

Abstract
Glial fibrillary acidic protein (GFAP) is the major intermediate filament of mature astrocytes in the mammalian CNS. Dominant gain of function mutations in GFAP lead to the fatal neurodegenerative disorder, Alexander disease (AxD), which is characterized by cytoplasmic protein aggregates known as Rosenthal fibers along with variable degrees of leukodystrophy and intellectual disability. The mechanisms by which mutant GFAP leads to these pleiotropic effects are unknown. In addition to astrocytes, GFAP is also expressed in other cell types, particularly neural stem cells that form the reservoir supporting adult neurogenesis in the hippocampal dentate gyrus and subventricular zone of the lateral ventricles. Here, we show that mouse models of AxD exhibit significant pathology in GFAP-positive radial glia-like cells in the dentate gyrus, and suffer from deficits in adult neurogenesis. In addition, they display impairments in contextual learning and spatial memory. This is the first demonstration of cognitive phenotypes in a model of primary astrocyte disease.
Introduction
Alexander disease (AxD) is a progressive and generally fatal disorder of the CNS, with a range of clinical phenotypes including cognitive and motor impairments, along with white matter deficits especially prominent in the frontal lobes (Messing et al., 2012). The most current classification system divides patients into two categories, with type I consisting of those with frontal predominance and early onset, and type II presenting with lesions concentrated in the hindbrain and cervical spinal cord, less pronounced leukodystrophy and cognitive deficits, and varied age of onset (Prust et al., 2011). The hallmark pathological feature in both groups is the presence of Rosenthal fibers, cytoplasmic protein aggregates of astrocytes, particularly in perivascular, subpial, and subependymal locations (Alexander, 1949; Iwaki et al., 1993). Nearly all cases of AxD result from heterozygous missense mutations in the astrocyte intermediate filament, GFAP (Brenner et al., 2001; 2009). Since this discovery, AxD has become a model system in which to explore the consequences of primary astrocyte dysfunction on other cells in the CNS.
To better understand how GFAP mutations cause disease, we have engineered mouse models with knockin mutations causing single amino acid changes orthologous to common human mutations at the endogenous Gfap locus (Hagemann et al., 2006). These models replicate some but not all of the pathology found in the human disease. AxD mice spontaneously increase expression of Gfap above normal levels, form the characteristic Rosenthal fibers within the cytoplasm of astrocytes, induce stress responses including Nrf2/ARE and small heat shock protein pathways, and activate microglia (Hagemann et al., 2012). However, myelination appears normal (Hagemann et al., 2006). Hence, these models offer a unique opportunity to test whether cognitive phenotypes resulting from astrocyte disease can occur in the absence of leukodystrophy.
To date, little attention has been given to the possibility that GFAP mutations associated with AxD impact neuronal development. Mature astrocytes regulate neurogenesis in the adult hippocampus (Song et al., 2002), and integration of new neurons in the dentate gyrus has a functional role in contextual learning, spatial memory, and pattern separation (Deng et al., 2010; Aimone et al., 2011; Sahay et al., 2011). Hippocampal astrocytes in mouse models of AxD are reactive, and chronic gliosis in other models of neurodegenerative disease contributes to the release of proinflammatory cytokines (Glass et al., 2010) that are known to disrupt adult neurogenesis (Kohman and Rhodes, 2013). However, GFAP is also expressed in radial glia-like stem cells of the dentate gyrus (Seri et al., 2001; Garcia et al., 2004), and it is conceivable that mutant GFAP exerts cell-autonomous effects in cells other than mature astrocytes.
In this study, we show that AxD mice perform poorly in fear conditioning and Morris water maze and are deficient in generating new neurons in the adult hippocampus. Neural stem cells that are normally quiescent appear to be mitotically activated, but proliferating progenitors are diminished in number and immature neurons are virtually absent. This is the first report to show cognitive deficits and disruption of adult neurogenesis in a model of human primary astrocyte disease.
Materials and Methods
Mice.
All animals were used in accordance with standards set by the Animal Care and Use Committee for the Graduate School at the University of Wisconsin, Madison. Gfap+/R236H mice (Hagemann et al., 2006) were maintained as heterozygotes in a 129S6 genetic background, unless otherwise noted.
EdU labeling.
To assess proliferation and differentiation of hippocampal progenitors in Gfap+/R236H mice, adult males (10 weeks of age) were injected intraperitoneally at 4 h intervals over a 12 h period (4 injections total) (Hayes and Nowakowski, 2002; Guo et al., 2011) with 50 mg/kg thymidine analog EdU (10 mg/ml PBS). Mice were collected at either 24 h or 28 d following the first injection to analyze proliferation and differentiation respectively. In both experiments, mice were anesthetized with 400 mg/kg avertin and transcardially perfused with PBS followed by 4% paraformaldehyde. Brains were removed, postfixed for 10–16 h, and cryoprotected in sucrose before sectioning with a sliding microtome.
Forty micrometer coronal sections were collected through the rostrocaudal extent of the dentate gyrus (−0.9 to −4.0 bregma), and every sixth section was analyzed. Tissues were permeabilized with 0.5% Triton X-100 in PBS with 3% BSA for 30 min and incorporation of EdU by cycling cells detected with an Alexa Fluor 488 Click-iT EdU Imaging Kit according to the manufacturer's protocol (Invitrogen C10337). Subsequent immunofluorescent staining was performed as described below.
Histology and immunofluorescence.
For EdU colabeling, tissue sections were blocked with 5% normal donkey serum before incubation with primary antibodies diluted in PBS with 1% BSA and 0.5% Triton X-100: rabbit anti-Ki67 (1:500, Leica NCL-Ki67p), mouse anti-NeuN (1:500, Millipore MAB377), rabbit anti-S100β (1:1000, Sigma HPAD15768), mouse anti-GFAP [1:1000, NeuroMab clone N206/8, University of California, Davis (UC Davis), Davis, CA], and rabbit anti-Iba1 (1:500, Wako 019–19741). Sections were incubated with primary antibodies at 4°C for 48 h and rinsed in PBS with 0.1% Triton X-100. Secondary antibodies Alexa Fluor 546 donkey anti-rabbit and anti-mouse IgG, and Cy5 goat anti-mouse (Invitrogen) were diluted 1:500 and incubated overnight at 4°C. Sections were rinsed with 0.1% Triton X-100 and mounted with ProLong Gold with DAPI (Invitrogen).
For other immunofluorescence, 30 μm sagittal sections were blocked and permeabilized in PBS with 5% normal donkey serum with 0.3% Triton X-100. Primary antibodies were diluted in 1% BSA with 0.3% Triton X-100: goat anti-Doublecortin (1:500, Dcx, Santa Cruz Biotechnology 8066), mouse anti-GFAP (1:1000, NeuroMab clone N206/8, UC Davis, CA), rabbit anti-Ki67 (1:500, Leica NCL-Ki67p), and goat anti-Sox2 (1:500, Santa Cruz Biotechnology17320) and incubated overnight at 4°C. Secondary antibodies were diluted 1:500 and incubated at room temperature for 2 h (Alexa Fluor 488 and 546 donkey anti-mouse, rabbit or goat). Tissues were rinsed with PBS between incubations and mounted with ProLong Gold with DAPI. Sections were imaged with a Nikon C1 confocal microscope. GFAP immunohistochemistry with eosin counterstaining was described previously (Hagemann et al., 2006).
Stereology.
For proliferation assays, EdU-positive cells in the subgranular zone of the dentate gyrus were counted from the left side of every sixth section (40 μm) and scored as Ki67 positive or negative. With the limited number of positive cells, all were counted except positive cells in the uppermost focal plane to avoid oversampling (Kempermann et al., 1997). Numbers were multiplied by 12 to account for sampling only one side and the section interval.
To assess differentiation at 28 d, tissues were labeled for EdU, NeuN, and S100β. EdU-positive cells throughout the granular layer were counted from both sides of every sixth section and identified as neurons, astrocytes, or unknown by confocal microscopy. Numbers were multiplied by 6 to account for section sampling.
Behavior.
Fear conditioning was performed in test cages from Coulbourn Instruments where 2 min after animals were placed in the cages, an 87 dB white noise cue (conditional stimulus) was given for 30 s. During the last 1.5 s, a 0.7 mA shock was delivered from the floor of the cage. After another 2 min, the mice were again exposed to the 30 s white noise paired with a 0.7 mA shock during the last 1.5 s. After 60 s the mice were removed from the test cage and returned to their home cage.
For context testing, mice were returned to the test cage 22 h later for 5 min while being imaged from above and analyzed with FreezeFrame2 software (Actimetrics). Mice were returned to their home cage for 2 h before being tested for cue conditioning. The test cages were altered with a white Plexiglas floor and gray covers for the walls. Vanilla extract was added below the floor to alter the scent. Cages were cleaned with isopropyl alcohol rather than ethanol (used during training and context testing) between mice. Mice were reintroduced to the altered context for 3 min before delivering the conditional cue: 87 dB white noise for the remaining 3 min. Again, FreezeFrame2 software was used for image analysis to measure freezing and also to control onset and duration of stimuli during training and testing. Images were captured at a rate of 4 per second and the motion index threshold was set at 1. A motion index <1 for 1 s (4 consecutive images) was considered freezing.
The Morris water maze was set in a 1.3-m-wide pool in water made opaque with white Crayola nontoxic paint. Visual stimuli of different colors and shapes were positioned around the room for spatial cues. The pool was virtually divided into four quadrants with EthoVision XT video tracking software (Noldus). A 10 cm platform was positioned 1 cm below the surface of the water in the trained quadrant. After an initial introduction to the platform and pool, mice were run through eight blocks of four training trials (two blocks per day). For each block mice were placed at the edge of the pool between each of the four quadrants (in random order) and allowed 60 s to find the platform. If they did not escape by the end of 60 s, mice were ushered to the platform before removal from the pool.
After the eighth training block, the platform was removed from the trained quadrant for the probe trial. Mice were placed at the edge of the pool between the quadrants opposite and to the right of the trained quadrant. Tracking software scored the number of times mice crossed virtual platforms positioned in each of the four quadrants during the 60 s probe trial as a measure of learning the maze.
Statistics.
For stereology, t tests were used to compare counts between genotypes. For fear conditioning results are shown as percentage freezing. For context this is the percentage time over the 5 min test. For cue conditioning, the percentage freezing during the first 3 min before the cue is initiated is subtracted from the percentage freezing during the cue as a baseline control. A one-tailed t test was used to compare freezing to context or cue between mice of the same gender to analyze effects of genotype (we hypothesized a priori that there would be impaired performance in the mutant mice and thus the difference would only be in one direction). Comparison of platform crossings during the probe trial of the Morris water maze were analyzed by repeated-measures ANOVA to detect a difference among quadrants, followed by Tukey's posttests to show a preference for the trained quadrant. Platform crossings in the trained quadrant were also compared by t test between genotypes.
Results
Gliosis and Rosenthal fibers in radial glia-like cells and lack of immature neurons in the dentate gyrus of Gfap+/R236H mice
In Gfap+/R236H mouse models of AxD, astrocytes in regions of adult neurogenesis, including those in the hippocampal dentate gyrus, have elevated GFAP and Rosenthal fibers (Hagemann et al., 2006). Reactive astrocytes are found throughout the hippocampus, and in the subgranular zone (SGZ) of the dentate gyrus, GFAP-positive cells with radial glia-like (RGL) morphology have thickened processes and a hypertrophic appearance (Fig. 1A). These cells show increased GFAP immunostaining and demonstrate Rosenthal fibers that appear as eosinophilic inclusions in the processes that extend through the granular layer (Fig. 1A, inset). The appearance of reactive glia in the dentate gyrus and more specifically in the SGZ suggests that both the hippocampal niche and the neural stem cell (NSC) population itself may be altered in Gfap+/R236H mice.
Figure 1.
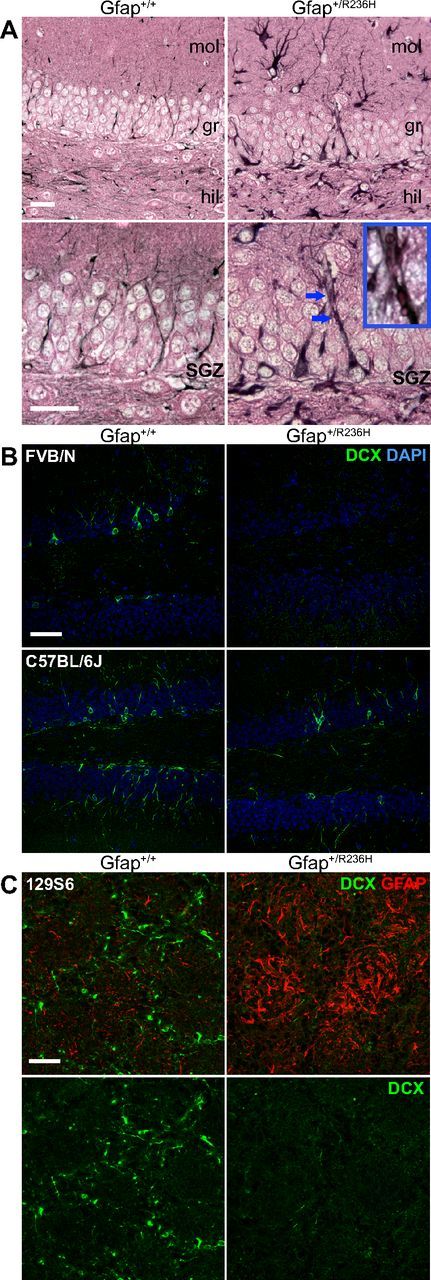
Hypertrophic radial-glia like cells with Rosenthal fibers and loss of immature neurons in Gfap+/R236H dentate gyrus. A, Immunohistochemical staining for GFAP (black, eosin counter stain) in the granular layer of the dentate gyrus shows intense hypertrophied radial glia-like cells in R236H mutants compared with wild-type mice, which in some cases have eosinophilic inclusion bodies that appear as Rosenthal fibers (bottom row, increased magnification, blue arrows, see inset; n = 4 per genotype, 3 months of age; scale bar, 25 μm; mol, dentate gyrus molecular layer; gr, granular layer; hil, hilus). B, Doublecortin (Dcx) immunofluorescent staining of immature neurons (green) in the dentate gyrus shows almost no staining in GFAP mutant mice compared with wild-type in the FVB/N background and reduced staining in C57BL/6J (n = 3 per genotype, FVB/N = 8 weeks, C57BL/6J = 12 weeks of age; scale bar, 50 μm). C, Dcx immunofluorescent staining (green) in olfactory bulb glomerular layer also shows a reduction of immature neurons in Gfap+/R236H mice with an increase in GFAP expression (red, top only). n = 4 per group, 129S6 at 12 weeks; scale bar, 50 μm.
To determine whether mutant GFAP and the resulting pathology disrupt hippocampal neurogenesis, we analyzed immature neurons expressing doublecortin (Dcx) in the granular layer of the dentate gyrus in adult Gfap+/R236H mice compared with their wild-type littermates in three different genetic backgrounds: 129S6, FVB/N, and C57BL/6J. We found almost no Dcx+ neurons in the dentate gyrus of Gfap+/R236H mice at 8 weeks of age in both 129S6 and FVB/N strains (Fig. 1B, FVB/N shown). This difference was not as pronounced in C57BL/6J until 3 months of age when the density of immature neurons appears reduced in this strain as well (Fig. 1B). Dcx+ neurons are also reduced in olfactory bulb glomeruli in Gfap+/R236H mice, suggesting deficits in SVZ neurogenesis (Fig. 1C). For this report we focus on the effects of mutant GFAP and gliosis on the hippocampal niche, the resulting deficits in adult neurogenesis and functional outcomes in the 129S6 genetic background.
Reduced hippocampal progenitor proliferation in Gfap+/R236H SGZ
Since doublecortin-positive immature neurons were mostly absent from Gfap+/R236H hippocampus, we sought to determine whether proliferating precursor cells were present in the mutant dentate gyrus. To identify dividing cells, adult mice were injected with the thymidine analog EdU using a saturating protocol to label all cells entering S phase (Guo et al., 2011) (Fig. 2A). After 24 h, nuclear EdU incorporation showed a reduced number of proliferating cells in the SGZ of Gfap+/R236H mice at 10 weeks of age (Fig. 2B,G). In wild-type Gfap+/+ mice, EdU+ progenitors were apparent as clusters of 2–8 cells throughout the rostrocaudal extent of the dentate gyrus (Fig. 2C), whereas Gfap+/R236H progenitors were usually by themselves or occasionally in pairs and reduced in number by 65% compared with wild type (Fig. 2D,E,G). These cells often had larger nuclei with atypical morphology compared with those in wild-type animals, and did not appear to be intermediate progenitor cells (IPCs; Fig. 2 compare D,C).
Figure 2.
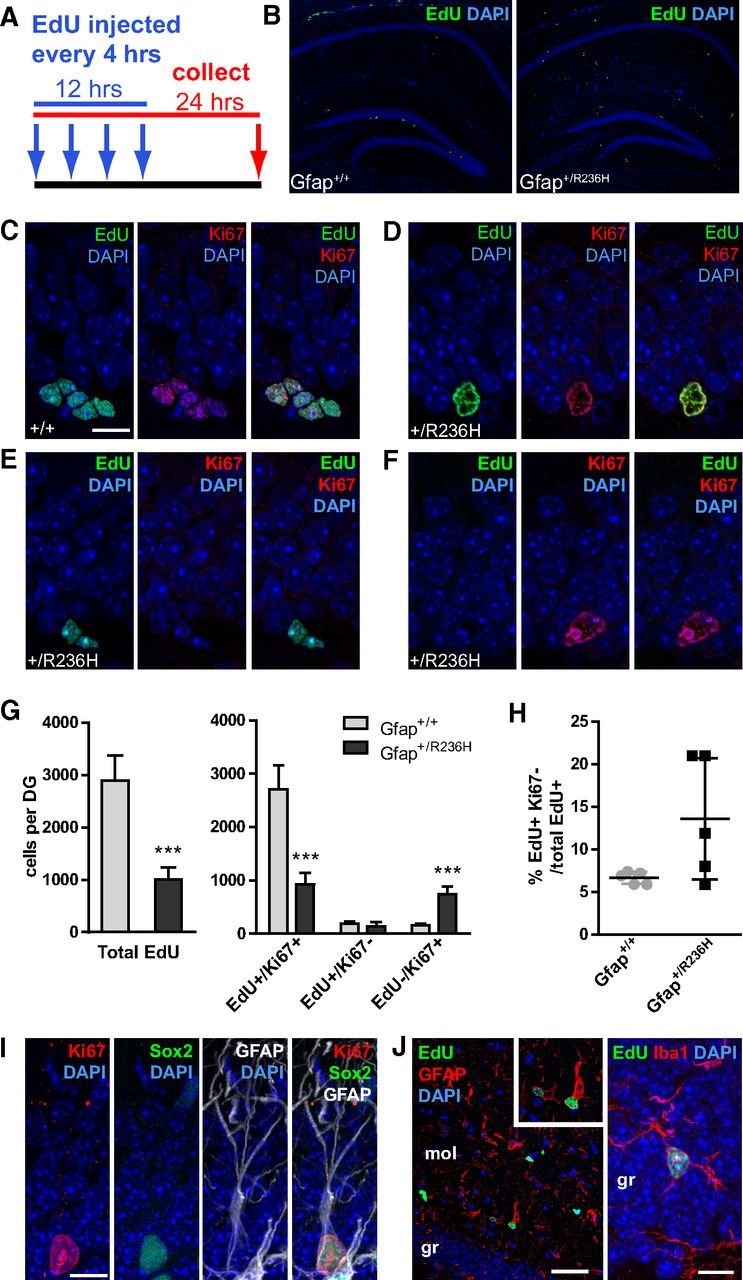
Reduced progenitor proliferation in SGZ of Gfap+/R236H dentate gyrus. A, EdU injection protocol and tissue collection time line for cell proliferation analysis. Arrows indicate injection time points every 4 h for saturated labeling of cycling progenitors followed by tissue collection 24 h after initial injection. B, EdU labeling of dividing cells in the hippocampus of Gfap+/+ mice shows proliferating progenitors in the subgranular zone of the dentate gyrus. In Gfap+/R236H mice, many cells were labeled with EdU outside of the dentate gyrus, with few cells labeled within the SGZ. C–F, EdU and Ki67 labeling in wild-type Gfap+/+ mice (C) shows clusters of dividing cells positive for both markers in the SGZ, whereas EdU+ cells in Gfap+/R236H mice (D–F) are often singular with a different nuclear morphology (D). EdU+/Ki67− cells that have left the cell cycle are apparent in both wild-type and mutant mice (E, Gfap+/R236H shown), and EdU−/Ki67+ cells are frequently apparent in Gfap+/R236H mice (F; scale bar (in C) C–F, 10 μm). G, The total number of cells demonstrating EdU incorporation in the SGZ throughout the rostrocaudal extension of the dentate gyrus (DG) was significantly decreased in Gfap+/R236H mice. The number of cells that were EdU+/Ki67− were similar in both groups, however, Gfap+/R236H mice had many more EdU−/Ki67+ cells than Gfap+/+ mice. Error bars =SD. ***p < 0.0001, two tailed t test between genotypes, n = 5 male mice at 10 weeks of age per genotype. H, The percentage of progenitors leaving the cell cycle (EdU+/Ki67−) was variable in Gfap+/R236H mice, but not significantly increased over that of wild-type mice. I, Immunostaining for Ki67, GFAP, and Sox2 shows colabeling of cells with RGL morphology in the SGZ of Gfap+/R236H mice (FVB/N at 8 weeks, n = 3; scale bar, 10 μm). J, EdU+ cells outside of the SGZ in Gfap+/R236H mice are often GFAP+ astrocytes and sometimes Iba1+ microglia (left scale bar, 50 μm; right scale bar, 10 μm; mol, dentate gyrus molecular layer; gr, granular layer).
To determine whether cells were leaving the cell cycle prematurely and potentially explain the reduced number of proliferating progenitors in Gfap mutant mice compared with wild-type, we colabeled for Ki67 to identify cells that were still cycling after the initial EdU label. Of cells that were EdU+ in Gfap+/+ mice, only 6.7% (± 0.7 SD) had left the cell cycle as demonstrated by lack of staining for Ki67 (Fig. 2H). In Gfap+/R236H mice the proportion of Ki67-negative cells was not significantly increased (Fig. 2E,H; 13.6 ± 7.1% SD, p = 0.063, t test). The actual numbers of EdU+/Ki67− cells were also similar between the two groups of mice (Fig. 2G, Gfap+/+ = 192.0 ± 31.8 SD; Gfap+/R236H = 140.4 ± 81.7 SD; p = 0.2244, t test). There was, however, a surprisingly high number of EdU−/Ki67+ cells in Gfap+/R236H compared with Gfap+/+ mice (Fig. 2F,G). Since Ki67 is present during all active phases of the cell cycle (G1, S, G2, and mitosis) and EdU incorporates during S-phase, this finding suggests that rather than leaving the cell cycle, cells in the SGZ are cycling abnormally and may be in cell cycle arrest.
To determine whether Ki67+ cells in the SGZ of Gfap+/R236H mice may be slowly dividing stem cells instead of transiently amplifying progenitors, we colabeled for the NSC markers Sox2 and GFAP. Nearly all Ki67+ cells were positive for Sox2 and GFAP in the SGZ of mutant mice (Fig. 2I). Similar Ki67+/Sox2+/GFAP+ cells were present but rare in Gfap+/+ mice.
In addition, there were EdU+ cells in Gfap+/R236H mice outside of the SGZ in the granular and molecular layer of the dentate gyrus, and throughout the hippocampus that were not present in Gfap+/+ mice (Fig. 2B). Many of these cells also stained for GFAP and are reactive astrocytes, some are microglia as indicated by Iba1 immunostaining (Fig. 2J), and some may be NG2 cells. These were not included in progenitor cell counts. Overall, these data show a marked reduction in the number of dividing cells in the SGZ, which would explain the lack of Dcx+-immature neurons in Gfap+/R236H mice. Abnormal cycling of the NSC population may also contribute to this deficit.
Abnormal neural progenitor differentiation in Gfap+/R236H dentate gyrus
Although Gfap mutant mice show reduced or abnormal progenitor proliferation within the SGZ, EdU+ cells are still present. To determine the fate of these cells, we evaluated their phenotype at 4 weeks postlabeling (EdU incorporation, Fig. 3A). In Gfap+/+ mice, 52% of EdU+ cells were also positive for NeuN, 21% were S100β+, showing normal production of new neurons and astrocytes, respectively, while the phenotype for the remaining EdU+ cells (26%) was undetermined (Fig. 3B–D,G). In Gfap+/R236H mice, there was a striking increase (2.5-fold) in the overall number of EdU label-retaining cells in the granular layer compared with wild-type mice (Fig. 3H); however, the majority of these cells (79%) were neither NeuN nor S100β positive and were not identified (Fig. 3F,G). The number of new granule cells (EdU+/NeuN+) was significantly reduced to <30% of those observed in wild-type mice (Fig. 3H), representing only 6% of total EdU labeled cells in mutant mice (Fig. 3G). However, EdU+/S100β+ astrocyte numbers were increased by nearly twofold in Gfap+/R236H mice (Fig. 3E,H).
Figure 3.

Abnormal differentiation of neural progenitors in Gfap+/R236H hippocampus. A, EdU injection protocol and tissue collection time line for cell fate analysis. Arrows indicate injection time points every 4 h for saturated labeling of cycling progenitors at 10 weeks of age, followed by tissue collection 28 d postinjection. B–D, Analysis of EdU-labeled cell types 4 weeks after injection shows both new neurons (B) and astrocytes (C) in the dentate gyrus of Gfap+/+ mice as shown by costaining with NeuN and S100β, respectively. Unidentified EdU+/NeuN−/S100β− cells were also apparent in Gfap+/+ mice (D). E, F, In Gfap+/R236H mice, this unidentified phenotype accounted for the majority of EdU-labeled cells (F), although many EdU+ cells were identified as S100β+ astrocytes (E). n = 5 male mice at 14 weeks of age per genotype. Scale bar, 10 μm. G, Gfap+/+ mice show a normal distribution of neuronal and astroglial EdU+ cell types in the granular layer of the dentate gyrus (DG), as well as a population of unidentified EdU+ cells at 4 weeks after EdU labeling. Gfap+/R236H mice show a lack of NeuN+ cells with the majority of EdU+ cells negative for either NeuN or S100β. H, The overall number of EdU labeled cells in the granular layer is increased in Gfap+/R236H mice at 4 weeks postlabel compared with Gfap+/+ mice. Although EdU+/NeuN+ neurons are depleted, EdU+/S100β+ astrocytes and especially NeuN−/S100β− cells are increased in mutant mice. *p < 0.05, **p < 0.01, ***p ≤ 0.001, two tailed t test, n = 5 male mice at 14 weeks of age per genotype. I, Immunolabeling shows the presence of EdU+/GFAP+/S100β− cells in the SGZ of Gfap+/R236H mice. Scale bar, 10 μm.
The general increase in label-retaining cells in Gfap+/R236H compared with Gfap+/+ mice after 1 month suggests that progenitors are not dividing at a normal rate (Fig. 3H). Since repeated division of intermediate progenitors dilutes EdU labeling, cells remaining positive at this time point are those that left the cell cycle shortly after the initial label (with no more than a few divisions) and survived to fully differentiate, or alternatively they are more slowly dividing cells. In Gfap+/R236H mice, most of the EdU+/NeuN−/S100β− cells 4 weeks postlabeling remain in the SGZ, suggesting they are not reactive glia that happen to be in the granular layer. Occasionally, EdU+ microglia are apparent in the granular layer (Fig. 2J), but are sparse and should not account for the total increase. To test whether these unidentified cells may be activated NSCs, as implied by the presence of Ki67+/Sox2+/GFAP+ cells shown earlier (Fig. 2I), we colabeled for S100β and GFAP. In Gfap+/R236H mice, many of the EdU-labeled cells in the SGZ were S100β negative and positive for GFAP (Fig. 3I), and while mature EdU+/S100β+/GFAP+ astrocytes were apparent in the granular layer, they were generally not in the SGZ. Although the identity of these cells as activated NSCs remains to be confirmed, the results show that mutant GFAP causes a marked deficit in the generation of new neurons and a possible shift toward gliogenesis.
Learning and memory deficits in Gfap+/R236H mice
To determine whether deficits in hippocampal neurogenesis have a functional impact on Gfap+/R236H mice, we tested both classical associative learning by fear conditioning and spatial learning and memory with the Morris water maze (Morris et al., 1982; Sutherland et al., 1982; Kim and Fanselow, 1992; Phillips and LeDoux, 1992; Logue et al., 1997; Saxe et al., 2006; Dupret et al., 2008; Imayoshi et al., 2008; Clelland et al., 2009; Deng et al., 2009). For fear conditioning, male and female mice between 9 and 10 weeks of age were conditioned with two trials of a cue stimulus paired with a brief shock (unconditioned stimulus, see Materials and Methods). The mice were then tested for a fear response (freezing) to the context or conditioned cue. Female Gfap+/R236H mice froze less often in response to context, but showed no difference from Gfap+/+ mice in response to the cue stimulus (Fig. 4A). Male mice at this age showed no effect of genotype to either the context or the cue stimulus. However, at one year of age both female and male mutant mice showed a decrease in contextual fear conditioning and male mice froze less often when presented with the cue stimulus (Fig. 4B).
Figure 4.

Behavioral tests for hippocampal-dependent tasks of learning and spatial memory show deficits in AxD mice. A, B, Fear conditioning in Gfap+/R236H mice shows a decrease in freezing in response to context in female mice at 10 weeks of age (A) and in both males and females at 13 months of age (B). At the later age, male AxD mice also show a significant decrease in conditioning to the cue (*p ≤ 0.05, **p ≤ 0.01, one tailed t test, n ≥ 10 per group). C, Performance in the Morris water maze is also affected, with both male and female mice (10 weeks) showing a lack of preference for the trained quadrant as indicated by the number of platform crossings. During the probe trial Gfap+/+ male and female mice crossed the platform region significantly more often in the trained quadrant than in any of the other quadrants. Female Gfap+/R236H mice showed no preference for the trained quadrant, and had significantly fewer trained platform crossings compared with Gfap+/+ mice. Male mutants showed a difference between the trained and opposite quadrant, but there was not a significant preference compared with the other two quadrants. p values derived from repeated-measures ANOVA with *Tukey's posttest to show significant difference with trained quadrant. †p < 0.05 Gfap+/+ vs Gfap+/R236H in trained quadrant (t test, n ≥ 11 per group).
To further evaluate hippocampal function, mice were tested in the Morris water maze at 10 weeks of age. After eight training blocks (two per day, see Materials and Methods), preference for the trained quadrant was assessed by platform crossings during the probe trial. This measure estimates how well the platform location has been encoded and is a better representation of spatial memory and hippocampal function related to neurogenesis compared with other measures such as latency and path length (Garthe and Kempermann, 2013). Gfap+/+ mice showed a clear preference for the trained quadrant, while both male and female Gfap+/R236H mice failed to show quadrant preference, and female mutant mice showed a significantly reduced number of trained platform crossings compared with wild-type females (Fig. 4C).
With the observed deficits in both immature and newborn neurons, the poor performance by Gfap mutant mice in hippocampal-dependent tasks of learning and memory correlates well with the reported association between adult neurogenesis and hippocampal function (Saxe et al., 2006; Dupret et al., 2008; Imayoshi et al., 2008; Clelland et al., 2009; Deng et al., 2009).
Discussion
The Gfap+/R236H mouse model of Alexander disease has extensive hippocampal pathology including protein aggregation, stress pathway activation, and reactive astrocytes and microglia (Hagemann et al., 2006, 2012). Here, we show that AxD mice also have deficits in hippocampal neurogenesis and perform poorly in tasks of learning and memory. Neural stem cells have elevated GFAP and appear to be in cell cycle arrest, progenitor proliferation is reduced, and immature neurons are diminished. Consequently, AxD mice show a lack in adult born granule cells in the dentate gyrus, while gliogenesis and new astrocytes appear to be increased. Finally, behavior tests also suggest deficits in spatial memory and hippocampal function. This is the first demonstration of neurogenic and cognitive deficits resulting from a primary disorder of astrocytes.
To better understand the effects of AxD pathology on neurogenesis, we evaluated progenitor proliferation and differentiation by labeling dividing cells with the thymidine analog EdU. The number of proliferating cells in the SGZ was reduced by 65% in Gfap+/R236H mice. The EdU-positive cells that are present are not found in clusters of recently divided progenitors as observed in Gfap+/+ mice, but rather as single cells or doublets, suggesting a slower replication cycle. Colabeling with Ki67 did not show an increase in progenitors exiting cell cycle (EdU+/Ki67−), however, there was a striking number of EdU−/Ki67+ cells in Gfap+/R236H mice that were not apparent in Gfap+/+ animals. Although NSCs cycle less frequently, their cycle length is slightly shorter than that of proliferating IPCs (Brandt et al., 2012). Since Ki67 labels all stages of cell cycle, these results indicate a population of cells in either a prolonged or arrested cycle. Given that there are similar numbers of EdU+/Ki67+ and EdU−/Ki67+-labeled cells, this block may not be complete. Further analysis shows numerous Ki67+/GFAP+/Sox2+ cells in the SGZ of mutants that are not prevalent in wild-type mice, suggesting these are abnormally activated NSCs. In a more severe model of AxD, Sosunov et al. (2013) found multinuclear hippocampal astrocytes and other nuclear abnormalities, indicating astrocytes are entering cell cycle but unable to divide. Our results suggest that mutant GFAP may cause inefficient or defective cell division in NSCs as well (Yasui et al., 1998).
In contrast to the reduced number of proliferating cells 24 h postlabel, cell fate analysis after 4 weeks shows more than twice as many EdU+ cells in mutant mice, the majority of which have not differentiated. The overall number of newborn neurons (EdU+/NeuN+) is markedly reduced in Gfap mutant mice, which correlates with the depletion of immature neurons, but newborn astrocytes (EdU+/S100β+) are increased by nearly twofold in the granular cell layer, showing that gliogenesis may be promoted. A shift toward glial fate could contribute to the deficit in neurogenesis; however, it is worth noting that EdU-labeled astrocytes are also evident in the hilus, molecular layer, and other regions of the hippocampus and CNS. These astrocytes are apparent at both 24 h and 4 weeks postlabeling, suggesting they did not migrate from the dentate gyrus, but are dividing because they are reactive (Buffo et al., 2008). This raises the question of whether the labeled astrocytes in the granular layer of the dentate gyrus originate from adult stem cells or are mature reactive astrocytes, and whether they are increased in number.
An increase in the proportion of EdU+/NeuN−/S100β− cells has been described before with the disruption of neurogenesis (Vallières et al., 2002; Kandasamy et al., 2010), but the fact that these undifferentiated cells outnumber the total number of EdU-retaining cells found in wild-type mice by twofold at this time point (4 weeks postlabel) is unusual in that fewer cells were labeled at 24 h compared with wild type. Since we have shown that in Gfap+/R236H mice, Ki67 labels GFAP+/Sox2+ cells that appear to be NSCs, we speculate that EdU+/NeuN−/S100β− cells in the SGZ may also be GFAP+ stem cells. Infrequently or abnormally dividing NSCs would retain the EdU label, and additional analysis shows the presence of EdU+/GFAP+/S100β− cells in the SGZ in Gfap+/R236H mice 4 weeks postlabel. Further investigation will determine the proportion of EdU+/NeuN−/S100β− cells that are positive for GFAP, whether these are radial NSCs, and whether this population is expanding or in cell cycle arrest.
Mature hippocampal astrocytes regulate neuronal fate specification in adult neurogenesis (Song et al., 2002; Barkho et al., 2006; Ashton et al., 2012; Wilhelmsson et al., 2012). In acute brain injury such as stroke, seizure, or trauma, neurogenesis generally increases; however, in transgenic models of neurodegenerative disease such as Alzheimer's, Huntington's, and Parkinson's with chronic reactive gliosis, the consequences are eventually negative with a decrease in adult-born granule cells and concomitant deficits in learning and memory (Zhao et al., 2008; Winner et al., 2011). It is well established that prolonged neuroinflammation generates a nonpermissive microenvironment for adult neurogenesis (Monje et al., 2003; Kohman and Rhodes, 2013), and astrocyte-specific overexpression of IL-β or IL-6 in transgenic mice activates microglia and has a similar effect in reducing progenitor proliferation and neurogenesis (Vallières et al., 2002; Wu et al., 2012). Gliosis is prominent in AxD, and Gfap+/R236H mice have elevated transcripts for IL-1β, IL-6, and TNFα in hippocampus (our unpublished data), each of which can independently inhibit neurogenesis (Vallières et al., 2002; Monje et al., 2003; Iosif et al., 2006; Koo and Duman, 2008; Keohane et al., 2010; Green et al., 2012; Wu et al., 2012; Kohman and Rhodes, 2013).
In addition to reactive gliosis, another common denominator between AxD and other neurodegenerative proteinopathies is that the mutant proteins in familial cases, including amyloid precursor protein (APP), presenilin, tau, α-synuclein, and huntingtin, are expressed in adult neural progenitor cells (Winner et al., 2011), and loss of function could also contribute to deficits in neurogenesis. However, knock-out of the culprit genes, including Gfap, often results in a different neurogenic phenotype or even enhanced neurogenesis (Larsson et al., 2004; Winner et al., 2012; Conforti et al., 2013). Gain of function in the form of toxic oligomers, protein aggregation, and the disruption of the ubiquitin-proteasome system may be a more likely mechanism underlying neurogenic deficits, especially since the major signaling pathways controlling neurogenesis, including Notch, Wnt/β-catenin, Hedgehog/Gli, and TGFβ/Smad, are all regulated by ubiquitin-targeted degradation (Inoue and Imamura, 2008; Tauriello and Maurice, 2010; Weinmaster and Fischer, 2011; Gulino et al., 2012; Moretti and Brou, 2013). Notch signaling, for example, has been implicated in driving NSC renewal and gliogenesis (Pierfelice et al., 2011). The activated Notch intracellular domain (NICD) is targeted for proteasome degradation by the Fbw7/SCF-E3 ubiquitin ligase (Wu et al., 2001). Deletion of Fbw7 causes NSC accumulation and increased astrogenesis while inhibiting neurogenesis in early development (Hoeck et al., 2010; Matsumoto et al., 2011). Adult NSCs express Notch1, and overexpression of activated Notch1 in GFAP-expressing cells causes increased cell cycle entry and NSC renewal at the expense of neurogenesis (Breunig et al., 2007). Hence, disruption of NICD degradation due to proteasome dysfunction in the NSC population could explain the increased numbers of Ki67+/GFAP+/Sox2+ SGZ cells observed in AxD mice.
Several studies using different approaches to deplete adult born dentate granule cells have shown that neurogenesis is associated with hippocampal-dependent learning (Saxe et al., 2006; Dupret et al., 2008; Imayoshi et al., 2008; Clelland et al., 2009; Deng et al., 2009). Here, we show that in addition to having deficits in hippocampal neurogenesis, Gfap+/R236H mice perform poorly in hippocampal-dependent behavioral tests, including contextual fear conditioning and Morris water maze. In fear conditioning, association between the unconditioned shock and conditioned cue is amygdala dependent, whereas relating the shock to context requires both amygdala and hippocampus (Phillips and LeDoux, 1992). Young adult female AxD mice are deficient in contextual fear conditioning, but show no difference in conditioning to the cue, indicating abnormal hippocampal function. In aged mice males and females show deficits in contextual conditioning, with male mice also showing decreased response to the cue stimulus, suggesting learning deficits in males may involve the amygdala. Why there would be a gender difference in fear conditioning is not clear. Early reports suggested the possibility of male predominance in AxD, particularly in juvenile patients (Li et al., 2005), but subsequent studies with larger patient populations show no gender bias, regardless of age group or classification (Prust et al., 2011). Both male and female Gfap+/R236H mice perform poorly in the Morris water maze at an early age, further demonstrating a deficit in hippocampal function (Morris et al., 1982; Sutherland et al., 1982; Kim and Fanselow, 1992; Phillips and LeDoux, 1992; Logue et al., 1997). Although there is controversy regarding the relevance of adult neurogenesis to water maze performance, by measuring platform crossings we show that AxD mice are unable to encode the goal position, a deficit specific to spatial memory and efficient pattern separation, hippocampal functions that likely depend on new neurons (Garthe and Kempermann, 2013).
Recently in a similar transgenic model of AxD (Tanaka et al., 2007), Lee et al. (2013) show that increased ATP release leads to suppression of GABAergic signaling and a reduced threshold for long-term potentiation. Generally enhanced LTP improves performance in hippocampal-dependent learning, but behavioral testing has not yet been reported for these mice. Hypertrophy and reactivity likely alter a number of the essential functions attributed to astrocytes, including the regulation of synapse formation and stability, gliotransmission, and neurotransmitter clearance (Halassa and Haydon, 2010), all of which could affect synaptic plasticity, learning, and memory. In addition, behavioral tests such as the Morris water maze rely on brain regions other than hippocampus, and astrocyte pathology in regions such as the entorhinal cortex or superior colliculus may also contribute to deficits in learning the maze (Redish, 1999; Garthe and Kempermann, 2013). Although we have not confirmed whether behavior deficits in the Gfap+/R236H knock-in model result from a lack of new neurons or a more direct interference of hippocampal function by reactive glia, it is clear that the mice have cognitive deficits as a result of mutations in Gfap and the associated pathology. Considering the lack of white matter pathology in the model, these deficits in standard learning paradigms suggest that gray matter astrocytes may also contribute to the cognitive deficits observed in human patients with AxD.
An important remaining question is whether stem cells expressing mutant GFAP produce fewer neuronal precursors, or whether progenitors are unable to thrive in a nonpermissive environment created by reactive niche astrocytes. Both mechanisms may play a role, and teasing out their effects will give a better understanding of the pathological process in AxD and the impact of chronic gliosis on neurogenesis and cognition in neurodegenerative disease.
Footnotes
This work was supported by the National Institutes of Health Grants R01-NS060120 and P01-NS42803 (to A.M.) and P30-HD03352 (to the Waisman Center), and by the Juanma Fund and the Rijkaart Fund. We thank Denice Springman for technical support and Dr. Xinyu Zhao for critical review and suggestions for this manuscript.
The authors have no competing financial interests.
References
- Aimone JB, Deng W, Gage FH. Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron. 2011;70:589–596. doi: 10.1016/j.neuron.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72:373–381. doi: 10.1093/brain/72.3.373. 3 pl. [DOI] [PubMed] [Google Scholar]
- Ashton RS, Conway A, Pangarkar C, Bergen J, Lim KI, Shah P, Bissell M, Schaffer DV. Astrocytes regulate adult hippocampal neurogenesis through ephrin-B signaling. Nat Neurosci. 2012;15:1399–1406. doi: 10.1038/nn.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkho BZ, Song H, Aimone JB, Smrt RD, Kuwabara T, Nakashima K, Gage FH, Zhao X. Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev. 2006;15:407–421. doi: 10.1089/scd.2006.15.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt MD, Hübner M, Storch A. Brief report: Adult hippocampal precursor cells shorten S-phase and total cell cycle length during neuronal differentiation. Stem Cells. 2012;30:2843–2847. doi: 10.1002/stem.1244. [DOI] [PubMed] [Google Scholar]
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- Brenner M, Goldman JE, Quinlan RA, Messing A. Alexander disease: a genetic disorder of astrocytes. In: Parpura V, Haydon PG, editors. Astrocytes in pathophysiology of the nervous system. Ed 1. New York: Springer; 2009. pp. 591–648. [Google Scholar]
- Breunig JJ, Silbereis J, Vaccarino FM, Sestan N, Rakic P. Notch regulates cell fate and dendrite morphology of newborn neurons in the postnatal dentate gyrus. Proc Natl Acad Sci U S A. 2007;104:20558–20563. doi: 10.1073/pnas.0710156104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD, Jr, Fragniere A, Tyers P, Jessberger S, Saksida LM, Barker RA, Gage FH, Bussey TJ. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325:210–213. doi: 10.1126/science.1173215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti P, Camnasio S, Mutti C, Valenza M, Thompson M, Fossale E, Zeitlin S, MacDonald ME, Zuccato C, Cattaneo E. Lack of huntingtin promotes neural stem cells differentiation into glial cells while neurons expressing huntingtin with expanded polyglutamine tracts undergo cell death. Neurobiol Dis. 2013;50:160–170. doi: 10.1016/j.nbd.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Deng W, Saxe MD, Gallina IS, Gage FH. Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci. 2009;29:13532–13542. doi: 10.1523/JNEUROSCI.3362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11:339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupret D, Revest JM, Koehl M, Ichas F, De Giorgi F, Costet P, Abrous DN, Piazza PV. Spatial relational memory requires hippocampal adult neurogenesis. PLoS One. 2008;3:e1959. doi: 10.1371/journal.pone.0001959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Garthe A, Kempermann G. An old test for new neurons: refining the Morris water maze to study the functional relevance of adult hippocampal neurogenesis. Front Neurosci. 2013;7:63. doi: 10.3389/fnins.2013.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green HF, Treacy E, Keohane AK, Sullivan AM, O'Keeffe GW, Nolan YM. A role for interleukin-1beta in determining the lineage fate of embryonic rat hippocampal neural precursor cells. Mol Cell Neurosci. 2012;49:311–321. doi: 10.1016/j.mcn.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Gulino A, Di Marcotullio L, Canettieri G, De Smaele E, Screpanti I. Hedgehog/Gli control by ubiquitination/acetylation interplay. Vitam Horm. 2012;88:211–227. doi: 10.1016/B978-0-12-394622-5.00009-2. [DOI] [PubMed] [Google Scholar]
- Guo W, Zhang L, Christopher DM, Teng ZQ, Fausett SR, Liu C, George OL, Klingensmith J, Jin P, Zhao X. RNA-binding protein FXR2 regulates adult hippocampal neurogenesis by reducing Noggin expression. Neuron. 2011;70:924–938. doi: 10.1016/j.neuron.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann TL, Connor JX, Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci. 2006;26:11162–11173. doi: 10.1523/JNEUROSCI.3260-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann TL, Jobe EM, Messing A. Genetic ablation of Nrf2/antioxidant response pathway in Alexander disease mice reduces hippocampal gliosis but does not impact survival. PLoS One. 2012;7:e37304. doi: 10.1371/journal.pone.0037304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes NL, Nowakowski RS. Dynamics of cell proliferation in the adult dentate gyrus of two inbred strains of mice. Brain Res Dev Brain Res. 2002;134:77–85. doi: 10.1016/S0165-3806(01)00324-8. [DOI] [PubMed] [Google Scholar]
- Hoeck JD, Jandke A, Blake SM, Nye E, Spencer-Dene B, Brandner S, Behrens A. Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nat Neurosci. 2010;13:1365–1372. doi: 10.1038/nn.2644. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Ohtsuka T, Takao K, Miyakawa T, Yamaguchi M, Mori K, Ikeda T, Itohara S, Kageyama R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat Neurosci. 2008;11:1153–1161. doi: 10.1038/nn.2185. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Imamura T. Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008;99:2107–2112. doi: 10.1111/j.1349-7006.2008.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, Jacobsen SE, Lindvall O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–9712. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki T, Iwaki A, Tateishi J, Sakaki Y, Goldman JE. Alpha B-crystallin and 27-kd heat shock protein are regulated by stress conditions in the central nervous system and accumulate in Rosenthal fibers. Am J Pathol. 1993;143:487–495. [PMC free article] [PubMed] [Google Scholar]
- Kandasamy M, Couillard-Despres S, Raber KA, Stephan M, Lehner B, Winner B, Kohl Z, Rivera FJ, Nguyen HP, Riess O, Bogdahn U, Winkler J, von Hörsten S, Aigner L. Stem cell quiescence in the hippocampal neurogenic niche is associated with elevated transforming growth factor-beta signaling in an animal model of Huntington disease. J Neuropathol Exp Neurol. 2010;69:717–728. doi: 10.1097/NEN.0b013e3181e4f733. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc Natl Acad Sci U S A. 1997;94:10409–10414. doi: 10.1073/pnas.94.19.10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keohane A, Ryan S, Maloney E, Sullivan AM, Nolan YM. Tumour necrosis factor-alpha impairs neuronal differentiation but not proliferation of hippocampal neural precursor cells: role of Hes1. Mol Cell Neurosci. 2010;43:127–135. doi: 10.1016/j.mcn.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Kohman RA, Rhodes JS. Neurogenesis, inflammation and behavior. Brain Behav Immun. 2013;27:22–32. doi: 10.1016/j.bbi.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. 2008;105:751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A, Wilhelmsson U, Pekna M, Pekny M. Increased cell proliferation and neurogenesis in the hippocampal dentate gyrus of old GFAP(−/−)Vim(−/−) mice. Neurochem Res. 2004;29:2069–2073. doi: 10.1007/s11064-004-6880-2. [DOI] [PubMed] [Google Scholar]
- Lee HU, Yamazaki Y, Tanaka KF, Furuya K, Sokabe M, Hida H, Takao K, Miyakawa T, Fujii S, Ikenaka K. Increased astrocytic ATP release results in enhanced excitability of the hippocampus. Glia. 2013;61:210–224. doi: 10.1002/glia.22427. [DOI] [PubMed] [Google Scholar]
- Li R, Johnson AB, Salomons G, Goldman JE, Naidu S, Quinlan R, Cree B, Ruyle SZ, Banwell B, D'Hooghe M, Siebert JR, Rolf CM, Cox H, Reddy A, Gutiérrez-Solana LG, Collins A, Weller RO, Messing A, van der Knaap MS, Brenner M. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol. 2005;57:310–326. doi: 10.1002/ana.20406. [DOI] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111:104–113. doi: 10.1037/0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Onoyama I, Sunabori T, Kageyama R, Okano H, Nakayama KI. Fbxw7-dependent degradation of Notch is required for control of “stemness” and neuronal-glial differentiation in neural stem cells. J Biol Chem. 2011;286:13754–13764. doi: 10.1074/jbc.M110.194936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci. 2012;32:5017–5023. doi: 10.1523/JNEUROSCI.5384-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Moretti J, Brou C. Ubiquitinations in the notch signaling pathway. Int J Mol Sci. 2013;14:6359–6381. doi: 10.3390/ijms14036359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037/0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Pierfelice T, Alberi L, Gaiano N. Notch in the vertebrate nervous system: an old dog with new tricks. Neuron. 2011;69:840–855. doi: 10.1016/j.neuron.2011.02.031. [DOI] [PubMed] [Google Scholar]
- Prust M, Wang J, Morizono H, Messing A, Brenner M, Gordon E, Hartka T, Sokohl A, Schiffmann R, Gordish-Dressman H, Albin R, Amartino H, Brockman K, Dinopoulos A, Dotti MT, Fain D, Fernandez R, Ferreira J, Fleming J, Gill D, et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology. 2011;77:1287–1294. doi: 10.1212/WNL.0b013e3182309f72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redish AD. Beyond the cognitive map. London: MIT; 1999. [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O'Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, Hen R, Drew MR. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103:17501–17506. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seri B, García-Verdugo JM, McEwen BS. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci. 2001;21:7153–7160. doi: 10.1523/JNEUROSCI.21-18-07153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Stevens CF, Gage FH. Astroglia induce neurogenesis from adult neural stem cells. Nature. 2002;417:39–44. doi: 10.1038/417039a. [DOI] [PubMed] [Google Scholar]
- Sosunov AA, Guilfoyle E, Wu X, McKhann GM, 2nd, Goldman JE. Phenotypic conversions of “protoplasmic” to “reactive” astrocytes in alexander disease. J Neurosci. 2013;33:7439–7450. doi: 10.1523/JNEUROSCI.4506-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland RJ, Kolb B, Whishaw IQ. Spatial mapping: definitive disruption by hippocampal or medial frontal cortical damage in the rat. Neurosci Lett. 1982;31:271–276. doi: 10.1016/0304-3940(82)90032-5. [DOI] [PubMed] [Google Scholar]
- Tanaka KF, Takebayashi H, Yamazaki Y, Ono K, Naruse M, Iwasato T, Itohara S, Kato H, Ikenaka K. Murine model of Alexander disease: analysis of GFAP aggregate formation and its pathological significance. Glia. 2007;55:617–631. doi: 10.1002/glia.20486. [DOI] [PubMed] [Google Scholar]
- Tauriello DV, Maurice MM. The various roles of ubiquitin in Wnt pathway regulation. Cell Cycle. 2010;9:3700–3709. doi: 10.4161/cc.9.18.13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallières L, Campbell IL, Gage FH, Sawchenko PE. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci. 2002;22:486–492. doi: 10.1523/JNEUROSCI.22-02-00486.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmaster G, Fischer JA. Notch ligand ubiquitylation: what is it good for? Dev Cell. 2011;21:134–144. doi: 10.1016/j.devcel.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U, Faiz M, de Pablo Y, Sjöqvist M, Andersson D, Widestrand A, Potokar M, Stenovec M, Smith PL, Shinjyo N, Pekny T, Zorec R, Ståhlberg A, Pekna M, Sahlgren C, Pekny M. Astrocytes negatively regulate neurogenesis through the Jagged1-mediated Notch pathway. Stem Cells. 2012;30:2320–2329. doi: 10.1002/stem.1196. [DOI] [PubMed] [Google Scholar]
- Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur J Neurosci. 2011;33:1139–1151. doi: 10.1111/j.1460-9568.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- Winner B, Regensburger M, Schreglmann S, Boyer L, Prots I, Rockenstein E, Mante M, Zhao C, Winkler J, Masliah E, Gage FH. Role of alpha-synuclein in adult neurogenesis and neuronal maturation in the dentate gyrus. J Neurosci. 2012;32:16906–16916. doi: 10.1523/JNEUROSCI.2723-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, Chui I, Deshaies RJ, Kitajewski J. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol Cell Biol. 2001;21:7403–7415. doi: 10.1128/MCB.21.21.7403-7415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MD, Hein AM, Moravan MJ, Shaftel SS, Olschowka JA, O'Banion MK. Adult murine hippocampal neurogenesis is inhibited by sustained IL-1beta and not rescued by voluntary running. Brain Behav Immun. 2012;26:292–300. doi: 10.1016/j.bbi.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y, Amano M, Nagata K, Inagaki N, Nakamura H, Saya H, Kaibuchi K, Inagaki M. Roles of Rho-associated kinase in cytokinesis; mutations in Rho-associated kinase phosphorylation sites impair cytokinetic segregation of glial filaments. J Cell Biol. 1998;143:1249–1258. doi: 10.1083/jcb.143.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]