

Abstract
The Patient-Reported Outcomes Safety Event Reporting (PROSPER) Consortium was convened to improve safety reporting by better incorporating the perspective of the patient. PROSPER comprises industry, regulatory authority, academic, private sector and patient representatives who are interested in the area of patient-reported outcomes of adverse events (PRO-AEs). It has developed guidance on PRO-AE data, including the benefits of wider use and approaches for data capture and analysis. Patient-reported outcomes (PROs) encompass the full range of self-reporting, rather than only patient reports collected by clinicians using validated instruments. In recent years, PROs have become increasingly important across the spectrum of healthcare and life sciences. Patient-centred models of care are integrating shared decision making and PROs at the point of care; comparative effectiveness research seeks to include patients as participatory stakeholders; and industry is expanding its involvement with patients and patient groups as part of the drug development process and safety monitoring. Additionally, recent pharmacovigilance legislation from regulatory authorities in the EU and the USA calls for the inclusion of patient-reported information in benefit–risk assessment of pharmaceutical products. For patients, technological advancements have made it easier to be an active participant in one’s healthcare. Simplified internet search capabilities, electronic and personal health records, digital mobile devices, and PRO-enabled patient online communities are just a few examples of tools that allow patients to gain increased knowledge about conditions, symptoms, treatment options and side effects. Despite these changes and increased attention on the perceived value of PROs, their full potential has yet to be realised in pharmacovigilance. Current safety reporting and risk assessment processes remain heavily dependent on healthcare professionals, though there are known limitations such as under-reporting and discordant perspectives between patient reports and clinician perceptions of adverse outcomes. PROSPER seeks to support the wider use of PRO-AEs. The scope of this guidance document, which was completed between July 2011 and March 2013, considered a host of domains related to PRO-AEs, including definitions and suitable taxonomies, the range of datasets that could be used, data collection mechanisms, and suitable analytical methodologies. PROSPER offers an innovative framework to differentiate patient populations. This framework considers populations that are prespecified (such as those in clinical trials, prospective observational studies and some registries) and non-prespecified populations (such as those in claims databases, PRO-enabled online patient networks, and social websites in general). While the main focus of this guidance is on post-approval PRO-AEs from both prespecified and non-prespecified population groups, PROSPER has also considered pre-approval, prespecified populations. The ultimate aim of this guidance is to ensure that the patient ‘voice’ and perspective feed appropriately into collection of safety data. The guidance also covers a minimum core dataset for use by industry or regulators to structure PRO-AEs (accessible in the online appendix) and how data, once collected, might be evaluated to better inform on the safe and effective use of medicinal products. Structured collection of such patient data can be considered both a means to an end (improving patient safety) as well as an end in itself (expressing the patient viewpoint). The members of the PROSPER Consortium therefore direct this PRO-AE guidance to multiple stakeholders in drug safety, including industry, regulators, prescribers and patients. The use of this document across the entirety of the drug development life cycle will help to better define the benefit–risk profile of new and existing medicines. Because of the clinical relevance of ‘real-world’ data, PROs have the potential to contribute important new knowledge about the benefits and risks of medicinal products, communicated through the voice of the patient.
Electronic supplementary material
The online version of this article (doi:10.1007/s40264-013-0113-z) contains supplementary material, which is available to authorized users.
Introduction
Current methods for safety reporting and risk assessment still rely heavily on healthcare professionals (HCPs). A way to improve the quantity and/or quality of safety information is to encourage patient-reported outcomes of adverse events (PRO-AEs), which are more patient focused and may have less formal data collection processes that do not rely on input from HCPs. Some HCPs such as pharmacists, however, might facilitate PRO-AE collection [1, 2]. HCPs will also retain a critical role in assessing the causality between adverse events (AEs) and drugs, especially for individual cases of severe and fatal AEs.
Because of the varied nature of patient populations (see Fig. 1), a range of different data collection tools, analytical approaches and methodologies may need to be deployed to meet different PRO-AE requirements. A classification based on whether or not the relevant patient population is prespecified (rather than just pre- or post-approval) provides a rational basis for further subdividing the safety populations [3].
Fig. 1.
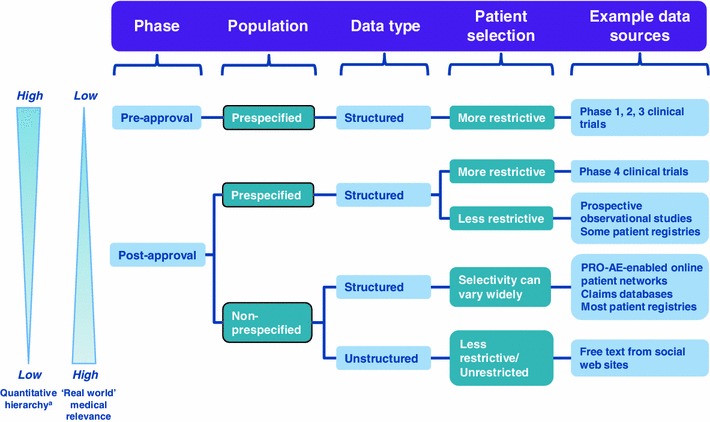
Classification of PRO-AEs. PRO-AE patient-reported outcome of adverse event. a Prespecified populations will have different analytics to non-prespecified populations, with the latter being lower in the quantitative hierarchy. Analytics used lower in this hierarchy can also be utilised for populations higher up
For instance, the dataset is more structured and the patient population is better defined in post-approval, prespecified populations (e.g. phase 4 clinical trials and prospective observational studies) than in post-approval, non-prespecified patient populations. However, even within the non-prespecified group there are variations—for example, some patient support websites use structured data templates to collect PRO-AEs, whereas most websites have only unstructured free text.
There are also distinctions between clinical trials, where the denominator is clear and there are specific goals around balancing safety and efficacy [4, 5], and safety surveillance systems that may have no clear denominator representing the total number of patients. There are also issues around either defining a cohort or opening up reporting to any patient, either voluntary or solicited.
Patient-reported outcome (PRO) data can be captured in a structured way using a suitable instrument, which is a specific reporting tool that through completion generates a structured dataset. This could be a questionnaire or diary along with supporting information such as training materials, scoring, etc.
The amount and nature of missing data, under-reporting and bias are substantially affected by the specific data collection approach taken. In the regulatory context, the approach to collecting PROs for AEs may differ depending on whether the aim is a comparative tolerability claim (i.e. a product having a more favourable tolerability profile compared with another) or just general documentation of AEs.
Tools used to capture PROs have become established in areas such as assessing the quality of care delivered to patients by providers and health-related quality of life (HRQoL) measures used in clinical trials. In these fields, PROs are based on methodologies that have been well validated. In the UK National Health Service (NHS), patient-reported outcome measures (PROMs) have been used to measure patient outcomes for some planned surgical interventions, such as hip and knee replacements, varicose veins and hernia [6], but the ability to collect PRO-AEs within this framework is currently relatively limited.
The use of PRO instruments for capturing AE data is not so well developed. Most progress has been made with the collection of pre-approval PRO-AEs in clinical trials, although there is also significant previous literature on voluntary reporting [3]. Some PRO questionnaires given to patients in trials cover potential adverse effects, and for certain conditions such as HIV, there has been systematic collection of symptoms from patients using checklists. The National Cancer Institute (NCI) in the USA is also developing a methodology for collecting PRO-AEs in oncology trials based on a patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). In the epidemiological context, AE outcome measures can be continuous variables or have binary outcomes.
This guidance will consider if methodologies used to collect pre-approval PRO-AEs could be modified for post-launch studies with prespecified populations. The successful use of PRO tools has suggested to some that monitoring of AEs reported by patients (as well as their care-givers and relatives, where appropriate) outside of the framework of clinical trials should be routinely considered in everyday practice, because it more closely represents the patients’ self-evaluation of HRQoL [7]. However, some signs or symptoms may be due to the disease rather than the drug, and it is important to distinguish these.
The use of PRO-AE instruments outside of clinical trials is also less well implemented. Whilst some countries have previously developed web-based systems to enable consumers to report post-approval suspected adverse reactions (ARs) directly to the regulatory authority [e.g. Food and Drug Administration (FDA) MedWatch, Medicines and Healthcare Products Regulatory Agency (MHRA) Yellow Card scheme, Australia TGA Blue Card scheme, Health Canada Med Effect] in an attempt to address under-reporting by HCPs, this approach has had only limited success in capturing safety data [8, 9]. Recent EU legislation means that there will be increased consumer reporting of this type across member states.
Similarly, there are currently only a few non-regulatory approaches aiming to capture PRO-AE data, including PRO-AE-enabled patient support websites [10]. Whilst popular with patients, these sites are primarily intended for the purposes of patient support and education rather than as PRO-AE instruments. The potential to mine AE data from unstructured text derived from online health forums is feasible, but needs further development before wider utilisation [11].
Patient support programmes (PSPs), which are developed by companies to assist patients and/or HCPs in better managing diseases and treatments, are already subject to AE reporting requirements. PSPs, however, are typically not specifically designed to collect PRO-AE data post launch and may represent missed opportunities to collect improved post-marketing safety data.
There is evidence [7] to suggest that collecting symptom data directly from patients can be beneficial, leading to an improvement in the accuracy and efficiency of symptomatic AE data gathering [12–14]. It is also clear that this is a growth area, particularly in the unregulated social network environment, and monitoring of this data is a concern for health authorities.
However, for industry, patients, academics and regulators to make best use of this information, guidelines are needed. The definitions, standards, methodologies and utility of PRO-AEs set out in this guidance will support and promote the use of PRO-AE instruments across the drug life cycle. This PRO-AE guidance may also be applied to other related situations, for example, the monitoring of medical devices used by patients or potentially the data generated by patient-worn devices (e.g. for the reporting of falls).
Definition of a PRO-AE
It is important to clearly define what is meant by PRO-AEs and to determine whether these are different from other forms of consumer reports. There are established definitions used by the European Medicines Agency (EMA) and the US FDA for PROs in general and for AEs [15–17]. A recommended definition for PRO-AEs based on these resources, and used in this document, is as follows:
A PRO-AE is any untoward medical occurrence, whether or not considered treatment/intervention related, that is reported or transmitted directly by the patient without interpretation by a clinician or anyone else. PRO-AEs may be collected by both structured and unstructured reports.
This definition includes information from patients directly reporting their own experiences (e.g. symptoms) and from patients transmitting data that they have received (e.g. laboratory test results). Patient-reported symptoms may be related to the drug or due to other factors, such as the underlying disease, that represent AEs rather than adverse drug reactions (ADRs). Although it can be argued that patients may not always be able to distinguish between symptoms, signs, safety events and ADRs, this should not matter unduly, provided that AEs and ADRs are distinguished and appropriately interpreted.
There is some debate about whether PROs should be covered by a narrower definition related only to the use of validated instruments, which includes just a subset of all patient self-reported data. For the purposes of this guidance, PROs encompass the full range of self-reporting, as the guidance proposes that an acceptable overarching framework can be created, and that approaches that are currently less validated may actually collect more clinically applicable information as newer analytic methods evolve.
Hence, PRO-AEs (whether structured or unstructured, and prespecified or non-prespecified) include signs and symptoms that are less severe and reflect tolerability, more severe ARs that reflect safety issues, as well as signs and symptoms that may be a precursor of more severe ARs (e.g. febrile angina preceding the onset of aplasia and systemic infection). In addition, abnormal laboratory values that could be a precursor for a potentially severe AR (e.g. neutropenia or liver injury) may be considered as patient-transmitted outcomes. Abnormal biological values may provide useful supporting information, although there could be heterogeneous reporting by patients.
There may be a limitation in adequately reporting some symptoms for which patients may not be aware, e.g. cognitive dysfunction or loss of memory, although in some cases a patient’s relative, carer or HCP may identify these to the patient, who subsequently reports them.
Scope
This guidance covers PRO-AEs from both the pre- and post-approval environments, as shown earlier in Fig. 1. It proposes definitions, methodologies, data collection tools, datasets and analytic approaches for PRO-AE reporting that are rigorous, transferable and consistent.
However, because the methods, tools and analytics for pre-approval PRO-AEs are already relatively well documented [18, 19], the main focus of this guidance will be on post-approval PRO-AEs from both prespecified and non-prespecified populations. As the adoption of PRO-AEs in clinical trials is well advanced, the transferability of knowledge gained in this area will also be examined.
The main purpose for collecting post-approval AE data is to enable companies and regulators to assess the ongoing safety profile of a product, under real-use conditions, using signal detection techniques. However, under-reporting, incomplete data, and reporting bias currently limit the utility of signal detection, and alternative methods of data collection such as PRO-AEs need to be explored.
The goal of analysing PRO-AE data is to add to the knowledge already provided by HCPs, when available, increasing the likelihood and speed of detection of signals. PRO-AEs can also be used complementary to traditional data on efficacy and safety for benefit–risk analysis, risk minimisation evaluation, and communication of safety issues to patients. The collection of symptom-based PROs is also important in many large post-approval studies to better define (in terms of frequency, description and evolution) symptoms that were previously reported in randomised clinical trials.
Analysis of non-prespecified populations gives access to much larger populations than can be achieved even from observational studies, as well as reports and perspectives on symptoms that are important to patients, but which are often not reported to HCPs or remain unreported by patients because of the sensitive nature of those symptoms. Thus, the collection and analysis of PRO-AEs may increase the ability of finding new safety concerns (especially rare safety issues that require large numbers of exposures to detect) and also broadens the types of data collected (events of concern specifically to the patient).
Although open questions may be used to elicit safety information in the latter population, there may be a significant risk of selection and reporting bias. Qualitative analysis can be difficult and might potentially lead to false signals if the population is heterogeneous and all relevant factors are not identified and analysed. Therefore, careful consideration must be given to both the questionnaire design and any subsequent analytics to reflect this (Sects. 5, 6). The same considerations apply to data collected from online patient communities.
Rationale for Wider Use of PRO-AEs
Problems with Current AE Data
Current safety reporting and risk assessment processes for both prespecified and non-prespecified populations are heavily dependent on input from HCPs and introduce a number of limitations, as shown in Table 1, although they still represent the current gold standard.
Table 1.
Problems with current AE data in prespecified and non-prespecified populations
| Subject | Prespecified populations | Non-prespecified populations |
|---|---|---|
| Sensitivity/representative data | Some current methods for detecting AEs in clinical trials lack sensitivity [20] A narrow and relatively small patient population is enrolled into clinical trials. Entries are usually made by the HCP and hence tend to be biased towards that perspective [21] |
Medical records in non-trial populations are often incomplete, possibly due to the patient not informing their HCP of any AEs. Entries are also usually made by the HCP and may differ from the patient perspective [7] |
| Early symptom detection | Symptoms might be identified earlier in the drug development life cycle if PRO-AEs were more commonly used in clinical trials | Labelled ARs tend to be under-reported by HCPs in non-trial situations since they are considered to be expected and therefore of lesser importance [22, 23] HCP are less likely to report non-serious events |
| Clinician detection of AEs | Clinicians can underestimate the importance of patients’ symptoms [24, 25] HCPs may concentrate on symptoms caused by drugs and not focus on capturing the significance of the symptoms to the patient’s holistic experience while using the drug product [7] HCP are more likely to report ARs that are known to be caused by drug effects (e.g. Stevens–Johnson Syndrome) HCP are less likely to report events that have a high background rate |
|
| HCP versus patient perspective | Clinicians may miss patient symptom-related AEs that patient self-reporting frequently captures [24, 25], which can result in preventable AEs Clinicians often miss baseline (pre-treatment) symptoms that patients may be experiencing—therefore these symptoms may subsequently incorrectly be attributed to drug Suboptimal patient-prescriber communication can lead to limited reporting of important safety issues [18] Discordance between patient and prescriber perspectives may exist, so the patient’s perspective might be devalued or overlooked completely [7] |
|
| Compatibility of verbatim reporting terms | Data incompatibilities when symptoms reported by patients are not controlled and mapped to accepted medical terms | Data incompatibilities also arising between PRO-AE data and HCP-recorded data captured in a regulatory system (e.g. MHRA Yellow Card scheme) or by a manufacturer [26] |
AE adverse event, AR adverse reaction, HCP healthcare professional, MHRA Medicines and Healthcare Products Regulatory Agency, PRO-AE patient-reported outcome of adverse event
Despite these shortcomings, there may still be a reluctance to adopt an approach that also includes PRO-AEs because of:
Regulatory constraints;
Concerns about feasibility;
Scepticism regarding the validity and reliability of using patient-reported AE data;
Higher data volumes needing review that might obscure or reduce focus on key safety concerns;
Added administrative requirements and cost;
Limitations of the available questionnaires/methods;
Industry concerns about the potentially high volume of relatively ‘minor’ AEs reported by patients, and the cost and practicality associated with processing them;
Privacy issues, including control of patient information collected and stored electronically.
However, these objections are not insurmountable, as shown in Table 2, and have been successfully dealt with in some clinical PRO safety systems, such as the NCI approach.
Table 2.
Potential solutions to objections against use of PRO-AE instruments
| Objection | Current situation and/or potential solution |
|---|---|
| Perceived regulatory constraints | Pre-approval—no regulatory requirement to prevent patients reporting potential AEs in clinical trials, e.g. the FDA mandates only that sponsors provide safety data during drug development and approval. In the EU Good Clinical Practice and the Clinical Trials Directive, the requirement is for the investigator to report AEs to the sponsor [18, 19] |
| Post-approval—currently, AEs in many regions have to be medically approved (not necessarily by the treating HCP) before they can be submitted as a serious case. There is increasing emphasis on patient reporting of AEs by regulatory authorities. Although EU member states now accept non-medically confirmed consumer reports, their collection and content may be less robust | |
| Concerns about feasibility and reproducibility in design |
Pre-approval—in numerous trials, instruments for outcome reporting by patients have been shown to be a valid and reliable way [18, 19] of collecting data on: • Symptoms • Health-related quality of life • Compliance with a drug regimen • Patient satisfaction with care |
| Post-approval—instruments for outcome reporting by patients have been used successfully to collect the types of data listed above | |
| Higher data volumes needing review that might obscure or reduce focus on key safety concerns; added administrative requirements and cost |
Pre-approval—most of the necessary infrastructure is already in place, since AE reporting is standard in clinical trials Incremental costs can be minimised by using widely available, inexpensive reporting technologies that rely on the internet or patients’ telephones A resource-efficient review process can be conducted, with prioritisation of the more serious safety issues. Regulators can encourage a proportionate risk approach |
| Post-approval—as for pre-approval. Regularly screening by a Marketing Authorisation Holder of internet and digital media under its management or responsibility for potential reports of suspected adverse reactions is required in EMA guidance [27]. Other specific monitoring might be accommodated as organisations already have resources to review relevant websites—all that would be required is additional prioritisation focused on, for example, the main patient support groups | |
| Limitations of the available questionnaires/methods | Pre-approval—there are multiple measures that could be immediately adopted or modified for this purpose, including the NCI PRO-CTCAE initiative [28] |
| Post-approval—the field of instrument development has advanced substantially in recent years, with standards that are now encoded in the FDA guidance on PRO development. The use of smartphone applications (‘apps’) for reporting is already underway in developing countries [29] and allows greater freedom in data capture |
AE adverse event, EMA European Medicines Agency, FDA Food and Drug Administration, HCP healthcare professional, NCI National Cancer Institute, PRO patient-reported outcome, PRO-CTCAE patient-reported outcomes version of the Common Terminology Criteria for Adverse Events
When Could PRO-AE Data Be Collected?
PRO-AE data could potentially be collected at all stages of drug development as well as post launch (see Fig. 2). Currently, the majority of PRO-AEs are collected in phases 2 and 3, often supporting more traditionally collected safety information.
Fig. 2.
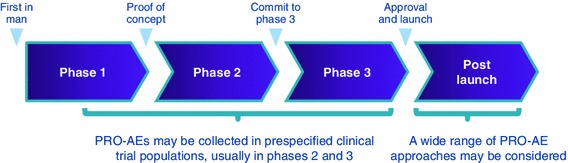
PRO-AEs are applicable at all stages of the medicinal product life cycle. PRO-AE patient-reported outcome of adverse event
A useful framework for subclassifying PROs would be to consider their impact on measuring benefit–risk balance at each stage along the drug development life cycle. For instance, it has been reported that PRO-AE data from pre-approval clinical trials have:
Better described the patient’s underlying health and functional status;
Led to the detection of potentially serious AE symptoms earlier than clinician reporting [19].
Similar advantages to collecting PRO-AEs from post-approval populations might be anticipated. A distinction can be drawn between PRO-AEs in specified populations (either pre- or post-launch, with both utilising similar methodologies) and those in patient populations not prespecified, which demand different methodologies.
Some of the PRO-AE measures that are well documented in the pre-approval space could also be adapted for non-prespecified, post-approval studies. However, issues with the reporting time frame, completeness of data, lack of medical confirmation, and the ability to accurately define the numerator and denominator at risk (i.e. number of subjects experiencing events as a proportion of total number of subjects) mean that different or additional approaches may be more appropriate for the post-approval, non-prespecified group.
Post-approval PRO measurements could therefore be used in:
Screening to detect signals in the general population and in observational cohorts;
Controlled trials for more targeted assessments;
Other PRO-AE-based approaches in post-authorisation studies, for instance, the proactive follow-up of patients after occurrence of an AE of special interest (which may include a medical confirmation of the detected events through the event-treating physician).
Benefits and Challenges of PRO-AE Data
Integrating the patient perspective into drug safety reporting not only improves the accuracy of the data collected but may also enhance the patient-centeredness of clinical research. In fields such as oncology, where symptoms are common and can substantially impair patients’ functioning and quality of life, information about patients’ experiences of AEs is essential for multiple stakeholders, including patients, drug developers, regulators, and payers. The benefits of PRO-AEs are summarised in Table 3.
Table 3.
Benefits of PRO-AEs to different stakeholders
| Stakeholder | PRO-AE benefits |
|---|---|
| Patients | Patients facing a treatment decision wish to know what they can expect in terms of symptoms, based on the prior experiences of a “patient like me.” However, there is a need to avoid patients having access to PRO-AE data from other patients in the pre-approval stage, as this could further exacerbate the placebo effect/bias results from clinical trials In reality, a patient may not have significant input into their treatment decision at the point of prescription. They may seek more information when they get home to know what to expect of their medicine as well as medication preferences |
| Drug developers | Developers want to understand how well patients will tolerate a product. This is particularly relevant with oral therapies for which compliance is strongly associated with symptomatic side effects. PRO-AEs can be useful in early-phase research towards identifying tolerated dose levels and in pivotal trials to compare tolerability between products from the patient perspective |
| Regulators | Regulators have long recognised the limitations of symptomatic adverse event information reported by clinicians in trials. A systematic, patient-reported approach would increase confidence in the fidelity of this information toward balancing risks and benefits, so long as the report was detailed and complete |
| Payers | Payers want to better understand the side effects of specific treatments, because it helps to predict the utilisation of healthcare services and determine the benefit–risk balance (i.e. value) of medicines |
| Healthcare professionals | PRO-AEs can provide the clinician with information of value on a subjective experience, which when combined with the physician perspective based on experience and training provides a more accurate understanding of the patient’s symptoms. The latter is improving the measurement of symptoms in clinical trials and practice |
PRO-AE patient-reported outcome of adverse event
Further advantages of PROs more generally, from a benefit–risk perspective, are shown in Table 4.
Table 4.
Further potential benefits of PROs in general
| Benefits |
|---|
| Pre-approval |
| • PRO end points in late-phase clinical trials support treatment benefit claims that describe a patient’s symptoms or ability to function |
| • Treatment modifications, symptom control and side-effect prevention techniques (risk minimisation) can be evaluated in late-phase clinical trials using PRO data |
| • Healthcare researchers and policymakers can use PRO data from late-phase trials to study the burden of disease on the targeted patient population |
| • While manufacturers and researchers use late-phase clinical trials to corroborate the safety and long-term effectiveness of their drug, incorporating PRO end points could ensure focus on patient-centred healthcare delivery |
| • Better dose finding taking into consideration the patient perspective |
| Post-approval |
| • Ability to be truly ‘patient-centric’—such data can then be used to guide improvements, providing a competitive advantage for a sponsor |
| • Potential ability to assess/evaluate how well risk minimisation activities are working |
| • The combination of drugs with the best benefit–risk profile can often vary among patient populations, so it becomes necessary to conduct the assessment from the patient’s perspective, using PRO measures, which can result in improved compliance |
| • Resource allocation, drug costs, and premium reductions can be made through the extraction of information from post-marketing surveillance data |
| • Valuable real-world safety data may be obtained from alternative sources, through which patients may be more willing to provide data than using standard channels |
| • Patient-reported AEs predict emergency room visits and utilisation of services |
AE adverse event, PRO patient-reported outcome
There are also potential public health downsides to patient adverse outcomes being disseminated without being put into an appropriate benefit–risk context, e.g. causing unnecessary alarm or leading to discontinuation of effective therapies. Safe and effective public communication of benefit–risk should form part of any organised collection of PRO-AEs, in both prespecified and non-prespecified populations.
However, how such communication takes place for unplanned, unstructured, spontaneously posted, web-based PRO-AEs is more challenging. It can be reasonably argued that meaningful analysis of such information offers the opportunity for objective communication of the actual risks, perhaps through regulators or other trusted third parties, thus representing a significant improvement on the current situation.
Other perceived disadvantages of PRO-AEs include costs, the possibility of swamping safety signal with noise, exacerbation of the placebo effect at pre-approval phases, and the possibility that the PRO-AE data is not independent of the AEs collected by physicians in pre- and post-approval phases, although most of these issues can be resolved with appropriate methodology. In fact, the two data sources might be highly correlated and therefore of low added value, although still of confirmatory benefit. However, it is rarely possible to match up patient information on reports in the safety database, usually because limited identifying information is provided for patients from non-standardised data collection schemes. There is a significant risk of having duplicates in the database for cases received from multiple sources.
This guidance does not attempt to discuss the merits of the newer forms of web-based communication of such information compared with the traditional processes, as this issue has a much wider scope than PRO-AEs and transcends the whole topic of better public communication of benefit–risk balance.
Evidence Supporting the Use of PRO-AE Data
There is now considerable evidence from clinical trials that patient and clinician reports of AEs, particularly those based on subjective symptoms during cancer treatment, can provide diverse yet complementary data. For example, it has been shown that including information gathered directly from study participants via PRO measures improved the predictive accuracy of clinician-reported CTCAEs [19, 30]. Although clinicians were able to predict survival well, patient reporting of symptomatic toxicities better reflected the patient’s underlying health state and functional status.
Patient reporting also appears to detect potentially serious AEs or their symptoms earlier than clinician reporting. This is well demonstrated by the timing and cumulative incidence of patient versus clinician reporting of severe diarrhoea in the irinotecan/fluorouracil/leucovorin arm of the NCI intergroup trial N9741 [31], in which many life-threatening gastrointestinal serious AEs were detected [32], as well as subjectively reported AEs in musculoskeletal and central nervous systems.
Evidence suggests that patients are best placed to report their subjective experiences, whereas HCPs contextualise that experience in terms of the disease [19]. Both views are valid and contribute towards improving the understanding of the treatment of AEs if recognised as such by regulators. Industry is thus incentivised to develop mechanisms that allow both datasets to be fully utilised [19].
For drugs already on the market, there is some evidence of increasing consumer activity in the reporting of AEs to regulators. However, the fastest rate of growth in potential consumer-reported AEs appears to be through non-regulatory, PRO-enabled patient websites [3].
Developing PRO Instruments for AEs
The FDA has produced guidance [16] and the EMA [17] has published a reflection paper on PRO measures in support of label claims. The FDA guidance states that PRO instruments can be used to measure important safety concerns if those concerns represent symptoms or signs that are best captured from the patient perspective.
A challenge remains of how to integrate PRO-AEs into a benefit–risk assessment, e.g. post-approval. Usually, PRO-AEs will be collected without PRO-FEs (favourable events). If there is lack of perceived efficacy, should patients report this as an AE? It is possible that a few years after marketing, the AE profile of the drug may be defined more accurately but without similar insight into the efficacy/effectiveness profile.
Hence, although the FDA guidance infers that PRO instruments should aim to measure the adverse consequences of treatment separately from the effectiveness of treatment, more recent methodologies would support efficacy (or effectiveness) data being collected from the same dataset as safety, and may in fact improve the utility of the information [33]. Although the context for the FDA and EMA documents is to develop PRO instruments to support labelling, there are some methods in these guidances that may apply to non-labelling objectives.
PRO-AE instruments, like PRO instruments in general, need to be validated and ‘fit for purpose’, i.e. practical for use by patients, to encourage reporting, and interpretable by a number of stakeholders (prescribers, regulators, industry, policymakers and, not least, patients themselves). The following subsections address:
A process for developing PRO-AE instruments;
Creating a suitable taxonomy;
Selecting appropriate technologies;
The availability of alternative data sources;
The relationship between PRO-AEs and AE data from other sources.
Process for Developing PRO-AE Instruments
A suitable framework for developing PRO-AE instruments should be used. An example is given here, which was adapted by the Patient-Reported Outcomes Safety Event Reporting (PROSPER) Consortium in a series of iterative discussions and reviews from an approach presented by the FDA in their guidance document [16]. The details associated with each stage are presented in Table 5, which may be iterative. Optional method-specific steps that may be needed are shown in Table 6.
Table 5.
Example approach for developing a PRO-AE instrument
| Stage | Brief description | Details |
|---|---|---|
| Stage 1 | Development of the conceptual framework | 1. Identification of concepts and domains that are to be measured 2. Identification of the purpose of the PRO instrument 3. Identification of the intended population |
| Stage 2 | Creation of the PRO instrument. [Criteria vary depending on whether developing a targeted measure to look at a particular AE (e.g. to compare tolerability in a phase 3 clinical trial) vs. general/generic screening questions. In the latter case, single items for each PRO-AE can be developed and broadly used] | 1. Generation of items 2. Choice of response options 3. Evaluation of patient understanding 4. Development of instructions and training 5. Identification of preliminary scoring of items and domains 6. Assessment of respondent and administrator burden 7. Confirmation of the conceptual framework and finalisation of the instrument |
| Stage 3 | Embedding of patient perspective [37] (This stage can be performed in parallel with Stage 2) |
1. Generate disease area candidate items (if required), in addition to generic items, with input from qualitative interviews with patients, and add those items to the core instrument for testing 2. Drop any candidate items that lack sensitivity 3. Validate the psychometric properties (validity, reliability, sensitivity) of the resulting instrument 4. Conduct cognitive debriefing interviews with patients to confirm the instrument’s ease of comprehension, relevance and acceptability |
| Stage 4 | Assessment of measurement properties | 1. Evaluation of reliability, e.g. what level of evidence (how large a patient sample) |
| 2. Assessment of validity | ||
| 3. Evaluation of ability to detect change | ||
| 4. Choice of methods for interpretation | ||
| a. Definition of responders | ||
| b. Definition of a minimum important difference | ||
| Stage 5 | Instrument deployment plan | 1. Determine format |
| 2. Determine timing | ||
| 3. Determine appropriate collection method | ||
| Stage 6 | Modification of instrument | 1. Revised measurement concept |
| 2. Application to a new population or condition | ||
| 3. Changed item content or instrument format | ||
| 4. Changed mode of administration | ||
| 5. Changed culture or language of application | ||
| 6. Test modified instrument with patients |
Some steps may be combined or shortened, particularly if adapting social media for non-structured, non-prespecified reports
AE adverse event, PRO patient-reported outcome, PRO-AE patient-reported outcome of adverse event
Table 6.
Additional steps that may be required in the example approach
| Additional step | Comment |
|---|---|
| The need to perform a quality check before passing from one stage to the next, to ensure each element in the stage has been completed satisfactorily | Need checklists to determine whether each stage is completed appropriately This intrinsic quality assurance should include a consideration of utility versus quality |
| The need to return to a previous stage and repeat the activities, possibly due to new information or a change in one or more criteria | Iteration is important, and in particular is essential in real-world usage |
| Usability testing of any software platform | |
| Specifying approach to minimising missing data |
A PRO initiative by the National Quality Forum in the USA has suggested that characteristics for a good PRO instrument for AEs and other usages include actionability, meaningfulness to both patient and HCP, facilitation of shared decision making between patient and HCP, and implementability [33].
Some instruments designed to capture adverse effects systematically are available, and have been shown to improve care [34, 35]. Nurses, as the professionals closest to patients, may be best placed to lead the profiling of patient problems. Ensuring that single pertinent questions capture clinically important symptoms efficiently requires cognitive input from both HCPs and patients [36].
Creating a Suitable Taxonomy
One issue with the development of PRO tools, and especially those used for collecting AE data, is the creation of a taxonomy by an iterative process. In one study, terms were aligned with the existing Medical Dictionary for Regulatory Activities (MedDRA) and refined with input from patients, including comprehension testing within which patient-reported symptoms were categorised and defined [7]. Once achieved, patient-reported symptoms and AEs were generally in agreement with those reported by clinicians and nurses. The PRO tool used in this study was a modified version of the CTCAE using 13 terms familiar to cancer patients. Cirillo et al. [7] have shown that a modified version of the dictionary used in trials can be utilised in the post-marketing setting by patients undergoing chemotherapy.
The development of a patient-friendly dictionary is an important consideration when developing PRO-AE tools to encourage reporting of PRO-AEs. An alternative approach being investigated by the World Health Organization (WHO)/Uppsala Monitoring Centre (UMC) consists of an anatomical interface to help patients identify appropriate AR terms. The electronic interface uses the image of a body, and clicking on a particular area drills down to present AR terms associated with that body area (WHO/UMC, unpublished communication).
Selecting Appropriate Technologies
In clinical trials, most PRO-AE reporting systems have been paper based. However, the NCI has developed a web-based platform for CTCAEs, as well as standard paper channels. The equivalency between web-based and other traditional channels is currently being tested. This initiative was undertaken to develop a system that would be used by patients to report adverse symptoms in cancer trials; generate useful data for investigators, regulators, clinicians, academic groups and patients; and be compatible with existing AE reporting systems [28].
For marketed drugs, there have been web-based systems allowing patients to report suspected ARs directly to the regulators in the UK, Denmark, Netherlands, US, Sweden, etc. Recent EU pharmacovigilance legislation has included a requirement for direct patient-reporting mechanisms to regulatory authorities.
The WHO/UMC is also developing a web-based system for patient self-reporting of AEs (WHO/UMC, unpublished communication). In contrast, some non-regulatory websites are not exclusively used to collect PRO-AEs, but are designed to be consumer friendly.
When developing PRO-AE instruments, there is a need to consider the most appropriate technology. However, deploying PRO-AE instruments through the internet or through smart technologies such as phones and tablets does require consideration to be given to:
Access to technology;
Data privacy and storage issues;
Appropriateness for the population;
Data transmission;
Cost;
Technical awareness of patients—since the dataset will be biased if only technically aware patients report;
Patient knowledge of the ability to report and patient willingness to report;
Data privacy and protection.
Availability of Alternative Data Sources
Data from existing patient sources including electronic patient records are routinely collected in primary care and may be suitable for AE screening, such as patient safety indicators, and to validate novel PRO-AE instruments.
The current FDA Sentinel Initiative is aiming to develop a national electronic safety monitoring system by augmenting existing safety monitoring systems. This has been achieved by leveraging multiple sources of electronic data through partnering with data holders and use of a common data model for healthcare systems, insurance companies and industry, with potential coverage of up to a third of the US population. Although currently not in Sentinel, large datasets such as this would be enhanced by systematically collected PRO-AE data.
The advantages of this approach include enhancing the active post-marketing monitoring of medical product safety to effectively and rapidly examine common outcomes (e.g. myocardial infarction, fractures) in large, real-world populations, with improved access to subgroups and special populations. Additionally, validated quantitative approaches have been developed for signal refinement using sequential monitoring of single time points with adjustments to minimise confounding, near real-time monitoring and an accessible library of tools/resources.
Relationship Between PRO-AEs and AE Data from Other Sources
There are various models that can be employed and the approach should be prespecified [19]:
Independent reporting, in which AE data gathered from the patient and the clinician are collected, analysed, and reported completely separately from each other. A subsequent comparison of the commonalities and differences between the patient-reported and clinician-reported data may provide valuable insights.
Merged reporting, in which AE data gathered from the patient and the clinician are collected separately but then merged and analysed together. However, this is a challenging goal, so in many situations, it is preferable to keep the two data pools of clinician- and patient-collected AEs separate to aid meaningful interpretation.
Collaborative reporting, in which AE data gathered from the patient is then provided to clinicians to inform their reporting. Specific trials might be needed to test the utility and validity of this concept.
The choice of approach depends on whether two specific pools of information, from patients or from medically confirmed sources, can be combined or whether, because they provide discrete data with different utility, they must be separately stored and analysed. Another factor is the impact of feedback of PRO-AE information on stakeholder behaviour (including prescribers and patients).
A formal comparison of these three approaches is planned as a part of the NCI’s PRO-CTCAE initiative [28]. In total, there are 124 questions that assess different attributes (e.g. presence, frequency, severity, interference) of 78 symptoms that are represented in both the CTCAE (version 4) and MedDRA AE lexicons. Using cognitive testing, these 124 questions have been extensively evaluated by cancer patients, and have been found to be comprehensible and to measure the symptom of interest. An electronic system provides a mechanism for scheduled periodic surveillance and collection of patient-reported symptom data, using the web or a touch-tone telephone. US validation study complete and reported at international meetings, with robust psychometric properties shown for most items. Linguistic adaptation to multiple languages is ongoing, and there are multiple ongoing trials assessing the feasibility of this approach [28].
Data Capture and Appropriate Datasets
As already shown in Fig. 1, it is envisaged that PRO-AE instruments will be used to collect different datasets at different stages of drug development. These data in turn will be used to measure different concepts and domains.
Because of this, an important part of PROSPER’s remit is to investigate what PRO-AE datasets might be appropriate when addressing different concepts and domains. Table 7 identifies some of the common AE data collection tools currently in use. European regulatory authorities are also developing standard, structured forms specifically for web-based reporting of suspected ARs by patients as well as HCPs.
Table 7.
Currently available datasets for adverse events data capture
| Dataset |
|---|
| • CIOMS I or MedWatch 3500 form (see Appendix: ESM 1 and 2) |
| • Minimum elements (identifiable patient, suspected drug, suspected adverse reaction, identifiable reporter) |
| • ICH E2B (see Appendix: ESM 3) |
| • Free text (linked to free text mining/coding) |
| • Additional specific datasets relevant to drug/class/disease |
CIOMS Council for International Organisations of Medical Sciences, ESM electronic supplementary material, ICH International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
However, tools such as CIOMS I, MedWatch 3500 and ICH E2B may not be suitable in their full form for use as PRO-AE instruments. These may require editing, with a focus on the PRO concepts/domains being measured, down to a core dataset suitable for PRO-AEs in non-prespecified patient populations. There may also be a need to add some further terms to optimise PROs for patient safety and benefit–risk analysis, e.g. severity as well as seriousness.
An attempt has been made to identify the minimum number of fields in the CIOMS I and MedWatch forms [see Appendix: electronic supplementary material (ESM) 1 and 2] that could provide valuable PRO-AE data. We have also identified those fields in the ICH E2B dataset that would provide the ‘gold standard’ in terms of PRO-AE data (see Appendix: ESM 3). The general aim should be to have enough fields completed by patients to allow easy linkage to individual case study reports (ICSRs) from health professionals, so that these reports may be used together as well as separately.
However, another approach would be to identify the PRO concept or end point that is being assessed and identify the minimum fields needed to determine this value. This may necessitate identifying new fields, e.g. the question “Has a report been submitted by your doctor or HCP?” could result in yes/no/I don’t know options being offered, and the “yes” option would mean that additional information may be required, such as HCP name.
Once a mechanism has been put in place to capture PRO-AEs, a key consideration is how to raise patient awareness to actively provide unsolicited PRO-AEs.
Guidance should also include a framework to define how data are managed, including any data privacy issues:
When data are collected, what are the rules of engagement?
What will be collected?
What will be received (by pharmaceutical companies/regulators)?
How will it be validated? The WHO/UMC has investigated this issue, and within its organisation, uses an instrument to score the validity of a report [38, 39].
How will duplicates be detected? For example, the ‘hit-miss model’.
Who performs these tasks is of less importance as long as the framework is clear about what needs to be done. However, if a pharmaceutical company is provided with the raw database, then this would trigger reporting responsibilities under current regulations. For the use of similar healthcare databases, personal details are of course already removed (i.e. anonymised databases) for reasons of confidentiality.
Analysis of PRO-AE Data
As already discussed, a useful way of classifying PRO-AEs is to relate them to the main population types under investigation, i.e. prespecified and non-prespecified. Further stratification can be provided by considering whether the AE data collected are structured or unstructured, and whether or not the patient selection method is more or less restrictive (see Fig. 1). Analyses of PRO-AEs from prespecified and non-prespecified patient populations need different methodologies, including definition of a core safety dataset to permit useful signal detection. Where possible, measuring baseline symptoms gives a useful additional perspective.
Patient-reported safety information that better reflects the ‘real world’, i.e. with a non-prespecified population and non-structured datasets, has increased medical relevance. However, this must be balanced against quantitative validity (see Fig. 1), at least against the current agreed standards of data from randomised, blinded and controlled trials.
This section considers how analytical robustness of patient-reported safety outcomes can be preserved within these more real-world settings, as well as more controlled settings such as pre-approval clinical trials.
Scope of the Evaluation
Prespecified Populations
In early clinical trials, the safety evaluation is exploratory in nature and is primarily used to delineate the toxicity boundaries of the dose-response curve (Table 8). However, direct PRO-AE measurements could be used at this stage to screen for unexpected reactions and, if their incidence and severity were confirmed in phase 3 trials, used to support filing/registration claims.
Table 8.
Analysis of safety data from different stages and different populations
| Population | Pre-approval | Post-approval |
|---|---|---|
| Prespecified (structured) |
In early clinical trials, the safety evaluation is exploratory and is only capable of detecting direct expressions of toxicity In later phases, where sample sizes are greater, the safety profile of a drug can be characterised more fully. Comparison with efficacy data informs the benefit–risk profile |
In post-approval studies, the safety profile can be further characterised. Comparison with efficacy or effectiveness data further informs the benefit–risk profile |
| Non-prespecified (structured and unstructured) |
Detection of low-probability signals from a larger, non-prespecified patient population Data on real-work safety profile (e.g. with concomitant medication and medical conditions) |
|
Although there are inherent limitations with pre-approval safety evaluations, such as small sample size, narrow eligibility criteria and limited duration of follow-up, their ability to detect adverse symptom events among study participants improves with the use of clear dataset definitions, methodology for collection, and agreed analytic approaches.
PRO-AE measurements could also be used for post-approval signal detection or for targeted assessments in phase 4 controlled trials.
As discussed in Sect. 5.5, safety data collected via PRO-AE instruments can potentially be analysed independently from or in combination with AE data from other sources. These data can also be used to inform the benefit–risk balance of a product by including in the analysis efficacy or effectiveness data, which may also include PROs.
Non-prespecified Populations
Post-approval safety data are useful for general screening to detect low-frequency safety signals through the exploitation of a larger user population, even from non-prespecified populations. In addition, other PRO-AE-based PASS study designs may benefit from proactive patient follow-up for occurrence of AEs of special interest.
Consequently, the main question is, can PRO-AE data be amalgamated with data from other sources? Low-frequency safety signals may potentially have a significant impact on the benefit–risk profile of a product.
Choice of Variables and Data Collection
In both the pre- and post-approval phases, the occurrence of serious AEs and treatment discontinuations due to AEs are particularly important to identify. If PRO-AE tools are utilised, a common AE dictionary that allows patient-reported data to be summarised and collated is important (Table 9). This might necessitate the development of a set of terms that are understood by the patient and can be mapped to higher level ‘medical’ terms, similar to the PRO-CTCAE dictionary developed by the NCI for oncology trials [28]. In addition, a severity scoring system for pre-agreed PRO-AEs can be developed. For example, in the case of a symptom like rash, questions will focus on its location on the body.
Table 9.
Data collection considerations
| Population | Pre-approval | Post-approval |
|---|---|---|
| Prespecified (structured) | If PRO-AE tools are used to collect clinical AEs, then the development of a patient-centric dictionary needs to be considered | A dataset suitable for the study needs to be established |
| Non-prespecified (structured) | Patient could potentially report on a social website AEs from a clinical trial where the website offers structured collection | Regulated consumer sites (e.g. MHRA-Yellow Card) have a well-defined dataset. Some patient support sites are also structured If PRO-AE tools are used to collect AEs, then the development of a patient-centric dictionary needs to be considered |
| Non-prespecified (unstructured) | Patient could potentially report on a social website AEs from a clinical trial | For PRO-AEs from non-regulated consumer sites, a minimum dataset needs to be agreed |
AE adverse event, MHRA Medicines and Healthcare Products Regulatory Agency, PRO-AE patient-reported outcome of adverse event
Prespecified Populations
In pre-approval clinical trials, data from objective laboratory tests, clinical variables (blood pressure, pulse, respiratory rate) and investigations (e.g. radiology) usually form a major part of the safety data. These types of data are reported directly and their transmission by patients would not normally be expected as part of a study. However, clinical AEs (diseases, signs, and symptoms) would be suitable for PRO-AE methods, although of these, a strong emphasis would be placed on patient-reported symptoms.
Non-prespecified Populations
A rapidly increasing quantity of patient-reported data is available on the internet. Unstructured data can present a particular challenge for extracting useful information, since the data are usually incomplete. PRO-AE-enabled websites allow a more structured approach, where the dataset can be designed pre-emptively, and so are far likelier to provide a higher quality of data.
Defining the Evaluable Population
Prespecified Populations
For pre- and post-approval clinical trials, the evaluable population for PRO-AE comparisons is usually defined as those subjects who received at least one dose of the investigational drug, as well as any comparator drug/placebo groups (Table 10). Safety variables should be collected as comprehensively as possible from these participants, including type of AE, severity, onset, and duration. Additional safety evaluations may be needed in specific subpopulations, such as females, the elderly, the severely ill, or those who have a common concomitant treatment.
Table 10.
Population groups
| Population | Pre-approval | Post-approval |
|---|---|---|
| Prespecified (structured) | In clinical trials, the evaluable population is usually subjects who received at least one dose of the investigational drug (or comparator/placebo) | |
| Non-prespecified (structured) | Patients could potentially report on a social website AEs from a clinical trial where the website offers structured collection | Currently, for regulated and non-regulated consumer sites, evaluable subjects require an identifiable patient, suspected drug, suspected AR and identifiable reporter as the minimum dataset In the current guidance, a core minimum dataset is proposed in the Appendix (see ESM) for non-regulated consumer sites and for PRO-AE tools |
| Non-prespecified (unstructured) | Patient could potentially report on a social website AEs from a clinical trial | For non-structured social networking sites, e.g. Facebook, a minimum dataset needs to be agreed. Comparator groups need particular consideration as any detection of safety signals will have to be done over and above the background rate for a ‘comparable’ population not receiving the drug/device. Identifying the comparator population will be further complicated in a self-reporting, social network environment |
AE adverse event, AR adverse reaction, ESM electronic supplementary material, PRO-AE patient-reported outcome of adverse event
All relevant safety variables will be evaluated, and the broad approach should be indicated in the protocol. All AEs should be reported, whether or not they are considered related to treatment. All available data in the study population should be accounted for in the evaluation. Definitions of measurement units and reference ranges of laboratory variables and other investigations should be made with care; if different units or different reference ranges appear in the same trial (e.g. if more than one laboratory is involved), then measurements should be appropriately standardised to allow a unified evaluation. Use of a toxicity grading scale should be prespecified and justified. Definition of PRO symptom severity scales, e.g. for pain, nausea, etc., can also be standardised between studies.
The incidence of an AE is usually expressed in the form of a proportion relating number of subjects experiencing events to the number of subjects at risk.
Non-prespecified Populations
Where PRO-AEs are being used in the post-approval phase with non-prespecified populations, the safety variables should be collected as comprehensively as possible from these subjects, including type of AE (using appropriate terms), severity, onset, and duration. Evaluable subjects are those for whom a retrospectively defined set of data elements is available. A suggested minimum dataset is provided in the Appendix (see the ESM), to which severity, onset and duration could also be added.
Statistical Evaluation Techniques
Prespecified Populations
Because, prior to approval, the range of possible adverse effects is very large, the assessment of unexpected PRO-AEs must be built into all protocols. As a result, the safety data (whether collected in the traditional manner or via a PRO-AE tool) are best addressed by applying descriptive statistical methods to the data, supplemented by calculation of confidence intervals wherever this aids interpretation. It is also valuable to make use of graphical presentations in which patterns of AEs are displayed both within treatment groups and within subjects. For example, presenting, in ascending order of risk, a series of AEs for the comparator and active drug together with their relative hazard ratio and confidence intervals is a good visual technique.
The calculation of p values can be useful to identify a specific difference or as a flagging device applied to a large number of safety variables to highlight differences worthy of further attention.
If hypothesis tests are used, statistical adjustments for multiplicity to quantify the Type I error are appropriate, but the Type II error is usually of more concern [40]. Care should be taken when interpreting putative statistically significant findings when there is no multiplicity adjustment.
In the majority of trials, investigators are seeking to establish that there are no clinically unacceptable differences in safety compared with either a comparator drug or placebo. As is the case for non-inferiority or equivalence evaluation of efficacy, the use of confidence intervals is preferred to hypothesis testing in this situation. In this way, the considerable imprecision often arising from low frequencies of occurrence is clearly demonstrated [40].
Non-prespecified Populations
There are a number of available approaches to aid the interpretation of PRO data, which could be adapted to PRO-AE data. They cover a spectrum of standard types of analyses for these types of data, including descriptive statistics, data mining/disproportionality, and multivariate analyses (Table 11).
Table 11.
Statistical methods suitable for post-approval, non-prespecified datasets
| Population | Post-approval |
|---|---|
| Non-prespecified (structured and unstructured) |
Descriptive statistics |
| • Numbers and percentages | |
| • Chi-squared test | |
| • Mean (e.g. known groups—the mean scores underlying particular clinical groups which can be used as a benchmark to compare other groups; normative and reference groups—customised benchmarks to compare other groups) | |
| • Minimal important difference (MID)—from the patient perspective, can be defined as “the smallest difference in score in the domain of interest which patients perceive as beneficial and which would mandate, in the absence of troublesome side effects and excessive cost, a change in the patient’s management” [44] | |
| • Incidence and/or prevalence | |
| • Relative risk or odds ratio | |
| Disproportionality—the occurrence of a drug–event pair at a higher frequency than would be expected from a statistically independent random occurrence | |
| A) Frequentist methods | |
| • Proportional reporting ratio (PRR)—a measure of the disproportionality | |
| • Reporting odds ratio (ROR) | |
| B) Bayesian methods | |
| • Bayesian confidence propagation neural network (BCPNN) | |
| • Multi-item gamma poisson shrinker (MGPS) | |
| Multivariate analysis | |
| • Linear regression | |
| • Logistic regression | |
| • Cox proportional hazard models |
Other approaches include comparing a PROMs score with known clinical parameters, such as days in hospital and illness severity [41], the proportion of patients whose PROMs scores improve or worsen after intervention [42], and cross-instrument calibration using advanced statistical analyses such as item response theory (IRT) [43]. Although not specific to PRO-AE evaluation, such techniques might assist the quantification and assessment of patient-reported benefit–risk profiles. For longitudinal data, comparison of patient-level AUCs, or cumulative incidence of worst severity scores may be performed.
The test for statistical significance will depend on the approach used for the data analysis. This may include p values, standard deviations, and/or confidence intervals.
The use of statistics based on disproportionality is also possible in non-prespecified populations with non-structured datasets, but more innovative approaches are needed for this group (see next section on data mining).
Data Mining Techniques
The use of data mining for signal detection during clinical trials is a relatively new concept and allows researchers to perform ad hoc and close-to-real-time data analysis, reducing time in the interchange between researchers, clinicians and statisticians [45].
Traditionally, analysis has been carried out by a systematic manual review of every report sent by physicians to pharmacovigilance experts. These reports are registered in pharmacovigilance database systems.
More recently, automated signal detection methods have been developed to supplement qualitative clinical methods (see Table 12 and ESM 4). While these automated methods cannot replace expert clinical reviewers, they can assist with the difficult task of screening huge numbers of drug–event combinations in databases for potential signals. Through commonly used methods that are based on an underlying model of statistical association, databases are scrutinised for a significant occurrence of disproportionalities or dependencies between drug–event pairs.
Table 12.
The use of data mining for different populations
| Population | Pre-approval | Post-approval |
|---|---|---|
| Prespecified (structured) | Data mining—possible but because of the low number of occurrences of each drug–event association, methods demonstrate low specificity | |
| Non-prespecified (structured) | Patient could potentially report on a social website AEs from a clinical trial where the website offers structured collection | Automated signal detection is well established and relies on data warehousing of large numbers of spontaneous reports and statistical analysis techniques |
| Data mining techniques include: | ||
| • Predictive modelling clustering or database segmentation | ||
| • Link analysis | ||
| • Deviation detection | ||
| Non-prespecified (unstructured) | Patient could potentially report on a social website AEs from a clinical trial | Data mining encounters problems with use of natural language and the interpretation of free text responses. Need advances in natural language processing for data mining free text, currently work in progress by WHO/UMC |
AE adverse event, UMC Uppsala Monitoring Centre, WHO World Health Organization
The WHO/UMC and the FDA are currently using automated detection algorithms based upon Bayesian analysis to achieve signal generation. These methods have a major role in prioritising signals by disproportionality, which are then clinically reviewed. The validity of the approach has been demonstrated in several papers and is now routinely practised [46]. Though the optimum algorithms are not established, there is much work being done in this area through the OMOP project [47–50].
Work has also been commenced on natural language processing to allow analysis of narratives and related terms [51].
MedDRA is the unified standard terminology currently used for recording and reporting adverse drug event data in most countries, but it may be necessary to extend/adapt it to include certain patient-reported terms being mapped to already accepted Preferred Terms (PTs)/Lower Level Terms (LLTs). In the meantime, verbatim terms reported by the patient can be reported directly, or grouped together into recognised symptom PTs.
Methods based on data warehousing and statistical analysis techniques provide various algorithms to identify trends or clusters of events, which may increase the accuracy and reliability of interpretation of safety data. The basic components are already available to companies for performing data mining of these safety databases for pharmacovigilance.
Statistical analysis system (SAS) tools are available for analytic processing, particularly to create complex analyses and reports. Utilising a suite of other SAS solutions enables companies to perform proactive pharmacovigilance. This suite may include data integration and tools to support clinical trials and drug development, which provides a standard, compliant environment for storage, retrieval, analysis, reporting and signal detection.
The signal detection process used by the WHO/UMC for analysis of VigiBase Centre consists of a combination of automated knowledge discovery methods [52], triage (prioritisation) algorithms and clinical review [53]. The knowledge discovery methods highlight drug–AR pairs with unexpectedly large numbers of reports relative to the average reporting ratios in the database. Triage algorithms use a combination of quantitative and qualitative information to focus attention on the key issues for follow-up [53]. Reports related to drug–AR pairs picked out by the triage algorithms are sent to an expert review panel, with pattern discovery methods often useful for profiling groups of reports and suggesting alternative explanations for increased reporting.
Hypotheses of suspected ARs first highlighted in automated knowledge discovery, which remain after clinical review, are then communicated to industry/regulators and published as appropriate [54, 55]. However, the risk of distortion from undiscovered data quality problems and the difficulty of obtaining complete, detailed information on reported AR incidents mean that signals of suspected ARs often remain tentative, even after clinical review [56].
A knowledge discovery process has been applied to the WHO/UMC VigiBase to resolve some of these shortcomings by examining the entire AR signal detection process [56]. The steps covered were data collection, cleaning and preparation; reduction and projection; data analysis and interpretation; and finally dissemination, incorporation into existing structures and action based on discovered knowledge. A duplicate detection method (the hit-miss model) that can reliably identify pairs of unexpectedly similar reports and a new measure for highlighting suspected drug–drug interaction have also been validated on the WHO/UMC database, which could be applied to compare PRO databases with overlapping medically confirmed spontaneous reports [56].
Examples of data mining techniques and related analytic approaches including predictive modelling, clustering or database segmentation, link analysis, and deviation detection are shown in ESM 4 in the Appendix (adapted from [57]).
Data Protection
Collection of identifiable patient data needs to meet the various international, regional (e.g. EU) and national data protection legal requirements, as appropriate. For example, real-world collection of identifiable patient data on the web, particularly when solicited as structured reports in the post-approval setting, must be done in a manner consistent with the relevant data protection legislation. The problem is complicated by the fact that the legislation would normally be enforceable in the country where the web collection tool originates, and also where any data is stored for safe harbour. Patients, of course, may be able to access and input their own data from other countries, and it is unclear to what extent these other countries’ data protection laws, if different from the enforceable country, need to be met.
Conclusions
The patient perspective is an essential component of drug safety profiles. Within this document, the PROSPER Consortium sets out an innovative framework for improving safety data during both clinical development and post-marketing phases by integrating the patient’s voice and perspective.
This comprehensive guidance suggests methods for including PRO-AEs as part of any structured questionnaire design, which may be used in both specified and non-prespecified populations. There is a valid distinction between the assessment and data mining of grouped, safety data, giving aggregate patterns of PRO-AE information, which do not necessarily require medical confirmation on an individual basis, as opposed to the assessment of single cases for which medical confirmation remains important.
The guidance proposes a minimum core dataset for use with unstructured data sources, details of which are provided in the Appendix (see the ESM). Various approaches to analytic methodology are considered, with the method of choice dependent on the specific patient-reported data being collected. For larger, non-prespecified populations and unstructured data, the quantitative methodology will require the use of more novel and emerging data mining approaches.
Patient-centeredness and patient safety are emerging as core elements of any responsive health system. As patients become more activated in their health and healthcare, they support the maxim of “Nothing about me, without me” and expect others involved in their care to engage in shared decision making, so that what matters to the patient is always included.
As stated at the outset of this document, the potential clinical utility and value of real-world information obtained from real patients taking real drugs is great [58]. Technological advances that were not available even a decade ago now offer unprecedented access for multidirectional communication between patients, clinicians, the private sector, industry and regulatory authorities.
We believe that the current guidance will help ensure that the voice of the patient will be heard, so that the safety profile and hence benefit–risk balance of new and existing medicines is better defined.
Glossary
Adverse event(AE): Any untoward medical occurrence in a patient or clinical trial subject administered a medicinal product, which does not necessarily have to have a causal relationship with this treatment [Dir 2001/20/EC 76 Art 2(m)]. An AE can therefore be any unfavourable and unintended sign (e.g. an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal product, whether or not considered related to the medicinal product [15].
Adverse reaction(AR): A response to a medicinal product that is noxious and unintended [DIR 2001/83/EC Art 1(11)]. ‘Response’ in this context means that a causal relationship between a medicinal product and an AE is at least a reasonable possibility.
Common Terminology Criteria for Adverse Events (CTCAE): A dictionary developed by the US NCI designed for use in clinical trials to aid clinicians in detecting and documenting AEs commonly encountered in oncology treatment.
Concept: The specific goal of measurement, i.e. the thing that is to be measured by the PRO instrument.
Data mining: The process of extracting meaningful patterns from large datasets using automated computational and statistical tools and techniques.
Disproportionality: Disproportionality analysis is an important tool for identifying new signals/patterns in spontaneous AE report databases. There are a variety of disproportionality measures (DPMs) used in safety signalling. DPMs are based on the ratio of the number of observed (O) cases and the number of expected (E) cases (i.e. O/E).
Domain: In a PRO instrument, a domain is a discrete concept within a multi-domain concept. All of the items within a single domain contribute to the measurement of the domain concept. The domain could be, but is not necessarily, considered a specific end point within the overall conceptual model.
Harm: Adverse impact on an individual’s health status.
Healthcare professional (HCP): Any qualified professional involved in the delivery of healthcare to the patient, whether or not involved in prescribing. Specifically, this term includes doctors, prescribers other than doctors, pharmacists, nurses, dieticians, and physiotherapists.
Health-related quality of life (HRQoL): A multi-domain concept that represents the patient’s overall perception of the impact of an illness, its treatment, and their overall state of health on their quality of life. This concept is used in FDA PRO effectiveness guidance (FDA, 2009).
Instrument: A structured means to capture PRO data, e.g. a questionnaire or diary along with supporting information such as training materials, scoring, etc. In the context of this guidance, this could be a specific reporting tool that through completion generates a structured dataset.
Item: An individual question within a PRO instrument that is evaluated by the patient.
Non-prespecified population: A group of patients where the number and type of patients included has not been pre-agreed.
Non-regulated consumer website: A website that is not set up with an aim to collect PRO-AEs, and where data posted will therefore be unstructured, and safety information posted is, at best, reviewed and collected.
Non-structured data: Data recorded without a data model.
Patient-reported outcome (PRO): The concept of any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else. For the purposes of this guidance, PROs are defined as encompassing the full range of self-reporting, ranging from those data collected via a structured and validated questionnaire to those collected in a free-text manner via spontaneous patient submissions (or in some cases from caregivers or surrogates).
Patient-reported outcome of adverse event(PRO-AE): A PRO-AE is any untoward medical occurrence, whether or not considered treatment/intervention related, which is reported or transmitted directly by the patient without interpretation by a clinician or anyone else. PRO-AEs may be collected by both structured and unstructured reports.
Patient-reported outcome measures (PROMs): An instrument, scale or single-item measure used to assess the PRO concept as perceived by the patient, obtained by directly asking the patient to self-report. The term has also been used in the UK-NHS, in the context of patient satisfaction, symptoms and functional status measures in relation to selected surgical procedures. In the context of epidemiology, AE outcome measures can be continuous variables or have binary outcomes.
Patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE): A specific subset from the US NCI CTCAE project taxonomy; work is in progress.
Patient support programme (PSP): Sometimes also called ‘patient assistance programmes’, these may comprise non-interventional observational study designs or medical enquiry/patient assistance services. Safety reporting processes should be clearly defined by the market authorisation holder before the programme commences, to ensure that serious and non-serious AR reports are captured, as appropriate, and that causality assessments and regulatory reporting are performed, where required.
Post-approval/post-authorisation: Occurring after a medicinal product has received regulatory approval (post-licensing).
Pre-approval/pre-authorisation: Occurring prior to a medicinal product receiving regulatory approval.
Prespecified population: Enrolment of a specific patient group in conformance with a pre-agreed number and patient type or profile. This profile may be restricted (with inclusion/exclusion criteria) or unrestricted (no specific inclusion/exclusion criteria, but the size and/or geography of the group could still be limited).
PRO-AE-enabled patient support website: A specific website (whether company sponsored or not) that uses structured data collection in a scientifically robust way and is designed for patient education, interaction and self-reporting of PRO-AEs via a structured data collection instrument.
PRO-enabled: Data collection processes/systems and technologies that are adapted for the collection of patient-reported outcomes.
Registry: A defined group of patients, whether having a specific exposure (e.g. taking one or more specific treatments) or having a specific disease (i.e. a disease registry). A registry may be prespecified (pre-agreed dataset) or non-prespecified (open enrolment).
Regulated consumer website: A website that is set up with an aim to collect PRO-AEs, and where data posted may be either structured or unstructured, but safety information posted is regularly reviewed and collected.
Risk: Defined by the UMC as “the probability of harm being caused; the probability (chance, odds) of an occurrence”. The EU defines “risks related to use of a medicinal product” as “any risk relating to the quality, safety or efficacy of the medicinal product as regards patients’ health or public health; and any risk of undesirable effects on the environment” [DIR 2001/83/EC Art 1(28)].
Social network site: Web-based services that allow individuals to construct a public or semi-public profile within a bounded system; articulate to a list of other users with whom they share a connection; and view and traverse their list of connections and those made by others within the system. Non-medical social network sites are not specifically designed for a treatment or disease, but are where PRO-AEs may be reported incidentally, often in an unstructured way (e.g. Facebook or Twitter).
Structured data: An organised dataset based on a data model, in contrast to unstructured data that consists of open text fields.
Validation: The process of evaluating a PRO instrument’s ability to measure a specific concept or collection of concepts accurately.
Voluntary enrolment: Where patients are able to enrol themselves, whether prespecified (usually in the post-approval setting) or non-prespecified.
Electronic supplementary material
Acknowledgments
The PROSPER Consortium wish to thank both the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) steering group and partners forum for their helpful review and constructive feedback in improving the guidance document. Facilitation, chairmanship, document drafting and consortium meeting support for the PROSPER group and guidance document were provided by Pope Woodhead and Associates, Ltd. and PatientsLikeMe®, Inc. The guidance does not support or endorse any specific technology, and where commercially available tools are quoted, these are only for illustration. Anjan K. Banerjee is also a part time Hon. Consultant Surgeon at ULH NHS Trust, and is in discussions with the UK Department of Health on PRO-AEs in the context of healthcare outcomes on a not-for-profit basis.
The writing group would also like to thank the following who contributed to PROSPER meetings and the related discussions: Willem Jan Atsma (Astellas), Laurent Auclert (Sanofi-aventis), Mary Baker MBE (European Brain Council), James Heywood (PatientsLikeMe), Steve Hobbiger (GlaxoSmithKline), Ken Hornbuckle (Lilly), David Jefferys (Eisai), Crispin Jenkinson (University of Oxford), Stefan Kaehler (Celgene), Paul Knights (Pfizer), David Lewis (Novartis), Rick Lilley (UCB), Rebecca Noel (Lilly), Niklas Norén (WHO/UMC), Sten Olsson (WHO/UMC), Sonja Pumpluen (Actelion), Jan Regenstrom (Scientific assessor, EMA), Jamie Robinson (Roche), Mette Stockner (Novo Nordisk), Dirk Teuwen (UCB), Sarah Trease (Roche), Spiros Vamvakas (Head of Scientific Advice, EMA), Steven Wojtanowski (Abbott Laboratories), and Amanda Worpole (European Federation of Neurological Associations).
Funding Disclosure
The consortium and guidance development were self-funded by Pope Woodhead and Associates, Ltd. and PatientsLikeMe®, Inc.
Conflict of Interest
Anjan K. Banerjee is employed by, and is a Director of, Pope Woodhead and Associates and has a conflict of interest related to holding Pope Woodhead and Associates stock, employment, and expert testimony, and has provided consultancy to multiple global pharmaceutical companies. Sally Okun is employed by PatientsLikeMe® and has a conflict of interest related to employment, holding PatientsLikeMe® stock grants received, and has provided consultancy to multiple global pharmaceutical companies. Paul Wicks is employed by PatientsLikeMe® and has a conflict of interest related to employment, holding PatientsLikeMe® stock grants received, and has provided consultancy to multiple global pharmaceutical companies. Dr. Smith is employed by AbbVie has a conflict of interest related to employment, holding AbbVie stocks, expert testimony, patents and royalties. Dr. Mayall is employed by Pope Woodhead and Associates and has a conflict of interest related to employment and has provided consultancy to multiple global pharmaceutical companies. Drs. Basch, Cleeland, Flamion and Edwards have no conflicts of interest that are directly relevant to the content of this manuscript.
Footnotes
On behalf of the Patient-Reported Outcomes Safety Event Reporting (PROSPER) Consortium.
References
- 1.Basch E. Systematic collection of patient-reported adverse drug reactions: a path to patient-centred pharmacovigilance. Drug Saf. 2013;36(4):277–278. doi: 10.1007/s40264-013-0026-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leone R, Moretti U, D’Incau P, Conforti A, Magro L, Lora R, et al. Effect of pharmacist involvement on patient reporting of adverse drug reactions: first Italian study. Drug Saf. 2013;36(4):267–276. doi: 10.1007/s40264-013-0028-8. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee AK, Ingate S. Web-based patient-reported outcomes in drug safety and risk management: challenges and opportunities? Drug Saf. 2012;35(6):437–446. doi: 10.2165/11632390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Calvert M, Blazeby J, Altman DG, Revicki DA, Moher D, Brundage MD. Reporting of patient-reported outcomes in randomized trials: the CONSORT PRO extension. JAMA. 2013;309(8):814–822. doi: 10.1001/jama.2013.879. [DOI] [PubMed] [Google Scholar]
- 5.Macefield RC, Avery KN, Blazeby JM. Integration of clinical and patient-reported outcomes in surgical oncology. Br J Surg. 2013;100(1):28–37. doi: 10.1002/bjs.8989. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee AK. Patient-reported outcome in colorectal health and disease. Colorectal Dis. 2012;14(12):1441–1444. doi: 10.1111/codi.12019. [DOI] [PubMed] [Google Scholar]
- 7.Cirillo M, Venturini M, Ciccarelli L, Coati F, Bortolami O, Verlato G. Clinician versus nurse symptom reporting using the National Cancer Institute-Common Terminology Criteria for Adverse Events during chemotherapy: results of a comparison based on patient’s self-reported questionnaire. Ann Oncol. 2009;20(12):1929–1935. doi: 10.1093/annonc/mdp287. [DOI] [PubMed] [Google Scholar]
- 8.Herxheimer A, Crombag R, Alves TL. Direct patient reporting of adverse drug reactions—a fifteen-country survey & literature review. 2010. http://www.haiweb.org/10052010/10_May_2010_Report_Direct_Patient_Reporting_of_ADRs.pdf. Accessed 22 Aug 2013.
- 9.van HF, Harmark L, Pal S, Olsson S, van GK. Experiences with adverse drug reaction reporting by patients: an 11-country survey. Drug Saf. 2012;35(1):45–60. [DOI] [PubMed]
- 10.Swan M. Emerging patient-driven health care models: an examination of health social networks, consumer personalized medicine and quantified self-tracking. Int J Environ Res Public Health. 2009;6(2):492–525. doi: 10.3390/ijerph6020492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chee BW, Berlin R, Schatz B. Predicting adverse drug events from personal health messages. 2013. http://www.ncbinlmnih.gov/pmc/articles/PMC3243174/. Accessed 22 Aug. 2013. [PMC free article] [PubMed]
- 12.Greenhalgh J, Abhyankar P, McCluskey S, Takeuchi E, Velikova G. How do doctors refer to patient-reported outcome measures (PROMS) in oncology consultations? Qual Life Res. 2013;22(5):939–950. doi: 10.1007/s11136-012-0218-3. [DOI] [PubMed] [Google Scholar]
- 13.Eschenhagen T, Force T, Ewer MS, de Keulenaer GW, Suter TM, Anker SD, et al. Cardiovascular side effects of cancer therapies: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2011;13(1):1–10. doi: 10.1093/eurjhf/hfq213. [DOI] [PubMed] [Google Scholar]
- 14.Buckman, RA, Berman, HK, Sridhar, SS, Joshua, AM. How much do the side effects of chemotherapy matter? Patients’ attitudes to side effects versus a potential loss of duration of remission. J Clin Oncol. 2010;28(Suppl 15):e19585.
- 15.European Medicines Agency (EMA). Guideline on good pharmacovigilance practices (GVP) Annex I—Definitions . 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Template_or_form/2013/05/WC500143294.pdf. Accessed 22 Sept 2013.
- 16.U.S. Food and Drug Administration (FDA). Guidance for industry. Patient-reported outcome measures: use in medical product development to support labeling claims. 2009. http://www.fda.gov/downloads/drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf. Accessed 23 Sept 2013. [DOI] [PMC free article] [PubMed]
- 17.Committee for Medicinal Products For Human Use (CHMP). Reflection paper on the regulatory guidance for the use of health related quality of life (HRQL) measures in the evaluation of medicinal products. 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003637.pdf. Accessed 22 Aug 2013.
- 18.Basch E. Patient-reported outcomes in drug safety evaluation. Ann Oncol. 2009;20(12):1905–1906. doi: 10.1093/annonc/mdp542. [DOI] [PubMed] [Google Scholar]
- 19.Basch E, Bennett A, Pietanza MC. Use of patient-reported outcomes to improve the predictive accuracy of clinician-reported adverse events. J Natl Cancer Inst. 2011;103(24):1808–1810. doi: 10.1093/jnci/djr493. [DOI] [PubMed] [Google Scholar]
- 20.The future of drug safety: promoting and protecting the health of the public. Washington DC: The National Academies Press; 2007. [Google Scholar]
- 21.Fromme EK, Eilers KM, Mori M, Hsieh YC, Beer TM. How accurate is clinician reporting of chemotherapy adverse effects? A comparison with patient-reported symptoms from the Quality-of-Life Questionnaire C30. J Clin Oncol. 2004;22(17):3485–3490. doi: 10.1200/JCO.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 22.Hazell L, Shakir SA. Under-reporting of adverse drug reactions: a systematic review. Drug Saf. 2006;29(5):385–396. doi: 10.2165/00002018-200629050-00003. [DOI] [PubMed] [Google Scholar]
- 23.Strom BL, Kimmel SE. Textbook of pharmacoepidemiology. 1. New Jersey: Wiley; 2006. [Google Scholar]
- 24.Pakhomov SV, Jacobsen SJ, Chute CG, Roger VL. Agreement between patient-reported symptoms and their documentation in the medical record. Am J Manag Care. 2008;14(8):530–539. [PMC free article] [PubMed] [Google Scholar]
- 25.Weingart SN, Gandhi TK, Seger AC, Seger DL, Borus J, Burdick E, et al. Patient-reported medication symptoms in primary care. Arch Intern Med. 2005;165(2):234–240. doi: 10.1001/archinte.165.2.234. [DOI] [PubMed] [Google Scholar]
- 26.Avery AJ, Anderson C, Bond CM, Fortnum H, Gifford A, Hannaford PC, et al. Evaluation of patient reporting of adverse drug reactions to the UK ‘Yellow Card Scheme’: literature review, descriptive and qualitative analyses, and questionnaire surveys. Health Technol Assess. 2011;15(20):1–234, iii–iv. [DOI] [PubMed]
- 27.European Medicines Agency (EMA). Guideline on good pharmacovigilance practices (GVP) Module VI—Management and reporting of adverse reactions to medicinal products 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129135.pdf. Accessed 22 Aug 2013.
- 28.National Cancer Institute. Patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). 2010. http://outcomes.cancer.gov/tools/pro-ctcae.html. Accessed 22 Aug 2013.
- 29.Lester RT, Ritvo P, Mills EJ, Kariri A, Karanja S, Chung MH, et al. Effects of a mobile phone short message service on antiretroviral treatment adherence in Kenya (WelTel Kenya1): a randomised trial. Lancet. 2010;376(9755):1838–1845. doi: 10.1016/S0140-6736(10)61997-6. [DOI] [PubMed] [Google Scholar]
- 30.Quinten C, Maringwa J, Gotay CC, Martinelli F, Coens C, Reeve BB, et al. Patient self-reports of symptoms and clinician ratings as predictors of overall cancer survival. J Natl Cancer Inst. 2011;103(24):1851–1858. doi: 10.1093/jnci/djr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basch E. The missing voice of patients in drug-safety reporting. N Engl J Med. 2010;362(10):865–869. doi: 10.1056/NEJMp0911494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg RM, Sargent DJ, Morton RF, Green E, Sanoff HK, McLeod H, et al. NCCTG Study N9741: leveraging learning from an NCI Cooperative Group phase III trial. Oncologist. 2009;14(10):970–978. doi: 10.1634/theoncologist.2009-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Quality Forum. Patient reported outcomes (PROs) workshop #1. Workshop Summary July 2012. http://www.qualityforum.org/Projects/n-r/Patient-Reported_Outcomes/Patient-Reported_Outcomes.aspx#t=2&s=&p=.
- 34.Jordan S. Managing adverse drug reactions: an orphan task. J Adv Nurs. 2002;38(5):437–448. doi: 10.1046/j.1365-2648.2002.02205.x. [DOI] [PubMed] [Google Scholar]
- 35.Jordan S, Knight J, Pointon D. Monitoring adverse drug reactions: scales, profiles, and checklists. Int Nurs Rev. 2004;51(4):208–221. doi: 10.1111/j.1466-7657.2004.00251.x. [DOI] [PubMed] [Google Scholar]
- 36.Gabe ME, Jordan SE. Development and clinical gains of nurse-led medication monitoring profiles. J Nurs Manag. 2013. doi:10.1111/jonm.12067. [DOI] [PubMed]
- 37.Wang XS, Williams LA, Eng C, Mendoza TR, Shah NA, Kirkendoll KJ, et al. Validation and application of a module of the M. D. Anderson Symptom Inventory for Measuring Multiple Symptoms in Patients with Gastrointestinal Cancer (the MDASI-GI) Cancer. 2010;116(8):2053–2063. doi: 10.1002/cncr.24920. [DOI] [PubMed] [Google Scholar]
- 38.Bergvall T, Nilsson J, Hill R, Noren GN, Lindquist M. Assessing systematic variability in the quality of individual case safety reports. Pharmacoepidemiol Drug Saf. 2010;19:184. [Google Scholar]
- 39.Lindquist M. Use of triage strategies in the WHO signal-detection process. Drug Saf. 2007;30(7):635–637. doi: 10.2165/00002018-200730070-00014. [DOI] [PubMed] [Google Scholar]
- 40.Committee for Proprietary Medicinal Products (CPMP). Points to consider on multiplicity issues in clinical trials 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003640.pdf. Accessed 22 Aug 2013.
- 41.Osoba D, King M. Meaningful differences. In: Fayers P, Hays R, editors. Assessing quality of life in clinical trials. Oxford: Oxford University Press; 1995. pp. 243–258. [Google Scholar]
- 42.Detmar SB, Muller MJ, Schornagel JH, Wever LD, Aaronson NK. Health-related quality-of-life assessments and patient-physician communication: a randomized controlled trial. JAMA. 2002;288(23):3027–3034. doi: 10.1001/jama.288.23.3027. [DOI] [PubMed] [Google Scholar]
- 43.Reeve BB, Fayers P. Applying item response theory modelling for evaluating questionnaire item and scale properties. In: Fayers P, Hays R, editors. Assessing quality of life in clinical trials. Oxford: Oxford University Press; 1995. pp. 55–73. [Google Scholar]
- 44.Jaeschke R, Singer J, Guyatt GH. Measurement of health status. Ascertaining the minimal clinically important difference. Control Clin Trials. 1989;10(4):407–415. doi: 10.1016/0197-2456(89)90005-6. [DOI] [PubMed] [Google Scholar]
- 45.White RW, Tatonetti NP, Shah NH, Altman RB, Horvitz E. Web-scale pharmacovigilance: listening to signals from the crowd. J Am Med Inform Assoc. 2013;20(3):404–408. doi: 10.1136/amiajnl-2012-001482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.CIOMS. Practical aspects of signal detection in pharmacovigilance. 1st ed. Geneva: CIOMS; 2010.
- 47.Ryan PB, Madigan D, Stang PE, Overhage JM, Racoosin JA, Hartzema AG. Empirical assessment of methods for risk identification in healthcare data: results from the experiments of the Observational Medical Outcomes Partnership. Stat Med. 2012;31(30):4401–4415. doi: 10.1002/sim.5620. [DOI] [PubMed] [Google Scholar]
- 48.Schuemie MJ. Methods for drug safety signal detection in longitudinal observational databases: LGPS and LEOPARD. Pharmacoepidemiol Drug Saf. 2011;20(3):292–299. doi: 10.1002/pds.2051. [DOI] [PubMed] [Google Scholar]
- 49.Noren GN, Hopstadius J, Bate A, Edwards IR. Safety surveillance of longitudinal databases: methodological considerations. Pharmacoepidemiol Drug Saf. 2011;20(7):714–717. doi: 10.1002/pds.2151. [DOI] [PubMed] [Google Scholar]
- 50.Caster O, Noren GN, Madigan D, Bate A. Large-scale regression-based pattern discovery: the example of screening the WHO global drug safety database. Stat Anal Data Min. 2010;3(4):197–208. [Google Scholar]
- 51.Noren GN, Bate A, Hopstadius J, Star K, Edwards IR. Temporal pattern discovery for trends and transient effects: its application to patient records. In: Proceedings of the Fourteenth International Conference on Knowledge Discovery and Data Mining SIGKDD 2008. New York: ACM; 2008. p. 963–71.
- 52.Bate A, Lindquist M, Edwards IR, Olsson S, Orre R, Lansner A, et al. A Bayesian neural network method for adverse drug reaction signal generation. Eur J Clin Pharmacol. 1998;54(4):315–321. doi: 10.1007/s002280050466. [DOI] [PubMed] [Google Scholar]
- 53.Stahl M, Lindquist M, Edwards IR, Brown EG. Introducing triage logic as a new strategy for the detection of signals in the WHO Drug Monitoring Database. Pharmacoepidemiol Drug Saf. 2004;13(6):355–363. doi: 10.1002/pds.894. [DOI] [PubMed] [Google Scholar]
- 54.Coulter DM, Bate A, Meyboom RH, Lindquist M, Edwards IR. Antipsychotic drugs and heart muscle disorder in international pharmacovigilance: data mining study. BMJ. 2001;322(7296):1207–1209. doi: 10.1136/bmj.322.7296.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanz EJ, De-las-Cuevas C, Kiuru A, Bate A, Edwards R. Selective serotonin reuptake inhibitors in pregnant women and neonatal withdrawal syndrome: a database analysis. Lancet. 2005;365(9458):482–487. doi: 10.1016/S0140-6736(05)17865-9. [DOI] [PubMed] [Google Scholar]
- 56.Noren GN. Statistical methods for knowledge discovery in adverse drug reaction surveillance. Sweden: Stockholm University; 2007.
- 57.Stang PE, Ryan PB, Racoosin JA, Overhage JM, Hartzema AG, Reich C, et al. Advancing the science for active surveillance: rationale and design for the Observational Medical Outcomes Partnership. Ann Intern Med. 2010;153(9):600–606. doi: 10.7326/0003-4819-153-9-201011020-00010. [DOI] [PubMed] [Google Scholar]
- 58.Egberts TC, Smulders M, de Koning FH, Meyboom RH, Leufkens HG. Can adverse drug reactions be detected earlier? A comparison of reports by patients and professionals. BMJ. 1996;313(7056):530–531. doi: 10.1136/bmj.313.7056.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.