was supported by an NMSS Career-Transition Fellowship to S.L.F.-M. as well as National Institutes of Health (NIH) NS30800 to R.H.M. We thank Dr. K. Nave (Max Plank Institute, Germany) for the CNP-Cre mice. We would also like to thank K. Wyatt and M. Hitomi for help with electron microscopy and J. Hecker for technical assistance.
The authors declare no competing financial interests.
References
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/S0022-510X(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Blakemore WF. Pattern of remyelination in the CNS. Nature. 1974;249:577–578. doi: 10.1038/249577a0. [DOI] [PubMed] [Google Scholar]
- Blakemore WF, Franklin RJ. Remyelination in experimental models of toxin-induced demyelination. Curr Top Microbiol Immunol. 2008;318:193–212. doi: 10.1007/978-3-540-73677-6_8. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28:7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe-Maricich SL, Karlo JC, Landreth GE, Miller RH. The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J Neurosci. 2011;31:843–850. doi: 10.1523/JNEUROSCI.3239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Oltrogge JH, Kemper R, Heilmann I, Bormuth I, Wolfer S, Wichert SP, Möbius W, Liu X, Lappe-Siefke C, Rossner MJ, Groszer M, Suter U, Frahm J, Boretius S, Nave KA. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J Neurosci. 2010;30:8953–8964. doi: 10.1523/JNEUROSCI.0219-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington EP, Zhao C, Fancy SP, Kaing S, Franklin RJ, Rowitch DH. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann Neurol. 2010;68:703–716. doi: 10.1002/ana.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, Bansal R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci. 2012;32:8855–8864. doi: 10.1523/JNEUROSCI.0137-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Bansal R. Sustained activation of ERK1/2 MAPK in oligodendrocytes and Schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. J Neurosci. 2013;33:175–186. doi: 10.1523/JNEUROSCI.4403-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery ND, Blakemore WF. Remyelination of mouse spinal cord axons demyelinated by local injection of lysolecithin. J Neurocytol. 1995;24:775–781. doi: 10.1007/BF01191213. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Ludwin SK, Maitland M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J Neurol Sci. 1984;64:193–198. doi: 10.1016/0022-510X(84)90037-6. [DOI] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prineas JW, Barnard RO, Revesz T, Kwon EE, Sharer L, Cho ES. Multiple sclerosis. Pathology of recurrent lesions. Brain. 1993;116:681–693. doi: 10.1093/brain/116.3.681. [DOI] [PubMed] [Google Scholar]
- Psachoulia K, Jamen F, Young KM, Richardson WD. Cell cycle dynamics of NG2 cells in the postnatal and ageing brain. Neuron Glia Biol. 2009;5:57–67. doi: 10.1017/S1740925X09990354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushton WA. A theory of the effects of fibre size in medullated nerve. J Physiol. 1951;115:101–122. doi: 10.1113/jphysiol.1951.sp004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Andrews JM, Waltz JM, Terry RD. Ultrastructural studies of multiple sclerosis. Lab Invest. 1969;20:444–454. [PubMed] [Google Scholar]
- Tyler WA, Gangoli N, Gokina P, Kim HA, Covey M, Levison SW, Wood TL. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. 2009;29:6367–6378. doi: 10.1523/JNEUROSCI.0234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Ferner AH, Wong AW, Denham M, Kilpatrick TJ, Murray SS. Extracellular signal-regulated kinase 1/2 signaling promotes oligodendrocyte myelination in vitro. J Neurochem. 2012;122:1167–1180. doi: 10.1111/j.1471-4159.2012.07871.x. [DOI] [PubMed] [Google Scholar]
- Ye P, Carson J, D'Ercole AJ. In vivo actions of insulin-like growth factor-I (IGF-I) on brain myelination: studies of IGF-I and IGF binding protein-1 (IGFBP-1) transgenic mice. J Neurosci. 1995;15:7344–7356. doi: 10.1523/JNEUROSCI.15-11-07344.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]