

Abstract
Anti-apoptotic Bcl-2 family proteins have been reported to play an important role in apoptotic cell death of human malignancies. The aim of this study was to delineate the mechanism of anti-apoptotic Bcl-2 family proteins in pancreatic cancer (PaCa) cell survival. We first analyzed the endogenous expression and subcellular localization of anti-apoptotic Bcl-2 family proteins in six PaCa cell lines by Western blot. To delineate the functional role of Bcl-2 family proteins, siRNA-mediated knock-down of protein expression was used. Apoptosis was measured by Cell Death ELISA and Hoechst 33258 staining. In the results, the expression of anti-apoptotic Bcl-2 family proteins varied between PaCa cell lines. Mcl-1 knock-down resulted in marked cleavage of PARP and induction of apoptosis. Down-regulation of Bcl-2 or Bcl-xL had a much weaker effect. Simultaneous knock-down of Bcl-xL and Mcl-1 strongly induced apoptosis, but simultaneous knock-down of Bcl-xL/Bcl-2 or Mcl-1/Bcl-2 had no additive effect. The apoptosis-inducing effect of simultaneous knock-down of Bcl-xL and Mcl-1 was associated with translocation of Bax from the cytosol to the mitochondrial membrane, cytochrome c release, and caspase activation. These results demonstrated that Bcl-xL and Mcl-1 play an important role in pancreatic cancer cell survival. Targeting both Bcl-xL and Mcl-1 may be intriguing therapeutic strategy in PaCa.
Keywords: Mcl-1, Bcl-xL, Bax, pancreatic cancer, apoptosis
1. Introduction
Of all gastrointestinal carcinomas, pancreatic cancer (PaCa) has the most unfavorable prognosis. Surgical resection is the only possible curative treatment for PaCa, but the 5-year survival rate of all patients is below 5 % [1]. One reason for the notoriously dismal prognosis of pancreatic cancer is the resistance to currently available chemotherapeutic drugs. Although gemcitabine has been the approved drug of choice in the treatment of pancreatic cancer, its efficacy is weak and the improvement of overall survival is marginal. Anti-apoptotic members of the Bcl-2 protein family are often over-expressed in human cancers and are thought to mediate the resistance to chemotherapeutic drugs. Thus, a better understanding of the anti-apoptotic mechanisms and the development of potent and non-toxic strategies to overcome the pro-survival mechanisms in PaCa cells are of utmost importance to improve the outcome of PaCa patients.
We previously reported that the polyphenol baicalein induced apoptosis through down-regulation of Mcl-1 in PaCa cells [2]. Mcl-1 is one of the anti-apoptotic Bcl-2 family proteins. The Bcl-2 family protein consists of anti-apoptotic proteins (Bcl-2, Bcl-xL, and Mcl-1) and pro-apoptotic proteins that include multi-domain molecules Bak and Bax, and the BH3-only proteins Bad, Bid, Bim, Puma, and Noxa [3]. Oligomerization of multi-domain molecules Bak and Bax is the key event on apoptosis; once oligomerization of Bak or Bax occurred, cytochrome c is released from the mitochondrial outer membrane to the cytosol, where it binds to apoptotic protease-activating factor-1 (APAF-1) and caspase-9, thereby activating the caspase activation cascade [4]. Since anti-apoptotic Bcl-2 family proteins can stabilize Bak or Bax activation, anti-apoptotic Bcl-2 family proteins are recognized as important mediators of cell death. Recently, the anti-apoptotic proteins Bcl-xL and Mcl-1, but not Bcl-2, have been reported to cooperatively protect cells from death stimuli in several cancer models [5, 6]. The importance of Bcl-xL and Mcl-1 on drug-induced apoptosis is increasingly recognized, but very little is known about their role in PaCa.
Here, we show that simultaneous knock-down of Bcl-xL and Mcl-1 robustly induced apoptosis through the Bax pathway in PaCa cells. These data provide the basis for future mechanistic studies on the role of Bcl-2 family proteins in PaCa and may help to develop novel therapeutic strategies aiming at the Bcl-xL/Mcl-1 pathway.
2. Materials and Methods
2.1. Reagents
Antibodies against caspase-3, cleaved caspase-3, caspase-7, cleaved caspase-7, PARP, cleaved-PARP, Bcl-2, Bcl-xL, Mcl-1, Bax, cytochrome c, cytochrome c oxidase subunit IV (cox-IV), and GAPDH were purchased from Cell Signaling Technology, Inc. (Danvers, MS). The pan-caspase inhibitor zVAD-fmk was from Calbiochem (Darmstadt, Germany). Other reagents were from common commercial sources.
Cell culture
The human PaCa cell lines, BxPC-3, HPAF-II, Capan-2, AsPC-1, MIA PaCa-2, and Panc-1 were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and cultured as described previously [7].
2.2. Transfection of siRNA
siGENOME SMARTpool against Bcl-2, Bcl-xL, Mcl-1, and Bax as well as siGENOME Non-Targeting Pool (#2) were purchased from Dharmacon (Lafayette, CO). One day before siRNA transfection, cells were seeded into 6-well tissue culture plates and incubated overnight at 37 °C without antibiotics. Cells were transfected with siRNA (final concentration of 20 nM) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. Five hours after transfection, medium was changed to serum free DMEM or RPMI and cells were incubated for another 24 hours.
2.3. DNA fragmentation assay
DNA fragmentation was measured as described previously [8]. Briefly, cells (5×105) were transfected with siRNA targeting Bcl-2, Bcl-xL, Mcl-1, and their combinations. DNA fragmentation was analyzed using the Cell Death Detection ELISAPlus kit (Roche) according to the manufacturer’s instructions. The extent of apoptosis was presented as a fold (mU of the treated cells/mU of the non-treated cells).
2.4. Hoechst 33258 staining
To assess changes in nuclear morphology during apoptosis, staining using fluorescent Hoechst 33258 (Invitrogen, Carlsbad, CA) was performed. Cells were seeded into a Lab-Tek®II Chamber Slide™ System (Nalgene Nunc International, Naperville, IL) and incubated overnight at 37 °C. Medium was changed to serum-free condition. Cells were transfected with Bcl-xL and Mcl-1 siRNA or control siRNA as described above, fixed with 4% paraformaldehyde in PBS for 30 min at room temperature, and stained with Hoechst 33258 for 15 min. Slides were mounted with VECTASHIELD (Vector Laboratories Inc., Burlingame, CA), and were observed under a fluorescence microscope (Nikon, Eclipse 90i, Tokyo, Japan). Apoptotic nuclei were identified by morphologic changes such as chromatin condensation and nuclear fragmentation.
2.5. Cell lysates
Total cell lysates were collected as described previously [9]. Protein concentration was measured by the BCA Protein Assay Kit (Pierce, Rockford, IL). For preparation of mitochondrial and cytosolic fractions, we used a mitochondria extraction kit (Pierce) according to the manufacturer’s instructions. Cells were re-suspended in lysis buffer, and then disrupted by 80 strokes with a Dounce homogenizer. Homogenates were centrifuged at 1,000 ×g for 10 min to pellet nuclei and cell debris. Supernatants were subsequently centrifuged at 16,000 ×g for 15 min, and the cytosolic fractions (supernatants) were collected. Pellets (heavy membranes enriched with mitochondria) were lysed in RIPA buffer. To determine the quality of cytosolic and mitochondrial separation, both fractions were assessed by immunoblotting for the mitochondrial marker cox-IV.
2.6. Western blot analysis
Western Blot was performed as described previously [10]. Briefly, equal amount of protein was fractionated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were incubated with 5% skim milk in TBS-T (0.1% Tween 20) for 1 hour at room temperature to block nonspecific binding and incubated with specific primary antibodies in TBS-T containing 3% skim milk overnight at 4 °C. Proteins were detected using horseradish peroxidase-conjugated secondary antibodies (Pierce). Protein-antibody complexes were visualized with the SuperSignal West Pico Chemiluminescent Substrate or with SuperSignal West Femto Chemiluminescent Substrate (Pierce).
2.7. Statistical analysis
Data are presented as means ± SD, and statistical comparisons were made using the Student’s t test for paired observations. Comparisons of more than two groups were made by a one-way ANOVA with post hoc Holm-Sidak analysis for pairwise comparisons and comparisons versus control. An alpha value of 0.05 was used to determine significant differences. All statistics were done in SigmaStat 3.1 (Systat Software, Inc.).
3. Results
3.1. Endogenous expression of anti-apoptotic Bcl-2 family proteins in PaCa
Since the anti-apoptotic Bcl-2 family proteins play a major role in the protection against DNA damage-induced apoptosis via the mitochondrial pathway [11, 12], we first sought to determine the endogenous expression of anti-apoptotic Bcl-2 family proteins in PaCa cell lines. Mitochondrial and cytosolic fractions were collected after 24h serum deprivation. Subcellular fractionation demonstrated that Bcl-2, Bcl-xL, and Mcl-1 proteins are mainly located at the mitochondrial membrane in untreated PaCa cells (Figure 1). The expression of anti-apoptotic protein (Bcl-2, Bcl-xL, and Mcl-1) varied between six PaCa cell lines. Bcl-2 was expressed in MIA PaCa-2 and Panc-1 cells, but virtually undetectable in other cell lines. Bcl-xL was almost equally expressed in six cell lines. Mcl-1 was detected in BxPC-3, MIA PaCa-2, Capan-2, and Panc-1 cells, and was weakly expressed in HPAF-II, but was not detected in AsPc-1 cells.
Figure 1.
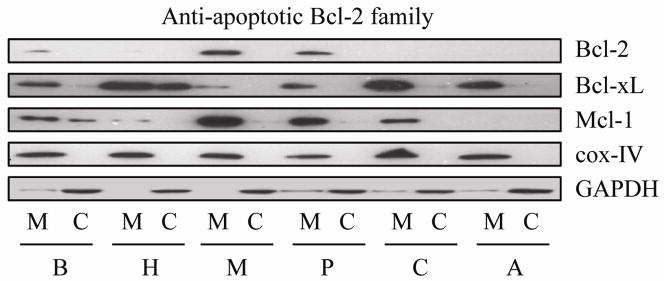
Endogenous expression of anti-apoptotic Bcl-2 family proteins. Mitochondrial membrane fraction and cytosolic fraction were separated as described in methods, after which anti-apoptotic Bcl-2 family protein expression of BxPC-3 (B), HPAF-II (H), MIA PaCa-2 (M), Panc-1 (P), Capan-2 (C), and AsPc-1 (A) cells were measured by Western blot. Cox IV (cytochrome oxidase IV) served as loading control for mitochondrial membrane fraction. Results are representative of three independent experiments.
3.2. Simultaneous knock-down of Bcl-xL and Mcl-1 strongly induces apoptosis in PaCa cells
We next investigated the importance of specific anti-apoptotic Bcl-2 family proteins on PaCa cell survival. First, we transfected PaCa cells with siRNA targeting Bcl-2, Bcl-xL, and Mcl-1. Transfection with siRNAs reduced their protein expression in three PaCa cell lines (Fig. 2A). Mcl-1 knock-down resulted in significant induction of DNA fragmentation in BxPC-3 and MIA PaCa-2 cells, but this effect was not detected in Panc-1 cells (Fig. 2B). On the other hand, down-regulation of Bcl-2 or Bcl-xL protein expression had no measurable effect on inducing apoptosis in PaCa cells. However, simultaneous knock-down of Bcl-xL and Mcl-1 very strongly induced DNA fragmentation in all cell lines (Fig. 2B). In contrast, simultaneous knock-down of Bcl-xL and Bcl-2 (data not shown) or Mcl-1 and Bcl-2 had no additional effect over Mcl-1 knockdown alone. We further confirmed that simultaneous knock-down of Bcl-xL and Mcl-1 induced apoptosis by detecting typical nuclear morphological changes, such as chromatin condensation and nuclear fragmentation, consistent with apoptosis (Fig. 2C). These results indicate that Bcl-xL and Mcl-1 cooperatively play a critical role in apoptotic cell death in PaCa cells. We next knock-downed Bcl-xL and Mcl-1 proteins in AsPC-1 cells, since the AsPc-1 cells have no detectable Mcl-1 protein. As shown in Fig. 2D, Bcl-xL knock-down resulted in significant induction of DNA fragmentation in AsPC-1 cells, but this effect was not detected by Mcl-1 knock-down. This result further indicate that Bcl-xL and Mcl-1 cooperatively play a role in apoptotic cell death.
Figure 2.

Simultaneous knock-down of Bcl-xL and Mcl-1 induces apoptosis in PaCa cells. BxPC-3, MIA PaCa-2, and Panc-1 cells were transiently transfected with siRNA against Mcl-1, Bcl-2, Bcl-xL for 24 h and the expression of these proteins was confirmed by Western blot (A). DNA fragmentation (B) and changes in nuclear morphology (C) after siRNA transfection were measured by Cell Death ELISA and Hoechst 33258 staining, respectively. Arrowheads indicate apoptotic cells characterized by morphologic changes such as chromatin condensation and nuclear fragmentation. Results are representative of three independent experiments and are presented as mean ± SD (bars). *p<0.05. **p<0.01. (D) AsPC-1 cells were transfected with siRNA against Mcl-1 and Bcl-xL for 24 h, after which DNA fragmentation was quantified by Cell Death ELISA. Results are representative of three independent experiments and are presented as mean ± SD (bars). *p<0.05.
3.3. The effect of Bcl-xL and/or Mcl-1 knock-down on gemcitabine-induced apoptosis in pancreatic cancer cells
It has been reported that Bcl-xL or Mcl-1 knock-down reinforces chemotherapeutic drug-induced apoptosis in several cancers, including PaCa [13, 14]. Hence, we examined whether Bcl-xL and/or Mcl-1 knock-down sensitizes PaCa cells to gemcitabine. As shown in Fig. 3A, gemcitabine (1 μg/ml) had apoptosis-inducing effect in BxPC-3 cells, but the effect was much weaker in MIA PaCa-2 cells. Knock-down of Bcl-xL had no effect on apoptosis in untreated MIA PaCa-2 cells, but slightly enhanced the cell death-inducing effects of gemcitabine in this cell line (Fig. 3A). Bcl-xL knock-down had no effect in untreated and gemcitabine-treated BxPC-3 cells. Mcl-1 knock-down alone showed a more robust apoptosis-inducing effect than Bcl-xL knock-down plus gemcitabine in both cells. However, Mcl-1 knock-down did not enhance gemcitabine toxicity in both cells. Simultaneous knock-down of Bcl-xL and Mcl-1 induced apoptosis substantially, and this effect was much more robust than gemcitabine plus Bcl-xL or Mcl-1 knock-down. However, again, adding gemcitabine to cells, in which Mcl-1 and Bcl-xL are downregulated, had no further effect on apoptosis (Fig. 3A). These effects were also confirmed by Western blot (Fig. 3B) detecting total and cleaved PARP.
Figure 3.

The effect of Bcl-xL and/or Mcl-1 knock-down on gemcitabine-induced apoptosis in PaCa.
Cells were transfected with Bcl-xL and/or Mcl-1 siRNA for 24h, after which cells were treated with 1 μg/ml gemcitabine for further 72h. DNA fragmentation and the cleavage of PARP were quantified by Cell Death ELISA (A) and Western blot (B), respectively. Results are representative of three independent experiments and are presented as mean ± SD (bars). *p<0.05.
3.4. The apoptosis inducing effect of simultaneous knock-down of Bcl-xL and Mcl-1 is mediated through mitochondrial cytochrome c release and caspase activation
Having demonstrated that simultaneous knock-down of Bcl-xL/Mcl-1 by siRNA strongly induced apoptosis in PaCa cells, we next investigated whether this effect was mediated by activation of caspases. As shown in Fig. 4A Bcl-xL or Mcl-1 knock-down induced cleavage of caspase-7 and PARP in BxPC-3 and MIA PaCa-2 cells. No effect on caspase-3 cleavage could be observed. However, Bcl-xL/Mcl-1 combined knock-down strongly induced cleavage of caspase-3, -7, and PARP in both cell lines, consistent with the results about DNA fragmentation (Fig. 2B, 2C). Furthermore, the pan-caspase inhibitor zVAD-fmk (100 μM) completely reversed Bcl-xL/Mcl-1 knock-down-induced DNA fragmentation as well as cleavage of PARP in both cell lines (Fig. 4B). Because it has been reported that activated caspase-3 cleaves Mcl-1 [15], we also measured Mcl-1 expression in this setting. Treatment with zVAD-fmk only very minimally increased Mcl-1 protein expression in Bcl-xL/Mcl-1 siRNA-treated BxPC-3 cells (but not in MIA PaCa-2 cells), indicating that Mcl-1 protein down-regulation is almost exclusively achieved by RNA interference and not by caspase-mediated cleavage. Since cytochrome c release into the cytosol is the key event in apoptosis, which leads to the activation of downstream caspases, we next investigated the release of cytochrome c. Bcl-xL/Mcl-1 knock-down induced cytochrome c release from the mitochondria (as evident by a decrease of mitochondrial cytochome c and an increase in cytosolic cytochrome c) in both cells (Fig. 4C).
Figure 4.

The apoptosis-inducing effect of simultaneous knock-down of Bcl-xL and Mcl-1 is mediated through mitochondria and caspases.
(A) BxPC-3 and MIA PaCa-2 cells were transiently transfected with Bcl-xL or/and Mcl-1 siRNA for 24h, after which the cleavage of caspase-3, -7, and PARP were measured by Western blot. Results are representative of three independent experiments. (B) Cells were treated with the pan-caspase inhibitor zVAD-fmk (100 μM) for 1 h, and then were transiently transfected with control or Bcl-xL/Mcl-1 siRNA for 24h, after which the extent of apoptosis was quantified by Cell Death ELISA. Results are representative of three independent experiments and are presented as mean ± SD (bars). *p<0.005. Furthermore, PARP cleavage and the expression of Bcl-xL and Mcl-1 were also measured by Western blot. (C) Cells were transfected with Bcl-xL and Mcl-1 siRNA for 24h, after which mitochondrial membrane and cytosolic fractions were separated as described in methods. Cytochrome c localization was measured by Western blot. Mitochondrial marker Cox-IV (cytochrome oxidase IV) was used as a loading control. Results are representative of three independent experiments. To make clear the translocation of proteins, each blots are measured by densitometry (right panel).
3.5. Bax knock-down diminishes the effect of simultaneous knock-down of Bcl-xL and Mcl-1 on apoptosis in pancreatic cancer cells
Since activation of the multi-domain molecule Bax (conformational change, mitochondrial translocation, and oligomerization) leads to cytochrome c release from mitochondria [16], we decided to investigate the effect of Bcl-xL/Mcl-1 knock-down on Bax activation. In this regard, we first measured the effect of Bcl-xL/Mcl-1 knock-down on Bax expression. As shown in Fig. 5A, Bcl-xL/Mcl-1 knock-down did not change Bax expression in whole cell lysates. Next, we investigated whether Bcl-xL/Mcl-1 knock-down induces Bax mitochondrial translocation. As shown in Fig. 5B, Bcl-xL/Mcl-1 knock-down clearly induced Bax translocation from the cytosol to the mitochondria in both cell lines. To further confirm the importance of Bax on apoptosis induced by Bcl-xL/Mcl-1 knock-down, we used siRNA against Bax. Bax knock-down partially reversed the induction of apoptosis by Bcl-xL/Mcl-1 knock-down (Fig. 5C), indicating that Bax activation may be responsible, at least in part, for the dramatic induction of apoptosis by the simultaneous knock-down of Bcl-xL and Mcl-1.
Figure 5.

Activation of Bax correlates with apoptosis induced by simultaneous knock-down of Bcl-xL and Mcl-1.
(A) Cells were transiently transfected with siRNA against Bcl-xL and Mcl-1 for 24 h, after which the Bax expression was measured by Western blot. Results are representative of three independent experiments. (B) Cells were transiently transfected with siRNA against Bcl-xL and Mcl-1 for 24 h, after which mitochondrial membrane and cytosolic fraction were separated as described in methods. Bax expression was then measured by Western blot. Results are representative of three independent experiments. To make clear the translocation of proteins, each blots are measured by densitometry (right panel). (C) Cells were first transfected with Bax siRNA for 24 h, after which cells were transfected with siRNA against Bcl-xL and Mcl-1 for further 24 h. The extent of apoptosis (left panel) and Bax protein expression (right panel) was measured. Results are representative of three independent experiments and are presented as mean ± SD (bars). *p<0.05.
4. Discussion
One reason for the dismal prognosis of patients with PaCa and the inefficiency of most chemo- and radio-therapeutic regimens is the strong resistance of PaCa cells to cell death. Anti-apoptotic Bcl-2 family proteins have been reported to protect cells from death stimuli in several cancer models. In this regard, we previously reported that Mcl-1 is an important factor in PaCa cell survival [2]. However, Mcl-1 knock-down failed to increase cell death in Panc-1 cells in this and a previous report [17], suggesting that additional factors may be important as well. In this study, we provide strong evidence that Mcl-1 and Bcl-xL, and not Bcl-2, play a critical role in PaCa cell death mechanisms. This result was consistent with previous reports that simultaneous knock-down of Bcl-xL and Mcl-1 by siRNA resulted in marked induction of apoptosis in chemo-resistant ovarian carcinoma and methothelioma cell lines [18, 19]. However, no report has to our knowledge described the efficacy of simultaneous knockdown of Bcl-2 family proteins in pancreatic cancer cells. Our data suggest that simultaneous Bcl-xL and Mcl-1 knockdown significantly induces apoptotic cell death in pancreatic cancer cells through a Bax-mediated mechanism. But, how does Bcl-xL and Mcl-1 knockdown induce Bax translocation? Bax is a strong multi-domain pro-apoptotic protein that resides in the cytoplasm as inactive monomer in healthy cells. Upon apoptotic stimuli, Bax undergoes conformational change leading to translocation from cytoplasm to mitochondria [20]. Similar to our results, it has been reported that Mcl-1 knock-down by siRNA induced Bax translocation to mitochondria in oral squamous cell carcinoma cells [21]. These results indicated that the loss of Mcl-1 itself can modulate the subcellular distribution of Bax. In this regard, activator BH3-only proteins, especially Bim and Bid, might play an important role. It has been reported that activator BH3-only proteins can bind both Bcl-xL and Mcl-1, and activator BH3-only proteins that were freed from Bcl-xL and Mcl-1 can directly activate Bax [22].
However, the effect of Bax knock-down on reversing apoptosis induced by Bcl-xL/Mcl-1 knockdown was only partial. Previous reports using BH3-mimetics (e.g. ABT-737 and obatoclax) showed that Mcl-1 and Bcl-xL cooperate to sequester the pro-apoptotic multidomain protein Bak and prevent its activation [23, 24]. Since we confirmed that Bak binds with Bcl-xL and Mcl-1 on the mitochondrial membrane in BxPC-3 and MIA Paca-2 cells [2], it is plausible that Bak, as well as Bax (as shown in the present study), might play an important role.
It has been reported that anti-apoptotic Bcl-2 family proteins control Bak and Bax activation by direct or indirect mechanisms in cooperation with BH3-only proteins [3]. In the indirect model, Bcl-xL and Mcl-1 bind to Bak not Bax in mouse embryo fibroblasts (MEFs) cells. Cytotoxic signals activate a combination of BH3-only proteins that can displace Bak from Bcl-xL and Mcl-1 to undergo conformational changes required for Bak activation. In the direct model, activator BH3-only proteins can directly bind and activate Bak or Bax. On the other hand, anti-apoptotic proteins also can bind to activator BH3-only proteins as well as sensitizer BH3-only proteins. Thus, the degree to which BH3-only proteins ultimately influences cellular apoptosis appears to be dependent on the delicate balance between anti-apoptotic proteins and the BH3-only proteins. In this regard, genetic knock-down of Bcl-xL and Mcl-1, as well as BH3-mimetic molecules, seems to be promising approach in PaCa cells. In this report, we showed the endogenous expression and localization of anti-apoptotic Bcl-2 family proteins. Bcl-xL and Mcl-1 were highly expressed in PaCa cell lines, while Bcl-2 was detected only in MIA PaCa-2 and Panc-1 cells. Similar to our results, the expression of Bcl-2, Bcl-xL, and Mcl-1 in human PaCa tissues was reported at 23–25%, 90%, and 90%, respectively [25]. This also suggests that Bcl-xL and Mcl-1, but not Bcl-2, may be an intriguing therapeutic target in PaCa.
Anti-apoptotic Bcl-2 family proteins have been reported to correlate with chemo-resistance in human malignancies [26, 27]. In contrast to our data, Wei and colleagues reported that Mcl-1 down-regulation enhanced the chemo-sensitivity to gemcitabine in PaCa cells [28]. However, our data demonstrated that gemcitabine could not augment the cell death induced by knockdown of Mcl-1 and/or Mcl-1/Bcl-xL. It may be caused by different levels of down-regulation of Mcl-1 protein in both studies. Simultaneous knock-down of Bcl-xL and Mcl-1 was clearly superior in inducing apoptosis than gemcitabine alone or a combination of gemcitabine with a knock-down of a single anti-apoptotic protein. These results indicate that Bcl-xL and Mcl-1 may be bone fide therapeutic targets in pancreatic cancer independent of gemcitabine and not only chemo-sensitizers.
Today, some therapeutic approaches have been reported aimed at abrogating the protective function of anti-apoptotic Bcl-2 family proteins. It has been described that certain BH3-mimetics can bind to and functionally inhibit Bcl-2 family proteins. The Bcl-2 antagonist ABT-737 targets Bcl-2 and Bcl-xL, but not Mcl-1, which may confer resistance to this agent. Down-regulation of Mcl-1 by siRNA or pharmaceutical drugs has been reported to potentiate ABT-737 lethality in human leukemia and melanoma cells [23, 29]. Similarly, Okumura et al. reported that Noxa induction sensitize Mcl-1 expressing human colorectal cancer cells to ABT-737 [30]. Another pan-Bcl-2 inhibitor, obatoclax (GX15-070) has also been reported to antagonize Mcl-1 and overcome Mcl-1 mediated resistance to apoptosis [31]. These observations are in accordance with our results showing that Bcl-xL and Mcl-1 cooperatively play a critical role in pancreatic cancer cell apoptosis. In addition to small molecule drugs, siRNA-based therapy is another promising therapeutic approach. Although the design and delivery system of siRNA has not been established yet [32], siRNA based therapy has made remarkable progress and siRNA-based therapy in vivo has been reported in recent years [33–35].
In summary, these findings reinforce the notion that disruption of a single anti-apoptotic protein, i.e. Bcl-xL or Mcl-1, may not be optimal for efficiently inducing cell death in pancreatic cancer cells. Instead, our data clearly suggest that reducing the expression of both anti-apoptotic proteins should be the goal.
Research Highlights.
Mcl-1 knock-down results in induction of apoptosis.
Down-regulation of Bcl-2 or Bcl-xL has a much weaker pro-apoptotic effect.
Simultaneous knock-down of Bcl-xL and Mcl-1 strongly induces apoptosis.
Bcl-xL and Mcl-1 knockdown leads to Bax translocation to the mitochondria.
Acknowledgments
This work was supported by the National Institutes of Health (P01 AT003960, P01 CA163200, R01CA122042), the Hirshberg Foundation for Pancreatic Cancer Research and JSPS KAKENHI Grant Number 24592036.
Financial support: This work was supported by the National Institutes of Health (P01AT003960, P01CA163200, R01CA122042), the Hirshberg Foundation for Pancreatic Cancer Research, and JSPS KAKENHI Grant Number 24592036.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hochster HS, Haller DG, de Gramont A, Berlin JD, Philip PA, Moore MJ, Ajani JA. Consensus report of the international society of gastrointestinal oncology on therapeutic progress in advanced pancreatic cancer. Cancer. 2006;107:676–685. doi: 10.1002/cncr.22036. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi H, Chen MC, Pham H, Angst E, King JC, Park J, Brovman EY, Ishiguro H, Harris DM, Reber HA, Hines OJ, Gukovskaya AS, Go VL, Eibl G. Baicalein, a component of Scutellaria baicalensis, induces apoptosis by Mcl-1 down-regulation in human pancreatic cancer cells. Biochim Biophys Acta. 2011;1813:1465–1474. doi: 10.1016/j.bbamcr.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13:7254–7263. doi: 10.1158/1078-0432.CCR-07-1598. [DOI] [PubMed] [Google Scholar]
- 4.Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta. 2011;1813:521–531. doi: 10.1016/j.bbamcr.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 5.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eibl G, Takata Y, Boros LG, Liu J, Okada Y, Reber HA, Hines OJ. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65:982–990. [PubMed] [Google Scholar]
- 8.Eibl G, Reber HA, Wente MN, Hines OJ. The selective cyclooxygenase-2 inhibitor nimesulide induces apoptosis in pancreatic cancer cells independent of COX-2. Pancreas. 2003;26:33–41. doi: 10.1097/00006676-200301000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Angst E, Reber HA, Hines OJ, Eibl G. Mononuclear cell-derived interleukin-1 beta confers chemoresistance in pancreatic cancer cells by upregulation of cyclooxygenase-2. Surgery. 2008;144:57–65. doi: 10.1016/j.surg.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi H, Li A, Dawson DW, Hines OJ, Reber HA, Eibl G. Cyclooxygenase-2 confers growth advantage to syngeneic pancreatic cancer cells. Pancreas. 2011;40:453–459. doi: 10.1097/MPA.0b013e31820b9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem. 2011;351:41–58. doi: 10.1007/s11010-010-0709-x. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee S, Choi M, Aboukameel A, Wang Z, Mohammad M, Chen J, Yang D, Sarkar FH, Mohammad RM. Preclinical studies of apogossypolone, a novel pan inhibitor of bcl-2 and mcl-1, synergistically potentiates cytotoxic effect of gemcitabine in pancreatic cancer cells. Pancreas. 2010;39:323–331. doi: 10.1097/MPA.0b013e3181bb95e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lei X, Huang Z, Zhong M, Zhu B, Tang S, Liao D. Bcl-XL small interfering RNA sensitizes cisplatin-resistant human lung adenocarcinoma cells. Acta Biochim Biophys Sin (Shanghai) 2007;39:344–350. doi: 10.1111/j.1745-7270.2007.00286.x. [DOI] [PubMed] [Google Scholar]
- 15.Weng C, Li Y, Xu D, Shi Y, Tang H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J Biol Chem. 2005;280:10491–10500. doi: 10.1074/jbc.M412819200. [DOI] [PubMed] [Google Scholar]
- 16.Er E, Oliver L, Cartron PF, Juin P, Manon S, Vallette FM. Mitochondria as the target of the pro-apoptotic protein Bax. Biochim Biophys Acta. 2006;1757:1301–1311. doi: 10.1016/j.bbabio.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 17.Huang S, Sinicrope FA. BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res. 2008;68:2944–2951. doi: 10.1158/0008-5472.CAN-07-2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brotin E, Meryet-Figuière M, Simonin K, Duval RE, Villedieu M, Leroy-Dudal J, Saison-Behmoaras E, Gauduchon P, Denoyelle C, Poulain L. Bcl-XL and MCL-1 constitute pertinent targets in ovarian carcinoma and their concomitant inhibition is sufficient to induce apoptosis. Int J Cancer. 2010;126:885–895. doi: 10.1002/ijc.24787. [DOI] [PubMed] [Google Scholar]
- 19.Varin E, Denoyelle C, Brotin E, Meryet-Figuière M, Giffard F, Abeilard E, Goux D, Gauduchon P, Icard P, Poulain L. Downregulation of Bcl-xL and Mcl-1 is sufficient to induce cell death in mesothelioma cells highly refractory to conventional chemotherapy. Carcinogenesis. 2010;31:984–993. doi: 10.1093/carcin/bgq026. [DOI] [PubMed] [Google Scholar]
- 20.Haneef J, Thankayyan PMSK, Sithul RH, Sreeharshan S. Bax translocation mediated mitochondrial apoptosis and caspase dependent photosensitizing effect of Ficus religiosa on cancer cells. PLoS One. 2012;7:e40055. doi: 10.1371/journal.pone.0040055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin JA, Jung JY, Ryu MH, Safe S, Cho SD. Mithramycin A Inhibits Myeloid Cell Leukemia-1 to Induce Apoptosis in Oral Squamous Cell Carcinomas and Tumor Xenograft through Activation of Bax and Oligomerization. Mol Pharmacol. 2012 Sep 27; doi: 10.1124/mol.112.081364. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 22.Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011;286:491–501. doi: 10.1074/jbc.M110.167148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Bélec L, Billot X, Acoca S, Purisima E, Wiegmans A, Cluse L, Johnstone RW, Beauparlant P, Shore GC. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyamoto Y, Hosotani R, Wada M, Lee JU, Koshiba T, Fujimoto K, Tsuji S, Nakajima S, Doi R, Kato M, Shimada Y, Imamura M. Immunohistochemical analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 expression in pancreatic cancers. Oncology. 1999;56:73–82. doi: 10.1159/000011933. [DOI] [PubMed] [Google Scholar]
- 26.Karnak D, Xu L. Chemosensitization of prostate cancer by modulating Bcl-2 family proteins. Curr Drug Targets. 2010;11:699–707. doi: 10.2174/138945010791170888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 28.Wei SH, Dong K, Lin F, Wang X, Li B, Shen JJ, Zhang Q, Wang R, Zhang HZ. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. 2008;62:1055–1064. doi: 10.1007/s00280-008-0697-7. [DOI] [PubMed] [Google Scholar]
- 29.Keuling AM, Felton KE, Parker AA, Akbari M, Andrew SE, Tron VA. RNA silencing of Mcl-1 enhances ABT-737-mediated apoptosis in melanoma: role for a caspase-8-dependent pathway. PLoS One. 2009;4:e6651. doi: 10.1371/journal.pone.0006651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okumura K, Huang S, Sinicrope FA. Induction of Noxa sensitizes human colorectal cancer cells expressing Mcl-1 to the small-molecule Bcl-2/Bcl-xL inhibitor, ABT-737. Clin Cancer Res. 2008;14:8132–8142. doi: 10.1158/1078-0432.CCR-08-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Bélec L, Billot X, Acoca S, Purisima E, Wiegmans A, Cluse L, Johnstone RW, Beauparlant P, Shore GC. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He S, Zhang D, Cheng F, Gong F, Guo Y. Applications of RNA interference in cancer therapeutics as a powerful tool for suppressing gene expression. Mol Biol Rep. 2009;36:2153–2163. doi: 10.1007/s11033-008-9429-7. [DOI] [PubMed] [Google Scholar]
- 33.Liang Y, Gao H, Lin SY, Goss JA, Brunicardi FC, Li K. siRNA-based targeting of cyclin E overexpression inhibits breast cancer cell growth and suppresses tumor development in breast cancer mouse model. PLoS One. 2010;5:e12860. doi: 10.1371/journal.pone.0012860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei M, Zhu L, Li Y, Chen W, Han B, Wang Z, He J, Yao H, Yang Z, Zhang Q, Liu B, Gu Q, Zhu Z, Shen K. Knocking down cyclin D1b inhibits breast cancer cell growth and suppresses tumor development in a breast cancer model. Cancer Sci. 2011;102:1537–1544. doi: 10.1111/j.1349-7006.2011.01969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takigami I, Ohno T, Kitade Y, Hara A, Nagano A, Kawai G, Saitou M, Matsuhashi A, Yamada K, Shimizu K. Synthetic siRNA targeting the breakpoint of EWS/Fli-1 inhibits growth of Ewing sarcoma xenografts in a mouse model. Int J Cancer. 2011;128:216–226. doi: 10.1002/ijc.25564. [DOI] [PubMed] [Google Scholar]