

Abstract
Severe sepsis is often accompanied by acute kidney injury (AKI) and albuminuria. Here we studied whether the AKI and albuminuria associated with lipopolysaccharide (LPS) treatment in mice reflects impairment of the glomerular endothelium with its associated endothelial surface layer. LPS treatment decreased the abundance of endothelial surface layer heparan sulfate proteoglycans and sialic acid, and led to albuminuria likely reflecting altered glomerular filtration perm-selectivity. LPS treatment decreased the glomerular filtration rate (GFR), while also causing significant ultrastructural alterations in the glomerular endothelium. The density of glomerular endothelial cell fenestrae was 5-fold lower whereas the average fenestrae diameter was 3-fold higher in LPS-treated than in control mice. The effects of LPS on the glomerular endothelial surface layer, endothelial cell fenestrae, GFR, and albuminuria were diminished in TNF receptor 1 (TNFR1) knockout mice, suggesting that these LPS effects are mediated by TNF-α activation of TNFR1. Indeed, intravenous administration of TNF decreased GFR and led to loss of glomerular endothelial cell fenestrae, increased fenestrae diameter, and damage to the glomerular endothelial surface layer. LPS treatment decreased kidney expression of vascular endothelial growth factor (VEGF). Thus, our findings confirm the important role of glomerular endothelial injury, possibly by a decreased VEGF level, in the development and progression of AKI and albuminuria in the LPS model of sepsis in the mouse.
Keywords: acute kidney injury, fenestrae, albuminuria, endothelial cells, endothelial surface layer, lipopolysaccharide
INTRODUCTION
Acute kidney injury (AKI) is a frequent and serious complication of sepsis. The incidence of AKI is about 40% in patients with severe sepsis and septic shock. Moreover, there is strong evidence that AKI in patients with severe sepsis is associated with a higher mortality rate.1 The high frequency and mortality of sepsis-associated AKI demand a better understanding of the pathophysiology of this disorder.2, 3
Cytokines released during sepsis cause some of the most frequent clinical features of this syndrome, such as hypoalbuminemia, edema, and reduced effective circulating volume, in part through their actions on endothelium.4-6 We and others have demonstrated, using the LPS model of sepsis, that the cytokine TNF-α plays a key, causative role in AKI through its action on renal endothelial TNFR1.7, 8 The injurious effect of TNF-α on renal ECs has been previously demonstrated.9
Vascular permeability in renal glomeruli is determined by the “glomerular filtration barrier” (GFB), which consists of the glomerular capillary endothelium, the podocytes, and their interposed basement membranes. The integrity of the GFB prevents the leak of albumin and other plasma proteins into the urine.10, 11 However, the effect of sepsis on the structure and function of the glomerular endothelium within the GFB has not been adequately investigated. Glomerular endothelial abnormalities have been suggested by the occurrence of albuminuria, the hallmark of GFB dysfunction, in patients with sepsis12, 13 and in animal models of acute endotoxemia such as those produced by lipopolysaccharide (LPS) and by Cecal Ligation and Puncture (CLP).14, 15
Endothelia have been classically divided into two main structural types: continuous and fenestrated endothelia. Sepsis-induced barrier dysfunction in continuous ECs such as pulmonary microvascular cells is believed to in part reflect disruption of inter-endothelial junctions (IEJs),16-20 even though the endothelial glycocalyx remains the dominant size-selective structure.21 Glomerular endothelial fenestrae are circular, transcellular pores 60–80 nm in diameter.22-25 These fenestrations, which occupy 20–50% of the endothelial surface,26 were initially thought to provide little restriction to the passage of albumin. However, Ryan and Karnovsky27 showed, using transmission electron microscopy, that albumin passes minimally through endothelial fenestrae and is largely confined to the glomerular capillary lumen under normal conditions. Now it is believed that a glycocalyx layer covering the fenestral domains of the glomerular EC luminal surface prevents or minimizes diffusion of plasma protein through endothelial fenestrae.22, 25, 28 The glycocalyx layer is formed from a complex set of varied EC membrane-associated macromolecules.29, 30 These include the very negatively charged glycoproteins bearing acidic oligosaccharides with terminal sialic acids, and negatively charged proteoglycans with their associated glycosaminoglycan (GAG) side chains such as heparan sulfate and chondroitin sulfate. In vivo, the glycocalyx is covered by a thicker “cell coat” composed of plasma proteins such as albumin and orosomucoid,31-34 and proteins and hyaluronan produced by the endothelium.35 The “cell coat” and the glycocalyx constitute the endothelial surface layer (ESL).11
In the present study we investigated the changes of glomerular endothelial fenestrae and ESL during severe experimental endotoxemia and TNF-induced AKI, and test the hypothesis that such changes may be related to signaling through TNFR1.
RESULTS
LPS induces AKI and increases urine concentration of albumin
We measured plasma urea levels as an indicator of glomerular filtration rate (GFR), and urine albumin-to-creatinine ratio to assess injury to the glomerular filtration barrier. In wild type (WT) mice, plasma urea levels increased from 28.8 ± 2.8 mg/dl to 112.5 ± 9.5 mg/dl (P < 0.01) 24 h after injection of LPS (10 mg/kg) (Figure 1a). LPS also induced significant weight loss (12.5 ± 1.1%, P < 0.01) compared to mice treated with normal saline (control) (2.6 ± 0.6%) (Figure 1c). The urinary albumin-to-creatinine ratio increased about 10-fold, from an initial value of 0.03 ± 0.01 to a 24 h value of 0.30 ± 0.06 (P < 0.05) (Figure 1b), despite the rapid decline in GFR.
Figure 1.

Endotoxemia induces TNFR1-dependent AKI and increases urine concentration of albumin, in association with decreased fenestral density and increased fenestral diameter of glomerular capillary endothelium. Effect of LPS (10 mg/kg for 24h) on (a) plasma BUN concentration, (b) urine albumin/creatinine ratio, (c) weight loss, (d) fenestral density, and fenestral diameter in wild type and Tnfr1−/− mice. Control, wild type treated with normal saline; WT LPS, wild type treated with LPS; TNFR1−/− LPS, Tnfr1−/− mice treated with LPS. Values are mean ± s.e.m. for 3-5 animals (a-c) 60-70 capillary loops (d) and 33-67 fenestrae (e). **P<0.01 vs. control. ##P<0.01 vs. wild-type mice injected with LPS.
Mice deficient in TNFR1 are resistant to LPS-induced AKI and albuminuria
TNF-α release into the circulation followed LPS administration, and Tnfr1−/− mice were resistant to LPS-induced AKI.7 We confirmed this finding and showed that plasma urea level was not elevated in Tnfr1−/− mice 24 h after LPS injection, despite similar LPS-induced weight loss in Tnfr1−/− and WT mice (Figure 1a and c). In addition to protection from a fall in GFR, Tnfr1−/− mice had reduced albuminuria in response to LPS. Tnfr1−/− mice had a urine albumin/creatinine ratio of only 0.03 ± 0.01 after LPS, significantly less than WT mice after LPS (0.30 ± 0.6, P < 0.05), and no different than WT control mice (Figure 1b). We did not compare Tnfr1−/− mice treated with normal saline with WT control mice, since previous data demonstrate similar baseline values of urinary albumin excretion and GFR in vehicle-treated WT and Tnfr1−/− mice.7, 36 Our results support the idea that TNF, acting through TNFR1, is a key mediator of LPS-induced AKI and albuminuria.
LPS-induced AKI is associated with changes in glomerular EC fenestration in normal but not Tnfr1−/− mice
Since transport of water across the glomerular capillary wall occurs predominantly through the endothelial fenestrae, a reduction in the diameter and/or density of endothelial fenestrae can reduce endothelial filtration area and glomerular ultrafiltration coefficient (Kf). To explore whether sepsis-induced acute renal failure is accompanied by morphological changes in glomerular fenestrae, and whether such changes require TNFR1, we compared the ultrastructural morphology of the glomerular endothelium in LPS-untreated and -treated WT mice with that of LPS-treated Tnfr1−/− mice. The glomerular capillary wall in control mice, as imaged by transmission electron microscopy, is shown lined with fenestrated endothelium, with fenestrae appearing circular when viewed en face in electron microscopic images (Figure 2a and d). However, LPS-treated WT mice show extensive detachment of glomerular ECs from their glomerular basement membranes (GBMs) (arrowheads, Figure 2b). The majority of glomerular ECs were often swollen, devoid of fenestrae, and detached from their GBMs (although intact fenestrae are evident at the bottom right of Figure 2b). The GBM itself and adjacent podocytes were normal without podocyte detachment or effacement (Figure 2b). However, in LPS-treated Tnfr1−/− mice, glomerular ECs appear normal, with minimal detachment from the GBMs (Figure 2c). Fenestral density per μm capillary length as measured in electron micrographs was 3.6±0.5 in the WT control mice, significantly higher than in the WT mice 24 h after the LPS injection (0.6±0.2). In contrast, fenestral density in the Tnfr1−/− mice 24 h post-LPS injection (3.2±0.3) was indistinguishable from that of WT control (Figure 1d). In en face electron microscopic images, the fenestral diameters were much larger in the LPS-treated mice (195±16.4 nm) than in saline-injected WT controls (64.2±2.4 nm; Figure 2e). The average diameter of the endothelial fenestrae in LPS-treated Tnfr1−/− mice was 75.5±2.5 nm, significantly smaller than in LPS-treated WT mice (Figure 1e). In conclusion, LPS treatment significantly increased size of glomerular EC fenestrae but decreased fenestral density, and both effects were completely prevented by absence of TNFR1. Even though LPS increased fenestral diameter, the fenestrated fraction along the glomerular capillary loop (average fenestral density/μm × average fenestral diameter in μm) was around 12%, much smaller than the 23% value in untreated WT mice.
Figure 2.

Transmission electron microscopic ultrastructure of glomerular endothelial cells in glomeruli of 8-wk old wild-type control mice (a, d) or wild-type mice injected with LPS (10 mg/kg for 24h) (b, e) or Tnfr1−/− mice injected with LPS (10 mg/kg for 24h; c f). (a-c) low magnification electron micrographs. Control, wild type treated with normal saline; WT LPS, wild type treated with LPS; TNFR1−/− LPS, Tnfr1−/− mice treated with LPS. Scale bar, 2 μm. (d-e) high magnification electron micrographs, with en face view of the fenestrae. Scale bar, 0.5 μm. Arrows indicate endothelial fenestrae. Arrowheads in (b) indicate endothelial cell detachment. CL, capillary lumen; F, fenestrate; P, podocyte foot processes; RBC, red blood cell.
Intravenous TNF injection causes AKI and similar changes in glomerular EC fenestration
To confirm the importance of circulating TNF acting alone, we injected recombinant TNF intravenously into mice. Injected TNF (2.5 μg) indeed not only decreased GFR, but also produced moderate tubular injury resembling that associated with LPS injection (Figure 3). This TNF-induced AKI corresponds to a serum level of TNF of 6.7±1.3 ng/ml measured 2 h after TNF injection, which falls in the same range as that 2 h after LPS challenge (3-10 ng/ml).37, 38 In contrast, AKI was not induced by low dose TNF (0.5 μg) yielding a serum TNF level of 0.6±0.3 ng/ml (Figure 3a). To explore whether TNF alone induces morphological changes in glomerular fenestrae similar to those of LPS-induced AKI, we compared the ultrastructural morphology of the glomerular endothelium in TNF-treated and matched control mice. The glomerular capillary wall in control mice, as imaged by transmission electron microscopy, was lined with fenestrated endothelium. Fenestrae viewed en face in electron microscopic images appeared circular (Figure 4a and c). In contrast, TNF-treated mice showed extensive loss of fenestrae (Figure 4b). En face electron microscopic images revealed fenestral diameters much larger in TNF-treated mice (141.5±10.7 nm) than in saline-injected controls (77.1±2.7 nm; Figure 4c and d). In conclusion, treatment with TNF alone had a similar effect as LPS on glomerular EC fenestrae; both significantly increased the size of glomerular EC fenestrae but decreased fenestral density.
Figure 3.
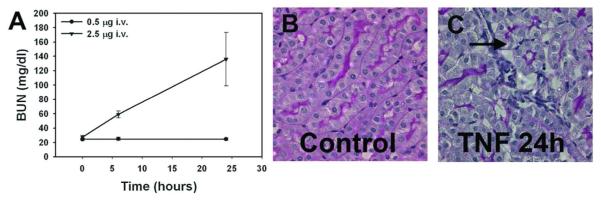
Intravenous injection of TNF-α recapitulates a form of ARF similar to that produced by LPS injection, with BUN elevation (A), pathological renal injury (B), and renal influx of neutrophils (C). Vacuolization and disruption of tubules is seen, with areas of leukocyte infiltration (arrow). Low dose intravenous TNF-α did not elevate BUN. BUN elevation required TNF-α levels similar to those achieved after LPS injection (3-10 ng/ml).
Figure 4.

Transmission electron microscopic ultrastructure of glomerular endothelial cells in glomeruli of wild-type mice 24 h after intravenous administration of saline (control, a and c) or 2.5 μg of TNF-α (b and d). (a, b) Loss of endothelial fenestrae in the glomerular capillaries in TNF-treated mice compared to controls. (c, d) high magnification electron micrographs, with en face view of the fenestrae, showing enlarged glomerular endothelial fenestrae in TNF-treated mice. Scale bar, 1 μm. Arrows indicate endothelial fenestrae.
Kidney VEGF level is decreased in LPS-induced AKI
VEGF is an important molecule known to induce fenestrae in vivo. It has been reported that kidney but not plasma VEGF protein levels significantly decreased 24 h after LPS injection, associated with increased circulation of soluble Flt-1.39 We examined the effect of LPS on the expression of VEGF in mouse kidneys. LPS treatment significantly decreased kidney VEGF mRNA levels measured by RT-PCR at 6 h and 24 h after injection (Figure 5a). Similarly, kidney VEGF protein levels were significantly decreased to 55.6 ± 3.8% of control levels (100.0 ± 7.7, P < 0.01) 24 h after LPS treatment (Figure 5b). We also investigated whether LPS affects the expression of the main VEGF receptor, VEGFR2, in glomerular ECs. In control kidneys, VEGFR2 was highly expressed in glomeruli as detected by immunofluorescence, but levels of neither VEGFR2 protein (Figure 6a and b) nor mRNA (Figure 6c) were significantly changed 24 h after LPS treatment (Figure 6c).
Figure 5.
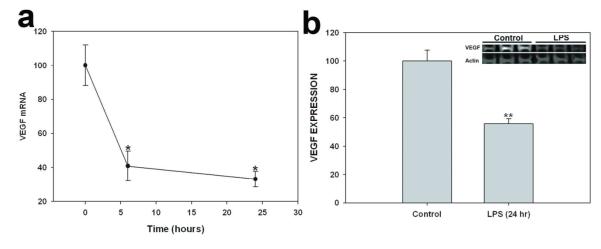
Effect of LPS on VEGF expression in kidney. (a) Time-dependent effect of LPS (10 mg/kg, 6h, 24h, 48h) on renal VEGF mRNA expression, as normalized to 18S mRNA expression. (g) Effect of LPS (10 mg/kg for 24h) on VEGF protein expression, as normalized to expression of β-actin protein. Top inset: Representative VEGF immunoblot of kidney lysate. Values are means ± s.e.m. for 4-6 animals. *P<0.05 vs. control. ** P<0.01 vs. control.
Figure 6.
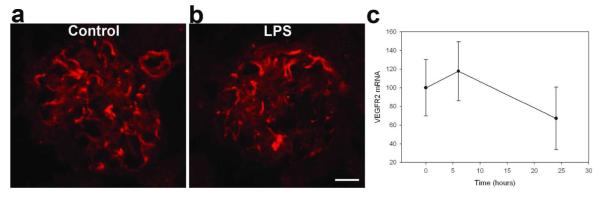
Effect of LPS on VEGFR2 expression in kidney. (a-b) Indirect immunofluorescence photomicrographs of frozen kidney cortex sections from wild-type control mice (a) and wild-type mice treated 24 h with 10/ mg/kg LPS (b), incubated with anti-VEGFR2 antibody. LPS did not change VEGFR2 expression in glomeruli. c) Time-dependent effect of LPS (10 mg/kg, 6h, 24h, 48h) on renal VEGFR2 mRNA expression, as normalized to 18S mRNA expression. Values are means ± s.e.m. for 4-6 animals. ANOVA shows no significant difference between the LPS and control groups. Scale bar 20 μm.
LPS and TNF-induced acute renal injury is associated with degradation of the glomerular ESL
To examine whether LPS-induced AKI is associated with damage of the glomerular ESL, kidney cryostat sections taken from mice 24 h after LPS or control injections were stained with WGA, a lectin which binds to negatively charged sugar residues of glycoproteins, such as sialic acid.40 WGA labeled glomerular ECs in both control and LPS-treated mice, as shown by co-staining with endothelial markers VE-Cadherin and CD31. LPS treatment decreased WGA staining of glomerular ECs (Figure 7a-f) by 33% relative to control glomeruli (P < 0.01; Figure 7o). We further confirmed that LPS injection disrupted the endothelial ESL by studying its effect on the most abundant proteoglycans (PGs) of the ESL, those containing heparan sulfate (HS) GAG chains. Some of these PGs are secreted and others are membrane-bound.41, 42 Immunostaining with anti-HS Ab mostly co-localized with VE-cadherin (data not shown), and again revealed substantial reduction in WT mice after LPS exposure (Figure 7m and n). TNF injection itself also reduced in WGA staining in glomerular ECs. (Figure 7j-l).
Figure 7.

Effect of LPS on glomerular ESL. (a-l) Glomerular ESL is shown by Alexa Fluor 594-WGA staining and co-staining for the endothelial junctional proteins VE-cadherin and CD31 in wild-type mice treated 24 h with 10 mg/kg LPS (d-f) and their matched controls (a-c), or in mice treated 24 h with 2.5 μg TNF intravenously (j-l) and their matched controls (g-i). Glomerular ESL is also shown by staining PGS containing HS GAG chains in wild-type control mice (m) and in wild-type mice 24 hrs after treatment with 10 mg/kg LPS (n). (o) Fluorescence intensity of glomerular WGA staining. Data (normalized to staining intensity of control mice) are expressed as mean ± s.e.m. **p<0.01 vs wild-type control. Scale bar 20 μm.
Both LPS and TNF increase glomerular heparanase expression
To identify changes to heparanase expression that might be responsible for LPS-induced ESL damage, heparanase localization and levels were examined by confocal microscopy and immunoblot. Heparanase was highly expressed in glomeruli, as shown by co-staining with nephrin (Figure 8). LPS treatment of mice dramatically increased glomerular loop staining of heparanase (Figure 8-4f). Immunoblot also revealed increased heparanase polypeptide levels in LPS-treated kidneys (279.6 ± 31.9%) compared with the control group (100.0 ± 13.8%, p < 0.01) (Figure 8g). TNF treatment similarly increased glomerular heparanase expression (data not shown).
Figure 8.

Effect of LPS on glomerular heparanase levels and expression. Indirect immunofluorescence photomicrographs of frozen kidney sections from wild-type control mice (a-c) and from wild-type mice treated 24 hrs with LPS (10 mg/kg), incubated with antibodies against Heparanase-1 (red; a, d) and against nephrin (green; b, e). (i) Top inset: Representative immunoblot of Heparanase-1 protein expression in kidney. Bar graph quantifying kidney heparanase-1 expression from immunoblot densitometry. Data normalized to “control” band are expressed as mean ± s.e.m. **p<0.01 vs wild-type control. Scale bar 20 μm.
Mice deficient in TNFR1 are resistant to LPS-induced increase of heparanase expression and degradation of glomerular ESL
Neither glomerular heparanase staining nor glomerular WGA staining changed significantly in LPS-treated Tnfr1−/− mice compared with control untreated mice, as shown in Figure S1. Immunoblot also confirmed unchanged heparanase protein levels in LPS-treated Tnfr1−/− kidneys as compared with the control group (data not shown).
LPS and TNF did not change expression of glomerular endothelial junction proteins VE-Cadherin and PECAM-1
To investigate whether the glomerular endothelial cell TJs were disrupted in LPS and TNF-induced endotoxemia, we examined localization and abundance of VE-cadherin, an endothelium-specific member of the cadherin family, and of PECAM-1 (CD31), an Ig-like cell adhesion molecule concentrated at sites of endothelial cell-cell contact.43 Confocal immunofluorescence studies on frozen kidney sections showed that levels of VE-cadherin and CD31 in glomerular ECs were not decreased in mice 24 h after treatment of mice with either LPS or TNF (Figure 8a-l).
DISCUSSION
Our results demonstrate that LPS and intravenous TNF itself induce similar forms of renal damage, including ultrastructural alterations of glomerular endothelial fenestrae and diffuse alteration of glomerular ESL components, together contributing to increased albumin permeability and decreased GFR. The absence of these changes in glomerular endothelial morphology in LPS-treated Tnfr1−/− mice, in parallel with GFR preservation, demonstrates a key role for TNF-mediated glomerular endothelial injury in LPS-induced AKI, and strongly suggests a key role in the syndrome of sepsis-induced AKI.
In this study, we demonstrate by TEM that LPS causes glomerular EC swelling and loss of fenestrae, without overt podocyte injury. Similar renal pathology has been noted in patients with preeclampsia.44 In patients with type 2 diabetes, loss of glomerular EC fenestration correlated with albuminuria and GFR reduction,45 although significant podocyte detachment was also observed in this report. Reduced numbers and increased diameters of glomerular EC fenestrae are quantifiable structural features of nephropathy in LPS-induced sepsis. Ours is the first study to demonstrate an association between loss of normal glomerular EC fenestration and declining GFR in an established endotoxin model of sepsis. A reduction in density of endothelial fenestrations with consequently reduced glomerular hydraulic permeability could be responsible for the decline in GFR. This is also the first study to demonstrate similar loss of fenestrae in AKI induced by intravenous administration of TNF.
The underlying mechanisms for the changes of glomerular endothelial fenestrae in sepsis were investigated. Knockout of TNFR1, which in kidney is predominantly expressed in the glomerular endothelium,8 prevented LPS-induced loss of endothelial fenestrae. TNF-α alone induced a similar loss of glomerular fenestrae, suggesting that the effects of LPS on glomerular fenestration are likely mediated by TNF-α acting through TNFR1. VEGF, one of the few known inducers of fenestrations, is expressed by podocytes.46 Glomerular ECs express VEGFR247, and the plasma level of VEGF has been directly associated with changes in glomerular EC fenestration.48, 49 TNF has been reported to down-regulate activity50 and expression of VEGFR2 in vitro.51, 52 However, we found that LPS treatment did not change glomerular VEGFR2 expression, whereas kidney levels of VEGF mRNA and protein were significantly decreased. Consistent with our finding, Yano et al. found that LPS administration in mice decreased kidney VEGF expression at 24 h with a concomitant increase in circulating soluble Flt-1.39 Karumanchi and coworkers have found that the soluble form of VEGF receptor-1 (sFlt-1) can account for the loss of glomerular fenestration observed in preeclampsia.53, 54 sFlt-1 blocks VEGF-A interaction with transmembrane VEGF receptors. Administration of sFlt-1 can lead to rapid loss of endothelial cell fenestrae, endothelial cell swelling, and proteinuria.55 The fact that sFlt-1 is increased in conditions such as experimental39 and clinical sepsis,56 type 2 diabetes,57 and preeclampsia, all characterized by loss of fenestrae in glomerular EC, strongly suggests that increased sFlt-1 and hence decreased kidney VEGF activity is the common mechanism underlying similar glomerular EC fenestral changes in distinct clinical settings. In addition, TNF-α treatment has been shown to increase circulation sFlt-1 in pregnant rats.58 Our finding that kidney VEGF mRNA level was decreased by LPS also suggests that a decreased production of VEGF by podocyte may contribute to the loss of fenestrae occurred in sepsis.
LPS-induced endotoxemia was also marked by reductions in two major components of the glomerular ESL, sialic acids as revealed by glomerular endothelial cell WGA staining, and by staining of PGs containing HS GAG chains. These changes were associated with loss of GFB perm-selectivity, as documented by albuminuria. While modest, this albuminuria developed despite a precipitous decrease in GFR, so fractional protein excretion was significantly abnormal. Glomerular ESL components rich in anions, especially sialic acids, may prevent the passage of anionic protein such as albumin into urine under physiological conditions, and thus are considered essential parts of the GFB.59-62 Singh et al.42 showed that the surface glycocalyx constitutes a barrier to protein in cultured human glomerular cells. Adembri et al.14 showed that massive disruption of the glomerular ESL occurred in albuminuria induced by CLP sepsis. Our experimental results support the idea that alterations of the glomerular ESL contribute to the albuminuria of sepsis, although coincident damage to tubular components cannot be excluded.15 These glomerular ESL changes occurred during LPS-induced sepsis and coincided with activation of a TNF-α-responsive heparanase in the glomerulus. Glomerular ECs subjected to injurious conditions such as diabetes secrete heparanase,63 an endo-beta-D-glucuronidase that specifically cleaves the heparan sulphate chain of PGs.64, 65 Therefore, the disruption of glomerular ESL during sepsis could be a result of sepsis-induced activation of glomerular heparanase. Consistent with our findings, a recent report in a sepsis model showed that pulmonary endothelial glycocalyx degradation involved the activation of endothelial heparanase and a loss of heparan sulfate.66
TNF-α can cause disruption of the endothelial glycocalyx in capillaries of cremaster muscle.67 It is likely that the mechanisms underlying glomerular ESL disruption and increased renal glomerular heparanase expression involve TNF-α activation of its receptor, TNFR1, since in Tnfr1−/− mice LPS did not induce degradation of the glomerular ESL nor increased heparanase activity. Indeed, intravenous administration of TNF alone caused similar glomerular ESL disruption, along with increased glomerular heparanase expression. Administration of TNF has also been shown to increase proteinuria.68
In conclusion, we have documented for the first time the concomitant degradation of glomerular ESL and loss of glomerular endothelial fenestration in LPS-induced endotoxemia in the mouse. We correlated quantitative structural changes in glomerular fenestration with the decline in GFR and albuminuria in endotoxemia. These data show that the pathological changes of the glomerular endothelium and glomerular ESL are likely mediated by TNF-α released during endotoxemia and acting through TNFR1, since the LPS-induced pathological changes were abolished in Tnfr1−/− mice and administration of TNF alone induced similar pathological changes. Our findings suggest an important role for these distinct glomerular endothelial injuries in the development of endotoxemia-induced AKI and albuminuria, and likely reflect mechanisms central to the pathogenesis of sepsis-associated AKI.
MATERIALS AND METHODS
LPS-induced acute endotoxemia
All animal experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee. 8 wk old male C57BL/6 wild-type and TNFR1-deficient (Tnfr1−/−; B6.129-Tnfrsf1atm1Mak/J; stock 002818) mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Tnfr1−/− mice were congenic on the C57BL/6J genetic background. Endotoxemia was induced by the administration of a single dose of LPS (10 mg/kg) as described.69 In brief, mice were given a single intraperitoneal injection of either Escherichia coli LPS (10 mg/kg in 0.1 mL 0.9 % normal saline) or 0.9 % normal saline (controls). Mice were also given 0.25 mL sterile saline as a series of subcutaneous injections every 12 h to minimize any contribution of volume depletion. Mice were sacrificed at 6, 24, or 48 h after injection. The kidneys were snap-frozen in liquid nitrogen and stored at −80 °C until extraction of total RNA or protein. For immunohistochemistry, kidneys were immediately embedded in TissueTek OCT compound (Fisher Scientific), frozen, and stored at −80°C. Analogous experiments were done in which TNF-α (R & D Systems), dissolved in sterile PBS, was injected by tail vein into wildtype mice.
Measurement of renal and blood parameters
Blood was obtained at 2, 6 and 24 h after TNF-α was administered as a single i.v. dose of 0.5 or 2.5 μg. Blood and spot urine was obtained at 24 h after LPS injection. TNF-α levels were determined from sera obtained 2 h after TNF admistration using a mouse TNF-α ELISA kit according to the manufacturer’s instructions. (eBioscience, San Diego, CA). Plasma concentration of urea were determined with a Beckman Coulter Synchron DXC600 autoanalyzer. Urine levels of albumin were determined using a commercially available mouse albumin ELISA (Bethyl labs, Montgomery, TX). Urine levels of creatinine were determined using Enzymatic Creatinine LiquiColor® Reagent (StanBio Lab, Boerne, TX).
Protein preparation and immunoblotting
Frozen kidney tissue was thawed and homogenized for western blot as described.69 Membranes were incubated overnight with polyclonal rabbit antibodies against heparanase-1 and VEGF (Abcam, Cambridge, MA). After being washed, the membranes were incubated for 2 h with the secondary antibody (800 nm goat anti-rabbit IgG, Li-Cor Biosciences, Lincoln, NE) and the protein bands were detected by an Odyssey infrared imager (Li-Cor Biosciences, ODY-1320). An actin control was performed for each membrane. Band density was measured with ImageJ (v1.44p, NIH, USA) and normalized to actin for each lane.
Immunofluorescence in kidney cryostat sections
Cryostat sections (4 μm) prepared from mice kidneys were fixed as described,69 and incubated at 4°C overnight with primary rabbit polyclonal antibody against heparanase-1, VEGFR2 (KDR antibody, Proteintech Group, Chicago, IL), or rat anti-Heparan Sulfate Proteoglycan (US Biological, Marblehead, MA), followed by incubation for 2 h at room temperature with secondary antibodies. Some cryostat sections immunostained as above were then either co-stained with rat antibodies to the endothelial marker VE-cadherin (Abcam, Cambridge, MA) and CD31 (BD Bioscience, San Jose, CA), or with goat polyclonal antibody against nephrin (Santa Cruz Biotechnology, Santa Cruz, CA). For wheat germ agglutinin (WGA) staining, cryostat sections were incubated with Alexa Fluor 594-conjugated WGA (Molecular Probes, Eugene, OR). The stained sections were then examined with a Fluoview 200 laser-scanning confocal microscope equipped with a 647-nm argon laser at ×20 and ×60 magnification. To quantify WGA expression, densitometric analysis of the intensity of the fluorescence signals was performed on digitized images of glomeruli using ImageJ software (National Institute of Health, NIH).
Transmission electron microscopic analyses of kidney tissue and assessments of glomerular endothelial fenestrae
Renal cortical tissue from control WT, LPS-treated (24 h) WT, TNF-treated WT, and LPS-treated (24 h) Tnfr1−/− mice (n = 4-6 for each group) was diced into 1-mm blocks, fixed overnight at 4°C by immersion in half-strength Karnovsky’s solution (2.5% glutaraldehyde / 2% paraformaldehyde) and then transferred into 0.1 M cacodylate buffer for storage (at 4°C). The tissue was embedded in Epon and sections were stained with uranyl acetate and lead citrate. Sections were examined using a Philips CM-10 electron microscope. Transmission electron microscopic (TEM) images of glomeruli from different groups of mice were obtained using the Gatan (Pleasanton, CA) Erlangshen ES1000W Model 785 CCD Camera. Glomerular EC fenestrae diameters were measured using Digital Micrograph software (Gatan). Sixty to seventy capillary loops were assessed per group.
Statistics
Data are presented as means +/− SEM, unless otherwise noted. The experimental and control groups were compared by two-tailed t-test. A P value < 0.05 was considered significant.
Supplementary Material
Figure S1. LPS fails to change expression of WGA and heparanse in glomeruli of Tnfr1−/− mice. Representative fluorescence photomicrographs of frozen kidney cortex sections from wild-type control mice (a-c) and Tnfr1−/− mice treated 24 h with 10 mg/kg LPS (d-e), incubated with antibodies against Heparanase-1 (green; a, d) and Alexa Fluor 594-WGA (red; b, e). Scale bar 20 μm.
ACKNOWLEDGMENTS
This work was supported by National Institute of Health Grants R01DK080863 (PNC).
Grants: This work was supported by National Institute of Health Grants R01DK080863 (PNC).
Footnotes
DISCLOSURE All the authors declared no competing interests.
REFERENCES
- 1.Oppert M, Engel C, Brunkhorst FM, et al. Acute renal failure in patients with severe sepsis and septic shock--a significant independent risk factor for mortality: results from the German Prevalence Study. Nephrol Dial Transplant. 2008;23:904–909. doi: 10.1093/ndt/gfm610. [DOI] [PubMed] [Google Scholar]
- 2.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 3.Muntner P, Warnock DG. Acute kidney injury in sepsis: questions answered, but others remain. Kidney Int. 2010;77:485–487. doi: 10.1038/ki.2009.517. [DOI] [PubMed] [Google Scholar]
- 4.Parrillo JE. The cardiovascular pathophysiology of sepsis. Annu Rev Med. 1989;40:469–485. doi: 10.1146/annurev.me.40.020189.002345. [DOI] [PubMed] [Google Scholar]
- 5.Czabanka M, Peter C, Martin E, et al. Microcirculatory endothelial dysfunction during endotoxemia--insights into pathophysiology, pathologic mechanisms and clinical relevance. Curr Vasc Pharmacol. 2007;5:266–275. doi: 10.2174/157016107782023389. [DOI] [PubMed] [Google Scholar]
- 6.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham PN, Dyanov HM, Park P, et al. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol. 2002;168:5817–5823. doi: 10.4049/jimmunol.168.11.5817. [DOI] [PubMed] [Google Scholar]
- 8.Al-Lamki RS, Wang J, Skepper JN, et al. Expression of tumor necrosis factor receptors in normal kidney and rejecting renal transplants. Lab Invest. 2001;81:1503–1515. doi: 10.1038/labinvest.3780364. [DOI] [PubMed] [Google Scholar]
- 9.Wu X, Guo R, Chen P, et al. TNF induces caspase-dependent inflammation in renal endothelial cells through a Rho- and myosin light chain kinase-dependent mechanism. Am J Physiol Renal Physiol. 2009;297:F316–326. doi: 10.1152/ajprenal.00089.2009. [DOI] [PubMed] [Google Scholar]
- 10.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 11.Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- 12.Roine I. Microalbuminuria: an index of severity in childhood meningitis. Pediatr Infect Dis J. 1993;12:584–588. [PubMed] [Google Scholar]
- 13.De Gaudio AR, Adembri C, Grechi S, et al. Microalbuminuria as an early index of impairment of glomerular permeability in postoperative septic patients. Intensive Care Med. 2000;26:1364–1368. doi: 10.1007/s001340000593. [DOI] [PubMed] [Google Scholar]
- 14.Adembri C, Sgambati E, Vitali L, et al. Sepsis induces albuminuria and alterations in the glomerular filtration barrier: a morphofunctional study in the rat. Crit Care. 2011;15:R277. doi: 10.1186/cc10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreiber A, Theilig F, Schweda F, et al. Acute endotoxemia in mice induces downregulation of megalin and cubilin in the kidney. Kidney international. 2012;82:53–59. doi: 10.1038/ki.2012.62. [DOI] [PubMed] [Google Scholar]
- 16.McKenzie JA, Ridley AJ. Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J Cell Physiol. 2007;213:221–228. doi: 10.1002/jcp.21114. [DOI] [PubMed] [Google Scholar]
- 17.Petrache I, Birukova A, Ramirez SI, et al. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28:574–581. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- 18.Koss M, Pfeiffer GR, 2nd, Wang Y, et al. Ezrin/radixin/moesin proteins are phosphorylated by TNF-alpha and modulate permeability increases in human pulmonary microvascular endothelial cells. J Immunol. 2006;176:1218–1227. doi: 10.4049/jimmunol.176.2.1218. [DOI] [PubMed] [Google Scholar]
- 19.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 20.Wyman TH, Bjornsen AJ, Elzi DJ, et al. A two-insult in vitro model of PMN-mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol Cell Physiol. 2002;283:C1592–1603. doi: 10.1152/ajpcell.00540.2001. [DOI] [PubMed] [Google Scholar]
- 21.Adamson RH, Lenz JF, Zhang X, et al. Oncotic pressures opposing filtration across non-fenestrated rat microvessels. J Physiol. 2004;557:889–907. doi: 10.1113/jphysiol.2003.058255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deen WM, Lazzara MJ, Myers BD. Structural determinants of glomerular permeability. Am J Physiol Renal Physiol. 2001;281:F579–596. doi: 10.1152/ajprenal.2001.281.4.F579. [DOI] [PubMed] [Google Scholar]
- 23.Deen WM, Lazzara MJ. Glomerular filtration of albumin: how small is the sieving coefficient? Kidney Int Suppl. 2004:S63–64. doi: 10.1111/j.1523-1755.2004.09216.x. 2004/10/16. [DOI] [PubMed] [Google Scholar]
- 24.Satchell SC, Braet F. Glomerular endothelial cell fenestrations: an integral component of the glomerular filtration barrier. Am J Physiol Renal Physiol. 2009;296:F947–956. doi: 10.1152/ajprenal.90601.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ballermann BJ, Stan RV. Resolved: capillary endothelium is a major contributor to the glomerular filtration barrier. J Am Soc Nephrol. 2007;18:2432–2438. doi: 10.1681/ASN.2007060687. [DOI] [PubMed] [Google Scholar]
- 26.Bulger RE, Eknoyan G, Purcell DJ, 2nd, et al. Endothelial characteristics of glomerular capillaries in normal, mercuric chloride-induced, and gentamicin-induced acute renal failure in the rat. J Clin Invest. 1983;72:128–141. doi: 10.1172/JCI110950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryan GB, Karnovsky MJ. Distribution of endogenous albumin in the rat glomerulus: role of hemodynamic factors in glomerular barrier function. Kidney Int. 1976;9:36–45. doi: 10.1038/ki.1976.5. [DOI] [PubMed] [Google Scholar]
- 28.Ohlson M, Sorensson J, Haraldsson B. A gel-membrane model of glomerular charge and size selectivity in series. Am J Physiol Renal Physiol. 2001;280:F396–405. doi: 10.1152/ajprenal.2001.280.3.F396. [DOI] [PubMed] [Google Scholar]
- 29.Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflugers Arch. 2000;440:653–666. doi: 10.1007/s004240000307. [DOI] [PubMed] [Google Scholar]
- 30.Curry FE, Adamson RH. Endothelial glycocalyx: permeability barrier and mechanosensor. Ann Biomed Eng. 2012;40:828–839. doi: 10.1007/s10439-011-0429-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curry FE, Rutledge JC, Lenz JF. Modulation of microvessel wall charge by plasma glycoprotein orosomucoid. Am J Physiol. 1989;257:H1354–1359. doi: 10.1152/ajpheart.1989.257.5.H1354. [DOI] [PubMed] [Google Scholar]
- 32.Haraldsson B, Rippe B. Orosomucoid as one of the serum components contributing to normal capillary permselectivity in rat skeletal muscle. Acta Physiol Scand. 1987;129:127–135. doi: 10.1111/j.1748-1716.1987.tb08047.x. [DOI] [PubMed] [Google Scholar]
- 33.Hjalmarsson C, Lidell ME, Haraldsson B. Beneficial effects of orosomucoid on the glomerular barrier in puromycin aminonucleoside-induced nephrosis. Nephrol Dial Transplant. 2006;21:1223–1230. doi: 10.1093/ndt/gfk050. [DOI] [PubMed] [Google Scholar]
- 34.Johnsson E, Haraldsson B. Addition of purified orosomucoid preserves the glomerular permeability for albumin in isolated perfused rat kidneys. Acta Physiol Scand. 1993;147:1–8. doi: 10.1111/j.1748-1716.1993.tb09466.x. [DOI] [PubMed] [Google Scholar]
- 35.Fridén V, Oveland E, Tenstad O, et al. The glomerular endothelial cell coat is essential for glomerular filtration. Kidney Int. 2011;79:1322–1330. doi: 10.1038/ki.2011.58. [DOI] [PubMed] [Google Scholar]
- 36.Chen CC, Pedraza PL, Hao S, et al. TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am J Physiol Renal Physiol. 2010;299:F1141–1150. doi: 10.1152/ajprenal.00344.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marino MW, Dunn A, Grail D, et al. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci U S A. 1997;94:8093–8098. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alexander JJ, Jacob A, Cunningham P, et al. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int. 2008;52:447–456. doi: 10.1016/j.neuint.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yano K, Liaw PC, Mullington JM, et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med. 2006;203:1447–1458. doi: 10.1084/jem.20060375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhavanandan VP, Katlic AW. The interaction of wheat germ agglutinin with sialoglycoproteins. The role of sialic acid. J Biol Chem. 1979;254:4000–4008. [PubMed] [Google Scholar]
- 41.Sörensson J, Björnson A, Ohlson M, et al. Synthesis of sulfated proteoglycans by bovine glomerular endothelial cells in culture. Am J Physiol Renal Physiol. 2003;284:F373–380. doi: 10.1152/ajprenal.00257.2002. [DOI] [PubMed] [Google Scholar]
- 42.Singh A, Satchell SC, Neal CR, et al. Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J Am Soc Nephrol. 2007;18:2885–2893. doi: 10.1681/ASN.2007010119. [DOI] [PubMed] [Google Scholar]
- 43.Albelda SM, Muller WA, Buck CA, et al. Molecular and cellular properties of PECAM-1 (endoCAM/CD31): a novel vascular cell-cell adhesion molecule. J Cell Biol. 1991;114:1059–1068. doi: 10.1083/jcb.114.5.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karumanchi SA, Maynard SE, Stillman IE, et al. Preeclampsia: a renal perspective. Kidney Int. 2005;67:2101–2113. doi: 10.1111/j.1523-1755.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 45.Weil EJ, Lemley KV, Mason CC, et al. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012 doi: 10.1038/ki.2012.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown LF, Berse B, Tognazzi K, et al. Vascular permeability factor mRNA and protein expression in human kidney. Kidney Int. 1992;42:1457–1461. doi: 10.1038/ki.1992.441. [DOI] [PubMed] [Google Scholar]
- 47.Robert B, Zhao X, Abrahamson DR. Coexpression of neuropilin-1, Flk1, and VEGF(164) in developing and mature mouse kidney glomeruli. Am J Physiol Renal Physiol. 2000;279:F275–282. doi: 10.1152/ajprenal.2000.279.2.F275. [DOI] [PubMed] [Google Scholar]
- 48.Kamba T, Tam BY, Hashizume H, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H560–576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- 49.Eremina V, Baelde HJ, Quaggin SE. Role of the VEGF--a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol. 2007;106:32–37. doi: 10.1159/000101798. [DOI] [PubMed] [Google Scholar]
- 50.Guo DQ, Wu LW, Dunbar JD, et al. Tumor necrosis factor employs a protein-tyrosine phosphatase to inhibit activation of KDR and vascular endothelial cell growth factor-induced endothelial cell proliferation. J Biol Chem. 2000;275:11216–11221. doi: 10.1074/jbc.275.15.11216. [DOI] [PubMed] [Google Scholar]
- 51.Patterson C, Perrella MA, Endege WO, et al. Downregulation of vascular endothelial growth factor receptors by tumor necrosis factor-alpha in cultured human vascular endothelial cells. J Clin Invest. 1996;98:490–496. doi: 10.1172/JCI118816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menon C, Iyer M, Prabakaran I, et al. TNF-alpha downregulates vascular endothelial Flk-1 expression in human melanoma xenograft model. Am J Physiol Heart Circ Physiol. 2003;284:H317–329. doi: 10.1152/ajpheart.00971.2001. [DOI] [PubMed] [Google Scholar]
- 53.Tjoa ML, Levine RJ, Karumanchi SA. Angiogenic factors and preeclampsia. Front Biosci. 2007;12:2395–2402. doi: 10.2741/2241. [DOI] [PubMed] [Google Scholar]
- 54.Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sugimoto H, Hamano Y, Charytan D, et al. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem. 2003;278:12605–12608. doi: 10.1074/jbc.C300012200. [DOI] [PubMed] [Google Scholar]
- 56.Skibsted S, Jones AE, Puskarich MA, et al. Biomarkers of Endothelial Cell Activation in Early Sepsis. Shock. 2013;39:427–432. doi: 10.1097/SHK.0b013e3182903f0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nandy D, Mukhopadhyay D, Basu A. Both vascular endothelial growth factor and soluble Flt-1 are increased in type 2 diabetes but not in impaired fasting glucose. J Investig Med. 2010;58:804–806. doi: 10.231/JIM.0b013e3181e96203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parrish MR, Murphy SR, Rutland S, et al. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am J Hypertens. 2010;23:911–916. doi: 10.1038/ajh.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salmon AH, Satchell SC. Endothelial glycocalyx dysfunction in disease: albuminuria and increased microvascular permeability. J Pathol. 2012;226:562–574. doi: 10.1002/path.3964. [DOI] [PubMed] [Google Scholar]
- 60.Obeidat M, Ballermann BJ. Glomerular endothelium: a porous sieve and formidable barrier. Exp Cell Res. 2012;318:964–972. doi: 10.1016/j.yexcr.2012.02.032. [DOI] [PubMed] [Google Scholar]
- 61.Galeano B, Klootwijk R, Manoli I, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–1594. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gelberg H, Healy L, Whiteley H, et al. In vivo enzymatic removal of alpha 2-->6-linked sialic acid from the glomerular filtration barrier results in podocyte charge alteration and glomerular injury. Lab Invest. 1996;74:907–920. [PubMed] [Google Scholar]
- 63.Kuwabara A, Satoh M, Tomita N, et al. Deterioration of glomerular endothelial surface layer induced by oxidative stress is implicated in altered permeability of macromolecules in Zucker fatty rats. Diabetologia. 2010;53:2056–2065. doi: 10.1007/s00125-010-1810-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hulett MD, Freeman C, Hamdorf BJ, et al. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nature medicine. 1999;5:803–809. doi: 10.1038/10525. [DOI] [PubMed] [Google Scholar]
- 65.Vlodavsky I, Friedmann Y, Elkin M, et al. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nature medicine. 1999;5:793–802. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]
- 66.Schmidt EP, Yang Y, Janssen WJ, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012 doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henry CB, Duling BR. TNF-alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am J Physiol Heart Circ Physiol. 2000;279:H2815–2823. doi: 10.1152/ajpheart.2000.279.6.H2815. [DOI] [PubMed] [Google Scholar]
- 68.Brennan DC, Yui MA, Wuthrich RP, et al. Tumor necrosis factor and IL-1 in New Zealand Black/White mice. Enhanced gene expression and acceleration of renal injury. J Immunol. 1989;143:3470–3475. [PubMed] [Google Scholar]
- 69.Eadon MT, Hack BK, Xu C, et al. Endotoxemia alters tight junction gene and protein expression in the kidney. Am J Physiol Renal Physiol. 2012;303:F821–830. doi: 10.1152/ajprenal.00023.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. LPS fails to change expression of WGA and heparanse in glomeruli of Tnfr1−/− mice. Representative fluorescence photomicrographs of frozen kidney cortex sections from wild-type control mice (a-c) and Tnfr1−/− mice treated 24 h with 10 mg/kg LPS (d-e), incubated with antibodies against Heparanase-1 (green; a, d) and Alexa Fluor 594-WGA (red; b, e). Scale bar 20 μm.