

Abstract
Metabolic reprogramming is a key event in tumorigenesis to support cell growth, and cancer cells frequently become both highly glycolytic and glutamine dependent. Similarly, T lymphocytes (T cells) modify their metabolism after activation by foreign antigens to shift from an energetically efficient oxidative metabolism to a highly glycolytic and glutamine-dependent metabolic program. This metabolic transition enables T cell growth, proliferation, and differentiation. In both activated T cells and cancer cells metabolic reprogramming is achieved by similar mechanisms and offers similar survival and cell growth advantages. Activated T cells thus present a useful model with which to study the development of tumor metabolism. Here, we review the metabolic similarities and distinctions between activated T cells and cancer cells, and discuss both the common signaling pathways and master metabolic regulators that lead to metabolic rewiring. Ultimately, understanding how and why T cells adopt a cancer cell-like metabolic profile may identify new therapeutic strategies to selectively target tumor metabolism or inflammatory immune responses.
Keywords: Cancer, Lymphocyte, Metabolism, Aerobic glycolysis
Review
The mid-twentieth century has been described as the ‘golden age of intermediary metabolism’ [1], with the work of Krebs, Lippman, Crane and others greatly advancing our understanding of cellular metabolic pathways. In the past decade interest in cell metabolism has been rejuvenated in several fields, especially cancer biology and lymphocyte immunology. In cancer biology, this renaissance has been driven by evidence that cancer metabolism presents an underexploited therapeutic target. Immunologists have been drawn to metabolic studies with the realization that the metabolism of T lymphocytes (T cells) is intimately tied to immunity [2]. Functionally, T cells and tumors have little in common; the former protects against invasive pathogens, the latter is a diseased tissue characterized by the accumulation of abnormal cells. However, both T cells and cancer cells have strong proliferative signals and undergo metabolic reprogramming during their respective life cycles, and there exist clear functional and mechanistic similarities between the reprogramming events in each cell type. These similarities make lymphocyte metabolic reprogramming a useful model with which to discover how and why tumors rewire their metabolism. The purpose of this review is to highlight and discuss the similarities and distinctions in how T cells and tumor cells solve similar metabolic problems.
T lymphocyte activation: a key lifestyle switch
Because of its inherently destructive nature, the immune system must be maintained in a quiescent state. To provide protection from pathogens, however, it must remain capable of rapid responses and effector function. This challenge is solved with a diverse pool of naïve lymphocytes that can quickly activate to produce a large, clonal pool of rapidly proliferating effector T cells. Naïve T cells express near-unique T cell antigen receptors (TCR) that are randomly generated through V(D)J recombination and pre-selected to recognize foreign antigens presented on major histocompatibility complexes (MHC). These naïve cells continually circulate the blood and lymphatic system sampling MHC-peptide complexes. Upon encounter with an antigen-presenting cell (APC) and cognate antigen, the T cell ceases to migrate, forming a prolonged contact with the APC. This induces sustained signaling through the TCR and other co-receptors, inducing T cell activation, proliferation and differentiation into effector cells. These effectors rapidly accumulate and migrate to sites of inflammation, ultimately clearing the invader [3].
Activation therefore simultaneously places T cells under several types of stress: they must proliferate rapidly; they must synthesize large amounts of effector proteins; and they have to prepare to enter a heterogeneous and potentially hypoxic, nutrient poor environment. Each of these stressors has a significant metabolic aspect reminiscent of the classic cancer metabolism paradigm: proliferation and anabolism require energy, biosynthetic building blocks and reducing equivalents, while nutrient stress and hypoxia both potentially limit metabolic flux by restricting metabolite access and oxygen. With similar metabolic demands and stresses, it is not surprising that these diverse cell types respond by adopting a similar metabolic profile.
A common metabolic solution: aerobic glycolysis
Three metabolic pathways are central to ATP production in proliferative lymphocytes and cancer cells: glycolysis, the tri-carboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS). While the TCA cycle does not directly generate ATP, it is inexorably linked to OXPHOS, providing several metabolic inputs to drive ATP production. In addition, intermediate metabolites from both the TCA cycle and glycolysis can be used as carbon sources for catabolic pathways producing cholesterol, lipids, ribose, and other biosynthetic molecules (Figure 1) [4]. Resting or non-proliferative cells often rely on mitochondrial lipid β-oxidation. Proliferative cells, in contrast, generally decrease lipid oxidation and instead conserve lipids to support cell growth [5].
Figure 1.

Major metabolic fates of glucose in highly proliferative cells. Glucose is taken into the cell by GLUT family transporters and then phosphorylated by hexokinases, trapping it within the cell as glucose-6-phosphate (G6P). G6P can be catabolized via glycolysis or used as a carbon donor for the synthesis of riboses via the pentose phosphate pathway (PPP). Catabolized G6P generates pyruvate plus small quantities of ATP, with much of the resultant pyruvate being converted to lactate by lactate dehydrogenase and then secreted through mono-carboxylic transporters (MCT). The remaining pyruvate is converted to acetyl-CoenzymeA (acetyl-CoA) by pyruvate dehydrogenase and used either as fuel for ATP production via the tri-carboxylic acid (TCA) cycle and oxidative phosphorylation or converted to fatty acids to generate structural lipids. At various points during glycolysis and the TCA cycle reaction intermediates can be removed to provide carbon for amino acid biosynthesis (not shown).
For mammalian cells that lack significant intracellular nutrient stores, extracellular glucose uptake represents a major carbon and energy source. Glucose is transported through facilitative glucose transporters and phosphorylated by hexokinases to initiate metabolic pathways and prevent its exit. Glucose-6-phosphate (G6P) is primarily metabolized through glycolysis or the pentose phosphate pathway (PPP). Glycolysis provides a small net ATP gain per glucose molecule consumed and yields pyruvate which can then either be: i) reduced to lactate by lactate dehydrogenase (LDH), concomitantly restoring NADH to NAD+, ii) converted to alanine by alanine aminotransferase, simultaneously converting glutamine to α-ketoglutarate, or iii) converted to acetyl-CoenzymeA (acetyl-CoA) in the mitochondria to be oxidized via the TCA cycle, generating large amounts of ATP via OXPHOS (respiration). Most non-proliferating cells utilize this latter pathway when oxygen is available in a process termed the Pasteur effect.
Not all cells, however, exhibit the Pasteur effect and cease lactate production under aerobic conditions. In the early 20th century, Otto Warburg observed that many tumor cells and tumor sections continued lactate secretion in the presence of oxygen [6]. This metabolic program is referred to as aerobic glycolysis, differentiating it from the obligatory fermentation of glucose to lactate that occurs under anaerobic conditions where no oxygen is available to fuel OXPHOS. Warburg postulated that the switch towards aerobic glycolysis arose from faults in respiration and that such defects were the primary cause of cancer [6,7]. While his observations stand, his proposed mechanism for aerobic glycolysis has now largely been discounted following studies demonstrating that cancer cells often have grossly normal respiratory function [8-10] and, indeed, can exhibit elevated rates of respiration [11]. Nevertheless, mitochondrial mutations are associated with some cancers and the relationships between aerobic glycolysis, mitochondrial function and tumorigenesis remain controversial [12].
Similar to his observations of aerobic glycolysis in cancer cells, in 1958 Warburg also found that stimulated leukocytes become highly glycolytic [13]. Subsequent reports in the 1970s to 1990s, using lectin-stimulated rat thymocytes and lymphocytes, also showed lymphocytes become glycolytic upon activation. Together, these studies demonstrated that resting lymphocytes obtain most of their ATP by OXPHOS of glucose, amino acids, and lipids. However, within hours of stimulation, lymphocytes begin to increase glucose uptake up to forty- or fifty-fold and to secrete most of the glucose-liberated carbon as lactate [14] (Figure 2). In parallel, lymphocytes increase oxygen consumption by around 60% [15-19]. These data have subsequently been confirmed using purified T cell populations stimulated with antibodies that trigger the TCR complex and associated co-receptors [20,21]. Importantly, this increase in aerobic glycolysis precedes and has been shown to be essential for the growth and proliferation of stimulated T cells [21-23].
Figure 2.

T cell activation results in metabolic reprogramming. Naïve T cells have an oxidative metabolism, using glucose, glutamine, and fatty acids as fuel sources. The majority of ATP is generated via oxidative phosphorylation. Following activation by stimulation of the T cell receptor and co-receptors, the cells adopt a metabolic profile that resembles the metabolism of many cancer cells, consuming large quantities of both glucose and glutamine but performing relatively little oxidative phosphorylation. The majority of glucose-derived carbon is secreted as lactate, with the remainder being used for biosynthesis.
Cancer cells and T cells are not metabolically unique, and the induction of aerobic glycolysis has also been reported during proliferation of other non-transformed cells. For example, a similar phenotype is also observed in both intestinal cells and fibroblasts during logarithmic growth [4,24]. However, few other cell types have shown such a distinct and acute induction of aerobic glycolysis from a near proliferative and metabolic standstill. T cell activation, therefore, provides a unique model to explore how and why metabolic rewiring occurs in cancer cells.
Aerobic glycolysis supports rapid proliferation
The metabolic needs of T cells change dramatically upon activation. Before encountering pathogens, resting T cells require only sufficient energy to support basal cellular needs and replacement biosynthesis. After activation, T cells undergo a transient period with little cell growth and then begin to rapidly grow and divide. T cells specific for a given MHC-antigen complex are rare [25,26], so clonal expansion must rapidly expand these small populations of hundreds of cells to the tens or hundreds of millions of cells necessary for protection. Remarkably, activated T cell doubling times of 4 to 6h have been observed in vitro[27], with even faster doubling rates reported in vivo[28,29]. Activated T cells, therefore, have a tremendous need for both ATP [30] and biosynthetic capacity to synthesize new proteins, lipids, and nucleic acids.
While a hallmark of cancer is cell cycle deregulation, there is little propensity for tumor cells to adopt increasingly rapid rates of cell division like activated T cells. Indeed, the majority of cells within a solid tumor may be in a state of G1 cell cycle arrest [31]. Extensive clinical studies have shown that although cell cycle length in tumors is more diverse than non-cancerous tissue, the median S-phase length across all tumor types is around 10 h [32] and, counter-intuitively, there is no clear relationship between proliferative ability and tumor aggressiveness [33]. Still, proliferation of cancer cells must exceed cell death to allow tumor growth. Thus, with the exception of an alternate glycolytic pathway in which tumor cells may bypass pyruvate kinase to convert phosphoenol pyruvate to pyruvate, and yield no net gain of ATP [34], activated T cells and tumor cells harness aerobic glycolysis to provide ATP and biosynthesis for proliferation.
Advantages of aerobic glycolysis: rapid ATP production
In contrast to OXPHOS, glycolysis is energetically inefficient, theoretically yielding only two molecules of ATP per glucose molecule consumed compared to up to thirty-six if fully oxidized. This is not a trivial issue as cancer cells have been shown to possess additional, unused respiratory capacity [8,35,36]. Thus, cancer cells do not increase glycolysis solely because their capacity for OXPHOS is saturated. Rather, aerobic glycolysis and basal OXPHOS provide sufficient energy to support the cell survival and growth demands of cancer cells and activated T cells.
One energetic advantage of adopting aerobic glycolysis as a primary metabolic program is the speed at which ATP can be regenerated. While OXPHOS yields more ATP than glycolysis, there is a trade-off between yield and speed [37,38]. Indeed, as described by Koppenol and Bounds [39], Warburg and colleagues observed this phenomena as early as 1923, reporting that for every one molecule of glucose oxidized by respiration, twelve are metabolized by glycolysis. Increased glycolysis can boost ATP production rate by two-thirds, provided cells are not concerned with efficiency. While wasteful, therefore, the speed of aerobic glycolysis offers a selective advantage both to tumor cells competing against other cells within the same environment [37,40], and to T cells racing to suppress invading pathogens.
Advantages of aerobic glycolysis: biosynthesis
Beyond ATP production, glycolysis and the TCA cycle form the nexus for many biosynthetic processes. Carbon intermediates derived from glycolysis and the TCA cycle are used for the generation of amino acids, lipids, cholesterol and nucleotides. A major function of aerobic glycolysis, therefore, is to provide sufficient intermediates to fuel biosynthesis for proliferation and growth. Indeed, increased glucose uptake can enhance T cell responses and growth in vivo as mice transgenically overexpressing the glucose transporter GLUT1 in T cells accumulate effector T cells with age [22,41] and GLUT1 overexpression is correlated with poor prognosis in a variety of cancers [42].
Rapid glucose uptake fuels both glycolysis and the PPP, each of which provides numerous metabolites to support cell growth. Glycolysis is a major source of serine synthesis as well as pyruvate that can either be converted to lactate to replenish NAD+ or can be transported into the mitochondria to enter the TCA cycle as acetyl-CoA. From the TCA cycle, citrate can exit to the cytosol to provide a basis for lipid synthesis [21,43]. Under hypoxic conditions, glutamine can undergo reductive carboxylation to provide a reverse flow through the TCA cycle as a source of lipogenesis in both cancer cells and in CD8+ T cells [44]. Notably, both tumor cells [45] and lectin-stimulated lymphocytes [46,47] perform extensive de novo synthesis of lipids, and only limited lipid β-oxidation. In addition to de novo lipogenesis, aggressive cancer cell lines and primary tumors also perform extensive lipid remodeling, in part due to elevated monoacylglycerol lipase activity [48]. Tumor lipid metabolism can be further enhanced by Akt-driven expression of the low-density lipoprotein receptor (LDLR), which increases cholesterol intake and promotes cell growth [49]. The relative importance of each of these pathways to lymphocyte lipid metabolism has yet to be determined.
The PPP provides nicotinamide adenine dinucleotide phosphate (NADPH) reducing potential and generates ribose sugars that can be directed into TCA cycle intermediates and into purine, pyrimidine and aromatic amino acid synthesis pathways. The PPP are strongly induced in T cell activation [21] and can be important in cancer; indeed U-C14 glucose tracer experiments have suggested that in some tumor types over 80% of the nucleotides in DNA and RNA are synthesized from glucose-derived carbon [50,51]. Upregulation of the PPP is facilitated, in part, by increased enzyme expression. Activated T cells increase expression of PPP enzymes and high levels of PPP enzyme activity have been reported in metastatic tumor cells [52]. For example, glioblastoma expression of the transketolase TKTL1, the key enzyme linking the PPP to glycolysis, directly correlates with tumor severity in the clinic [53].
NADPH is a critical reducing agent in the synthesis of fatty acids and cholesterol as well as maintaining cellular redox status and control reactive oxygen species (ROS) produced by OXPHOS [54]. While some degree of ROS is beneficial for both T cell activation [55] and tumor development [56], excessive ROS leads to oxidative organelle damage and the induction of apoptosis. Strategies that drive cancer cells to increase the OXPHOS-glycolysis ratio, for example by increasing pyruvate dehydrogenase activity to drive mitochondrial conversion of pyruvate to acetyl-CoA, decrease both proliferation and growth [57]. Similarly, glucose restriction of activated lymphocytes induces an increase in OXPHOS, a drop in glycolysis, and an inhibition of proliferation [20,58]. In proliferating cells efficient OXPHOS should, therefore, be balanced by high PPP flux to prevent overloading the demand for NADPH.
Advantages of aerobic glycolysis: adaptation to the environment
Glycolysis and the TCA cycle are amphibolic and supply both ATP and intermediates to multiple pathways to potentially support cells under stress conditions. Indeed, we have shown that high rates of glycolysis can be protective against apoptosis [59,60]. A high rate of metabolic flux makes it thermodynamically less costly to redirect intermediates down different pathways, that is, high metabolic flux permits rapid rerouting of metabolites [61-63]. This control sensitivity may permit a faster response to specific nutrient deprivation as cells enter potentially nutrient-poor environments. This may explain why the rate of glucose consumption in both activated T cells and many tumor types appears in excess of that required to meet either the biosynthetic or energetic demands of the cell [64].
Further, glycolysis is not oxygen dependent, and so adopting a glycolytic metabolism can prepare cells for entry or survival in a hypoxic environment. Even after vascularization, solid tumors feature extensive hypoxic domains [65]. Similarly, lymph nodes [66], spleen [67], tumors, dermal/surgical wounds [68] and other regions frequented by activated lymphocytes contain extensive areas of low oxygen tension. Adaption of a highly glycolytic metabolism with low oxygen dependency may help both tumors and lymphocytes survive and proliferate during low oxygen availability.
Common mechanisms drive glycolytic reprogramming in T cells and tumors
Transporter expression and izozyme switching
A limiting step in glucose metabolism is the rate at which glucose can be captured and trapped within the cell. There are two major glucose transporter families, the Na+/glucose linked transporter (SGLT) symporters, and the GLUT family of passive transporters. Fourteen mammalian GLUT family transporters have been identified [69] and the major glucose transporters in lymphocytes appear to be GLUT1 and GLUT3, the expression levels of which increase significantly following activation [70]. Facilitated diffusion of glucose by the GLUTs requires a glucose gradient across the extracellular membrane. This so-called glucose sink is maintained by hexokinase phosphorylation of intracellular glucose. Following T cell activation, hexokinase activity increases significantly [71] and T cells undergo a switch in HK isozyme expression from HKI to HKII [72,73]. While both HKI and HKII both feature two potential catalytic domains, in HKI one of these is non-functional, thus HKII has a higher Km for both glucose and ATP compared to HKI [74]. Second, signals from the TCR and co-receptors drive HKI and HKII to bind mitochondria at porin (ATP-exporting) complexes [75]. This close coupling of HK and mitochondria provides HKII with access to a large pool of ATP.
Following lectin stimulation, lymphocytes also switch expression of other glycolytic isozymes. This includes induction of pyruvate kinase M2 (PKM2), LDH-A4, and enolase I [21,73]. These changes in expression are associated with increases in maximal glycolytic enzyme activity [16,72], and the relieving of allosteric inhibition that would otherwise limit glycolytic flux. One example of this is the regulation of the glycolytic enzyme 6-phosphofructo-1-kinase (PFK1), a key regulatory enzyme in glycolysis (Figure 3). PFK1 is allosterically inhibited by ATP and allosterically activated by fructose-2,6-bisphosphate (F26P2). F26P2 is generated by the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB), and in naïve lymphocytes PFKFB isoform 2 predominates. However, following activation T cells express large quantities of PFKFB isoform 3 [76,77]. PFKFB3 has a very low phosphatase activity compared to PFKFB2 [78], and so this isozyme switch enhances PFK1 flux by both increasing F26P2 and depleting ATP.
Figure 3.
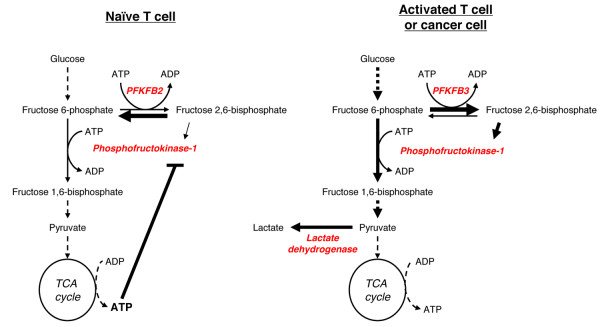
Glycolytic isozyme switching promotes high rates of glycolysis. Activated T cells, cancer cells and other highly proliferative cells express different glycolytic isozymes in comparison to quiescent cells, increasing glycolytic flux. One key step in glycolysis is the phosphorylation of fructose 6-phosphate by phosphofructokinase-1 (PFK-1). PFK-1 is allosterically activated by fructose 2,6-bisphosphate and allosterically inhibited by ATP. Both activated T cells and tumor cells express isoform 3 of the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB). In contrast, naïve T cells express PFKFB isoform 2. PFKFB3 differs from PFKFB2 in that it has low phosphatase activity, leading to the accumulation of fructose 2,6-bisphosphate and localized depletion of ATP. This results in increased PFK-1 activity and higher rates of glycolysis.
Cancer cells also show a general increase in glycolytic enzyme activity and expression of specific isozymes. This includes expression of HKII, LDH-A and PFKFB3 [52,79,80]. Tumor cells express PKM2, but there is now strong evidence that this is largely in the metabolically inactive, dimeric form, rather than the active tetramer [81]. In many tumor cells PKM2 activity is further inhibited by direct tyrosine phosphorylation and by the binding of phosphotyrosine containing peptides, both of which restrict cofactor binding. Reduced PKM2 activity enhances aerobic glycolysis and tumor growth [82,83]. Cascades of tyrosine phosphorylation are central to T cell activation; however, it has yet to be determined if these cascades result in PKM2 inhibition. Recent reports in tumor cells have demonstrated that PKM2 can be selectively degraded in an acetylation-dependent fashion at times of high glucose availability [84], allowing additional glycolytic intermediates to be used for biosynthesis. Phosphoenol-pyruvate flux through a non-ATP generating pathway may then sustain rapid pyruvate generation while preventing ATP-driven feedback inhibition of glycolysis [34]. This regulatory loop for PKM2 may represent a further mechanism to maintain high rates of glycolytic flux, but this has yet to be examined in activated lymphocytes.
Beyond glucose metabolism: glutamine
Glutamine has multiple metabolic fates, being used for ATP regeneration, anaplerosis of the TCA cycle, and redox regulation. Within the cell glutamine is readily converted to glutamate by glutaminase. Glutamate is used together with cysteine and glycine to generate glutathione, is used for lipid synthesis through reductive carboxylation under hypoxia, and is a major nitrogen donor during purine and pyrimidine synthesis. Naïve lymphocytes utilize glutamine as a primary oxidative fuel for ATP generation. Following T cell activation, cMyc greatly increases the expression of glutaminolysis enzymes and the rate of glutamine uptake [15,21]. After conversion to glutamate, glutamate dehydrogenase generates α-ketoglutarate to support the TCA cycle. Notably, while the early stages of lymphocyte activation do not require glutamine, subsequent proliferation and the expression of effector cytokines following TCR stimulation correlate directly with glutamine availability [85-87], and there is clinical evidence to suggest that glutamine availability can be a limiting factor in lymphocyte activation during inflammatory responses [88-90].
Many tumor types exhibit high rates of glutamine consumption relative to non-transformed cells [91-93]. Cancers driven by oncogenic cMyc, for example, become highly dependent on glutamine [94,95] and can be exquisitely sensitive to glutamine deprivation [96]. Other tumors, however, can exhibit little sensitivity to glutamine deprivation [93,97-99]. This resistance to glutamine deprivation may relate to the induction of glutamine synthase in these cells, and so although less dependent on exogenous glutamine, they still exhibit high rates of glutamate flux. Also, expression of pyruvate carboxylase can allow glucose-derived pyruvate to convert to oxaloacetate to support the TCA cycle and maintain export of citrate for lipid synthesis through anapleurosis [100]. Given these potential differences, activated T cells may represent a better metabolic model for primarily glutamine-dependent tumors.
Common signaling events drive metabolic reprogramming
The cancer metabolism phenotype is ultimately initiated by oncogenic signaling events that induce metabolic gene expression and stimulate aerobic glycolysis. Importantly, T cell receptor and co-receptor engagement are now well understood and activate many of these same signaling pathways (see Smith-Garvin et al., 2009, for a detailed review [101]). Briefly, the TCR is associated with several CD3 accessory chains and when the TCR is engaged, tyrosine phosphorylation of accessory chains recruits kinases and scaffold proteins. This recruitment, along with co-stimulation, triggers localized stimulation of three signaling pathways: calcium flux, MAPK (ERK/p38) signaling, and phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3) signaling. Autocrine and paracrine cytokine signaling loops induce further PI(3,4,5)P3 and MAPK activation, along with JAK/STAT signaling. Notably, several of the downstream targets of these pathways regulate key metabolic regulators, with mutations in components downstream of these pathways strongly implicated in oncogenesis. Identifying the specific signaling pathways in activated T cells that induce metabolic reprogramming is therefore informative in identifying the oncogenes involved in driving the same processes in tumors.
PI3K, PTEN, Akt and mTORC1
PI(3,4,5)P3 is generated by phosphatidylinositol-3-kinase (PI3K) and depleted by phosphatases such as the tumor suppressor, PTEN (phosphatase and tensin homologue deleted on chromosome 10). Both sides of this signaling equilibrium can impact cancer, as activating PI3K and disrupting PTEN mutations frequently promote constitutive signaling through PI(3,4,5)P3-dependent pathways [102]. Of the downstream targets for PI(3,4,5)P3 signaling, the best described is Akt, an established metabolic regulator in both tumors and lymphocytes. In hematopoietic cells and naïve T cells, the expression of a constitutively active Akt leads to increased GLUT1 surface localization, improved coupling of HKII to the mitochondria and increased rates of glycolysis [20,103,104]. Similarly, in tumor models Akt drives cells towards aerobic glycolysis and makes cells highly dependent on exogenous glucose for survival [105].
Akt promotes aerobic glycolysis by direct phosphorylation and activation of glycolytic enzymes, such as PFK2 [106], by phosphorylation of TBC1D1/4 to regulate GLUT1 trafficking, and by regulating several transcription factors (reviewed in detail by Manning and Cantley, 2007) [107]. Further, Akt is able to activate mTORC1 (mammalian target of rapamycin complex 1) via phosphorylation of upstream regulators PRAS40 and TSC2. mTORC1 is a key driver of anabolic metabolism. Indeed, activating the PI3K/Akt pathway can be considered a key regulator of glucose metabolism in both T cells and cancer [108]. Inhibition of this pathway in T cells is potently immunosuppressive and leads to generation of tolerant or regulatory T cells rather than effectors. Given the frequency of cancer-associated mutations in this pathway, delineating how PI(3,4,5)P3 signaling leads to metabolic reprogramming in lymphocytes may provide a unique opportunity to understand the regulation of cancer metabolism.
MAPK and HIF1α
The multifactorial roles of the mitogenic ras-MAPK signaling pathways in cancer have been extensively reviewed recently [109-111]. MAPK have multiple roles in metabolic regulation in both tumors [112] and during T cell activation [71,87]. One mechanistic role of recent interest is MAPK regulation of hypoxia inducible factor 1α (HIF1α). HIF1α is a heterodimeric transcription factor that induces gene expression in response to hypoxia. HIF1α induces the expression of many glycolytic genes, and HIF1α can be a key mediator of the Pasteur effect in normal cells [113]. HIF1α protein levels are elevated without the need for hypoxia by PI(3,4,5)P3 signaling through mTOR and other pathways. Activated T cells and many tumor cells, therefore, can exhibit elevated levels of HIF1α. MAPK, however, also play a key role in enhancing HIF1α transcriptional ability, by enhancing HIF1α interactions with transcriptional co-factors [114].
HIF1α is not strongly expressed in normal tissues under normoxic conditions and presents a potential therapeutic target to selectively suppress tumor glucose metabolism. In support of this strategy, several studies have reported that HIF1α null tumor xenografts show reduced growth, while overexpression of xenograft HIFα promotes increased growth [115]. Curiously, and in contrast to these data, HIF1α−/− T cells exhibit normal proliferative and initial metabolic responses to TCR and co-receptor stimulation [116,117]. Instead, the impact of HIF1α loss is only apparent when activated HIF1α−/− T cells are subsequently skewed to different cell fates. HIF1α−/− CD4+ T cells are unable to form interleuken-17 (IL-17) producing T helper cells, which are highly glycolytic. Instead, HIF1α−/− T cells become immunosuppressive regulatory T cells in which lipid metabolism, not glycolysis, is the major metabolic program [41,117]. The role of HIF1α in metabolic regulation is therefore limited during T cell activation. Determining the signaling context by which T cell skewing directs HIF1α regulation of metabolism may, however, be informative in determining how HIF1α functions in tumors.
JAK/STATs and the PIM kinases
T cell activation induced metabolism is maintained by sustained signaling from IL-2 and other cytokines acting on common gamma chain (γc) cytokine receptor complexes. This effect is in part mediated by direct and STAT5 driven PI(3,4,5)P3/Akt signaling [118,119]. However, additional STAT driven, Akt-independent, signaling events also play a role. Of note, JAK/STAT3 signaling in lymphocytes induces the expression of the PIM family of kinases, which themselves can promote glycolytic metabolism [120].
PIM kinases are constitutively active [121] and are potent oncogenes, being induced by, and synergizing with, the transcription factor cMyc in several cancer types [122]. In addition, persistent STAT3 signaling is common in many tumor types. While oncogenic STAT3 mutations have not been reported, aberrant STAT3 signaling can arise from inactivation of the STAT-suppressing suppressor of cytokine signaling (SOCS) proteins or by elevated activation of JAKs [123]. The γc-receptor-directed maintenance of activated T cell metabolism, therefore, potentially presents a useful tool with which to study the role of STAT-driven, PIM-mediated, regulation of metabolism. Unfortunately, the PIMs share substrate specificity with Akt [120], and are inhibited by the classical PI3K inhibitor LY294002, a compound historically used to study Akt function [124]. The specific role of PIM kinases in metabolic reprogramming is hence unclear. Studies of activated, PIM-null T cells [125] may help resolve this issue.
Calcium signaling and AMPK
Immediately after TCR activation there is a coordinated flux of calcium from intracellular stores and also an increase in mitochondrial calcium uptake [126]. These changes stimulate the calcium-activated mitochondrial dehydrogenases that drive the TCA cycle [127]. In addition, calcium flux downstream of the TCR causes a short term phosphorylation of AMP activated protein kinase (AMPK) [128], a master metabolic regulator that promotes catabolic pathways when the ATP-AMP ratio falls. AMPK is activated by binding of AMP and when phosphorylated by CaMKKβ or the tumor suppressor LKB1 [129]. While the metabolic impact of AMPK activation by the TCR has yet to be fully defined, calcium-induced AMPK activity during T cell activation may help to promote an initial phase of oxidative and ATP- generating metabolism. This could prepare T cells to enter a rapid growth phase and to resist the stress of nutrient-deficient conditions. The latter role may be particularly important as AMPK-null T cells show only a limited metabolic phenotype under nutrient-rich conditions, but fail to respond to metabolic stress in vitro[130]. In vivo, nutrients are potentially limiting in lymph nodes or inflamed tissues, and TCR-induced activation of AMPK may be important to maintain ATP levels and maximize survival, so that T cells can proceed to a later phase in which AMPK activity is reduced and rapid cell growth begins.
Although misregulation of calcium signaling can be important in tumorigenesis [131], direct regulation of tumor metabolism by calcium has not been studied in detail. Indeed, the role of AMPK in cancer metabolism is still controversial. While LKB1 has an established role as a tumor suppressor, LKB1 has a variety of substrates and how LKB1 tumor suppression relates to AMPK activation is unclear. AMPK activation has been proposed as being anti-tumorigenic, as it suppresses cell cycle progression and can oppose Akt activity by suppressing mTORC1 [132]. Recent data, however, indicates that transient AMPK activation in response to energy stress can promote tumor survival by maintaining NADPH homeostasis [133]. Understanding how AMPK activation supports activated T cells in vivo in times of metabolic stress may provide new clues as to the role of AMPK in tumor metabolism.
Limitations of T cells as a model for tumor metabolism
Metabolic reprogramming in activated T cells is a useful model to study the metabolic changes that occur during tumorigenesis. Indeed, many of the pathways are similar and approaches to disrupt cancer metabolism can also be quite immunosuppressive. However, the two systems have some significant differences that may provide useful insight into novel anti-cancer therapies.
T Cell metabolic reprogramming is both transient and reversible
Following activation, T cells can differentiate into effector, regulatory and memory T cells that have differing metabolic profiles [2,117,134]. Activated T cells are, therefore, metabolically flexible and not fixed into a specific metabolic program. Unlike cancer cells with specific oncogenic mutations, T cell metabolism is dependent on signaling pathways triggered by the local environment. Indeed, even once T cell functional and metabolic fate has been defined there is a degree of reversibility and plasticity, for example, lipid-dependent regulatory T cells can be redirected to form highly glycolytic, IL-17-producing cells by altering the cytokine environment [41,135]. In contrast, tumor cells are largely fixed on one metabolic route that is dictated by irreversible mutations in upstream signaling pathways. Thus, cancer cells have less metabolic flexibility than T cells and the response of each cell type to inhibition of specific metabolic pathways may lead to distinctly different outcomes.
Activated T cells are not tumorigenic
Despite the metabolic and other similarities between stimulated T cells and a cancer cell undergoing aerobic glycolysis, activated T cells are not cancerous. Instead, following clearance of an infection the vast majority of activated T cells will die due to activation-induced cell death or from cytokine neglect. Both activated T cells and tumor cells are kept alive by a precarious balance of pro- and anti-apoptotic BH3 domain-containing proteins. In lymphocytes this balance is maintained by cytokine signaling through Akt and other pathways, and, in addition, by glycolytic flux [136-139]. Within tumors this balance is maintained both by glycolytic flux and oncogenic signaling. Understanding how activated T cells die following the loss of glycolytic flux and cytokine signals may provide insight into how anti-metabolites kill, or fail to kill, cancer cells.
Tumor cells are metabolically and genetically diverse
It is becoming evident that while the phenomena of aerobic glycolysis is common to many tumors, different cancer cells, potentially even within the same tumor, are metabolically diverse. Even within cell lines established from the same type of tumor there exists significant metabolic variation [140,141]. This heterogeneity can be representative of cancer stage or subtype, as in prostate and breast cancer. Given the strong dependence of T cells on glutamine, activated T cells represent a better model for glutamine-addicted tumors, for example those driven by oncogenic Myc [21,95], than more glucose dependent tumors, for example those driven by Met [141]. More importantly, activated T cells themselves become metabolically diverse as they differentiate into specific effector or regulatory subsets [41]. These T cell differentiation pathways are regulated by specific signaling events and it will be interesting to determine if distinct T cell subtypes may represent specific cancer types or stages. This is an important consideration as the sensitivity of tumor cells to metabolic inhibitors varies depending on the oncogenes involved [142].
Conclusions
Cancer cells and activated T cells adopt comparable metabolic profiles to cope with similar environmental and proliferative stressors. Given that both T cell activation and tumorigenesis often resort to the same signaling pathways to induce this metabolic rewiring, T cell activation offers a useful model with which to study the mechanics of metabolic reprogramming. While cancer metabolism is inherently more diverse and susceptible to selective pressures, T cells have the significant advantage in a laboratory setting of being quiescent and non-cycling prior to activation, aiding in the delineation of cell signaling and cell cycle effects.
The aerobic glycolysis and glutamine dependency of cancer cells have been identified as potential novel targets for cancer therapy, and so developing an improved understanding of how these metabolic programs arise is of clinical importance. However, given the close similarity between activated T cell and tumor metabolic reprogramming, consideration must be given to the impact drugs targeting these pathways will have on T cells. T cell metabolism and T cell survival are intertwined, and the loss of anti-tumor T cells may negate many of the benefits of drugs targeting tumor metabolism. This is especially significant in the context of recent data indicating that metabolic suppression of activating T cells skews them toward an immunosuppressive phenotype, which may suppress anti-tumor immune responses [41].
Abbreviations
acetyl-CoA: acetyl-CoenzymeA; AMPK: AMP activated protein kinase; APC: antigen-presenting cell; F26P2: fructose-2,6-bisphosphate; G6P: glucose-6-phosphate; HIF1α: hypoxia inducible factor 1α; HK: hexokinase; JAK: Janus kinase; LDH: lactate dehydrogenase; LDLR: low-density lipoprotein receptor; MAPK: mitogen-activated protein kinase; MCT: mono-carboxylic transporters; MHC: major histocompatibility complexes; mTORC1: mammalian target of rapamycin complex 1; NADPH: nicotinamide adenine dinucleotide phosphate; OXPHOS: oxidative phosphorylation; PFK1: 6-phosphofructo-1-kinase; PFKFB: 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase; PI(3,4,5)P3: phosphatidylinositol-3,4,5-trisphosphate; PI3K: phosphatidylinositol-3-kinase; PKM2: pyruvate kinase M2; PPP: pentose phosphate pathway; PTEN: phosphatase and tensin homologue deleted on chromosome 10; ROS: reactive oxygen species; SGLT: sodium/glucose linked transporter; SOCS: suppressor of cytokine signaling; STAT: signal transducer and activator of transcription; TCA: tri-carboxylic acid; TCR: T cell antigen receptor; TKTL1: transketolase 1; γc: common gamma chain.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ANM and JCR wrote the manuscript. Both authors read and approved the final manuscript.
Contributor Information
Andrew N Macintyre, Email: a.n.macintyre@duke.edu.
Jeffrey C Rathmell, Email: jeff.rathmell@duke.edu.
Acknowledgements
We thank the members of the Rathmell laboratory for insightful discussions and comments. Funding: National Institutes of Health Grants R01 AI063345 and R01 HL108006 (to JCR); the American Asthma Foundation (JCR is the Bernard Osher Fellow of the American Asthma Foundation); the Lupus Research Institute (JCR); and the Leukemia and Lymphoma Society (JCR).
References
- Khorana HG. Chemical Biology; Selected Papers of H. Gobind Khorana. Singapore: World Scientific Publishing Co; 2000. [Google Scholar]
- Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. 2012;33:168–173. doi: 10.1016/j.it.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G, Lane PJ, Jenkinson EJ. Generating intrathymic microenvironments to establish T-cell tolerance. Nat Rev Immunol. 2007;7:954–963. doi: 10.1038/nri2187. [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281:37372–37380. doi: 10.1074/jbc.M608372200. [DOI] [PubMed] [Google Scholar]
- Warburg O, Posener K, Negelein E. In: On Metabolism of Tumors. Warburg O, editor. London: Constable; 1930. Ueber den stoffwechsel der tumoren. [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313:459–465. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]
- Vaupel P, Mayer A. Availability, not respiratory capacity governs oxygen consumption of solid tumors. Int J Biochem Cell Biol. 2012;44:1477–1481. doi: 10.1016/j.biocel.2012.05.019. [DOI] [PubMed] [Google Scholar]
- Weinhouse S. On respiratory impairment in cancer cells. Science. 1956;124:267–269. doi: 10.1126/science.124.3215.267. [DOI] [PubMed] [Google Scholar]
- Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond) 2010;7:7. doi: 10.1186/1743-7075-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Gawehn K, Geissler AW. [Metabolism of leukocytes] Z Naturforsch B. 1958;13B:515–516. [PubMed] [Google Scholar]
- Coloff JL, Mason EF, Altman BJ, Gerriets VA, Liu T, Nichols AN, Zhao Y, Wofford JA, Jacobs SR, Ilkayeva O, Garrison SP, Zambetti GP, Rathmell JC. Akt requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J Biol Chem. 2011;286:5921–5933. doi: 10.1074/jbc.M110.179101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder PC. Fuel utilization by cells of the immune system. Proc Nutr Soc. 1995;54:65–82. doi: 10.1079/PNS19950038. [DOI] [PubMed] [Google Scholar]
- Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem. 1994;269:31484–31490. [PubMed] [Google Scholar]
- Roos D, Loos JA. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res. 1973;77:127–135. doi: 10.1016/0014-4827(73)90561-2. [DOI] [PubMed] [Google Scholar]
- Buttgereit F, Brand MD, Muller M. ConA induced changes in energy metabolism of rat thymocytes. Biosci Rep. 1992;12:381–386. doi: 10.1007/BF01121501. [DOI] [PubMed] [Google Scholar]
- Hedeskov CJ. Early effects of phytohaemagglutinin on glucose metabolism of normal human lymphocytes. Biochem J. 1968;110:373–380. doi: 10.1042/bj1100373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/S1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munyon WH, Merchant DJ. The relation between glucose utilization, lactic acid production and utilization and the growth cycle of L strain fibroblasts. Exp Cell Res. 1959;17:490–498. doi: 10.1016/0014-4827(59)90069-2. [DOI] [PubMed] [Google Scholar]
- Obar JJ, Khanna KM, Lefrancois L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity. 2008;28:859–869. doi: 10.1016/j.immuni.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WT, Pasos G, Cecchini L, Mittler JN. Continued antigen stimulation is not required during CD4(+) T cell clonal expansion. J Immunol. 2002;168:1682–1689. doi: 10.4049/jimmunol.168.4.1682. [DOI] [PubMed] [Google Scholar]
- Lawrence CW, Braciale TJ. Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J Immunol. 2004;173:1209–1218. doi: 10.4049/jimmunol.173.2.1209. [DOI] [PubMed] [Google Scholar]
- Yoon H, Kim TS, Braciale TJ. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One. 2010;5:e15423. doi: 10.1371/journal.pone.0015423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit F, Burmester GR, Brand MD. Bioenergetics of immune functions: fundamental and therapeutic aspects. Immunol Today. 2000;21:192–199. doi: 10.1016/s0167-5699(00)01593-0. [DOI] [PubMed] [Google Scholar]
- Tay DL, Bhathal PS, Fox RM. Quantitation of G0 and G1 phase cells in primary carcinomas. Antibody to M1 subunit of ribonucleotide reductase shows G1 phase restriction point block. J Clin Invest. 1991;87:519–527. doi: 10.1172/JCI115026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rew DA, Wilson GD. Cell production rates in human tissues and tumours and their significance. Part II: clinical data. Eur J Surg Oncol. 2000;26:405–417. doi: 10.1053/ejso.1999.0907. [DOI] [PubMed] [Google Scholar]
- Rew DA, Wilson GD. Cell production rates in human tissues and tumours and their significance. Part 1: an introduction to the techniques of measurement and their limitations. Eur J Surg Oncol. 2000;26:227–238. doi: 10.1053/ejso.1999.0781. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, Cantley LC. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolkova K, Bellance N, Scandurra F, Genot E, Gnaiger E, Plecita-Hlavata L, Jezek P, Rossignol R. Mitochondrial bioenergetic adaptations of breast cancer cells to aglycemia and hypoxia. J Bioenerg Biomembr. 2010;42:55–67. doi: 10.1007/s10863-009-9267-x. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Thierbach R, Voigt A, Drewes G, Mietzner B, Steinberg P, Pfeiffer AF, Ristow M. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J Biol Chem. 2006;281:977–981. doi: 10.1074/jbc.M511064200. [DOI] [PubMed] [Google Scholar]
- Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292:504–507. doi: 10.1126/science.1058079. [DOI] [PubMed] [Google Scholar]
- Stucki JW. The optimal efficiency and the economic degrees of coupling of oxidative phosphorylation. Eur J Biochem. 1980;109:269–283. doi: 10.1111/j.1432-1033.1980.tb04792.x. [DOI] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL. The Warburg effect and metabolic efficiency: re-crunching the numbers. Science. 2009. E-letter.
- Schuster S, Kreft JU, Schroeter A, Pfeiffer T. Use of game-theoretical methods in biochemistry and biophysics. J Biol Phys. 2008;34:1–17. doi: 10.1007/s10867-008-9101-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airley RE, Mobasheri A. Hypoxic regulation of glucose transport, anaerobic metabolism and angiogenesis in cancer: novel pathways and targets for anticancer therapeutics. Chemotherapy. 2007;53:233–256. doi: 10.1159/000104457. [DOI] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, Dwyer MA, Nelson ER, Pollizzi KN, Ilkayeva O, Giguere V, Zuercher WJ, Powell JD, Shinohara ML, McDonnell DP, Rathmell JC. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci USA. 2011;108:18348–18353. doi: 10.1073/pnas.1108856108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookhtens M, Kannan R, Lyon I, Baker N. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Physiol. 1984;247:R146–153. doi: 10.1152/ajpregu.1984.247.1.R146. [DOI] [PubMed] [Google Scholar]
- Nathaniel D, Mellors A. Mitogen effects on lipid metabolism during lymphocyte activation. Mol Immunol. 1983;20:1259–1265. doi: 10.1016/0161-5890(83)90154-2. [DOI] [PubMed] [Google Scholar]
- de Bittencourt PI H Jr, Peres CM, Yano MM, Hirata MH, Curi R. Pyruvate is a lipid precursor for rat lymphocytes in culture: evidence for a lipid exporting capacity. Biochem Mol Biol Int. 1993;30:631–641. [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, Babic I, Tanaka K, Dang J, Iwanami A, Gini B, Dejesus J, Lisiero DD, Huang TT, Prins RM, Wen PY, Robins HI, Prados MD, Deangelis LM, Mellinghoff IK, Mehta MP, James CD, Chakravarti A, Cloughesy TF, Tontonoz P, Mischel PS. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer discovery. 2011;1:442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros LG, Puigjaner J, Cascante M, Lee WN, Brandes JL, Bassilian S, Yusuf FI, Williams RD, Muscarella P, Melvin WS, Schirmer WJ. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997;57:4242–4248. [PubMed] [Google Scholar]
- Horecker BL, Domagk G, Hiatt HH. A comparison of C14-labeling patterns in deoxyribose and ribose in mammalian cells. Arch Biochem Biophys. 1958;78:510–517. doi: 10.1016/0003-9861(58)90375-8. [DOI] [PubMed] [Google Scholar]
- Board M, Humm S, Newsholme EA. Maximum activities of key enzymes of glycolysis, glutaminolysis, pentose phosphate pathway and tricarboxylic acid cycle in normal, neoplastic and suppressed cells. Biochem J. 1990;265:503–509. doi: 10.1042/bj2650503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volker HU, Hagemann C, Coy J, Wittig R, Sommer S, Stojic J, Haubitz I, Vince GH, Kammerer U, Monoranu CM. Expression of transketolase-like 1 and activation of Akt in grade IV glioblastomas compared with grades II and III astrocytic gliomas. Am J Clin Pathol. 2008;130:50–57. doi: 10.1309/6H9844AMMET82DBJ. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhri G, Clark IA, Hunt NH, Cowden WB, Ceredig R. Effect of antioxidants on primary alloantigen-induced T cell activation and proliferation. J Immunol. 1986;137:2646–2652. [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Brand KA, Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. 1997;11:388–395. doi: 10.1096/fasebj.11.5.9141507. [DOI] [PubMed] [Google Scholar]
- Mason EF, Zhao Y, Goraksha-Hicks P, Coloff JL, Gannon H, Jones SN, Rathmell JC. Aerobic glycolysis suppresses p53 activity to provide selective protection from apoptosis upon loss of growth signals or inhibition of BCR-Abl. Cancer Res. 2011;70:8066–8076. doi: 10.1158/0008-5472.CAN-10-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Coloff JL, Ferguson EC, Jacobs SR, Cui K, Rathmell JC. Glucose metabolism attenuates p53 and puma-dependent cell death upon growth factor deprivation. J Biol Chem. 2008;283:36344–36353. doi: 10.1074/jbc.M803580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsholme EA, Board M. Application of metabolic-control logic to fuel utilization and its significance in tumor cells. Adv Enzyme Regul. 1991;31:225–246. doi: 10.1016/0065-2571(91)90015-e. [DOI] [PubMed] [Google Scholar]
- Newsholme EA, Crabtree B, Ardawi MS. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci Rep. 1985;5:393–400. doi: 10.1007/BF01116556. [DOI] [PubMed] [Google Scholar]
- Newsholme P, Newsholme EA. Rates of utilization of glucose, glutamine and oleate and formation of end-products by mouse peritoneal macrophages in culture. Biochem J. 1989;261:211–218. doi: 10.1042/bj2610211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curi R, Newsholme P, Pithon-Curi TC, Pires-de-Melo M, Garcia C, Homem-de-Bittencourt Junior PI, Guimaraes AR. Metabolic fate of glutamine in lymphocytes, macrophages and neutrophils. Braz J Med Biol Res. 1999;32:15–21. doi: 10.1590/s0100-879x1999000100002. [DOI] [PubMed] [Google Scholar]
- Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- Ohta A, Diwanji R, Kini R, Subramanian M, Sitkovsky M. In vivo T Cell Activation in Lymphoid Tissues is Inhibited in the Oxygen-Poor Microenvironment. Front Immunol. 2011;2:27. doi: 10.3389/fimmu.2011.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, Sitkovsky MV. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol. 2001;167:6140–6149. doi: 10.4049/jimmunol.167.11.6140. [DOI] [PubMed] [Google Scholar]
- Hopf HW, Hunt TK, West JM, Blomquist P, Goodson WH 3rd, Jensen JA, Jonsson K, Paty PB, Rabkin JM, Upton RA, von Smitten K, Whitney JD. Wound tissue oxygen tension predicts the risk of wound infection in surgical patients. Arch Surg. 1997;132:997–1004. doi: 10.1001/archsurg.1997.01430330063010. discussion 1005. [DOI] [PubMed] [Google Scholar]
- Thorens B, Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298:E141–145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Maianu L, Melbert BR, Garvey WT. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: a role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol Dis. 2004;32:182–190. doi: 10.1016/j.bcmd.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Marko AJ, Miller RA, Kelman A, Frauwirth KA. Induction of glucose metabolism in stimulated T lymphocytes is regulated by mitogen-activated protein kinase signaling. PLoS One. 2010;5:e15425. doi: 10.1371/journal.pone.0015425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanovic S, Wielburski A, Nelson BD. Effect of phorbol myristate acetate and concanavalin A on the glycolytic enzymes of human peripheral lymphocytes. Biochim Biophys Acta. 1988;970:1–6. doi: 10.1016/0167-4889(88)90215-7. [DOI] [PubMed] [Google Scholar]
- Marjanovic S, Eriksson I, Nelson BD. Expression of a new set of glycolytic isozymes in activated human peripheral lymphocytes. Biochim Biophys Acta. 1990;1087:1–6. doi: 10.1016/0167-4781(90)90113-G. [DOI] [PubMed] [Google Scholar]
- Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- Nista A, De Martino C, Malorni W, Marcante ML, Silvestrini B, Floridi A. Effect of lonidamine on the aerobic glycolysis of normal and phytohemagglutinin-stimulated human peripheral blood lymphocytes. Exp Mol Pathol. 1985;42:194–205. doi: 10.1016/0014-4800(85)90027-9. [DOI] [PubMed] [Google Scholar]
- Bosca L, Mojena M, Diaz-Guerra JM, Marquez C. Phorbol 12,13-dibutyrate and mitogens increase fructose 2,6-bisphosphate in lymphocytes. Comparison of lymphocyte and rat-liver 6-phosphofructo-2-kinase. Eur J Biochem. 1988;175:317–323. doi: 10.1111/j.1432-1033.1988.tb14199.x. [DOI] [PubMed] [Google Scholar]
- Colombo SL, Palacios-Callender M, Frakich N, De Leon J, Schmitt CA, Boorn L, Davis N, Moncada S. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc Natl Acad Sci USA. 2010;107:18868–18873. doi: 10.1073/pnas.1012362107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara R, Kato M, Okamura N, Nakagawa T, Komada Y, Tominaga N, Shimojo M, Fukasawa M. Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J Biochem. 1997;122:122–128. doi: 10.1093/oxfordjournals.jbchem.a021719. [DOI] [PubMed] [Google Scholar]
- Bassalyk LS, Ljubimova NV. Hexokinase isoenzymes in the diagnosis of gastric and esophageal neoplasms. Neoplasma. 1987;34:319–324. [PubMed] [Google Scholar]
- Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z, Mitchell R, Bucala R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002;62:5881–5887. [PubMed] [Google Scholar]
- Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J, Chen J. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:18–186. doi: 10.1038/452018a. [DOI] [PubMed] [Google Scholar]
- Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan KL, Lei QY. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horig H, Spagnoli GC, Filgueira L, Babst R, Gallati H, Harder F, Juretic A, Heberer M. Exogenous glutamine requirement is confined to late events of T cell activation. J Cell Biochem. 1993;53:343–351. doi: 10.1002/jcb.240530412. [DOI] [PubMed] [Google Scholar]
- Yaqoob P, Calder PC. Cytokine production by human peripheral blood mononuclear cells: differential senstivity to glutamine availability. Cytokine. 1998;10:790–794. doi: 10.1006/cyto.1998.0358. [DOI] [PubMed] [Google Scholar]
- Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, Frauwirth KA. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185:1037–1044. doi: 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AL. Therapeutic considerations of L-glutamine: a review of the literature. Alternative medicine review: a journal of clinical therapeutic. 1999;4:239–248. [PubMed] [Google Scholar]
- Wilmore DW, Shabert JK. Role of glutamine in immunologic responses. Nutrition. 1998;14:618–626. doi: 10.1016/S0899-9007(98)00009-4. [DOI] [PubMed] [Google Scholar]
- Newsholme P. Why is L-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J Nutr. 2001;131:2515S–2522S. doi: 10.1093/jn/131.9.2515S. discussion 2523S-2514S. [DOI] [PubMed] [Google Scholar]
- Kovacevic Z, Morris HP. The role of glutamine in the oxidative metabolism of malignant cells. Cancer Res. 1972;32:326–333. [PubMed] [Google Scholar]
- Eagle H. Nutrition needs of mammalian cells in tissue culture. Science. 1955;122:501–514. doi: 10.1126/science.122.3168.501. [DOI] [PubMed] [Google Scholar]
- Kallinowski F, Runkel S, Fortmeyer HP, Forster H, Vaupel P. L-glutamine: a major substrate for tumor cells in vivo? J Cancer Res Clin Oncol. 1987;113:209–215. doi: 10.1007/BF00396375. [DOI] [PubMed] [Google Scholar]
- Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci USA. 2012;109:8983–8988. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178:93–105. doi: 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung HN, Marks JR, Chi JT. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011;7:e1002229. doi: 10.1371/journal.pgen.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CL, Wasa M, Souba WW, Abcouwer SF. Regulation of glutamine synthetase in human breast carcinoma cells and experimental tumors. Surgery. 1997;122:451–463. doi: 10.1016/S0039-6060(97)90039-8. [DOI] [PubMed] [Google Scholar]
- Colquhoun A, Newsholme EA. Aspects of glutamine metabolism in human tumour cells. Biochem Mol Biol Int. 1997;41:583–596. doi: 10.1080/15216549700201621. [DOI] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrg3199. [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol. 2003;33:2223–2232. doi: 10.1002/eji.200324048. [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. 1997;272:17269–17275. doi: 10.1074/jbc.272.28.17269. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter C, Waldmann H, Cobbold SP. mTOR signalling and metabolic regulation of T cell differentiation. Curr Opin Immunol. 2010;22:655–661. doi: 10.1016/j.coi.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Aksamitiene E, Kiyatkin A, Kholodenko BN. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: a fine balance. Biochem Soc Trans. 2012;40:139–146. doi: 10.1042/BST20110609. [DOI] [PubMed] [Google Scholar]
- Davies C, Tournier C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans. 2012;40:8–89. doi: 10.1042/BST20110641. [DOI] [PubMed] [Google Scholar]
- del Barco Barrantes I, Nebreda AR. Roles of p38 MAPKs in invasion and metastasis. Biochem Soc Trans. 2012;40:79–84. doi: 10.1042/BST20110676. [DOI] [PubMed] [Google Scholar]
- Gehart H, Kumpf S, Ittner A, Ricci R. MAPK signalling in cellular metabolism: stress or wellness? EMBO Rep. 2010;11:834–840. doi: 10.1038/embor.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, Laderoute K, Johnson RS. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol. 2001;21:3436–3444. doi: 10.1128/MCB.21.10.3436-3444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard MA, Ohh M. The role of hypoxia-inducible factors in cancer. Cellular and molecular life sciences: CMLS. 2007;64:2170–2180. doi: 10.1007/s00018-007-7082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood. 2008;111:2101–2111. doi: 10.1182/blood-2007-06-096297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish GH, Sinclair LV, Cantrell DA. Differential regulation of T-cell growth by IL-2 and IL-15. Blood. 2006;108:600–608. doi: 10.1182/blood-2005-12-4827. [DOI] [PubMed] [Google Scholar]
- Beharry Z, Mahajan S, Zemskova M, Lin YW, Tholanikunnel BG, Xia Z, Smith CD, Kraft AS. The Pim protein kinases regulate energy metabolism and cell growth. Proc Natl Acad Sci USA. 2011;108:528–533. doi: 10.1073/pnas.1013214108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian KC, Wang L, Hickey ER, Studts J, Barringer K, Peng C, Kronkaitis A, Li J, White A, Mische S, Farmer B. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J Biol Chem. 2005;280:6130–6137. doi: 10.1074/jbc.M409123200. [DOI] [PubMed] [Google Scholar]
- Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11:23–34. doi: 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- Vera J, Rateitschak K, Lange F, Kossow C, Wolkenhauer O, Jaster R. Systems biology of JAK-STAT signalling in human malignancies. Prog Biophys Mol Biol. 2011;106:426–434. doi: 10.1016/j.pbiomolbio.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Jacobs MD, Black J, Futer O, Swenson L, Hare B, Fleming M, Saxena K. Pim-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by LY294002. J Biol Chem. 2005;280:13728–13734. doi: 10.1074/jbc.M413155200. [DOI] [PubMed] [Google Scholar]
- Fox CJ, Hammerman PS, Thompson CB. The Pim kinases control rapamycin-resistant T cell survival and activation. J Exp Med. 2005;201:259–266. doi: 10.1084/jem.20042020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Kummerow C, Junker C, Becherer U, Hoth M. Morphological changes of T cells following formation of the immunological synapse modulate intracellular calcium signals. Cell Calcium. 2009;45:109–122. doi: 10.1016/j.ceca.2008.07.003. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Tamas P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, Cantrell DA. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med. 2006;203:1665–1670. doi: 10.1084/jem.20052469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, Jones RG. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. 2011;187:4187–4198. doi: 10.4049/jimmunol.1100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkash J, Asotra K. Calcium wave signaling in cancer cells. Life Sci. 2010;87:587–595. doi: 10.1016/j.lfs.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. J Pathol. 2011;223:283–294. doi: 10.1002/path.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S-M, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. 2008;84:949–957. doi: 10.1189/jlb.0108024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plas DR, Rathmell JC, Thompson CB. Homeostatic control of lymphocyte survival: potential origins and implications. Nat Immunol. 2002;3:515–521. doi: 10.1038/ni0602-515. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6:683–692. doi: 10.1016/S1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286:42626–42634. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, Chen X, Bishop JM. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15:157–170. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Dickman KG, Zong WX. Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J Biol Chem. 2010;285:7324–7333. doi: 10.1074/jbc.M109.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]