

Abstract
Memantine and ketamine, voltage- and activation-dependent channel blockers of N-methyl-d-aspartate (NMDA) receptors (NMDARs), have enjoyed a recent resurgence in clinical interest. Steady-state pharmacodynamic differences between these blockers have been reported, but it is unclear whether the compounds differentially affect dynamic physiologic signaling. In this study, we explored nonequilibrium conditions relevant to synaptic transmission in hippocampal networks in dissociated culture and hippocampal slices. Equimolar memantine and ketamine had indistinguishable effects on the following measures: steady-state NMDA currents, NMDAR excitatory postsynaptic current (EPSC) decay kinetics, progressive EPSC inhibition during repetitive stimulation, and extrasynaptic NMDAR inhibition. Therapeutic drug efficacy and tolerability of memantine have been attributed to fast kinetics and strong voltage dependence. However, pulse depolarization in drug presence revealed a surprisingly slow and similar time course of equilibration for the two compounds, although memantine produced a more prominent fast component (62% versus 48%) of re-equilibration. Simulations predicted that low gating efficacy underlies the slow voltage–dependent relief from block. This prediction was empirically supported by faster voltage-dependent blocker re-equilibration with several experimental manipulations of gating efficacy. Excitatory postsynaptic potential–like voltage commands produced drug differences only with large, prolonged depolarizations unlikely to be attained physiologically. In fact, we found no difference between drugs on measures of spontaneous network activity or acute effects on plasticity in hippocampal slices. Despite indistinguishable synaptic pharmacodynamics, ketamine provided significantly greater neuroprotection from damage induced by oxygen glucose deprivation, consistent with the idea that under extreme depolarizing conditions, the biophysical difference between drugs becomes detectable. We conclude that despite subtle differences in voltage dependence, during physiologic activity, blocker pharmacodynamics are largely indistinguishable and largely voltage independent.
Introduction
Memantine and ketamine are activation-dependent and voltage-dependent N-methyl-d-aspartate (NMDA) receptor (NMDAR) channel blockers. Memantine is neuroprotective and is used to enhance cognition in dementia (Ditzler, 1991; Lipton, 2005). Ketamine is an anesthetic and analgesic with psychotomimetic effects and abuse potential (Krystal et al., 1994), but it has drawn recent attention as a fast-acting antidepressant (Zarate et al., 2006; Aan Het Rot et al., 2012). The drugs are pharmacologically similar, but the literature suggests small steady-state differences. Because physiologic activity is not inherently steady-state, we explored cellular and receptor-level drug differences under dynamic conditions.
Ketamine and memantine both require channel opening for access to and for exit from the NMDAR channel (Parsons et al., 1993; Lipton, 2005; Johnson and Kotermanski, 2006; Gilling et al., 2009; Kotermanski et al., 2009). During sustained, steady-state agonist presentation, memantine may display slightly faster kinetics of block than ketamine (Gilling et al., 2009). Further, memantine has been found to bind two sites with differing dependence on channel opening, leading to partial trapping upon channel closure. Meanwhile, ketamine exhibits full trapping (Blanpied et al., 1997; Gilling et al., 2009; Kotermanski et al., 2009), although there is some evidence for two ketamine sites (Orser et al., 1997). Contrary to agonist presentation in most previous studies, physiologic, synaptic agonist presentation is brief and transient (Lester et al., 1990; Clements et al., 1992). This non–steady-state profile of agonist presentation could influence activation-dependent channel blockers and accentuate any pharmacodynamic differences. Memantine might be a selective antagonist for extrasynaptic NMDAR populations (Okamoto et al., 2009; Xia et al., 2010; Wroge et al., 2012). However, whether this effect is secondary to the more sustained agonist presentation at extrasynaptic versus synaptic receptors is unclear, and whether ketamine shares any extrasynaptic receptor selectivity has not been explored.
Both drugs are also voltage dependent. At sustained positive potentials, memantine appears to exhibit weaker block (more voltage dependence) than ketamine (Gilling et al., 2009). Depolarization, like agonist presentation, is rarely sustained during physiologic activity except under pathophysiologic conditions. How transient depolarization interacts with NMDAR blocker actions is unclear. Sufficiently rapid dissociation during excitatory postsynaptic potentials (EPSPs) could preserve transient synaptic transmission while blocking low-level, tonic extrasynaptic activation (Lipton, 2005; Gilling et al., 2009). On the other hand, if drug re-equilibration is slow relative to EPSP duration, drugs may have largely voltage-independent effects except under the most extreme conditions.
Our study focused on nonequilibrium agonist presentation and rapid voltage changes applicable to synaptic transmission. In a number of paradigms, the drugs exhibit remarkable similarities. The sole distinction observed was a difference in the apparent voltage dependence of the drugs in whole-cell, voltage-pulse experiments. Simulations revealed that the apparent voltage dependence could result from differences in channel opening and closing rates while blocker is bound and need not involve changes in microscopic voltage dependence of drug action. The difference between drugs did not manifest under most physiologic conditions because of the low open probability of the NMDAR channel (Rosenmund et al., 1995; Chen et al., 1999), which did not allow sufficient opportunity for dissociation of either drug during transient, synaptic-like depolarizations. The drugs also behaved similarly in their effects on spontaneous network activity and induction of plasticity. Differences emerged only under extreme depolarization during oxygen and glucose deprivation, during which memantine proved the weaker neuroprotectant.
Materials and Methods
Cell Cultures.
Hippocampal cultures were prepared as either mass cultures or microcultures (as indicated in figure legends) from postnatal day 1 to 3; male and female rat pups were anesthetized with isoflurane, under protocols consistent with National Institutes of Health guidelines and approved by the Washington University Animal Studies Committee. Methods were adapted from earlier descriptions (Huettner and Baughman, 1986; Bekkers et al., 1990; Tong and Jahr, 1994; Mennerick et al., 1995). Hippocampal slices (500-μm thickness) were digested with 1 mg ml−1 papain in oxygenated Leibovitz L-15 medium (Life Technologies, Gaithersburg, MD). Tissue was mechanically triturated in modified Eagle’s medium (Life Technologies) containing 5% horse serum, 5% fetal calf serum, 17 mM d-glucose, 400 μM glutamine, 50 U ml−1 penicillin, and 50 μg ml−1 streptomycin. Cells were seeded in modified Eagle’s medium at a density of ∼650 cells mm−2 as mass cultures (onto 25-mm cover glasses coated with 5 mg ml−1 collagen or 0.1 mg ml−1 poly-d-lysine with 1 mg ml−1 laminin) or 100 cells mm−2 as “microisland” cultures (onto 35-mm plastic culture dishes coated with collagen microdroplets on a layer of 0.15% agarose). Cultures were incubated at 37°C in a humidified chamber with 5% CO2/95% air. Cytosine arabinoside (6.7 μM) was added 3 to 4 days after plating to inhibit glial proliferation. The following day, half of the culture medium was replaced with Neurobasal medium (Life Technologies) plus B27 supplement (Life Technologies).
Human Embryonic Kidney Cell Transfection.
Human embryonic kidney cell line (HEK293) cells were grown and split when 50–75% confluent in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 1 mM glutamine with penicillin or streptomycin. One day before transfection, cells were split and plated directly on 35-mm plastic dishes. When 30–50% confluent, cells were transferred to Opti-Med transfection media and transfected using Lipofectamine 2000 with glutamate receptor subunits GluN1a and GluN2A or GluN2B in a 1:3 ratio. GluN subunit DNA constructs were gifts of Drs. Elias Aizenman and Jon Johnson (University of Pittsburgh). Green fluorescent protein (0.05 µg) was used as a transfection efficiency marker. After 2–4 hours, transfection medium was removed and replaced with culture medium containing 100 µM ketamine to prevent excitotoxicity (Boeckman and Aizenman, 1996). For recordings performed 48–72 hours after transfection, medium was removed and washed repeatedly with ketamine-free recording solution. Repetitive agonist application before data collection and interleaved experimental conditions ensured complete removal of blocker before experiments.
Electrophysiology.
Whole-cell recordings were performed at room temperature from neurons cultured for 5–10 days (depending on the experiment) using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA) and Digidata 1440A converter using Clampex 10.1 software. Young cells were favored for biophysical experiments to minimize voltage-clamp errors, and older, synaptically mature cells were used for excitatory postsynaptic current (EPSC) measurements. green fluorescent protein–positive HEK cells were recorded 2–5 days post transfection. For recordings, cells were transferred to an extracellular (bath) solution containing (in mM): 138 NaCl, 4 KCl, 2 CaCl2, 10 glucose, 10 HEPES, and 0.05 d-2-amino-5-phosphonovalerate (d-APV), pH 7.25, adjusted with NaOH. Solutions were perfused over the cells using a gravity-driven local perfusion system from a common tip. The estimated solution exchange times were <100 millisecond (10–90% increase), estimated from junction current rises at the tip of an open patch pipette. For synaptic recordings, these solutions contained (in mM) 0.01 glycine, 0.001 NBQX (2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline), and 0.025 bicuculline methobromide and contained no d-APV. For exogenous NMDA application during voltage perturbations, 0.25 mM CaCl2 was used (in APV-free perfusion solutions) to minimize Ca2+-dependent NMDAR desensitization (Zorumski et al., 1989), and 250 nM tetrodotoxin was added to prevent network activity. During network activity studies (Fig. 9), all blockers were eliminated and 2 mM Ca2+, 1 mM Mg2+, and 1 µM glycine were used. During HEK cell recordings, antagonists and tetrodotoxin were omitted. Unless otherwise noted, exogenous NMDA concentration was 300 µM. In experiments using tricine, 10 mM tricine was added to external solutions, and pH was adjusted to 7.25. The tip resistance of patch pipettes was 3–6 MΩ when filled with an internal solution containing (in mM): 130 potassium gluconate, 0.5 CaCl2, 5 EGTA, 4 NaCl, and 10 HEPES at pH 7.25, adjusted with KOH. Potassium was used as the main cation for autaptic stimulation to preserve action potential waveform, but in experiments examining current responses to exogenous agonists, cesium methanesulfonate or cesium gluconate was used in place of potassium gluconate to block potassium channels and improve spatial voltage clamp quality. Holding voltage was typically −70 mV unless otherwise noted. Access resistance (8–10 MΩ) was compensated 80%–100% for EPSC measurements. For evoked EPSCs, cells were stimulated with 1.5 millisecond pulses to 0 mV from −70 mV to evoke autaptic transmitter release (Mennerick et al., 1995).
Fig. 9.

Network effects of memantine and ketamine. (A) A neuron was voltage clamped at −70 mV in a blocker-free external solution containing 1 µM glycine. Network activity was measured as AMPA-driven spontaneous EPSCs onto the neuron over 60-second intervals. Network activity was allowed to stabilize for 2 minutes before baseline data collection, and drugs were allowed 60 seconds of equilibration before recording. Memantine and ketamine (10 µM) were interleaved between saline recordings, and the presentation order was reversed from cell to cell. (B) Synaptic activity was quantified as synaptic charge exceeding a threshold of 7.5 pA over the 60 seconds of recording time. Activity in drug was compared with the average activity during baseline and washout conditions. At 2 µM, no significant effect of either drug on synaptic activity (n = 6) was seen. However, at 10 µM, both drugs depressed activity (*P < 0.05, unpaired t test to normalized baseline), but depression did not significantly differ between drugs (n = 12). (C) MEA recordings from cultures in the presence of control media (baseline) or 10 µM memantine or ketamine. Representative raster plots are shown from one experiment using sibling cultures for memantine and ketamine. (D) Overall statistics for array-wide and network properties summarized and normalized to baseline. Asterisks indicate significant differences (P < 0.05, two-tailed unpaired t test) from baseline (indicated by dotted gray line). Memantine and ketamine did not significantly differ from each other in any parameter tested (n = 10, two-tailed, unpaired t test). Bursts were defined as described in Materials and Methods.
Artificial EPSP (αEPSP) waveforms were generated in Excel using the following equation:
Vmax was set to values varying from 0 to +70 mV, yielding a maximum depolarization to −20 mV from the holding potential of −90 mV, and τ was either 30 milliseconds (for short αEPSPs) or 300 milliseconds (for long αEPSPs). The resulting data were converted to a command waveform in Clampfit 10.1. Voltage-gated sodium currents were suppressed with 250 nM tetrodotoxin for this experiment.
For network activity analysis, mass cultures were seeded on multielectrode arrays (MEAs; Multichannel Systems) (Mennerick et al., 2010). Recordings were performed 15 days in vitro. For figures and statistics, effects of drugs over a 30-minute recording period were compared with the average of activity (30 minutes) before drug administration and after drug washout. Array-wide spike detection rate was measured as the total number of spikes across the entire array in each second of recording. Bursts were defined as three or more spikes on a single contact with an interspike interval less than a critical duration of 150 milliseconds (Wagenaar et al., 2006). Burst duration was the interval between the first spike and the last spike in a burst.
Hippocampal Slices.
Slices were harvested from 28- to 32-day-old male albino rats under isoflurane anesthesia (Tokuda et al., 2010). For electrophysiology, hippocampal slices were transferred to a submerged recording chamber with continuous bath perfusion of artificial cerebrospinal fluid (124 NaCl, 5 KCl, 2 MgSO4, 2 CaCl2, 1.25 NaH2PO4, 22 NaHCO3, 10 glucose, bubbled with 95% O2/5% CO2) at 2 ml/min at 30°C. Extracellular recordings were obtained from the apical dendritic layer of the CA1 region elicited with 0.1-millisecond constant current pulses through a bipolar stimulating electrode placed in stratum radiatum. EPSPs were monitored using a half-maximal stimulus based on a baseline input-output curve. After establishing a stable baseline, long-term potentiation (LTP) was induced by applying a single 100 Hz × 1 second high-frequency stimulus using the same intensity stimulus as used for monitoring. Long-term depression (LTD) was induced with 1-Hz low-frequency stimulation for 15 minutes. An input-output curve was repeated 60 minutes after induction protocols for statistical comparisons of changes in EPSP slopes at half-maximal intensity. Signals were digitized and analyzed using pCLAMP software (Molecular Devices, Union City, CA).
Oxygen Glucose Deprivation.
Culture medium in mass cultures (13–14 days in vitro) was exchanged for minimum essential medium (catalog no. 11090-081; Life Technologies) with no added glutamine or glucose, supplemented with 10 μM glycine immediately before hypoxia exposure in a commercially available chamber (Billups-Rothenberg, Inc., Del Mar, CA), humidified, and saturated with 95% nitrogen and 5% CO2 at 37°C for 2.5 hours. Drugs were added directly to culture medium before oxygen-glucose deprivation (OGD). The gas exchange followed the specifications of the chamber manufacturer (flow of 20 l/min for 4 minutes to achieve 100% gas exchange). After OGD, cells were returned to their original medium and incubated under standard culture conditions until the cell death assay (24 hours later). We used Hoechst 33342 (5 μM) to identify all nuclei and propidium iodide (3 μM) for 30 minutes to stain nuclei of cells with compromised membranes. Five 10× microscope fields were quantified per condition per experiment, yielding >100 total neurons for each condition. Ratios of healthy neurons were quantified as the fraction of propidium iodide–negative neuronal nuclei to total neuronal nuclei. Automated cell-counting algorithms (ImageJ Software; NIH, Bethesda, MD) were used for cell counts. Toxicity experiments were treated as a dependent sample design, in which sibling cultures plated in identical media lost and exposed to hypoxia at the same time were compared by repeated measures statistics.
Simulations.
NEURON software (Carnevale and Hines, 2006) was used to generate simulations of voltage step perturbations. The kinetic scheme used for the NMDAR, and rate constants were adapted from previous work (Blanpied et al., 1997).
Data Analysis.
Electrophysiology data acquisition and analysis were performed primarily using pCLAMP 10 software (Molecular Devices). MEA data were analyzed with Igor Pro (Wavemetrics, Lake Oswego, OR). All electrophysiologic measurements were processed with Microsoft Excel and are presented as mean ± S.E.M. Statistical significance was determined using a Student’s one-tailed or two-tailed t test, unless indicated otherwise, with a Bonferroni correction for multiple comparisons where appropriate, and significance was taken as P < 0.05. One-tailed tests were reserved for later experiments on network function and toxicity, after a hypothesis about the direction of difference between memantine and ketamine emerged based on the difference in voltage dependence between drugs. Data plotting, statistical analysis, and curve fitting to the Hill equation were performed with SigmaPlot (Systat, San Jose, CA). Figure preparation was performed in SigmaPlot and Adobe Photoshop (San Jose, CA). Decay time constants were measured using standard exponential fitting functions using a least-squares minimization algorithm or a Chebyshev transform in pCLAMP software.
Materials.
All drugs were obtained from Sigma-Aldrich (St. Louis, MO) except for d-APV and NBQX (Tocris Bioscience, Minneapolis, MN). Culture media were obtained from Life Technologies (Grand Island, NY).
Results
Indistinguishable Modulation of NMDAR EPSCs by Memantine and Ketamine at Constant Voltage.
To aid the choice of ketamine and memantine concentrations for our studies, we examined drug effects during sustained NMDA application in cultured dissociated hippocampal neurons (Fig. 1, A and B). Consistent with other studies (Gilling et al., 2009; Kotermanski and Johnson, 2009; Traynelis et al., 2010), both drugs behaved similarly at equimolar concentration. Although memantine’s IC50 was 2.1 μM and ketamine’s was 1.5 μM, these values were statistically indistinguishable (Fig. 1C). The values are likely a slight overestimate because block did not always reach a steady state at low antagonist concentrations. However, in subsequent experiments, equimolar concentrations inevitably yielded indistinguishable steady-state effects at negative membrane potentials, even with prolonged blocker application.
Fig. 1.

Memantine and ketamine have indistinguishable IC50s (A and B). Inhibition of NMDAR current was evaluated for memantine (A) and ketamine (B) as indicated in dissociated hippocampal cultures at day in vitro 9 and 10. Increasing concentrations of the drugs (0.1, 1, 10, 100 μM) were applied to hippocampal neurons in the constant presence of 30 μM NMDA. Peak current is truncated for clarity. (C) Concentration-response curves, with IC50 values estimated from fits to the Hill equation (nN = 6). Memantine IC50: 2.1 µM; ketamine IC50: 1.5 µM.
As use-dependent NMDAR antagonists, both memantine and ketamine require NMDAR channel activation to block the channel. Thus, the temporal parameters of NMDAR activation strongly affect the blocking ability of memantine and ketamine. During steady-state receptor activation, the blocker equilibrates with its binding site, and kinetic differences between blockers may be masked. Steady-state agonist presentation also poorly recapitulates physiologic events where agonist presentation is brief and NMDAR activation is transient (Lester et al., 1990; Clements et al., 1992). To investigate whether memantine and ketamine exhibit kinetic differences that result in differential modulation of the NMDAR during synaptic activity, we measured evoked NMDA-EPSC amplitude and decay kinetics in the presence of drug. Drugs were applied using an interleaved protocol, and EPSCs were allowed to recover to baseline between drug applications. With 20-second drug preapplication that persisted through EPSC stimulation, we found that both drugs equivalently accelerated EPSC decay (Fig. 2A). As previously observed, NMDAR EPSC kinetics exhibited biexponential decay (Lester et al., 1990). Both time constants of decay were accelerated in the presence of memantine or ketamine, and the contribution of the fast component of decay increased. No detectable difference was found between equimolar memantine and ketamine in any of these properties (Fig. 2B).
Fig. 2.

Memantine and ketamine effects on NMDAR EPSCs are indistinguishable. (A) Autaptic EPSCs from solitary neurons 10–12 days in vitro were measured in the presence of saline, memantine (10 μM), and ketamine (10 μM). Memantine and ketamine sweeps alternated with saline (5 sweeps) to restore EPSC to baseline. Two or three replicate sweeps for each condition were averaged. Biexponential fits for the three conditions (blue, saline; red, memantine; green, ketamine) are overlaid on the raw traces. Inset shows scaled traces on an expanded time scale to indicate the fit of the initial fast component and the similarity of drug effects. (B) Parameters from biexponential curve fitting of the decay phase of the EPSC. Memantine and ketamine both significantly accelerated the biexponential decay kinetics of the EPSC (fast and slow τ and the relative contribution, *P < 0.05, Student’s t test), but effects did not differ between drugs (n = 8).
If memantine and ketamine differ in their ability to dissociate from closed channels, the drugs may have differing effects on peak EPSC amplitudes during intermittent, repetitive synaptic agonist presentation. To test this, we evoked NMDAR EPSCs continuously at a frequency of 0.04 Hz to allow complete recovery from presynaptic, frequency-dependent facilitation and depression (Mennerick et al., 1995). We found that the time course of progressive block by ketamine and memantine during this continuous stimulation was indistinguishable (Fig. 3). The similarity of progressive block by memantine and ketamine suggests similar binding rates for the two drugs at −70 mV.
Fig. 3.

Memantine and ketamine reach maximal synaptic block with similar time course. (A) Memantine (10 μM) block of autaptic NMDAR EPSCs generated with 0.04-Hz stimulation. Indicated are saline (black), first memantine application (dark red), and final memantine sweep (bright red). (B) Identical protocol with 10 μM ketamine: dark green is first application, bright green is final sweep. (C) Amplitudes of successive sweeps are graphed as a percentage of initial baseline amplitude. Memantine and ketamine did not differ in the degree or rate of block over successive applications (n = 5, P > 0.05, Student’s t test). Memantine data are reproduced from Wroge et al. (2012).
Memantine has been described as a selective blocker of extrasynaptic NMDARs over synaptic NMDARs compared with the very slowly reversible channel blocker MK-801 [(5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate] (Xia et al., 2010). However, recent studies have suggested that apparent selectivity may result from differences in time course of agonist presentation to extrasynaptic versus synaptic NMDARs rather than memantine affinity for extrasynaptic populations (Wroge et al., 2012). Memantine has not previously been compared with channel blockers of similar kinetics. Because memantine and ketamine exhibited similar actions at synaptic NMDARs (Figs. 2 and 3), we hypothesized that they may equivalently inhibit extrasynaptic populations. We pharmacologically enriched extrasynaptic NMDARs by stimulating autaptic EPSCs at 0.04 Hz in the presence of MK-801 until maximum synaptic block was reached (Fig. 4, A1 and B1). We then activated the resulting enriched extrasynaptic NMDARs with 30 µM NMDA and challenged these remaining NMDARs with 2 µM memantine or ketamine. We were unable to distinguish the degree of block during 10-second coapplication (Fig. 4, A2 and B2; 77% ± 3% inhibition for memantine, 75% ± 2% inhibition for ketamine; n = 6, P > 0.05). As another approach, we used established protocols to block synaptic NMDARs in neuronal networks using 15-minute bath incubation with MK-801 (10 μM) and bicuculline (50 μM) (Hardingham and Bading, 2002; Wroge et al., 2012). We have shown that this protocol blocks NMDAR EPSCs without blocking AMPAR EPSCs (Wroge et al., 2012). Memantine and ketamine also similarly blocked the enriched extrasynaptic population isolated in this manner (71% ± 4% inhibition for memantine, 76% ± 3% for ketamine; P = 0.36). Our results demonstrate that memantine and ketamine act indistinguishably from each other at enriched extrasynaptic populations; thus, memantine is not unique in its effects on extrasynaptic NMDARs.
Fig. 4.

Memantine and ketamine block extrasynaptic NMDARs equally. (A1 and B1) Synaptic NMDARs were blocked by the slowly reversible open-channel blocker MK-801 (10 µM) during stimulation of autaptic EPSCs (0.04 Hz). (A2 and B2) Isolated extrasynaptic NMDARs were activated by 30 µM NMDA and challenged with an IC50 concentration of memantine (A2, 2 µM) or ketamine (B2, 2 μM). The relative block achieved by memantine or ketamine at the end of the application compared with steady-state control NMDA response measured immediately before drug application did not differ between the two drugs (see Results for values, P > 0.05 Student’s t test, n = 6).
We noted that inhibition by 2 µM memantine or ketamine at enriched extrasynaptic receptor populations in Fig. 4 appeared to be stronger than expected from Fig. 1. This could result from selectivity of both drugs for extrasynaptic receptor populations. However, when we tested the total receptor population, memantine inhibited NMDAR currents by 70% ± 2% (n = 8), statistically indistinguishable from an effect on MK-801–isolated extrasynaptic receptors in a matched comparison group with similar experimental conditions (72% ± 4%, n = 6; P = 0.6).
A second factor distinguishing isolated extrasynaptic receptor activation from total receptor activation is the number of activated receptors and therefore the level of Ca2+ influx. Since the overall Ca2+ load is expected to be lower with isolated extrasynaptic receptors (representing ∼30% of the total receptor pool), desensitization could be reduced and blocker effects could be altered. To test whether external calcium concentration affects blocker potency, we measured memantine (2 μM) block of steady-state NMDA responses in high (2 mM) Ca2+ or low (0.25 mM) Ca2+. We found that both conditions resulted in indistinguishable block (2 mM Ca2+: 72 ± 2% versus 0.25 mM Ca2+: 70% ± 2%, n = 8, P > 0.05). These data also suggest that external calcium itself has little effect on blocker potency. Thus, similar to our previous conclusions (Wroge et al., 2012), we find little evidence for drug selectivity for extrasynaptic receptors.
Re-Equilibration in Response to Voltage Pulses Reveals Subtle Differences between Drugs.
Memantine and ketamine, as positively charged channel blockers, display strong voltage sensitivity (Gilling et al., 2009). Previous studies suggest that memantine may exhibit slightly more steady-state voltage dependence than ketamine (Gilling et al., 2009). To explore kinetic differences that might underlie such effects, we recorded NMDA responses during a voltage pulse from −70 mV to +50 mV in the presence and absence of either memantine or ketamine. Although both drugs equivalently inhibited NMDA responses at −70 mV, memantine exhibited weaker inhibition than ketamine during a 10-second pulse to +50 mV (Fig. 5, A and B).
Fig. 5.

Memantine exhibits a significantly larger fast component of voltage-dependent re-equilibration after a voltage-pulse depolarization. (A1 and A2) 300 μM NMDA alone (black) or during steady-state block current of 10 μM memantine (A1, red) or 10 μM ketamine (A2, green) during a voltage step from −70 to +50 mV. Drugs were interleaved on the same cell, with NMDA application to relieve block. Traces are displayed after digital baseline saline subtraction. Gray line indicates 0 pA. Memantine and ketamine effects are shown as a percentage of baseline NMDA control (obtained by digital ratio) below original traces. (B) The percent block of NMDA currents achieved by both memantine and ketamine at −70 mV (before the voltage step) were identical, but inhibition at +50 mV was significantly lower for memantine than ketamine (n = 14 *P < 0.05). Two-way analysis of variance (ANOVA) with replication and Bonferroni correction for multiple comparisons). (C) The relaxation from block at −70 mV to the new steady state at +50 mV was fit with a biexponential curve. Memantine traces showed a significantly stronger fast component compared with ketamine (n = 14,*P < 0.05, Student t test). n.s., not significant.
We examined the time course of the approach toward a new (reduced) steady-state level of inhibition at +50 mV (time course of re-equilibration) by generating time-resolved inhibition plots (Frankiewicz et al., 1996) created by dividing the leak-subtracted current trace in the presence of blocker by the leak-subtracted trace in the presence of NMDA alone and displayed as a percentage. The time course of blocker re-equilibration was biexponential for both drugs (Fig. 5, A1 and A2, lower panels), and the fast and slow time constants were indistinguishable (Fig. 5, A and C). However, memantine’s fast component of re-equilibration was slightly but significantly more prominent than ketamine’s (Fig. 5C, right). Consistent with the faster kinetics of memantine, weighted time constants for the return pulse to −70 mV also differed (0.8 ± 0.07 seconds for memantine, 1.3 ± 0.06 seconds for ketamine; P < 0.05).
In summary, voltage-pulse relaxation analyses uncovered two interesting aspects of drug actions that we pursued further in subsequent experiments. First, the data above yielded blocker re-equilibration weighted time constants of 1.2 ± 0.2 seconds for memantine and 2.1 ± 0.6 seconds for ketamine at +50 mV. This slow overall time course of re-equilibration for both drugs was surprising and seems in contrast to the previous suggestion that fast kinetics may permit normal synaptic transmission and therapeutic tolerability (Lipton, 2005). We explored the underlying mechanism of slow voltage dependence below. Second, memantine exhibited slightly faster voltage-dependent blocker re-equilibration as a result of a more prominent fast component. We explored the physiologic and pathophysiologic relevance of this difference in subsequent studies.
Low Channel Gating Efficacy Limits Time Course of Blocker Re-Equilibration.
Prior work examining voltage-dependent channel blockers has found that the time course of re-equilibration in response to voltage pulses can provide direct estimates of drug binding and unbinding rate constants (Adams, 1977; Jin et al., 2009). However, the slow re-equilibration we observed yields kon and koff values for memantine and ketamine that are inconsistent with those derived by other methods (Blanpied et al., 1997; Gilling et al., 2009). NMDA receptor channels have a low gating efficacy at full agonist occupancy with peak open probability (Popen) 0.04–0.35, depending on experimental conditions and subunit composition (Rosenmund et al., 1995; Chen et al., 1999). In contrast, nicotinic and glycine receptors possess peak Popen near 0.9 (Jin et al., 2009). Because channel opening is required for blocker binding and dissociation (Chen and Lipton, 1997), we reasoned that the slow blocker re-equilibration might result from this low Popen rather than slow drug binding/dissociation kinetics. A low gating efficacy might limit the rate of memantine and ketamine re-equilibration after a step perturbation of voltage.
To test this possibility, we simulated a voltage-pulse protocol. We used a kinetic scheme from previous work (Blanpied et al., 1997) and previously established rate constants (Fig. 6A). The scheme accommodates previous observations of low agonist efficacy and gating of the blocked channel (Blanpied et al., 1997) but neglects desensitization and multiple open and closed states (Popescu and Auerbach, 2003). Although simplified, the model has fewer free parameters than more complex models, and it has proven useful in prior analyses. Thus, it represents a useful heuristic tool for our subsequent experimental tests. Simulation in the absence of blocker resulted in a Popen of 0.03, similar to experimentally derived values for hippocampal neurons (Rosenmund et al., 1995). Because NMDAR channel gating exhibits only weak inherent voltage sensitivity (Jahr and Stevens, 1987; Clarke and Johnson, 2008), voltage dependence of gating was not incorporated into the model, and Popen remained constant at both −70 and +50 mV, but the change in driving force was simulated (Fig. 6B, black trace).
Fig. 6.

Simulations demonstrate that low Popen slows voltage-dependent re-equilibration of blocker. (A) Kinetic scheme used in the simulations. Rate constants were adapted from Blanpied et al. (1997) and were ka+ = 2 µM−1 s−1, ka-= 40 sec−1, β = 5.2 sec−1, α = 130 sec−1, kon = 14.9 µM−1 s−1, koff = 7.6 sec−1, β′ = 0.3 sec−1, α′ = 35.4 sec−1, ka+′ = 0.2 µM−1 s−1, ka-′ = 0.02 sec−1, [NMDA] = 300 µM, [blocker] = 10 µM. A2R* and A2R*B represent the open channel in its unblocked and blocked state, respectively. (B) Output of the simulation replicating a voltage jump in the presence (gray) and absence (black) of a voltage-dependent open-channel blocker of the NMDAR. Voltage dependence of the open channel blocker was achieved by decreasing koff by e-fold per 31.5 mV (Kotermanski and Johnson, 2009). Colored traces are as depicted in the legend. See Materials and Methods for more details of the simulation. Simulation output (representing channels in the A2R* state) was normalized to initial NMDA-only amplitude (3.8% of receptors for the black, gray, and blue traces; 28.6% for the red trace). Changing blocker dissociation (blue) reduced steady-state block but did not appreciably alter the re-equilibration kinetics at +50 mV. By contrast, accelerating channel opening (red) sped re-equilibration kinetics during wash on/off at −70 mV and after the pulse to +50 mV. (C) Calculated rate constants derived from tau of re-equilibration at positive potentials at varying efficacies, plotted as a function of Popen (altered by incrementing β and β′ in10-fold steps). Simulation output (black line) is compared with simulation input (gray line). Popen needed to approach 1 (0.97) to retrieve values of the same order of magnitude of simulation input. Calculated rate constants for memantine from our data in Fig. 5 are also plotted as red lines. Horizontal lines are for reference purposes and are not a function of Popen.
We simulated the effect of 10 µM blocker with binding properties given in Fig. 6B legend (gray trace). The voltage dependence of the blocker was simulated by altering its koff value e-fold per 31.5 mV (Kotermanski and Johnson, 2009). Inhibition at −70 mV was relieved after a pulse to +50 mV with a single-exponential τ of 1.4 seconds. To test the effect of gating efficacy on re-equilibration, we altered efficacy by increasing β and β′ 10-fold. This manipulation caused the relaxation time constant during a step to +50 mV to decrease approximately 10-fold to 163 milliseconds (Fig. 6B, red trace, and C). A further increase in efficacy (1000-fold increase in opening rate) yielded a Popen > 0.9, and the resulting relaxation time constant was consistent with drug binding kinetics [Fig. 6C, τ = 0.002 second, equivalent to 1/(kon + koff)]. Although performed with a simplified gating scheme, these simulations demonstrate in principle that inefficient gating rate limits the re-equilibration of drug. Previous investigations have verified the assumptions of high Popen for voltage-pulse relaxation analysis of other channels (Adams, 1977; Jin et al., 2009), but binding and dissociation rate constants for low Popen NMDARs cannot be directly derived from whole-cell, voltage-pulse relaxations.
Another possible explanation for the slow time course of unblock could be slow dissociation kinetics of the drugs. We simulated the effect of altering koff for the blocker 10-fold, which resulted in substantially less steady-state block but little change in the time course of re-equilibration at positive voltages (Fig. 6B, blue trace). We conclude, therefore, that drug unbinding kinetics likely does not explain the slow time constants.
Experimentally Increasing Gating Efficacy Speeds Blocker Re-Equilibration.
Our modeling predicts that increasing Popen should speed re-equilibration of channel blockers at positive potentials. To test this in our neuronal population, we manipulated levels of extracellular glycine. As a necessary coagonist, glycine levels control NMDAR gating. We found that decreasing bath glycine levels from 10 to 0.2 μM significantly slowed memantine re-equilibration (10 μM versus 0.2 μM glycine: weighted τ 0.97 ± 0.15 seconds versus 2.8 ± 0.3 seconds; n = 8, P < 0.05).
To test this further, we took advantage of the fact that diheteromeric GluN1/GluN2A NMDARs have higher efficacy than diheterometic GluN1/GluN2B receptors (Chen et al., 1999). These subunits are present in nearly all NMDARs in cultured hippocampal neurons (Tovar and Westbrook, 1999). We heterologously expressed GluN2A or GluN2B, along with GluN1, subunits in HEK293 cells and measured the time course of memantine re-equilibration at +50 mV in the presence of 300 µM NMDA. As expected, if gating kinetics rate limits blocker behavior, we found that memantine re-equilibration at positive potentials was significantly faster in NMDARs containing GluN2A subunits (468 ± 67 millseconds, weighted τ) than in those containing GluN2B subunits (709.3 ± 94.4 millseconds weighted τ; P < 0.05, Student’s t test, n = 20–22) (Fig. 7, A and B). Ketamine retained its property of slightly slowed voltage-dependent re-equilibration compared with memantine at both GluN2 isoforms (770 ± 146 milliseconds for GluN2A, 1727 ± 334 millseconds for GluN2B; P < 0.05 at each). Steady-state inhibition for memantine at −70 mV of GluN2A-containing versus GluN2B-containing receptors was 93% ± 1% versus 98 ± 1% (P < 0.05, n = 20–22). The slight difference we found is consistent with previous work showing that memantine is weakly selective for GluN2B receptors with an IC50 ∼2-fold greater for GluN2A over GluN2B populations (Kotermanski and Johnson, 2009). When memantine and ketamine were compared on the same GluN2A- or GluN2B-bearing cell, both drugs displayed a weak preference for GluN2B-containing receptors (GluN2A versus GluN2B: memantine: 90% ± 2% versus 97% ± 2%; P < 0.05, and ketamine: 92% ± 2% versus 98% ± 2%; P < 0.05, n = 7), once again highlighting very similar effects of the two channel blockers.
Fig. 7.

Experimentally increasing Popen speeds re-equilibration. (A and B) GluN2A-containing NMDA receptors exhibit faster re-equilibration than GluN2B-containing receptors. (C1 and D1) 10 mM Tricine potentiates NMDA currents in GluN1/GluN2A-transfected HEK cells but not GluN1/GluN2B cells. (C2 and D2) Tricine speeds re-equilibration of memantine at positive voltages in GluN2A-containing but not GluN2B-containing receptors. See Results for quantification.
As a more direct and sensitive test of the hypothesis that gating efficacy dictates the slow re-equilibration, we took advantage of selective inhibition of GluN2A-containing receptors by low (nanomolar) concentrations of ambient zinc, which decreases open probability of GluN2A receptors, likely by affecting gating efficacy (Paoletti et al., 1997; Erreger and Traynelis, 2008; Gielen et al., 2009; Amico-Ruvio et al., 2011). We buffered ambient zinc in our extracellular bath solution using 10 mM tricine to relieve the effects of contaminating Zn2+ and found that NMDA responses from GluN2A, but not GluN2B, currents were significantly potentiated (71 ± 13% increase in GluN2A, n = 8, P < 0.05 versus 9% ± 2%; n = 8, P > 0.05 in GluN2B; Fig. 6, C1 and D1). Correspondingly, we found acceleration of memantine re-equilibration time course at +50 mV in the presence of tricine in GluN2A receptors (487 ± 134 milliseconds in the absence of tricine versus 135 ± 26 milliseconds in the presence of tricine, n = 8, P < 0.5; Fig. 7, C2 and D2). Blocker re-equilibration at GluN2B NMDARs was not significantly affected by tricine (545 ± 61 ms in the absence of tricine versus 630 ± 135 milliseconds in the presence of tricine, n = 8; Fig. 7, C2 and D2). Taken together, our data strongly suggest that the low Popen of the fully liganded NMDAR rate limits blocker re-equilibration, effectively trapping memantine and ketamine in the channel despite high agonist concentration.
Memantine and Ketamine Show Little Unblocking during Mild-to-Moderate Artificial EPSPs.
We found that memantine exhibited a stronger fast component of relaxation (62% versus 48%) after a pulse to +50 mV compared with ketamine (Fig. 5). Because both blockers re-equilibrate slowly after strong depolarization, it is unclear what impact this subtle drug difference would have during EPSPs. Synaptic depolarizations are briefer and smaller than the voltage pulses to +50 mV and therefore may not elicit the drug difference observed in Fig. 5. To explore the size and duration of EPSPs necessary to elicit a difference between drug actions, we designed a voltage-command waveform that mimicked EPSPs of different amplitude and duration (see Materials and Methods). By examining current responses in the presence of 300 µM NMDA alone and in the presence of NMDA plus 2 µM memantine or ketamine, we created inhibition plots analogous to those in Figs. 5 and 7. We found that at mild to moderate depolarizations (up to a peak voltage of −55 mV), memantine and ketamine displayed little unblock and acted very similarly during short-duration αEPSPs (Fig. 8C1). Memantine unblock diverged strongly from that of ketamine only with strong, prolonged αEPSPs (compare Fig. 8, C1 and C2). These data suggest that weak synaptic activity is insufficient to relieve synaptic memantine or ketamine block. However, stronger activity reveals drug differences consistent with observations from voltage-pulse experiments. It is possible that the prolonged strong αEPSPs in this experiment mimic the effects of high-frequency nervous system activity. If so, we might expect that spontaneous network activity or high-frequency stimulation used to induce LTP might be differentially affected by memantine and ketamine. These ideas were tested in subsequent experiments.
Fig. 8.

Differences in voltage dependence are detectable only with large, broad depolarizations. (A1 and A2) Hippocampal neurons were voltage clamped using an αEPSP voltage command waveform of short (A1, τ = 30 milliseconds) or long (A2, τ = 300 milliseconds) duration. Cells were bathed in saline, NMDA alone (300 μM), then memantine (2 μM) plus NMDA. In each condition, from a Vm of –90 mV, consecutive sweeps simulated EPSPs of increasing amplitude (Vm at maximum: −72.5, −55, −37.5,−20 mV). Leak currents in saline were subtracted from each experimental condition. Traces represent percent inhibition of the NMDA-only current during the smallest depolarization (–72.5 mV, dark red) and largest (−20 mV, bright red), so upward excursion of the traces represents relief from drug-induced inhibition. (B1 and B2) Same protocol for ketamine (2 μM) (traces, dark green: −72.5 mV, bright green: −20 mV). (C1 and C2) Peak block at each potential for each EPSP duration for memantine and ketamine. Colored circles correspond to the conditions represented by the traces in A and B. No significant interaction was found between drug and voltage with the brief αEPSPs [C1, two-way analysis of variance (ANOVA)]. However a significant interaction emerged with prolonged αEPSPs, with memantine exhibiting more unblock than ketamine on strong depolarization (C2, *P < 0.05, drug by voltage interaction, two-way ANOVA, n = 5 to 6). n.s., not significant.
Memantine and Ketamine Suppress Network Activity Similarly but Diverge in Neuroprotective Ability with Extreme Depolarization.
Our αEPSP data suggest that memantine may more effectively escape the NMDAR channel during strong depolarization than does ketamine, but it is unclear whether physiologic activity achieves sufficiently prolonged, strong depolarization to reveal a drug difference, even with temporal summation of EPSPs and associated spiking. Furthermore, continuous agonist application inherent in the design of Fig. 8 may have facilitated drug dissociation during depolarization. To test directly whether the subtle difference in voltage dependence, apparent under the controlled conditions of Fig. 8, is detectable under conditions of physiologically transient depolarization and agonist presentation, we took a two-pronged approach. We examined the network effects of the channel blockers using both single-cell recording and network activity analysis using multielectrode arrays. First, using whole-cell recording in hippocampal cultures, we measured AMPAR-mediated, network-driven EPSCs as a measure of network activity (Fig. 9, A and B). Although NMDAR function was inhibited in the recorded cell by bath Mg2+ and voltage clamp to −70 mV, NMDARs in the surrounding network were free to contribute to spontaneous activity. We found that a low concentration (2 μM) of memantine and ketamine had negligible effects on network activity and did not differ from each other (Fig. 9B). In part, the weak effects could be due to the presence of physiologic Mg2+ and glycine concentrations (1 mM and 1 μM, respectively). Given that the weak effects of the antagonists may render differences indiscernible, we increased the antagonist concentration to 10 µM. At this concentration, both drugs suppressed network activity by ∼30% (memantine: 25% ± 6%, ketamine: 32% ± 7%, n = 12; Fig. 9B), but again we could detect no differences between the drugs (P > 0.05 paired t test; Fig. 9B).
As a second approach, we measured network activity using MEAs to monitor spiking across the network. MEA recordings permitted us to examine network synchrony and spatial relationships not captured by single-cell recordings. These recordings were performed at 37°C and with divalent cations present to mimic physiologic conditions. We recorded network activity in the presence of 10 µM memantine or ketamine relative to baseline activity recorded before drug application and after drug washout (Fig. 9C). Both drugs strongly suppressed network activity as measured by array-wide spike detection rate and measures of bursting (Fig. 9D). Effects of the two drugs covaried indistinguishably across all measured parameters.
To explore effects in more intact networks, we examined the effects of ketamine and memantine on synaptic plasticity in hippocampal slices. At concentrations up to 10 µM, neither drug acutely affected the induction of LTP elicited by 100 Hz stimulation for 1 second (Fig. 10A). On the other hand, both drugs at concentrations of 1 and 10 µM inhibited LTD induction, evoked by prolonged 1-Hz low-frequency stimulation (Fig. 10B). The difference in effects between the two forms of NMDAR-induced plasticity could result from differences in stimulation parameters or other factors (see Discussion). Regardless, these results extend the profile of similar pharmacodynamics between the two drugs to well-established protocols of plasticity induction and highlight another nonsteady-state condition (repetitive stimulation) in which the drugs behave quite similarly.
Fig. 10.

Memantine and ketamine’s effects on LTP and LTD induction. (A) LTP induction [100 Hz for 1 second, arrow, high-frequency stimulus (HFS)] was unaffected by 10 µM memantine or ketamine (preapplied for 15 minutes as indicated). (B) LTD induction (1 Hz for 15 minutes white bar) was blocked by both drugs (10 µM, preapplied for 15 minutes). Drugs were administered for the durations shown by the black bars. LTP and LTD in control slices are shown by black circles (A) and white triangles (B). Six slices were tested for each experimental condition in A and B. Insets show representative fEPSP waveforms.
Although the biophysical differences between memantine and ketamine were not evident during three assays of physiologic and stimulated activity, extreme conditions that evoke prolonged NMDAR activation might reveal differences between drugs as predicted by Fig. 8. To test this, we challenged cultured hippocampal neurons with OGD and evaluated the neuroprotective effects of memantine and ketamine. In this system, synaptic NMDARs play a dominant role in mediating excitotoxicity (Wroge et al., 2012). At the same concentration (10 µM) used for network studies, ketamine was significantly more neuroprotective than memantine was (Fig. 11). We conclude that under extreme conditions of pathophysiologic NMDAR activation, a pharmacodynamic difference between memantine and ketamine appears.
Fig. 11.
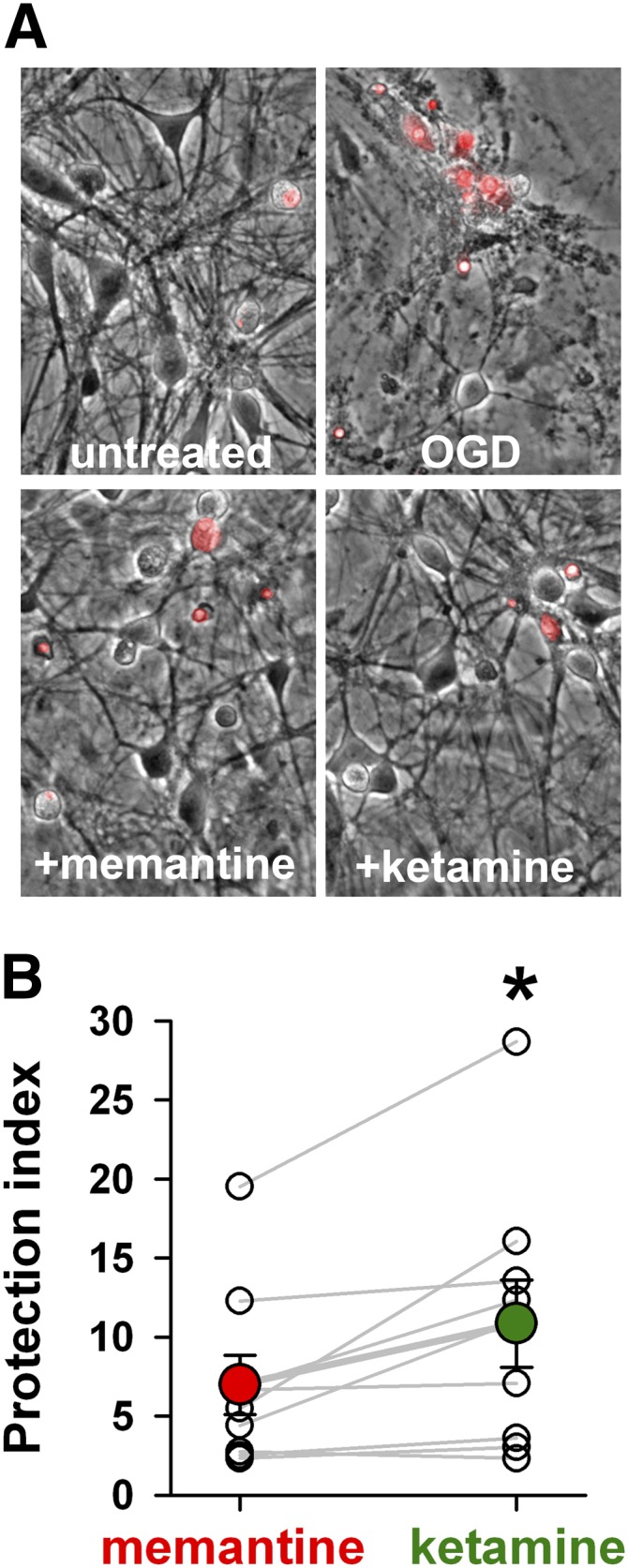
Ketamine promotes stronger neuroprotection than memantine against OGD. (A) Representative fields from DIV13-15 hippocampal cultures exposed to 2.5 hours OGD, allowed to recover for 24 hours, then assayed with propidium iodide (red, 3 μM) to stain nuclei of compromised neurons. Propidium iodide image is superimposed on a phase contrast image. Drugs were evaluated at 10 µM and were present during OGD only. (B) Protection index of memantine and ketamine was calculated by normalizing survival in all OGD conditions to survival of an untreated sibling dish and plotting drug effects relative to OGD alone. Open circles and gray lines indicate the results of individual experiments using sibling cultures treated at the same time (n = 9). Colored circles are averages across all experiments. Ketamine neuroprotection was significantly greater than that with memantine (*P = 0.01, paired t test).
Discussion
Memantine and ketamine exhibit remarkably different clinical properties despite similar activity as NMDAR activation-dependent channel blockers. Our results suggest only slight pharmacodynamic differences in voltage dependence between drugs, even under nonsteady-state conditions, that do not become relevant until cells are challenged with pathophysiologic depolarization and NMDAR activation (OGD). Thus, pharmacokinetic and dosing differences may be more likely than pharmacodynamic differences to explain clinical differences (but see Kotermanski et al., 2013).
We aimed to determine whether the two drugs differ in nonequilibrium synaptic actions, which have not been explored in detail previously. Similarities in steady-state block may belie kinetic differences that could become more evident during non–steady-state conditions of physiologic activity. By analogy, the well-known calcium chelators EGTA and BAPTA [1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid] have similar Kd values but differ 100-fold in their kon and koff values. Differences between these buffers are not easily appreciated under equilibrium conditions. However, the faster buffer BAPTA much more effectively depresses synaptic vesicle release during nonequilibrium, depolarization-elicited Ca2+ influx (Rozov et al., 2001). In the case of memantine and ketamine, by contrast, steady-state similarities do not mask non–steady-state dissimilarities.
One pharmacodynamic difference may be a selective inhibition by memantine of extrasynaptic receptors (Okamoto et al., 2009; Xia et al., 2010). However, we found that memantine and ketamine have indistinguishable actions even at extrasynaptic NMDARs. Previous experiments arguing for a selective effect of memantine on extrasynaptic receptors compared memantine with the slowly dissociating open-channel blocker MK-801 (Xia et al., 2010), but our results demonstrate that memantine holds no extrasynaptic preference over ketamine. Any preference for extrasynaptic receptors is likely to result from the slow, sustained agonist presentation of ambient neurotransmitter at extrasynaptic receptors, rather than drug preference for certain receptor populations (Wroge et al., 2012).
A second hypothesis to explain memantine’s therapeutic profile involves its rapid kinetics coupled with its strong voltage dependence. These properties are proposed to allow memantine to dissociate rapidly during transient synaptic depolarization and preserve synaptic transmission (Parsons et al., 1993; Frankiewicz et al., 1996; Lipton, 2005). During sudden voltage changes, contrary to our expectations and previous results (Frankiewicz et al., 1996), we found that memantine and ketamine both equilibrated slowly, suggesting little relief from block during synaptic depolarization and agonist presentation.
Our conclusion that the efficiency of channel gating affects the time course of relief from block during depolarization has implications for prior work investigating blocker kinetics. Past estimates of memantine binding kinetics differ by several orders of magnitude across different studies (Blanpied et al., 1997; Gilling et al., 2009). Our results provide a partial resolution. Studies using voltage pulses (Frankiewicz et al., 1996) or offset currents after drug washout (Gilling et al., 2009) will be susceptible to errors in kon/koff estimates resulting from low gating efficacy. Some studies of other cell types have found that memantine re-equilibrates at positive potentials much faster than we observed (Frankiewicz et al., 1996). Although we do not have a complete explanation for this discrepancy, one possibility is that the receptors from different neuronal populations used in previous work exhibited higher efficacy, yielding faster apparent drug re-equilibration (Fig. 6).
Despite slow overall kinetics, memantine yielded faster voltage-dependent relief from block than ketamine and more steady-state relief from block with strong depolarization. What is the basis for this difference between drugs? We suggest that it may reflect differences in channel gating while drug is bound. Although the model that we used does not capture many complexities of NMDAR channel gating (Popescu and Auerbach, 2003), it allowed us to test whether differences in gating can in principle explain experimentally observed drug differences. We used the baseline blocking parameters from Fig. 6 to represent ketamine-like block. From this baseline, we attempted to recapitulate features of memantine block by doubling β′ (opening rate of blocker-bound NMDAR). We first doubled α′ to maintain identical steady-state block at −70. This led to a speeding of time course of re-equilibration but no change in steady-state block at +50 mV (not shown). However, if α′ was increased by somewhat less, the simulated steady-state block at −70 mV was still very similar to baseline (likely experimentally indistinguishable; Fig. 12), the time course of re-equilibration was accelerated, and the apparently stronger steady-state voltage dependence of memantine was recapitulated (Fig. 12). Thus, by altering channel opening and closing with blocker bound, the two most important differences between memantine and ketamine in our experiments were captured. Note that the apparent voltage-dependent drug differences were mimicked without adjusting the microscopic voltage dependence (e-fold per 31.5 mV for both drugs) or microscopic kinetics of drug binding. Although this explanation for drug differences remains hypothetical, it is attractive in its simplicity and its reliance on the previously proposed nature of blocker action (Blanpied et al., 2005).
Fig. 12.
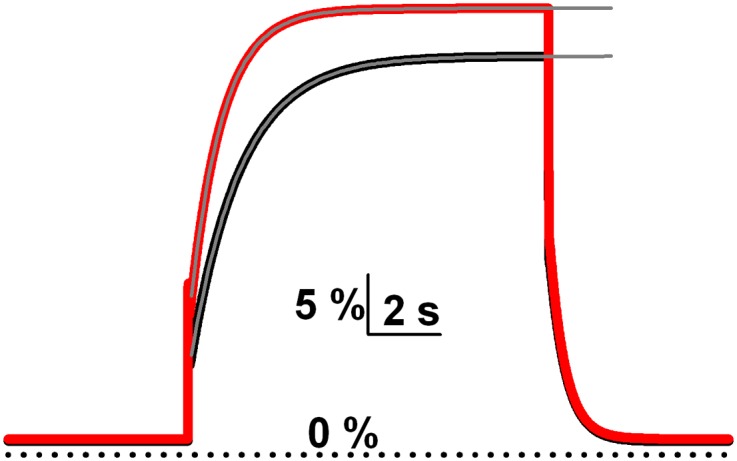
Altering receptor gating while blocker is bound recapitulates experimentally observed differences between memantine and ketamine. Simulation output depicted as percent NMDA response in the presence of blocker using the kinetic scheme in Fig. 6 (0% at dotted line represents full block) for baseline (black, β′: 0.3 sec−1, α′: 35.4 sec−1, same as Fig. 6) and adjusted rate constants (red, β′: 0.6 sec−1, α′: 55.4 sec−1). Adjusted kinetic values were reached by doubling β′ and then manipulating α′ to yield similar steady-state block at −70 mV. Faster kinetics of gating in the blocked states resulted in a faster time course of re-equilibration (gray lines represent exponential fits; τ = 883 milliseconds for red 1393 milliseconds for black) and increased apparent steady-state voltage dependence, similar to the experimental difference between memantine (faster, more voltage dependence) and ketamine (slower, weaker voltage dependence).
This difference in voltage dependence of the drugs was the sole difference detected, and we investigated its potential physiologic significance with four experiments: artificial EPSPs, two measures of network activity, and plasticity induction. These results suggest that acute effects of the drugs are indistinguishable acting on neuronal circuits in dissociated culture and brain slices. This conclusion holds for a wide range of temperatures (22°C for spontaneous EPSCs, 30°C for slices, and 37°C for MEA recordings) and under conditions in which divalent cation concentrations were near physiologic 1 to 2 mM (all studies of physiologic activity and toxicity). Thus, although we might expect voltage-dependent differences between memantine and ketamine to accumulate during spontaneous or evoked high-frequency activity, differences were not evident in our assays of network spiking or plasticity induction.
The differential effects on LTD versus LTP of both drugs could have several explanations. Differential effects on LTD could reflect selective inhibition of extrasynaptic NMDARs (Xia et al., 2010), which have been preferentially associated with LTD (Papouin et al., 2012). However, our previous work and results herein suggest little selectivity for extrasynaptic versus synaptic receptors in hippocampal preparations when agonist presentation is held constant (Wroge et al., 2012; but see Wild et al., 2013). LTD also appears to preferentially involve GluN2B-containing receptors (Liu et al., 2004; Izumi et al., 2005, 2006). The slight preference of both ketamine and memantine for GluN2B-containing receptors (Fig. 7) could therefore participate in the differential block of LTD. Another contributor could be differences in induction protocols themselves (900 pulses over 900 seconds versus 100 pulses over 1 second). LTD induction is associated with less postsynaptic charge transfer than LTP induction (Dudek and Bear, 1992; Bear, 1995). This would seem to favor block during LTP induction. However, NMDAR responses during LTD are associated with less overall membrane depolarization, a condition favoring block. Tonic receptor activation (Sah et al., 1989; Povysheva and Johnson, 2012) and associated accumulated block may also be greater during the longer blocker incubation associated with LTD. Finally, LTP inhibition may require a greater fractional block of receptors than does LTD inhibition. Previous work showed that 4–6 hours of memantine incubation is required to inhibit LTP, with an IC50 of ∼12 µM (Frankiewicz et al., 1996). It is unclear why such a long incubation should be required, as much briefer incubation was sufficient in our studies to block LTD, and blocker effects on spontaneous activity in MEA recordings inevitably equilibrated by <15 minutes. It is possible that channel block achieved early in incubation in the Frankiewicz et al. (1996) study resulted in complicated downstream signaling cascades, similar to the effects seen with ketamine (Autry et al., 2011) and other NMDAR antagonists (Zorumski and Izumi, 2012). Regardless of the multifactorial nature of the differential effects of blockers on plasticity, the indistinguishable actions of the drugs during plasticity induction emphasize their remarkable similarity under varied conditions.
Despite statistical indistinguishability in most assays, ketamine proved more neuroprotective than did memantine against OGD damage. We previously showed that during in vitro OGD under our conditions, damage is mainly NMDAR-mediated (Hogins et al., 2011; Wroge et al., 2012). In the present experiments, we predicted that the sustained depolarization and receptor activation produced by OGD would reveal the difference in voltage dependence predicted from experiments described in Figs. 5 and 8. Indeed, memantine proved slightly less neuroprotective, presumably because of the slightly faster and more complete block relief with depolarization. The result is ironic since memantine is the clinically indicated neuroprotectant. If memantine is the better clinical neuroprotectant, off-target effects or other in vivo factors may explain the discrepancy with our results.
In summary, we find that the clinically important drugs memantine and ketamine are largely pharmacologically indistinguishable under both steady-state and nonequilibrium conditions. A slight difference in voltage dependence is the sole characteristic that distinguishes the drugs. Only under severe depolarizing conditions of OGD was this biophysical difference revealed as a neuroprotective difference between drugs.
Acknowledgments
The authors thank Ann Benz for technical help with the cultures, Hong-Jin Shu and Andy Linsenbardt for help with HEK cell transfections, and laboratory members for critical discussions. The authors also thank Drs. Elias Aizenman and Jon Johnson (University of Pittsburgh) for the kind gifts of GluN subunits.
Abbreviations
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid
- d-APV
d-2-amino-5-phosphonovalerate
- EPSC
excitatory postsynaptic current
- EPSP
excitatory postsynaptic potential
- αEPSP
artificial EPSP
- HEK293
human embryonic kidney cell line 293
- LTD
long-term depression
- LTP
long-term potentiation
- MEA
multielectrode array
- MK-801
(5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate
- NBQX
2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline
- NMDA
N-methyl-d-aspartate
- NMDAR
NMDA receptor
- OGD
oxygen glucose deprivation
- Popen
open probability
Authorship Contributions
Participated in research design: Emnett, Eisenman, Izumi, Zorumski Mennerick.
Conducted experiments: Emnett, Eisenman, Taylor, Izumi, Mennerick.
Performed data analysis: Emnett, Eisenman, Taylor, Izumi, Mennerick.
Wrote or contributed to the writing of the manuscript: Emnett, Eisenman, Taylor, Izumi, Zorumski, Mennerick.
Footnotes
This work was supported by the Bantly Foundation; the National Institutes of Health National Institute of General Medical Sciences [Grant T32GM008151]; the National Institutes of Health National Institute of Mental Health [Grants MH078823 and MH077791]; and the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grant AA01743].
References
- Aan Het Rot M, Zarate CA, Jr, Charney DS, Mathew SJ. (2012) Ketamine for depression: where do we go from here? Biol Psychiatry 72:537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PR. (1977) Voltage jump analysis of procaine action at frog end-plate. J Physiol 268:291–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amico-Ruvio SA, Murthy SE, Smith TP, Popescu GK. (2011) Zinc effects on NMDA receptor gating kinetics. Biophys J 100:1910–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM. (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF. (1995) Mechanism for a sliding synaptic modification threshold. Neuron 15:1–4 [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Richerson GB, Stevens CF. (1990) Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc Natl Acad Sci USA 87:5359–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. (1997) Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol 77:309–323 [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Clarke RJ, Johnson JW. (2005) Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J Neurosci 25:3312–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckman FA, Aizenman E. (1996) Pharmacological properties of acquired excitotoxicity in Chinese hamster ovary cells transfected with N-methyl-D-aspartate receptor subunits. J Pharmacol Exp Ther 279:515–523 [PubMed] [Google Scholar]
- Carnevale NT, Hines ML. (2006) The Neuron Book, Cambridge University Press, Cambridge [Google Scholar]
- Chen HS, Lipton SA. (1997) Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol 499:27–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Luo T, Raymond LA. (1999) Subtype-dependence of NMDA receptor channel open probability. J Neurosci 19:6844–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RJ, Johnson JW. (2008) Voltage-dependent gating of NR1/2B NMDA receptors. J Physiol 586:5727–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. (1992) The time course of glutamate in the synaptic cleft. Science 258:1498–1501 [DOI] [PubMed] [Google Scholar]
- Ditzler K. (1991) Efficacy and tolerability of memantine in patients with dementia syndrome. A double-blind, placebo controlled trial. Arzneimittelforschung 41:773–780 [PubMed] [Google Scholar]
- Dudek SM, Bear MF. (1992) Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci USA 89:4363–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, Traynelis SF. (2008) Zinc inhibition of rat NR1/NR2A N-methyl-D-aspartate receptors. J Physiol 586:763–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankiewicz T, Potier B, Bashir ZI, Collingridge GL, Parsons CG. (1996) Effects of memantine and MK-801 on NMDA-induced currents in cultured neurones and on synaptic transmission and LTP in area CA1 of rat hippocampal slices. Br J Pharmacol 117:689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P. (2009) Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459:703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilling KE, Jatzke C, Hechenberger M, Parsons CG. (2009) Potency, voltage-dependency, agonist concentration-dependency, blocking kinetics and partial untrapping of the uncompetitive N-methyl-D-aspartate (NMDA) channel blocker memantine at human NMDA (GluN1/GluN2A) receptors. Neuropharmacology 56:866–875 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. (2002) Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta 1600:148–153 [DOI] [PubMed] [Google Scholar]
- Hogins J, Crawford DC, Jiang X, Mennerick S. (2011) Presynaptic silencing is an endogenous neuroprotectant during excitotoxic insults. Neurobiol Dis 43:516–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW. (1986) Primary culture of identified neurons from the visual cortex of postnatal rats. J Neurosci 6:3044–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Auberson YP, Zorumski CF. (2006) Zinc modulates bidirectional hippocampal plasticity by effects on NMDA receptors. J Neurosci 26:7181–7188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Nagashima K, Murayama K, Zorumski CF. (2005) Acute effects of ethanol on hippocampal long-term potentiation and long-term depression are mediated by different mechanisms. Neuroscience 136:509–517 [DOI] [PubMed] [Google Scholar]
- Jahr CE, Stevens CF. (1987) Glutamate activates multiple single channel conductances in hippocampal neurons. Nature 325:522–525 [DOI] [PubMed] [Google Scholar]
- Jin X, Covey DF, Steinbach JH. (2009) Kinetic analysis of voltage-dependent potentiation and block of the glycine α 3 receptor by a neuroactive steroid analogue. J Physiol 587:981–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Kotermanski SE. (2006) Mechanism of action of memantine. Curr Opin Pharmacol 6:61–67 [DOI] [PubMed] [Google Scholar]
- Kotermanski SE, Johnson JW. (2009) Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J Neurosci 29:2774–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotermanski SE, Johnson JW, Thiels E. (2013) Comparison of behavioral effects of the NMDA receptor channel blockers memantine and ketamine in rats. Pharmacol Biochem Behav 109:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotermanski SE, Wood JT, Johnson JW. (2009) Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J Physiol 587:4589–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214 [DOI] [PubMed] [Google Scholar]
- Lester RAJ, Clements JD, Westbrook GL, Jahr CE. (1990) Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature 346:565–567 [DOI] [PubMed] [Google Scholar]
- Lipton SA. (2005) The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res 2:155–165 [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304:1021–1024 [DOI] [PubMed] [Google Scholar]
- Mennerick S, Chisari M, Shu HJ, Taylor A, Vasek M, Eisenman LN, Zorumski CF. (2010) Diverse voltage-sensitive dyes modulate GABAA receptor function. J Neurosci 30:2871–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Que J, Benz A, Zorumski CF. (1995) Passive and synaptic properties of hippocampal neurons grown in microcultures and in mass cultures. J Neurophysiol 73:320–332 [DOI] [PubMed] [Google Scholar]
- Okamoto S-i, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, et al. (2009) Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 15:1407–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orser BA, Pennefather PS, MacDonald JF. (1997) Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology 86:903–917 [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J. (1997) High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci 17:5711–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. (2012) Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150:633–646 [DOI] [PubMed] [Google Scholar]
- Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. (1993) Patch clamp studies on the kinetics and selectivity of N-methyl-D-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan). Neuropharmacology 32:1337–1350 [DOI] [PubMed] [Google Scholar]
- Popescu G, Auerbach A. (2003) Modal gating of NMDA receptors and the shape of their synaptic response. Nat Neurosci 6:476–483 [DOI] [PubMed] [Google Scholar]
- Povysheva NV, Johnson JW. (2012) Tonic NMDA receptor-mediated current in prefrontal cortical pyramidal cells and fast-spiking interneurons. J Neurophysiol 107:2232–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Feltz A, Westbrook GL. (1995) Synaptic NMDA receptor channels have a low open probability. J Neurosci 15:2788–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. (2001) Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol 531:807–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA. (1989) Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246:815–818 [DOI] [PubMed] [Google Scholar]
- Tokuda K, O’Dell KA, Izumi Y, Zorumski CF. (2010) Midazolam inhibits hippocampal long-term potentiation and learning through dual central and peripheral benzodiazepine receptor activation and neurosteroidogenesis. J Neurosci 30:16788–16795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Jahr CE. (1994) Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron 12:51–59 [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 19:4180–4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Pine J, Potter SM. (2006) An extremely rich repertoire of bursting patterns during the development of cortical cultures. BMC Neurosci 7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild AR, Akyol E, Brothwell SL, Kimkool P, Skepper JN, Gibb AJ, Jones S. (2013) Memantine block depends on agonist presentation at the NMDA receptor in substantia nigra pars compacta dopamine neurones. Neuropharmacology 73:138–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroge CM, Hogins J, Eisenman L, Mennerick S. (2012) Synaptic NMDA receptors mediate hypoxic excitotoxic death. J Neurosci 32:6732–6742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. (2010) Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci 30:11246–11250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. (2006) A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856–864 [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Izumi Y. (2012) NMDA receptors and metaplasticity: mechanisms and possible roles in neuropsychiatric disorders. Neurosci Biobehav Rev 36:989–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorumski CF, Yang J, Fischbach GD. (1989) Calcium-dependent, slow desensitization distinguishes different types of glutamate receptors. Cell Mol Neurobiol 9:95–104 [DOI] [PMC free article] [PubMed] [Google Scholar]