

Abstract
Astrocytic glutamate transporter-1 (GLT-I) is critical to control the bulk of glutamate uptake and, thus, to regulate synaptic plasticity and excitotoxicity. GLT-I glutamate uptake is driven by the sodium gradient implemented by Na+/K+-ATPases (NKAs) and the α2 subunit of NKA (NKA-α2) is actually linked to GLT-I to regulate astrocytic glutamate transport. We recently found that adenosine A2A receptors (A2ARs), which control synaptic plasticity and neurodegeneration, regulate glutamate uptake through unknown mechanisms. Here we report that A2AR activation decreases NKA activity selectively in astrocytes to inhibit glutamate uptake. Furthermore, we found a physical association of A2ARs with NKA-α2s in astrocytes, as gauged by coimmunoprecipitation and in situ proximity ligation assays, in the cerebral cortex and striatum, two brain regions where A2ARs inhibit the astrocytic glutamate uptake. Moreover, the selective deletion of A2ARs in astrocytes (using Gfa2-A2AR-KO mice) leads to a concurrent increase of both astrocytic glutamate uptake and NKA-α2 levels and activity in the striatum and cortex. This coupling of astrocytic A2ARs to the regulation of glutamate transport through modulation of NKA-α2 activity provides a novel mechanism linking neuronal activity to ion homeostasis controlling glutamatergic activity, all of which are processes intricately associated with the etiology of several brain diseases.
Introduction
Glutamate is the most abundant neurotransmitter, mediating nearly 80% of synaptic transmission in the brain (Benarroch, 2010). To manage the rapid extracellular buildup and prevent the harmful consequences of overstimulating glutamate receptors, an efficient transport system dynamically regulates the extracellular glutamate levels, thus preventing glutamate accumulation and “spillover” between neighboring synapses (Dunlop, 2006). The astroglial-specific glutamate transporter-I subtype (GLT-I) is the dominant glutamate transporter in the adult brain. This transporter's importance is underscored by the impact of modifying GLT-I activity on synaptic plasticity as well as on neurodegeneration (Sattler and Rothstein, 2006). GLT-Is are Na+-dependent transporters, relying on the Na+ electrochemical gradient generated by Na+/K+-ATPases (NKAs) to drive glutamate uptake (Anderson and Swanson, 2000). NKAs comprise a class of ubiquitous plasma membrane enzymes responsible for maintaining the membrane potential of cells using the energy of adenosine triphosphate (ATP) hydrolysis (Reinhard et al., 2013). A functional NKA consists of a catalytic α-subunit harboring the ATP-binding sites and a smaller β-subunit required for full enzymatic activity and also functioning as an anchoring protein (Aperia, 2007). In the brain, three different α-subunit isoforms are present in a cell-specific manner: the low-affinity α1 is present in all cell types, the high-affinity α2 isoform is restricted to astrocytes, and the high-affinity α3 isoform is expressed exclusively in neurons (Benarroch, 2011). Thus, it is not surprising that NKA activity and specifically the α2 isoform has emerged as a robust modulator of glutamate uptake in astrocytes, as heralded by the observations that (1) ATP depletion leads to a reversal of glutamate uptake (Longuemare et al., 1999); (2) inhibitors of NKA, such as ouabain, impair glutamate transporter activity (Pellerin and Magistretti, 1997; Rose et al., 2009; Genda et al., 2011) and lead to glutamate transporter clustering and redistribution (Nakagawa et al., 2008; Nguyen et al., 2010); and (3) the α2 subunit of NKA colocalizes and physically associates in the same protein complex with glutamate transporters (Cholet et al., 2002; Rose et al., 2009; Genda et al., 2011).
We have previously shown that adenosine, a classical and ubiquitous modulator of synaptic transmission (Fredholm et al., 2005), by activating astrocytic adenosine A2A receptors (A2ARs), controls the uptake of glutamate through a dual mechanism (Matos et al., 2012b): a long-term activation of A2AR triggers a cAMP/protein kinase A-dependent decrease of the expression of GLT-I and glutamate-aspartate transporter (GLAST) before the reduction of the levels and activity of both transporters (Matos et al., 2012b), whereas the acute short-term activation of astrocytic A2ARs decreases the activity of glutamate transporters through an unknown mechanism that might depend on the physical proximity of A2ARs and GLT-I (Matos et al., 2012b). We have now tackled the mechanism of A2AR-mediated inhibition of the astrocytic glutamate transport, which was found to depend on a physical association and modulation by A2ARs of NKA-α2 in astrocytes. This provides the first demonstration that A2ARs control ion homeostasis in astrocytes, paving the way to understand the broad neuroprotective impact of A2AR antagonists in different brain disorders (Gomes et al., 2011).
Materials and Methods
Animals.
Initial experiments were performed using adult (2–3 months old) male C57BL/6 mice. We also used glial fibrillary acidic protein (GFAP) gene promoter-driven A2AR conditional knock-out (Gfa2-A2AR-KO) mice, which were generated using the Cre/loxP system, as previously described (Matos et al., 2012b). The Gfa2-Cre line was obtained from David Gutmann (Department Neurology, Washington University School of Medicine, St. Louis, Missouri) using the gfa2 transgene construct (Bajenaru et al., 2002). The transgene construct consists of the 2.2 kb fragment of the human GFAP promoter (Gfa2; obtained from M. Brenner, National Institute of Neurological Disorders and Stroke) coupled to the encephalomyocarditis virus IRES and to a cDNA encoding the nucleus-targeted Cre recombinase (for details, see Lee et al., 2006, 2008). The 55 bp segment of the gfa2 promoter, spanning bp 21488 to 21434 with respect to the RNA start site, has been shown to contain a 45 bp sequence spanning bp 21443 to 21399 required for silencing expression in neurons. Thus, the specific Gfa2 promoter, in opposition to other GFAP promoter constructs, has been elegantly shown as astrocyte-specific in all CNS regions (Lee et al., 2008). Briefly, both transgenic Gfa2-cre mice (Bajenaru et al., 2002) and mice carrying the “floxed” A2AR gene (A2Aflox/flox; Bastia et al., 2005) were back-crossed for 10–12 generations to C57BL/6 mice (Charles River). Gfa2-cre mice were then crossed with nontransgenic (no cre) A2Aflox/flox mice to generate Gfa2-A2AR-KO and Gfa2-A2AR-WT mice. Animals were maintained in a controlled environment (23 ± 2°C; 12 h light/dark cycle; ad libitum access to food and water) and handled according to the Animal Care and Use Committee at Boston University School of Medicine and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (1982).
Preparation of total membranes.
Mice were killed by decapitation after deep anesthesia with isoflurane and cortical and striatal brain tissue was collected and homogenized in sucrose (0.32 m) solution [containing 1 mm EDTA, 10 mm HEPES, 1 mg/ml bovine serum albumin (BSA; Sigma-Aldrich), pH 7.4] at 4°C. The homogenates were centrifuged at 3000 × g for 10 min at 4°C and the resulting supernatants were centrifuged again at 14,000 × g for 10 min at 4°C. The pellets were washed in Krebs-HEPES-Ringer solution (140 mm NaCl, 1 mm EDTA, 10 mm HEPES, 5 mm KCl, 5 mm glucose, pH 7.4) at 4°C and further centrifuged at 14,000 × g for 10 min at 4°C. The pellets were resuspended in RIPA buffer (150 mm NaCl, 1.0% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mm Tris, pH 8.0) with protease inhibitor mixture (CLAPS, composed of 10 μg/ml chymostatin, leupeptin, antipain, and pepstatin A; Sigma-Aldrich. The protein content was then measured with the bicinchoninic acid (BCA) assay (Thermo Scientific).
Preparation of gliosomes and synaptosomes.
After the homogenization of the brain tissue (cortex or striatum), purified synaptosomes and gliosomes were obtained using a discontinuous Percoll gradient (2, 6, 15, and 23% v/v of Percoll in a medium containing 0.32 m sucrose and 1 mm EDTA, pH 7.4), as previously described (Matos et al., 2012b). The layers between 2 and 6% of Percoll (gliosomal fraction) and between 15 and 23% of Percoll (purified presynaptic nerve terminals, i.e., synaptosomal fraction) were collected, washed in 10 ml of HEPES buffered medium (140 mm NaCl, 5 mm KCl, 5 mm NaHCO3, 1.2 mm NaH2PO4, 1 mm MgCl2, 10 mm glucose, 10 mm HEPES, pH 7.4) and further centrifuged at 22,000 × g for 15 min at 4°C to remove myelin components and postsynaptic material from the gliosomal and synaptosomal fractions, respectively. Crude synaptosomes were prepared after consecutive differential centrifugations of the brain homogenate in sucrose solution and in a 45% Percoll solution at 4°C (Canas et al., 2009). The fractions were resuspended either in Krebs buffer containing (in mm) 132 NaCl, 4 KCl, 1.2 Na2HPO4, 1.4 MgCl2, 6 glucose, 10 HEPES, 1 CaCl2, pH 7.4) or in N-methylglucamine (NMG) buffer, which is identical to Krebs buffer except that NaCl is isosmotically replaced by NMG.
NKA activity assay.
NKA activity in synaptosomes and gliosomes was measured using a high-sensitivity colorimetric ATPase assay kit following the manufacturer's instructions (Innova Biosciences). Gliosomes or synaptosomes (20 μg) were incubated with the reaction buffer containing 100 mm Tris, 1 mm ATP, and 5 mm MgCl2, pH 7.4, in the absence or in the presence of ouabain (0.01 μm–2 mm), [[6-amino-9-(N-ethyl-β-d-ribofuranuronamidosyl)-9H-purin-2 yl]amino]ethyl]benzene propanoic acid hydrochloride (CGS 21680; 30–100 nm) and/or 2-(2-furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine (SCH 58261; 50 nm) for 30 min, at 37°C. The amount of inorganic phosphate (Pi) released was quantified colorimetrically at 630 nm, as previously described (Sarkar, 2002; Nguyen et al., 2010) and the protein content measured with the BCA assay. The specific activity of NKA was calculated by subtracting the ouabain-insensitive activity from the overall activity (in the absence of ouabain) and expressed as μmol Pi liberated from ATP by 1 μg of protein (μmol Pi/μg protein).
[3H]d-aspartate uptake.
The uptake of the nonmetabolizable glutamate analog [3H]d-aspartate is a validated readout of the activity of glutamate transporters (Anderson and Swanson, 2000) and was performed as previously described (Matos et al., 2012a, b). Briefly, the gliosomal or synaptosomal fractions were diluted in Krebs or NMG buffer and equilibrated at 37°C for 10 min. Triplicates (150 μl) of each fractions were added to 150 μl of Krebs or NMG medium containing a final concentration of 50 nm [3H]d-aspartate (11.3 Ci/mmol; PerkinElmer). The mixtures were incubated for 10 min at 37°C and the reaction terminated by rapid vacuum filtration over Whatman GF/C glass microfiber filters (GE Healthcare) and further washed three times with ice-cold NMG buffer. Filters were dried overnight, drenched in 2 ml of liquid scintillation mixture (PerkinElmer), and counted on a LKB Wallac 1219 liquid scintillation counter (Wallac). The specific uptake of [3H]d-aspartate was calculated by subtraction from the total uptake of the nonspecific uptake measured in a Na+-free medium (NMG).
Drug treatments.
The selective A2AR agonist CGS 21680 (Tocris Bioscience), the A2AR antagonist SCH 58261 (Tocris Bioscience), and the NKA inhibitor ouabain octahydrate (Tocris Bioscience) were added to synaptosomes and gliosomes to reach final concentrations of 100 nm, 50 nm, and 1 mm (or other when specified), respectively, at 30 min before the [3H]d-aspartate uptake and the NKA activity assays, as previously described (Matos et al., 2012a, b).
Coimmunoprecipitation.
Coimmunoprecipitation was performed as previously described (Ciruela et al., 2006). Briefly, total membranes from the cortex or striatum were prepared as described above and washed in PBS (140 mm NaCl, 3 mm KCl, 20 mm Na2HPO4, 1.5 mm KH2PO4, pH 7.4) before centrifugation at 14,000 × g for 10 min at 4°C. The pellets were resuspended in the immunoprecipitation buffer (IPB; containing 20 mm Tris, pH 7.0, 100 mm NaCl, 2 mm EDTA, 2 mm EGTA, 50 mm NaF, 1 mm sodium orthovanadate, 1 μm okadaic acid, 0.1 mm PMSF, and 1:1000 protease inhibitor mixture) with 1% Triton X-100, sonicated for 30 s on ice and further spun down for 10 min to remove insoluble materials. A sample was collected for determining protein concentration using the BCA assay, another was stored at −20°C as input (positive control), and the rest was processed for immunoprecipitation at a dilution of 0.5 mg/ml. Protein A Sepharose beads were incubated with the sample for 1 h at 4°C under rotation to preabsorb any protein that nonspecifically bound to the protein A Sepharose beads. The supernatant was recovered by centrifugation and 3 μg of anti-A2AR antibody (Millipore) or irrelevant IgG (for negative control) were added and incubated for 3 h at 4°C under rotation. To pool-down the immune complexes, the samples were incubated with protein A Sepharose beads for 2 h at 4°C and centrifuged. The pellets were washed twice in IPB with 1% Triton X-100, three times in IPB with 1% Triton X-100 and 500 mm NaCl, and twice in IPB. The immunoprecipitates were resolved by SDS-PAGE buffer, and Western blots were performed with anti-NKA-α2 isoform or anti-GLT-I/EAAT2 antibodies (see Western blot).
Western blot.
Western blotting of gliosomal or synaptosomal extracts was performed as previously described (Canas et al., 2009; Matos et al., 2012a). Incubation with the primary antibodies, namely anti-A2AR (1:200; Millipore), anti-GLT-I/EAAT2 (1:1000; Millipore), anti-NKA-α2 isoform (1:200; Millipore), and anti-β-actin (1:5000; Sigma-Aldrich), all diluted in Tris-buffered saline (TBS; 137 mm NaCl, 20 mm Tris-HCl, pH 7.6) with 0.1% Tween and 3% BSA (fatty acid free), was performed overnight at 4°C. After washing, the membranes were revealed using an enhanced chemifluorescence kit (GE Healthcare) and visualized under a fluorescence LAS-4000 digital imaging system (Fujifilm). The densiometric analysis of protein bands was performed using Quantity One software version 4.4.1 (Bio-Rad).
Immunohistochemistry.
Immunohistochemistry in brain slices was performed as described previously (Canas et al., 2009). After a transcardiac perfusion, the brains were postfixed overnight in PBS with 4% paraformaldehyde and cryopreserved in PBS containing 25% sucrose. The frozen brains were sectioned (30 μm coronal slices) with a Leica CM3050S cryostat (Leica Microsystems). The sections corresponding to cortex and striatum were permeabilized, blocked, and incubated overnight at room temperature in the presence of goat polyclonal anti-NKA-α2 isoform antibody (1:500) and mouse monoclonal anti-GLT-I/EAAT2 (1:1000) antibody. The sections were subsequently incubated with donkey anti-mouse and anti-goat secondary antibody conjugated with a fluorophore (Alexa Fluor 488 or Alexa Fluor 555, 1:200; Invitrogen) for 2 h at room temperature. After rinsing, the sections were mounted on slides and allowed to dry. Vectashield mounting medium with DAPI (Vector Laboratories) was applied as well as the cover glass. All sections were examined under a fluorescence Nikon eclipse E600 microscope, with SPOT software 4.7 (Diagnostic Instruments).
In situ proximity ligation assay.
The proximity ligation assay (PLA) was performed as previously described (Söderberg et al., 2006; Augusto et al., 2013) in brain sections from Gfa2-A2AR-KO and WT littermates prepared as described for immunohistochemistry. The sections were rinsed in TBS (0.1 m Tris, pH.7.4, and 0.9% w/v NaCl) and blocked with TBS with 10% fetal bovine serum and 0.5% Triton X-100 for 2 h at room temperature. Subsequently, the slices were incubated with goat polyclonal anti-NKA-α2 isoform antibody (1:500) and rabbit polyclonal anti-A2AR antibody (1:500) overnight at room temperature. After washing in TBS with 0.2% Triton X-100, the slices were incubated for 2 h at 37°C with the PLA secondary probes anti-rabbit Plus and anti-goat Minus (1:5; Olink Bioscience) under gentle agitation. Afterward, the slices were washed twice with Duolink II Wash Buffer A (Olink Bioscience) and incubated with the ligation-ligase solution (Olink Bioscience) for 30 min at 37°C. After a new rinse, the slices were incubated with DNA polymerase (1:40; Olink Bioscience) in the amplification solution (Olink Bioscience) for 100 min at 37°C. After several washes in consecutive decreasing concentrations of SSC buffers (Olink Bioscience), the slices were mounted on slides and allowed to dry. The coverslips were applied with Duolink Mounting Medium (Olink Bioscience). Fluorescence images were acquired on an Axiovert 200M inverted confocal microscope (Carl Zeiss Microscopy) using a 40× numerical aperture objective. The images were then analyzed and the PLA puncta signals quantified with ImageJ software. A threshold was selected manually to discriminate PLA puncta from background fluorescence. The built-in macro “Analyze Particles” was then used to count all objects in the thresholded image. Objects larger than 5 μm2 were rejected, thereby effectively removing nuclei. The remaining objects were counted as A2AR- NKA-α2 PLA-positive puncta.
Statistical data analysis.
Data are expressed as absolute or arbitrary values or percentages of values obtained in control conditions or conditions mentioned in the figures legends, and are presented as means ± SEM. Parametric ANOVA was used to determine statistically significant differences, with the indicated post hoc test. All data were analyzed using Prism software (Version 5.0, GraphPad).
Results
Activation of A2ARs decreases NKA activity in gliosomes
Since A2ARs control the uptake of glutamate by the astrocytic glutamate transporters GLT-I (Matos et al., 2012b) and the efficiency of glutamate transporters depend on the sodium gradient ensured by the activity of NKA (Benarroch, 2011), we tested the impact of A2AR activation on the activity of NKA in astrocytes and neurons. We first prepared gliosomes (astrocyte-enriched plasmalemmal vesicles) and synaptosomes (enriched nerve terminals) from the cerebral cortex of adult mice and challenged them with the selective A2AR agonist CGS 21680 and/or the A2AR antagonist SCH 58261 before determining NKA activity, assessed as the ouabain-sensitive ATP hydrolysis (Fig. 1). Activation of A2ARs in cortical gliosomes by CGS 21680 (at 100 nm, but not at lower concentrations of 30–50 nm) led to a 66.0 ± 4.0% decrease (n = 4, p < 0.01) of NKA activity in comparison with nontreated gliosomes (Fig. 1A); this effect was prevented (n = 4, p < 0.05) by the preadministration of SCH 58261 (50 nm; Fig. 1B). In contrast, CGS 21680 (100 nm) induced a 93.0 ± 13.0% increase (n = 4, p < 0.01) of the NKA activity in synaptosomes, which was prevented by SCH 58261 (n = 4, p < 0.01; Fig. 1A,B). A similar trend was observed in the striatum (Fig. 1C), another brain area where the A2AR modulation of glutamate uptake in astrocytes has been documented (Pintor et al., 2004). Thus, in striatal gliosomes, CGS 26180 (100 nm) decreased NKA activity by 36.0 ± 8.4% (n = 3, p < 0.05), an effect prevented by SCH 58261 (50 nm; n = 3, p < 0.05); in contrast, 100 nm CGS 26180 tended to increase (57.0 ± 27.0%, n = 3; p > 0.05) NKA activity in striatal synaptosomes (Fig. 1C).
Figure 1.

Activation of A2ARs leads to a selective decrease of the activities of both NKA and glutamate transporters in gliosomes but not in synaptosomes from either the cerebral cortex or striatum. Gliosomes and synaptosomes from brain cortex or striatum were incubated without or with the A2AR-selective agonist CGS 21680 (30–100 nm) and/or antagonist SCH 58261 (50 nm). A, The activation of A2ARs by CGS 21680 in cortical gliosomes (open symbols) reduces NKA activity, whereas it increases NKA activity in synaptosomes (closed symbols). B, C, These opposite effects of CGS 21680 (100 nm) on NKA activity were prevented by SCH 58261 in cortical gliosomes and synaptosomes (B) and in striatal gliosomes (C). D, E, The activation of A2ARs with CGS 21680 (30–100 nm) inhibited [3H]d-aspartate uptake both in cortical gliosomes and in synaptosomes (D) and SCH 58261 prevented this effect of CGS 21680 (100 nm; E). F, A2AR activation by CGS 21680 (100 nm) also inhibited [3H]d-aspartate uptake in striatal gliosomes, whereas no significant effects were observed in striatal synaptosomes. NKA activity was determined by subtracting the total ATPase activity from the ATPase activity in the presence of membrane ATPase inhibitor ouabain and was expressed as micromole Pi liberated from ATP by 1 μg of protein (μmol Pi/μg protein), whereas the specific uptake of [3H]d-aspartate was calculated by subtracting the uptake activity from the uptake activity in the presence of Na+-free buffer NMG and was expressed as nanomoles of [3H]d-aspartate retained per milligrams of gliosome protein per minute. Data are mean ± SEM of at least three independent experiments done in triplicate. Statistical differences were gauged using the Tukey's post hoc test applied after one-way ANOVA with *p < 0.05 and **p < 0.01, when compared with nontreated conditions.
Comparison of the effect of A2ARs on Na+/K+-ATPase activity and on d-aspartate uptake in gliosomes and synaptosomes
To explore a possible link between NKA activity and glutamate uptake, we began by comparing the impact of CGS 21680 and of SCH 58261 on NKA activity and on [3H]d-aspartate uptake in gliosomes and synaptosomes from either the cerebral cortex or of the striatum. As shown in Figure 1D, CGS 21680 (50–100 nm) inhibited [3H]d-aspartate uptake both in cortical gliosomes (79.2 ± 3.2% at 100 nm, n = 4; p < 0.001) as well as in cortical synaptosomes (26.4 ± 7.2% at 100 nm, n = 4; p < 0.05). This CGS 21680-induced inhibition was prevented by SCH 58261 in both cortical gliosomes (n = 4; p < 0.01) and cortical synaptosomes (n = 4; p < 0.01; Fig. 1E). A similar profile of A2AR-mediated inhibition of [3H]d-aspartate uptake was observed in gliosomes from the striatum (Fig. 1F).
Overall, these results (Fig. 1) show a parallel effect of A2ARs controlling NKA activity and the uptake of [3H]d-aspartate in gliosomes, whereas there is a qualitative dissociation between the impact of A2ARs on the activity of NKA and on glutamate uptake in synaptosomes, as would be expected since both NKA and glutamate transporter isoforms are different in astrocytes and in neurons.
Low concentrations of Na+/K+-ATPase-inhibitor ouabain blunt the A2AR-mediated inhibition of d-aspartate uptake in astrocytes
To strengthen the link between NKA activity and glutamate uptake in astrocytes, we next analyzed the concentration-dependent effect of the NKA inhibitor ouabain both on NKA activity (Fig. 2A) and on [3H]d-aspartate uptake (Fig. 2B) in gliosomes from the cerebral cortex of adult mice, where the uptake of [3H]d-aspartate was nearly twice greater than in striatal gliosomes (Fig. 1, compare E, F) and where NKA and [3H]d-aspartate uptake were similarly modulated by A2ARs (Fig. 1, compare A, D). Ouabain caused a bimodal but parallel impact on the activities of both NKA (Fig. 2A) and of glutamate transporters (Fig. 2B) in cortical gliosomes. Thus, a low ouabain concentration (0.1 μm) induced a 40.0 ± 5.0% increase (n = 4, p < 0.05) of NKA activity compared with nontreated gliosomes, in agreement with previous reports (Lichtstein et al., 1985; Gao et al., 2002; Antolović, 2006) and a low/moderate concentration of ouabain (1 μm) had no effect on NKA activity. Meanwhile, moderate/higher concentrations (10–100 μm) inhibited NKA activity (n = 4, p < 0.05), and a higher concentration (2 mm) of ouabain caused a 73.0 ± 11.2% inhibition (n = 4, p < 0.01) of NKA activity (Fig. 2A). In accordance with the key NKA-mediated control of GLT-I activity, a low ouabain concentration (0.1 μm) increased [3H]d-aspartate uptake by 26.1 ± 4.1% (n = 4, p < 0.05), a low/moderate concentration (1 μm) had no effect on [3H]d-aspartate uptake, a moderate/higher concentration (10 μm) inhibited (n = 4, p < 0.05) [3H]d-aspartate uptake, and a higher concentration (2 mm) inhibited [3H]d-aspartate uptake by 75.0 ± 9.0% (n = 4, p < 0.001; Fig. 2B), as previously observed (Pellerin and Magistretti, 1997; Rose et al., 2009).
Figure 2.
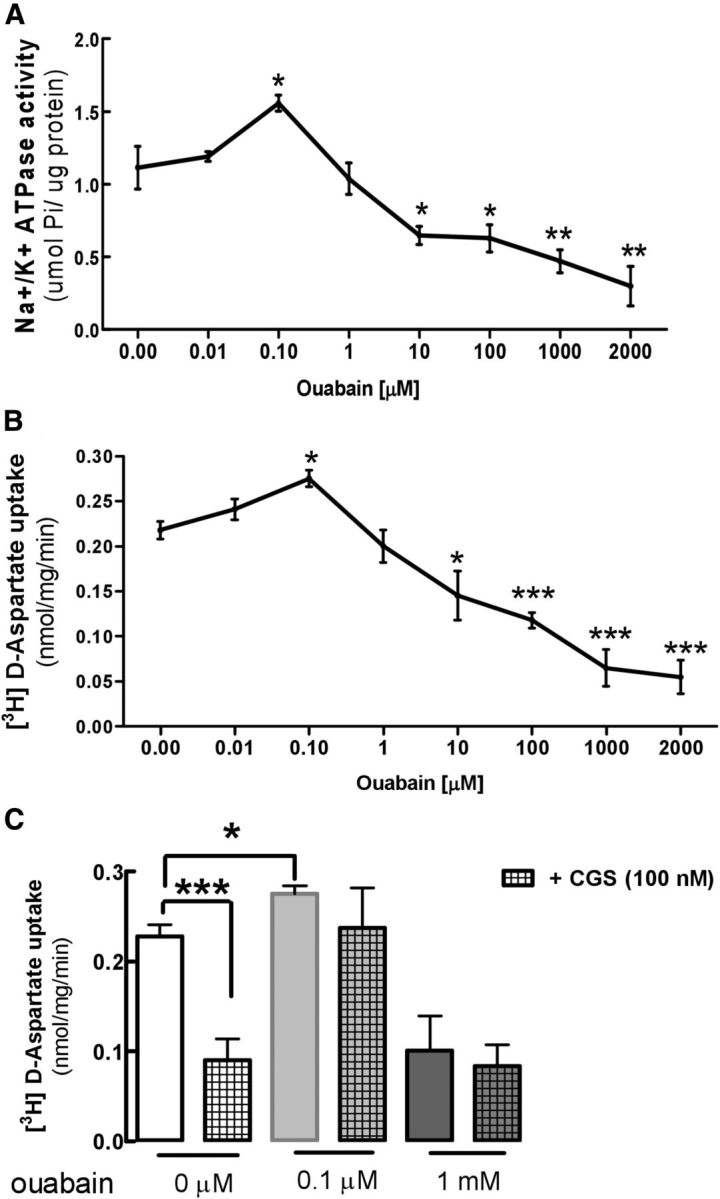
The NKA-inhibitor ouabain has a parallel impact on the activities of NKA and of glutamate transport and blunt the impact of A2ARs on [3H]d-aspartate uptake in cortical gliosomes. A, Concentration-dependent inhibition of NKA activity by ouabain in cerebral cortical gliosomes from WT mice. Ouabain at 0.1 μm enhanced NKA activity, but at >10 μm inhibited NKA activity. NKA activity was expressed as micromole Pi liberated from ATP by 1 μg of protein (μmol Pi/μg protein). B, Concentration-dependent inhibition of [3H]d-aspartate uptake in cerebral cortical gliosomes from WT mice. Ouabain at 0.1 μm enhanced [3H]d-aspartate uptake, but at >100 μm inhibited [3H]d-aspartate uptake. The specific uptake of [3H]d-aspartate was expressed as nanomoles of [3H]d-aspartate retained per milligram of gliosome protein per minute. C, Acute (30 min) incubation of cerebral cortical gliosomes with the A2AR-selective agonist CGS 21680 (100 nm) decreased [3H]d-aspartate uptake, an effect no longer observed upon perturbation of the activity of NKA by preincubation with either a low (0.1 μm) or a high (1 mm) concentration of ouabain. Data are the mean ± SEM of five independent experiments done in triplicate. Statistical difference was assessed using a two-way ANOVA analysis. *p < 0.05, **p < 0.01, ***p < 0.001, comparison with control-nontreated condition.
We next analyzed how a low and high concentration of ouabain affected the A2AR-induced inhibition of the astrocytic glutamate uptake. As depicted in Figure 2C, activation of A2ARs in cortical gliosomes with 100 nm CGS 21680 decreased [3H]d-aspartate uptake by 61.0 ± 1.1% (n = 5, p < 0.001), and this effect of CGS 21680 was blunted in the presence of either a low (0.1 μm) or a high (1 mm) concentration of ouabain. In fact, in the presence of 0.1 μm ouabain, the effect of CGS 21680 on [3H]d-aspartate uptake was the same as that occurring in the presence of 1 mm ouabain, and thus was no longer significant (Fig. 2C). These data show that the perturbation of NKA activity blunts the ability of A2ARs to control glutamate uptake, which suggests that astrocytic A2ARs may require NKA activity to rapidly modulate glutamate uptake. However, because NKA activity provides the driving force for glutamate uptake (among several other transport systems) in astrocytes, NKA activity may not be linearly related to GLT-I activity and, when affected with ouabain, will always influence the driving force of glutamate uptake and thus will indirectly alter the effects of CGS 21680 on glutamate uptake. Thus, it is difficult for activity studies or pharmacological studies to provide unequivocal evidence for this A2AR–NKA–GLT-I relationship.
Na+/K+ATPase activity is increased selectively in astrocytes from Gfa2-A2AR-KO mice
To better understand the association between A2ARs and NKAs to control astrocytic glutamate uptake, we next used Gfa2-A2AR-KO mice (Matos et al., 2012b) to investigate how the selective deletion of A2ARs in astrocytes affects NKA and GLT-I activities in astrocytes and neurons. As portrayed in Figure 3, gliosomes collected from the cortex (Fig. 3A) or striatum (Fig. 3B) of Gfa2-A2AR-KO mice displayed a significantly higher NKA activity than gliosomes collected from WT littermates (58.1 ± 9.0%, n = 4, p < 0.05 in the cortex; 33.1 ± 6.0%, n = 4, p < 0.05 in the striatum). In contrast, NKA activity was not significantly different in cortical (n = 4, p = 0.94) or striatal (n = 4, p = 0.24) synaptosomes from Gfa2-A2AR-KO or Gfa2-A2AR-WT mice. A similar analysis of the activity of glutamate transporters revealed that [3H]d-aspartate uptake was significantly increased (62.0 ± 7.2%, n = 4, p < 0.001) in cerebral cortical gliosomes, but not in synaptosomes (n = 4, p > 0.05), from Gfa2-A2AR-KO mice compared with WT littermates (Fig. 3C). Similarly, [3H]d-aspartate uptake was also selectively increased (44.0 ± 9.0%, n = 4, p < 0.01) in striatal gliosomes, but not in synaptosomes (n = 4, p > 0.05), from Gfa2-A2AR-KO mice compared with WT littermates (Fig. 3D). The observed parallel modification of NKA and glutamate uptake activities selectively in gliosomes of Gfa2-A2AR-KO mice is further suggestive of an astrocyte-selective coupling between A2ARs and NKAs to control glutamate uptake.
Figure 3.

NKA activity and glutamate uptake are increased in parallel selectively in gliosomes from the cortex or striatum of Gfa2-A2AR-KO mice. A–D, Gliosomes and synaptosomes from Gfa2-A2AR-KO mice and from corresponding WT littermates were prepared before the NKA activity (A, B) and the [3H]d-aspartate uptake (C, D) assays. The increased NKA activity was restricted to gliosomes from GFAP-A2AR-KO mice (black columns), particularly in the cortex (A) but also in the striatum (B), compared with WT mice (white columns). [3H]d-aspartate uptake was also selectively increased in gliosomes from the cortex (C) and striatum (D). Data are mean ± SEM of at least four independent experiments. Statistical differences were gauged using the Tukey's post hoc test applied after one-way ANOVA with *p < 0.05, **p < 0.01, and ***p < 0.001.
GLT-I and NKA-α2 immunoreactivities are increased in Gfa2-A2AR-KO mice
As a first step toward testing the hypothesis that A2ARs, NKA-α2s, and glutamate transporters might be physically associated in astrocytes, we compared the density and distribution of GLT-Is and NKA-α2s in the cerebral cortex and striatum from Gfa2-A2AR-KO mice and WT littermates (Fig. 4). Western blot analysis showed that the density of GLT-Is was significantly increased in the cortex (138.1 ± 4.4%; n = 6, p < 0.001) and striatum (121.1 ± 2.0%; n = 6, p < 0.01) of Gfa2-A2AR-KO compared with Gfa2-A2AR-WT mice (Fig. 4A,E). Notably, the density of NKA-α2s was also significantly increased in the cortex (156.0 ± 9.0%; n = 6, p < 0.001) and striatum (124.0 ± 7.0%; n = 6, p < 0.05) of Gfa2-A2AR-KO compared with WT mice (Fig. 4B,F).
Figure 4.
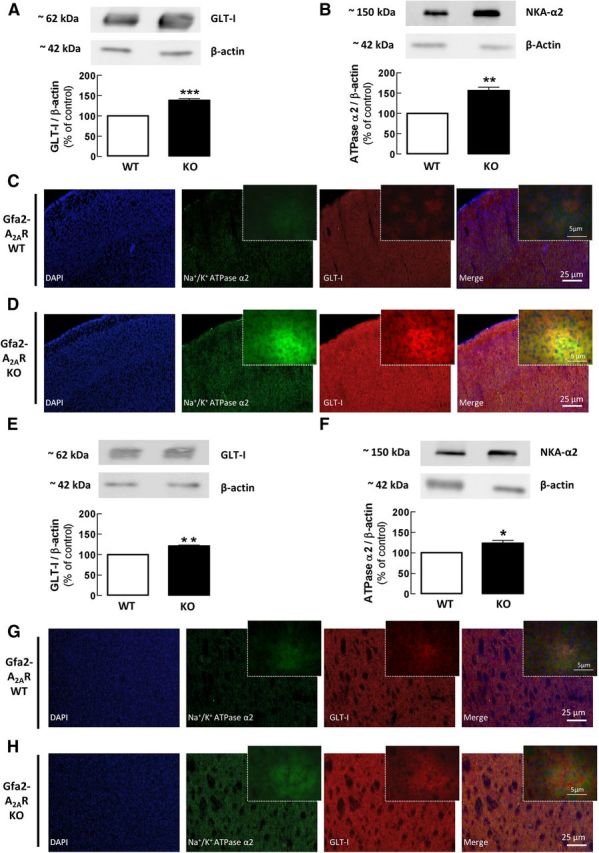
GLT-I and NKA-α2 immunoreactivities are increased in Gfa2-A2AR-KO mice. A, B, E, F, Western blot analysis of total membranes showed that the density of GLT-I (A, E) and of NKA-α2 (B, F) was significantly increased in the cortex (A, B) and striatum (E, F) of Gfa2-A2AR-KO versus Gfa2-A2AR-WT mice. The bars represent the relative immunoreactivity obtained with each primary antibody normalized with anti-β actin (reference) immunoreactivity and were expressed as percentage of WT littermates. C, D, G, H, The immunohistochemical data show the immunoreactivity of GLT-I and NKA-α2 in the cortex (A–D) and in the striatum (E–H) of Gfa2-A2AR-KO (D, H) and Gfa2-A2AR-WT (C, G) littermates with corresponding higher amplifications displayed in the upper right corner of each image. Data are mean ± SEM of at least six independent experiments. Statistical differences were gauged using the Tukey's post hoc test applied after one-way ANOVA with *p < 0.05, **p < 0.01 and ***p < 0.001, comparison with naive WT littermates. Scale bars: C, D, G, H, 25 μm; inset, 5 μm.
Immunohistochemical analysis confirmed the Western blot results, showing an increased immunoreactivity of both GLT-Is and NKA-α2s in the frontal cortex (Fig. 4C,D) and dorsal striatum (Fig. 4G,H) of Gfa2-A2AR-KO compared with Gfa2-A2AR-WT mice (n = 6). These observations are in agreement with the reported superimposable ultrastructural distribution of the α2 subunit of NKA and GLT-I (Cholet et al., 2002; Rose et al., 2009; Genda et al., 2011; Bauer et al., 2012) and further suggest that astrocytic A2ARs are key modulators of this coupling.
A2ARs are physically associated with NKA-α2s
Previous coimmunoprecipitation studies revealed a closed association between GLT-I and NKA-α2 (Rose et al., 2009; Genda et al., 2011; Bauer et al., 2012), forming a protein complex at the plasma membrane of astrocytes to ensure the maintenance of the electrochemical Na+ gradient required for glutamate uptake during neuronal activity. Since we have also shown a close association between A2ARs and glutamate transporters (Matos et al., 2012b), we next sought to test whether A2ARs and NKA-α2s might also copurify in the cerebral cortex or striatum. The pull-down of A2ARs from cortical and striatal homogenates was followed by a Western blot analysis of the A2AR-immunoprecipate with the anti-NKA-α2 antibody (Fig. 5, IP) or with an anti-IgG antibody as a negative control (Fig. 5, CTR−), while confirming the presence of NKA-α2 in the input sample in nonimmunoprecipitated membranes (Fig. 5, CTR+) and the presence of A2ARs in the input and pull-down samples (Fig. 5, upper lanes, WB). As depicted in Figure 5, we observed a close association between NKA-α2s and A2ARs in the brain extracts from Gfa2-A2AR-WT mice (n = 3; Fig. 5A,B, lower lanes, IP), which was highly decreased in both cortical (Fig. 5A) or striatal extracts (Fig. 5B) from Gfa2-A2AR-KO mice (n = 3), in comparison with the WT littermates. These data provide strong evidence of a close association between A2ARs and NKA-α2s in astrocytes, which is absent in Gfa2-A2AR-KO mice.
Figure 5.
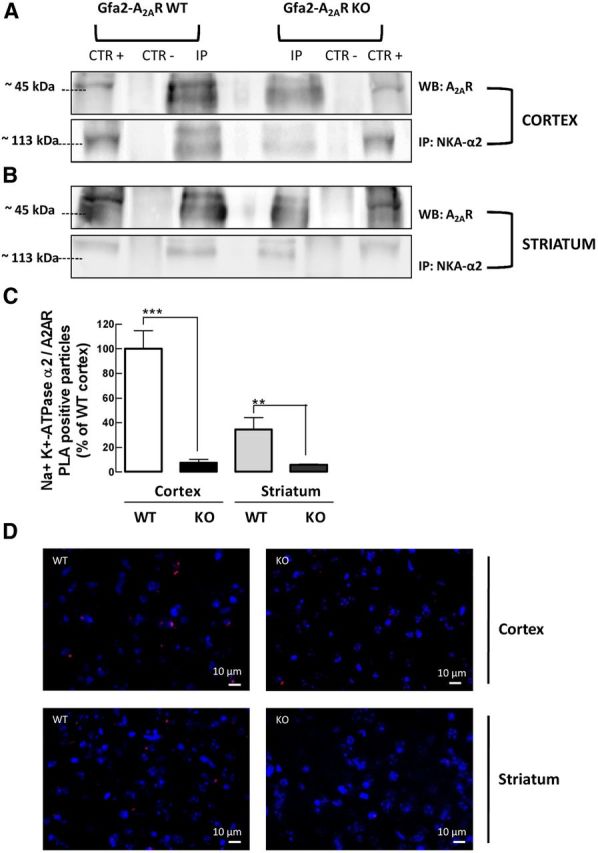
A2ARs are physically associated with NKA-α2s and this coupling is abrogated in Gfa2-A2AR-KO mice. A, B, Immunoprecipitation of A2ARs from cerebral cortical (A) or striatal (B) total membranes from Gfa2-A2AR-KO mice and Gfa2-A2AR-WT littermates with anti-A2AR antibody (IP) or lack of A2AR pull-down with IgG (CTR−), followed by Western blot analysis with anti-NKA-α2 antibody, revealed an association between NKA-α2s and A2ARs in the WT immunoprecipitate (IP), which was absent in Gfa2-A2AR-KO mice. The presence of NKA-α2s in the input sample was confirmed in noncoimmunoprecipitated membranes (CTR+) in the lower (IP) lanes. The presence of A2ARs was confirmed by Western blot analysis in the upper lanes (WB). C, D, A PLA assay further corroborated the close proximity (≤16 nm) between astrocyte A2ARs and NKA-α2s in the cortex and striatum from Gfa2-A2AR-WT mice, which was blunted in Gfa2-A2AR-KO mice. D, Representative confocal images of the PLA assay showing distinct bright red spots in the cortex and striatum from WT mice, corresponding to the amplification products between DNA probes linked to the anti-A2AR and anti-NKA-α2 antibodies. C, D, Data are mean ± SEM of at least three independent experiments. Statistical differences were gauged using the Tukey's post hoc test applied after one-way ANOVA with **p < 0.01 and ***p < 0.001. Scale bars: 10 μm.
Next, using an in situ PLA, we attempted to confirm the existence of A2AR and NKA-α2 complexes in cortical and striatal brain slices of Gfa2-A2AR-KO or Gfa2-A2AR-WT littermates (Söderberg et al., 2006; Augusto et al., 2013). PLA is an antibody-based method in which the A2AR and NKA-α2 proteins were first immunolabeled with primary antibodies and then with secondary antibodies conjugated to complementary oligonucleotides, which can only ligate and be amplified if the A2AR and NKA-α2 antibody molecules are in close proximity (<16 nm) to be identified as fluorescent A2AR-NKA-α2 puncta (Söderberg et al., 2006). Figure 5C illustrates the existence of A2AR-NKA-α2-positive signals in both the cerebral cortex and striatum with a higher A2AR-NKA-α2 cross-linking signal in the cortex than in the striatum (35.0 ± 10.0% of cortical-positive signals, n = 3), possibly reflecting the different density of astrocytes in the two brain areas (Kálmán and Hajós, 1989; Taft et al., 2005) or an eventual different density of A2ARs in astrocytes in these two brain regions. The specific association between A2ARs and NKA-α2s in astrocytes is further consolidated by the sharp and significant decrease of the A2AR-NKA-α2-positive signals in the cortex (93.0 ± 3.0%, n = 3, p < 0.001) and in the striatum (82.3 ± 27.0% decrease, n = 3, p < 0.01) of Gfa2-A2AR-KO mice compared with WT littermates (Fig. 5C,D).
Discussion
The present results provide the first direct evidence of the colocalization and functional interaction between A2ARs and Na+/K+-ATPases (NKA-α2s) specifically in astrocytes in the mouse adult brain. This physical association and control of NKA activity by A2ARs provides a novel mechanism by which A2ARs regulate astrocytic glutamate uptake. This was concluded based on a combination of parallel neurochemical assays of NKA activity and [3H]d-aspartate uptake, coupled to pharmacological manipulations of A2AR and NKA activity and further confirmed by coimmunoprecipitation and PLA assays, all validated though the comparative study of Gfa2-A2AR-KO and WT mice.
The key role of NKA controlling astrocytic glutamate transport is well established, as heralded by the ability of the NKA inhibitor ouabain to impair glutamate uptake (Pellerin and Magistretti, 1997; Cholet et al., 2002; Rose et al., 2009; Nguyen et al., 2010). Notably, this involves a physical association among NKA-α2, GLT-I, and GLAST, as evidenced by their colocalization, copurification, and coimmunoprecipitation (Cholet et al., 2002; Rose et al., 2009; Genda et al., 2011; Bauer et al., 2012) and by the reversed ability of glutamate transporters to modulate NKA activity (Gegelashvili et al., 2007). In parallel, we had previously documented the colocalization and functional interaction between A2AR and GLT-I in astrocytes (Matos et al., 2012a, b). The present demonstration that A2ARs physically associate with NKA-α2s suggests the existence of a macromolecular complex encompassing A2ARs, NKA-α2s, and GLT-Is in astrocytic membranes, in accordance with the role of NKAs as a docking station of molecular signaling hubs (Reinhard et al., 2013) and the versatility of A2ARs to interact with different neurotransmitters receptors, enzymes, and anchoring proteins (Burgueño et al., 2003; Ferré et al., 2007; Zezula and Freissmuth, 2008; Navarro et al., 2009). This ability of A2ARs to control NKA-α2s provides a novel mechanism to understand how the acute A2AR activation decreases glutamate uptake by astrocytes; thus, A2AR activation not only triggers a cAMP-PKA-dependent pathway to decrease the expression of astrocytic glutamate transporters, but also triggers a rapid inhibition of astrocytic glutamate transport (Matos et al., 2012b). Albeit the modification of glutamate uptake in astrocytes upon selective A2AR elimination from astrocytes may result from a short-term and/or long-term regulation (Matos et al., 2012b), the observed parallel modification of NKA and glutamate uptake activities selectively in gliosomes of Gfa2-A2AR-KO mice further suggests an astrocyte-selective coupling between A2ARs and NKAs to regulate glutamate uptake. The molecular mechanism operated by A2ARs to control NKAs may involve a direct conformational control of NKAs (Arystarkhova and Sweadner, 2005) as a result of the observed physical association between A2ARs and NKA-α2s, which would allow understanding the opposite impact of A2ARs on astrocytic NKA-α2 activity (inhibition) and neuronal NKA-α3 activity (stimulation). Whereas in astrocytes A2ARs selectively couple with NKA-α2s to control glutamate uptake mainly operated through GLT-Is, neither of these A2AR targets are present in neurons (Benarroch, 2010, 2011) and the mechanism by which A2ARs control neuronal (putatively) NKA-α3 activity is still unresolved, although it seems unrelated to the control of glutamate clearance since, in contrast to gliosomes, neuronal A2ARs modulate in an opposite manner NKA (facilitation) and glutamate uptake (inhibition). This is in agreement with the predominant role of astrocytes rather than neurons to remove extracellular glutamate (Danbolt, 2001; Sattler and Rothstein, 2006).
The selective interaction and colocalization of NKA-α2s with A2ARs to mediate the fast control of glutamate uptake provides new insights to understand important neurobiological processes, including synaptic plasticity, cognition, and neurodegeneration, that are influenced by the abnormal functioning of either glutamate transporters (Dunlop, 2006; Benarroch, 2010) or NKA-α2s (De Fusco et al., 2003; Moseley et al., 2007; Benarroch, 2011) and which are controlled by A2ARs (Chen et al., 2007; Gomes et al., 2011). Thus, modification of glutamate uptake biases synaptic plasticity and affects cognition (Huang and Bergles, 2004; Tzingounis and Wadiche, 2007; Bechtholt-Gompf et al., 2010); similarly, NKA-α2 gene mutations have been associated with impaired spatial learning, epilepsy, and anxiety (Lingrel et al., 2007; Moseley et al., 2007; Benarroch, 2011). Our finding of the direct interaction between A2ARs and NKA-α2s controlling GLT-I activity provides the tentative explanation that the A2AR-mediated control of synaptic plasticity (Costenla et al., 2011), working memory (Zhou et al., 2009; Wei et al., 2011), and memory impairment in animal models of Alzheimer's disease (Canas et al., 2009; Cunha and Agostinho, 2010) may involve an A2AR-mediated control of glutamate uptake by astrocytes (Matos et al., 2012a). This corresponds to a shift from neurons to astrocytes as the main cellular site of action of A2ARs to control different brain pathologies. In fact, the predominant localization of A2ARs in medium spiny neurons (Schiffmann et al., 2007) and in synapses throughout the brain (Rebola et al., 2005) has prompted researchers to point to neuronal-based mechanisms as responsible for A2AR-mediated neuroprotection (Chen et al., 2007; Gomes et al., 2011), whereas the role of A2ARs in astrocytes (Boison et al., 2010) has received less attention. The presently reported ability of A2ARs to control astrocytic NKA activity implies a tight regulation by A2ARs of ionic homeostasis (see below) in astrocytes (Türközkan et al., 1996; Leite et al., 2011) indirectly controlling glutamatergic neurotransmission, which may provide the explanation for the broad spectrum of neuroprotection of A2AR antagonists in diverse brain regions against a variety of brain insults (Chen et al., 2007; Gomes et al., 2011). The observed quantitative differences between A2AR/NKA-α2/glutamate transporters in the striatum and cortex suggest a qualitatively general control of NKA-α2s and GLT-Is by A2ARs, but also indicates quantitative differences between different brain regions, probably related to different expression of astrocytic A2ARs and/or the different astrocyte-neuron interplay in controlling the extracellular glutamate levels in different brain regions.
It is worth noting that, while A2ARs similarly affected both NKA and GLT-I activities in astrocytes, A2AR agonists affected those activities differently, with a slight variance in potency. This may result either from an ability of A2ARs to allosterically control the NKA-α2–GLT-I complex in a manner independent of NKA activity or to the fact that the impact of A2AR-mediated control of NKA activity in astrocytes may actually override the importance of the control of glutamate uptake so that minor changes of NKA-α2 activity have a disproportional impact on GLT-I activity. NKA-α2 has a prime role in maintaining Na+ and K+ gradients, which provide the driving force for multiple cellular functions, such as regulation of cell volume, pH, energization of the resting membrane potential, and Na+-coupled secondary transport of H+, Ca2+, and glucose across the astrocytic plasma membrane (Aperia, 2007; Kirischuk et al., 2012). Thus the regulation of astrocytic NKA-α2s by A2ARs suggests a potential ability of A2ARs to affect each of these astrocytic processes and thus influence a variety of neurobiological processes. For instance, NKA-α2 activity controls the extracellular K+ homeostasis to regulate neuronal depolarization, synaptic fidelity, and the signal-to-noise ratio of synaptic transmission (Wang et al., 2012), which may well underlie the ability of A2ARs to control synaptic plasticity and the salience of information encoding in neuronal networks (Cunha, 2008). Also, the control of extracellular K+ and pH by astrocytic NKA-α2 (Obara et al., 2008; Benarroch, 2011) may provide novel mechanistic insights for the ability of A2ARs to control abnormal excitability characteristic of animal models of epilepsy (El Yacoubi et al., 2008). Additionally, the control by A2ARs of astrocytic ion homeostasis may also be involved in the control of glucose and lactate metabolism, in accordance with the impact of caffeine (an adenosine receptor antagonist) and A2ARs on brain metabolism (Hammer et al., 2001; Duarte et al., 2009).
Notably, our novel key observation that A2ARs physically associate with and inhibit NKA-α2 also prompts a novel mechanism to link metabolic control with ion homeostasis in astrocytes. Thus, NKA activity is the chief controller of ion homeostasis at the cost of considerable energetic support. As NKA activity consumes ATP, it generates adenosine, and this local metabolic imbalance then feeds back to curtail excessive activity of NKA-α2 and control ion homeostasis through the activation of astrocytic A2ARs. Thus, this novel observation that A2ARs regulate NKA-α2 activity points to the hitherto unrecognized possibility that the impact of A2ARs and of caffeine consumption on brain dysfunction may involve a primary target on astrocytic ion homeostasis that indirectly affects synaptic function and viability. Interestingly, we observed an opposite A2AR modulation of NKA activity in gliosomes and synaptosomes, which suggests a complex and potential “fine-tuning” modulation of NKA activity in astrocytes and neurons to affect cognition, mood, and neurodegeneration processes. However, future work is required to understand what might be the physiopathological impact of the A2AR-mediated control of NKA activity in neurons.
In conclusion, we provide molecular and functional evidence showing the physical association of A2ARs and NKA-α2s and the ability of A2ARs to decrease NKA-α2 activity. This was shown to constitute the mechanism by which the acute manipulation of A2ARs controls the transport of glutamate by astrocytes as an example of the possible importance of this novel A2AR–NKA-α2 molecular hub to understand the neuroprotective impact of caffeine and A2AR antagonists on diverse neurological conditions.
Footnotes
This work was supported by the Portuguese Foundation for Science and Technology (PTDC/SAU-NSC/122254/2010), the National Institutes of Health (Grant NS041083-07), and Defense Advanced Research Projects Agency (Grant 09-68-ESR-FP-010). M.M. and E.A. acknowledge their FCT/FSE (Fundação para a Ciência e a Tecnolgia/European Social Fund) fellowships (SFRH/BD/36289/2007, SFRH/BD/47824/2008).
References
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. doi: 10.1002/1098-1136(200010)32:1<1::AID-GLIA10>3.0.CO%3B2-W. [DOI] [PubMed] [Google Scholar]
- Antolović R. Low nanomolar concentrations of ouabain may induce higher activity of the Na+/K+-ATPase in human erythrocytes. Veterinarski Arhiv. 2006;76:489–495. [Google Scholar]
- Aperia A. New roles for an old enzyme: Na, K-ATPase emerges as an interesting drug target. J Intern Med. 2007;261:44–52. doi: 10.1111/j.1365-2796.2006.01745.x. [DOI] [PubMed] [Google Scholar]
- Arystarkhova E, Sweadner KJ. Splice variants of the gamma subunit (FXYD2) and their significance in regulation of the Na, K-ATPase in kidney. J Bioenerg Biomembr. 2005;37:381–386. doi: 10.1007/s10863-005-9475-y. [DOI] [PubMed] [Google Scholar]
- Augusto E, Matos M, Sévigny J, El-Tayeb A, Bynoe MS, Müller C, Cunha RA, Chen JF. Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J Neurosci. 2013;33:11390–11399. doi: 10.1523/JNEUROSCI.5817-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastia E, Xu YH, Scibelli AC, Day YJ, Linden J, Chen JF, Schwarzschild MA. A crucial role for forebrain adenosine A2A receptors in amphetamine sensitization. Neuropsychopharmacology. 2005;30:891–900. doi: 10.1038/sj.npp.1300630. [DOI] [PubMed] [Google Scholar]
- Bauer DE, Jackson JG, Genda EN, Montoya MM, Yudkoff M, Robinson MB. The glutamate transporter, GLAST, participates in a macromolecular complex that supports glutamate metabolism. Neurochem Int. 2012;61:566–574. doi: 10.1016/j.neuint.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtholt-Gompf AJ, Walther HV, Adams MA, Carlezon WA, Jr, Ongür D, Cohen BM. Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology. 2010;35:2049–2059. doi: 10.1038/npp.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Glutamate transporters: diversity, function, and involvement in neurologic disease. Neurology. 2010;74:259–264. doi: 10.1212/WNL.0b013e3181cc89e3. [DOI] [PubMed] [Google Scholar]
- Benarroch EE. Na+, K+-ATPase: functions in the nervous system and involvement in neurologic disease. Neurology. 2011;76:287–293. doi: 10.1212/WNL.0b013e3182074c2f. [DOI] [PubMed] [Google Scholar]
- Boison D, Chen JF, Fredholm BB. Adenosine signaling and function in glial cells. Cell Death Differ. 2010;17:1071–1082. doi: 10.1038/cdd.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgueño J, Blake DJ, Benson MA, Tinsley CL, Esapa CT, Canela EI, Penela P, Mallol J, Mayor F, Jr, Lluis C, Franco R, Ciruela F. The adenosine A2A receptor interacts with the actin-binding protein alpha-actinin. J Biol Chem. 2003;278:37545–37552. doi: 10.1074/jbc.M302809200. [DOI] [PubMed] [Google Scholar]
- Canas PM, Porciúncula LO, Cunha GM, Silva CG, Machado NJ, Oliveira JM, Oliveira CR, Cunha RA. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by β-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci. 2009;29:14741–14751. doi: 10.1523/JNEUROSCI.3728-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Sonsalla PK, Pedata F, Melani A, Domenici MR, Popoli P, Geiger J, Lopes LV, de Mendonça A. Adenosine A2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog Neurobiol. 2007;83:310–331. doi: 10.1016/j.pneurobio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Cholet N, Pellerin L, Magistretti PJ, Hamel E. Similar perisynaptic glial localization for the Na+, K+-ATPase alpha 2 subunit and the glutamate transporters GLAST and GLT-1 in the rat somatosensory cortex. Cereb Cortex. 2002;12:515–525. doi: 10.1093/cercor/12.5.515. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortés A, Canela EI, López-Giménez JF, Milligan G, Lluis C, Cunha RA, Ferré S, Franco R. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci. 2006;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costenla AR, Diógenes MJ, Canas PM, Rodrigues RJ, Nogueira C, Maroco J, Agostinho PM, Ribeiro JA, Cunha RA, de Mendonça A. Enhanced role of adenosine A2A receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur J Neurosci. 2011;34:12–21. doi: 10.1111/j.1460-9568.2011.07719.x. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Different cellular sources and different roles of adenosine: A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity. Neurochem Int. 2008;52:65–72. doi: 10.1016/j.neuint.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Agostinho PM. Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. J Alzheimers Dis. 2010;20(Suppl 1):S95–S116. doi: 10.3233/JAD-2010-1408. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, Ballabio A, Aridon P, Casari G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33:192–196. doi: 10.1038/ng1081. [DOI] [PubMed] [Google Scholar]
- Duarte JM, Carvalho RA, Cunha RA, Gruetter R. Caffeine consumption attenuates neurochemical modifications in the hippocampus of streptozotocin-induced diabetic rats. J Neurochem. 2009;111:368–379. doi: 10.1111/j.1471-4159.2009.06349.x. [DOI] [PubMed] [Google Scholar]
- Dunlop J. Glutamate-based therapeutic approaches: targeting the glutamate transport system. Curr Opin Pharmacol. 2006;6:103–107. doi: 10.1016/j.coph.2005.09.004. [DOI] [PubMed] [Google Scholar]
- El Yacoubi M, Ledent C, Parmentier M, Costentin J, Vaugeois JM. Evidence for the involvement of the adenosine A2A receptor in the lowered susceptibility to pentylenetetrazol-induced seizures produced in mice by long-term treatment with caffeine. Neuropharmacology. 2008;55:35–40. doi: 10.1016/j.neuropharm.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Quiroz C, Luján R, Popoli P, Cunha RA, Agnati LF, Fuxe K, Woods AS, Lluis C, Franco R. Adenosine receptor heteromers and their integrative role in striatal function. ScientificWorldJournal. 2007;7:74–85. doi: 10.1100/tsw.2007.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- Gao J, Wymore RS, Wang Y, Gaudette GR, Krukenkamp IB, Cohen IS, Mathias RT. Isoform-specific stimulation of cardiac Na/K pumps by nanomolar concentrations of glycosides. J Gen Physiol. 2002;119:297–312. doi: 10.1085/jgp.20028501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegelashvili M, Rodriguez-Kern A, Sung L, Shimamoto K, Gegelashvili G. Glutamate transporter GLAST/EAAT1 directs cell surface expression of FXYD2/gamma subunit of Na, K-ATPase in human fetal astrocytes. Neurochem Int. 2007;50:916–920. doi: 10.1016/j.neuint.2006.12.015. [DOI] [PubMed] [Google Scholar]
- Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O'Donnell JC, Spruce LA, Xiao R, Guo W, Putt M, Seeholzer S, Ischiropoulos H, Robinson MB. Co-compartmentalization of the astroglial glutamate transporter, GLT-I, with glycolytic enzymes and mitochondria. J Neurosci. 2011;31:18275–18288. doi: 10.1523/JNEUROSCI.3305-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta. 2011;1808:1380–1399. doi: 10.1016/j.bbamem.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Hammer J, Qu H, Håberg A, Sonnewald U. In vivo effects of adenosine A2 receptor agonist and antagonist on neuronal and astrocytic intermediary metabolism studied with ex vivo 13C MR spectroscopy. J Neurochem. 2001;79:885–892. doi: 10.1046/j.1471-4159.2001.00622.x. [DOI] [PubMed] [Google Scholar]
- Huang YH, Bergles DE. Glutamate transporters bring competition to the synapse. Curr Opin Neurobiol. 2004;14:346–352. doi: 10.1016/j.conb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Kálmán M, Hajós F. Distribution of glial fibrillary acidic protein (GFAP)-immunoreactive astrocytes in the rat brain. I. Forebrain. Exp Brain Res. 1989;78:147–163. doi: 10.1007/BF00230694. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Parpura V, Verkhratsky A. Sodium dynamics: another key to astroglial excitability? Trends Neurosci. 2012;35:497–506. doi: 10.1016/j.tins.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Lee Y, Su M, Messing A, Brenner M. Astrocyte heterogeneity revealed by expression of a GFAP-LacZ transgene. Glia. 2006;53:677–687. doi: 10.1002/glia.20320. [DOI] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia. 2008;56:481–493. doi: 10.1002/glia.20622. [DOI] [PubMed] [Google Scholar]
- Leite MR, Wilhelm EA, Jesse CR, Brandão R, Nogueira CW. Protective effect of caffeine and a selective A2A receptor antagonist on impairment of memory and oxidative stress of aged rats. Exp Gerontol. 2011;46:309–315. doi: 10.1016/j.exger.2010.11.034. [DOI] [PubMed] [Google Scholar]
- Lichtstein D, Samuelov S, Bourrit A. Characterization of the stimulation of neuronal Na+, K+-ATPase activity by low concentrations of ouabain. Neurochem Int. 1985;7:709–715. doi: 10.1016/0197-0186(85)90069-5. [DOI] [PubMed] [Google Scholar]
- Lingrel JB, Williams MT, Vorhees CV, Moseley AE. Na, K-ATPase and the role of alpha isoforms in behavior. J Bioenerg Biomembr. 2007;39:385–389. doi: 10.1007/s10863-007-9107-9. [DOI] [PubMed] [Google Scholar]
- Longuemare MC, Rose CR, Farrell K, Ransom BR, Waxman SG, Swanson RA. K+-induced reversal of astrocyte glutamate uptake is limited by compensatory changes in intracellular Na+ Neuroscience. 1999;93:285–292. doi: 10.1016/S0306-4522(99)00152-9. [DOI] [PubMed] [Google Scholar]
- Matos M, Augusto E, Machado NJ, dos Santos-Rodrigues A, Cunha RA, Agostinho P. Astrocytic adenosine A2A receptors control the amyloid-β peptide-induced decrease of glutamate uptake. J Alzheimers Dis. 2012a;31:555–567. doi: 10.3233/JAD-2012-120469. [DOI] [PubMed] [Google Scholar]
- Matos M, Augusto E, Santos-Rodrigues AD, Schwarzschild MA, Chen JF, Cunha RA, Agostinho P. Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia. 2012b;60:702–716. doi: 10.1002/glia.22290. [DOI] [PubMed] [Google Scholar]
- Moseley AE, Williams MT, Schaefer TL, Bohanan CS, Neumann JC, Behbehani MM, Vorhees CV, Lingrel JB. Deficiency in Na, K-ATPase α isoform genes alters spatial learning, motor activity, and anxiety in mice. J Neurosci. 2007;27:616–626. doi: 10.1523/JNEUROSCI.4464-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Otsubo Y, Yatani Y, Shirakawa H, Kaneko S. Mechanisms of substrate transport-induced clustering of a glial glutamate transporter GLT-I in astroglial-neuronal cultures. Eur J Neurosci. 2008;28:1719–1730. doi: 10.1111/j.1460-9568.2008.06494.x. [DOI] [PubMed] [Google Scholar]
- Navarro G, Aymerich MS, Marcellino D, Cortés A, Casadó V, Mallol J, Canela EI, Agnati L, Woods AS, Fuxe K, Lluís C, Lanciego JL, Ferré S, Franco R. Interactions between calmodulin, adenosine A2A, and dopamine D2 receptors. J Biol Chem. 2009;284:28058–28068. doi: 10.1074/jbc.M109.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KT, Buljan V, Else PL, Pow DV, Balcar VJ. Cardiac glycosides ouabain and digoxin interfere with the regulation of glutamate transporter GLAST in astrocytes cultured from neonatal rat brain. Neurochem Res. 2010;35:2062–2069. doi: 10.1007/s11064-010-0274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara M, Szeliga M, Albrecht J. Regulation of pH in the mammalian central nervous system under normal and pathological conditions: facts and hypotheses. Neurochem Int. 2008;52:905–919. doi: 10.1016/j.neuint.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake stimulates Na+, K+-ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J Neurochem. 1997;69:2132–2137. doi: 10.1046/j.1471-4159.1997.69052132.x. [DOI] [PubMed] [Google Scholar]
- Pintor A, Galluzzo M, Grieco R, Pèzzola A, Reggio R, Popoli P. Adenosine A2A receptor antagonists prevent the increase in striatal glutamate levels induced by glutamate uptake inhibitors. J Neurochem. 2004;89:152–156. doi: 10.1111/j.1471-4159.2003.02306.x. [DOI] [PubMed] [Google Scholar]
- Rebola N, Canas PM, Oliveira CR, Cunha RA. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience. 2005;132:893–903. doi: 10.1016/j.neuroscience.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Reinhard L, Tidow H, Clausen MJ, Nissen P. Na+, K+-ATPase as a docking station: protein-protein complexes of the Na+, K+-ATPase. Cell Mol Life Sci. 2013;70:205–222. doi: 10.1007/s00018-012-1039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose EM, Koo JC, Antflick JE, Ahmed SM, Angers S, Hampson DR. Glutamate transporter coupling to Na, K-ATPase. J Neurosci. 2009;29:8143–8155. doi: 10.1523/JNEUROSCI.1081-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar PK. A quick assay for Na+-K+-ATPase specific activity. Z Naturforsch C. 2002;57:562–564. doi: 10.1515/znc-2002-5-628. [DOI] [PubMed] [Google Scholar]
- Sattler R, Rothstein JD. Regulation and dysregulation of glutamate transporters. Handb Exp Pharmacol. 2006;175:277–303. doi: 10.1007/3-540-29784-7_14. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Fisone G, Moresco R, Cunha RA, Ferré S. Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol. 2007;83:277–292. doi: 10.1016/j.pneurobio.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Taft JR, Vertes RP, Perry GW. Distribution of GFAP+ astrocytes in adult and neonatal rat brain. Int J Neurosci. 2005;115:1333–1343. doi: 10.1080/00207450590934570. [DOI] [PubMed] [Google Scholar]
- Türközkan N, Bilighan A, Cayci B, Doðulu F, Aykol S. The effects of 2-chloroadenosine and deoxycoformycin on the ATP level, Na-K ATPase activity in experimental brain ischemia of gerbil. Neurol Res. 1996;18:345–348. doi: 10.1080/01616412.1996.11740434. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, Takano T, Bekar L, Nedergaard M. Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+ Sci Signal. 2012;5:ra26. doi: 10.1126/scisignal.2002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CJ, Singer P, Coelho J, Boison D, Feldon J, Yee BK, Chen JF. Selective inactivation of adenosine A2A receptors in striatal neurons enhances working memory and reversal learning. Learn Mem. 2011;18:459–474. doi: 10.1101/lm.2136011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zezula J, Freissmuth M. The A2A-adenosine receptor: a GPCR with unique features? Br J Pharmacol. 2008;153(Suppl 1):S184–S190. doi: 10.1038/sj.bjp.0707674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou SJ, Zhu ME, Shu D, Du XP, Song XH, Wang XT, Zheng RY, Cai XH, Chen JF, He JC. Preferential enhancement of working memory in mice lacking adenosine A2A receptors. Brain Res. 2009;1303:74–83. doi: 10.1016/j.brainres.2009.09.082. [DOI] [PubMed] [Google Scholar]