

SUMMARY
Neutrophil degranulation plays an important role in acute innate immune responses and is tightly regulated because the granule contents can cause tissue damage. However, this regulation remains poorly understood. Here we identify the complex of STK24 and CCM3 as being an important regulator of neutrophil degranulation. Lack of either STK24 or CCM3 increases the release of a specific granule pool without affecting other neutrophil functions. STK24 appears to suppress exocytosis by interacting and competing with UNC13D C2B domain for lipid binding, whereas CCM3 has dual roles in exocytosis regulation. While CCM3 stabilizes STK24, it counteracts STK24-mediated inhibition of exocytosis by recruiting STK24 away from the C2B domain through its Ca2+-sensitive interaction with UNC13D C2A domain. This STK24/CCM3-regulated exocytosis plays an important role in protection of kidneys from ischemia-reperfusion injury. Together, these findings reveal a previously unknown function of the STK24 and CCM3 complex in the regulation of ligand-stimulated exocytosis.
INTRODUCTION
Serine/threonine protein kinase (STK) 24 [also known as mammalian sterile 20-like kinase (MST) 3], MST4, and STK25 belong to the germinal center kinase (GCK) III sub-family of sterile-20 kinases (Pombo et al., 2007). These GCKIII sub-family kinases have been implicated in regulating a number of cellular functions (Schinkmann and Blenis, 1997; Huang et al., 2002; Irwin et al., 2006; Lu et al., 2006; Wu et al., 2008; Lorber et al., 2009; Fidalgo et al., 2010; Wu et al., 2011) and interact with CCM3 [also known as programmed cell death 10 (PDCD10)] (Rual et al., 2005; Fidalgo et al., 2010; Zheng et al., 2010; Ceccarelli et al., 2011; Kean et al., 2011). Mutations in the CCM3 gene as well as in two other structurally unrelated genes, CCM1 and CCM2, were found in the familial forms of Cerebral Cavernous Malformations (CCM) (Faurobert and Albiges-Rizo, 2010; Cavalcanti et al., 2012). CCM is a vascular pathological condition that affects the vasculature of the central nervous system and results in stroke, seizure and cerebral hemorrhage with a high prevalence. CCM3 has two domains---an N-terminal domain and a C-terminal focal adhesion targeting-homology (FATH) domain. Global Ccm3 disruption resulted in embryonic lethality probably due to defects in the cardiovascular system (He et al., 2010). STK24 and STK25 also appear to function in the same pathway as CCM3 in cardiovascular development (Voss et al., 2009; Fidalgo et al., 2010; Zheng et al., 2010; Yoruk et al., 2012). However, the underlying biochemical mechanisms for how GCKIII kinases or CCM3 regulate these functions are still poorly understood.
Exocytosis occurs in every cell and is a process by which a cell directs the contents (secreted proteins, membrane proteins, and lipids) of secretory vesicles toward extracellular space. Neutrophils play important roles in innate immunity and utilize a regulated exocytic process of degranulation to execute some of their functions. Degranulation results in the releases of various proteases and other cytotoxic agents, including matrix metalloproteinases (MMPs) and myeloperoxidase (MPO) (Lacy and Eitzen, 2008). These granule contents are antimicrobial, but can also cause tissue damage and organ failure during ischemia-reperfusion that occurs in stroke or organ transplantation and during adapted immune responses in chronic inflammation and viral infections (Lacy and Eitzen, 2008).
Exocytosis is accomplished by the fusion of secretory vesicles with the plasma membrane through the assembly of the SNARE complex. Before membrane fusion, additional proteins mediate and regulate the initial interaction between the vesicles and the acceptor membrane. They include the Rab family of small GTPases, the exocyst, and numerous other regulatory proteins (Sugita, 2008; He and Guo, 2009; Sudhof and Rizo, 2011; Jahn and Fasshauer, 2012). Among these regulatory proteins is the UNC13 (Munc13) family of proteins, which consists of UNC13A-D (Koch et al., 2000; Feldmann et al., 2003). UNC13D (also known as Munc13-4), which is expressed at high levels in hematopoietic cells, contains two separate C2 domains (C2A and C2B) and two Munc13-homology domains (MHD1 and MHD2) (Fukuda, 2005). UNC13D binds to RAB27, through which it is tethered to vesicles, and also binds to syntaxins and DOC2α (Higashio et al., 2008; Boswell et al., 2012). Syntaxins are components of SNARE complexes, which are involved in membrane fusion, whereas DOC2α is an exocytosis regulator. Both human and mouse genetic evidence has established an important role of UNC13D in the regulation of exocytosis in cytotoxic T cells, mast cells, platelets, and neutrophils (Feldmann et al., 2003; Crozat et al., 2007; Brzezinska et al., 2008; Pivot-Pajot et al., 2008; Ren et al., 2010; Elstak et al., 2011). UNC13D is involved in granule tethering to plasma membranes through the binding of its C2B domain to membrane lipids during granule docking, and priming SNARE-mediated fusion (Menager et al., 2007; de Saint Basile et al., 2010; Elstak et al., 2011; Boswell et al., 2012).
Exocytic vesicles or granules exist in different forms, many of which are released in a controlled manner, often by extracellular stimuli. Ligand-stimulated exocytosis, which neutrophil degranulation is a form of, plays important roles in various philological and pathophysiological processes. A great deal of knowledge has been gained on how ligands stimulate exocytosis particularly through Ca2+ over the years (Pang and Sudhof, 2010; Parekh, 2011; Yamashita, 2012). There appear to be at least two pools of exocytic granules that correspond to the two phases of exocytosis observed upon ligand stimulation. One pool is docked/primed and readily releasable. The other one is non-readily releasable or reserved, which is not docked/primed before stimulation and has to undergo docking/priming upon stimulation (Sugita, 2008; Pores-Fernando and Zweifach, 2009; Seino et al., 2011). In our investigation of the roles of STK24 and CCM3 in regulation of neutrophil functions, we unexpectedly discovered the STK24 and CCM3 complex as being a regulator of ligand/Ca2+-stimulated exocytosis that regulates the second phase of degranulation in neutrophils. We elucidated the mechanisms by which STK24 and CCM3 regulate the exocytic process through their interactions with UNC13D and demonstrated that CCM3 and STK24-regulated exocytosis play an important role in tissue injury protection. These results together have revealed previously unidentified physiological and cellular roles of STK24 and CCM3 and provided mechanistic insights into ligand-regulated degranulation/exocytosis.
RESULTS
STK24-deficiency enhances degranulation
The Stk24 gene is expressed at a high level in human neutrophils (Payton et al., 2009), and its protein was readily detected in mouse neutrophils (Fig. 1A). To investigate the importance of Stk24 in neutrophils, a Stk24 mutant mouse line was acquired from the International Gene Trap Consortium. In this mouse line, the Stk24 gene was disrupted by an insertion, resulting in a hypomorphic allele (Fig. S1A). The mice homozygous for this hypomorphic allele, designated as Stk24h/h, were viable and showed no gross phenotypes. The STK24 protein content in the Stk24h/h neutrophils is greatly reduced (by more than 90%) (Fig. 1A). STK24 deficiency did not appear to affect the numbers of neutrophils or other leukocytes including monocytes or lymphocytes in the peripheral blood (Fig. 1B). Additionally, STK24 deficiency did not cause significant alterations in the ability of the neutrophils to chemotax (Fig. 1C–E), produce superoxide (Fig. 1F), or phagocytose (Fig. 1G), compared to those isolated from the wildtype (WT) littermates. However, we found that Stk24h/h neutrophils released significantly greater amounts of myeloperoxidase (MPO) (Fig. 1H) and matrix metalloproteinases (MMPs) (Fig. 1I) than the WT cells did in response to the stimulation of chemoattractant fMLP or MIP2. The MPO protein contents in WT and Stk24h/h neutrophils are similar (Fig. 1A). Thus, these results suggest that the lack of STK24 may result in enhanced neutrophil degranulation. In support of this conclusion, expression of STK24-GFP fusion protein in the Stk24h/h neutrophils reduced MPO release (Fig. 1J). Because expression of a kinase-dead mutant of STK24 (K53R) at a level similar to that of WT STK24-GFP (Fig. S1B) also led to a similar reduction in MPO release (Fig. 1J), STK24 probably regulates degranulation through a kinase activity-independent mechanism.
Figure 1. STK24-deficiency increases neutrophil degranulation.

A. Verification of STK24-deficiency in neutrophils. Neutrophils were isolated from WT and Stk24h/h mice and analyzed by Western analysis using antibodies specific to STK24, MPO or Gβ2. See also Figure S1A.
B. Lack of an effect of STK24-deficiency on the number of circulating leukocytes. PMN, neutrophils; MN, monocytes; and LY, lymphocytes. The data are presented as Means ± SD
C–E. Lack of an effect of STK24-deficiency on chemotactic activities. Chemotactic responses of neutrophils were examined using a Dunn Chamber containing an fMLP gradient. The data are presented as Means ± SD
F. Lack of an effect of STK24-deficiency on superoxide production upon stimulation by 1 μM fMLP.
G. Lack of an effect of STK24-deficiency on phagocytosis of opsonized E. coli. The data are presented as Means ± SD
H & I. Increased secretion of MMP and MPO from Stk24h/h neutrophils. Neutrophils were stimulated with 500 nM fMLP or 10 nM MIP2. The experiments were repeated three times. The data are presented as Means ± SD (*, P<0.05, Student’s t-test).
J. Suppression of MPO secretion by STK24 expression in Stk24h/h neutrophils. Stk24h/h neutrophils were transfected with GFP, GFP-STK24 (STK) or GFP-STK24-K53R (KR) plasmids, and GFP positive cells were isolated by FACS. Cells were then stimulated with 500 nM fMLP and assayed for MPO secretion. The data are presented as Means ± SD (*, P<0.05, Student’s t-test; n=3). See also Figure S1B.
STK24 regulates the release of a specific pool of granules
Immunostaining of mouse neutrophils with an anti-STK24 antibody revealed a punctate pattern of STK24 subcellular localization, suggesting that STK24 may primarily be localized with granules (Fig. 2A). Co-staining with an anti-MPO antibody showed that most of the MPO-positive granules were STK24-positive, whereas about a quarter of STK24-positive granules were MPO positive. Because neutrophils are known to contain different types of granules, a fraction of which contains MPO (Lacy and Eitzen, 2008), these results suggest that STK24 may be involved in the regulation of different types of granules. This concept is consistent with the finding that STK24-deficiency is also associated with increased release of MMP (Fig. 1I), which is in a different type of granules from those containing MPO (Lacy and Eitzen, 2008). Using confocal microscopy we compared the number of MPO-positive granules in WT and STK24-deficient neutrophils and found no significant difference (data not shown). We also compared the number of MPO-positive granules in close proximity (<200 nm) to plasma membranes as detected by the total internal reflection fluorescence (TIRF) microscopy (Fig. S2A) and again observed no significant difference between WT and Stk24h/h neutrophils (Fig. 2B).
Figure 2. STK24 regulates the release of the reserved pool of granules.

A. Colocalization of STK24 and MPO in mouse neutrophils. Mouse neutrophils were stained for STK24 and MPO and detected by a confocal microscope. Single optical sections are shown.
B. Lack of an effect of STK24-deficiency on the number of MPO-containing granules in resting neutrophils. Mouse neutrophils were stained for MPO and imaged using a TIRF microscope. The number of granules from 25 WT and mutant cells was counted and is presented as Means ± SD (Student’s t-test, n>50). See also Figure S2A.
C–D. STK24-deficiency increases granule fusion events upon a follow-up stimulation. Individual WT and STK24-deficient neutrophils expressing VAMP8-pHluorin were stimulated with one puff of fMLP (10 μM) and CB (10 μM) from a micropipette (initial stimulation). Four minutes later, the neutrophils were stimulated with a follow-up puff (follow-up stimulation). Time-lapsed images were acquired using a TIRF microscope. Representative image series of single granule fusions are shown in (C). The image time interval is 200 milliseconds. The durations of individual granule fusions for the initial and follow-up stimulations (D) are quantified, and the numbers of fusion events in per neutrophil after the initial stimulations and follow-up stimulations (E) are counted. Data are presented as Means ± SD (*, P<0.05, Student’s t-test; n=9). See also Figure S2B.
F. Time-dependent effects of STK24-deficiency on MPO release. WT and STK-deficient neutrophils were treated with 500 nM fMLP and assayed for MPO secretion at different time points. Data are presented as Means ± SD (*, P<0.05; ^, P>0.1, Student’s t-test, n=3). See also Figure S2C.
To further investigate the degranulation process, we visualized and compared the fusion processes of individual granules in the WT and Stk24h/h neutrophils using live-cell TIRF microscopy. We have previously used the fusion protein of the v-SNARE VAMP2 (vesicle-associated membrane protein 2) with a pH-sensitive fluorescent protein, pHluorin, to monitor the exocytosis of GLUT4 storage vesicles (Xu et al., 2011). The fluorescence of pHluorin becomes much brighter when transferred from an acidic (inside vesicles) to a neutral environment (after vesicle fusion with the plasma membrane) (Miesenbock et al., 1998; Jankowski et al., 2001; Sankaranarayanan and Ryan, 2001). Although VAMP2 is not associated with MPO-positive vesicles (data not shown), VAMP8 is (Fig. S2B). We expressed VAMP8-pHluorin in neutrophils and locally stimulated each individual neutrophil by fMLP using a micropipette. Shortly after the stimulation, there was an increase in the number of “flashes” as the result of a sudden increase in fluorescence upon granule fusion and dissipation of fluorescence due to lateral diffusion of the VAMP molecules in the plasma membrane after granule fusion (Fig. 2C). Quantitative analysis of fusion events revealed no significant differences in either the total number of fusion events or the kinetics of each fusion event between WT or Stk24h/h neutrophils after the initial stimulation (Fig. 2D, E).
Two types of secretory granules have been described. One is docked/primed and readily releasable. The other is non-readily releasable or reserved, which is not docked/primed before stimulation and has to undergo docking/priming upon activation (Sugita, 2008; Pores-Fernando and Zweifach, 2009; Seino et al., 2011). We thus investigated whether STK24-deficiency might affect the granule release from this reserved pool. We first examined the time course of MPO release upon uniform stimulation. Although there was no significant difference in the MPO release between WT and Stk24h/h neutrophils after 1 min stimulation, significant differences were observed after longer stimulations (Fig. 2F), suggesting that STK24 may be involved in regulating the release of the reserved pool of granules. To further corroborate this conclusion, we performed sequential stimulations in the live TIRF experiment using micropipettes to transiently stimulate the cells. When the cells were stimulated at 4 minutes after the initial stimulation, we observed a significant increase in the number of granule fusion events in STK24-deficient neutrophils compared to that in WT neutrophils in response to this follow-up stimulation (Fig. 2E), even though there was no significant difference in the fusion kinetics between WT and mutant neutrophils (Fig. 2D). When the follow-up stimulations were given 20 min after the initial stimulations, the differences in fusion events became less apparent (Fig. S2C). We interpret these results to suggest that STK24 primarily suppresses the release of the reserved pool of granules without apparent role in the release of docked granules.
CCM3 regulates ligand-stimulated exocytosis
STK24 forms a complex with CCM3 (Rual et al., 2005; Zheng et al., 2010; Ceccarelli et al., 2011; Kean et al., 2011). We confirmed the interaction in a co-precipitation assay (data not shown). In mouse neutrophils, GFP-STK24 is also colocalized with CCM3-mCherry (Fig. 3A). To investigate whether CCM3 has a role in neutrophil degranulation, we generated myeloid-specific CCM3-deficient mice (Ccm3m/m) by crossing floxed CCM3 mice with the B6.129-Lyzstm1(cre)Ifo/J mice (Fig. 3B). CCM3-deficient neutrophils also released greater amounts of MPO and MMP in response to chemoattractants than those of WT neutrophils (Fig. 3C, D). Our data also confirmed a previous observation that CCM3 stabilized STK24 (Fidalgo et al., 2010), because the amount of STK24 protein was reduced by more than 60% in CCM3-deficient neutrophils compared to that in WT neutrophils (Fig. 3B). We also examined VAMP8-positive granule fusion in CCM3-deficient neutrophils using live-cell TIRF microscopy (Fig. 3E). Similar to what we observed with Stk24h/h neutrophils, CCM3-deficiency increased the number of fusion events in response to the follow-up stimulations compared to WT, without affecting the initial responses or the fusion kinetics (Fig. 3F–H). Thus, we concluded that CCM3 and STK24 function similarly in neutrophil degranulation. Because neutrophils isolated from Stk24h/+ mice showed enhanced MPO secretion (Fig. S3A), the enhanced exocytosis phenotype of CCM3-deficiency, which resulted in more than one half reduction in STK24 protein content (Fig. 3B), may at least in part be due to reduced STK24 protein content. Consistent with this conclusion, overexpression of STK24 or CCM3, but not a CCM3 mutant (Fig. S3B) that cannot bind to STK24 (Fig. S3C), was able to revert CCM3-deficiency-induced increase in MPO release. This conclusion is further supported by the observation that overexpression of CCM3 in wildtype neutrophils, which led to an increase in STK24 protein, reduced MPO release (Fig. S3D, E).
Figure 3. CCM3-deficiency Increases neutrophil degranulation.

A. Colocalization of CCM3 and STK24 in mouse neutrophils. A representative neutrophil expressing GFP-STK24 and CCM3-mCherry was detected by a confocal microscope. Single optical sections are shown.
B. Validation of CCM3-deficiency in Ccm3m/m neutrophils. Ccm3m/m neutrophils were analyzed by Western using an antibody to CCM3, STK24, or Gβ2.
C, D. Increased secretion of MMP and MPO from Ccm3m/m neutrophils. Neutrophils were stimulated with 500 nM fMLP or 10 nM MIP2. The data are presented as Means ± SD (*, P<0.05, n=3, Student’s t-test). See also Figure S3.
E–H. Effects of CCM3-deficiency on granule fusion. Cells were treated and analyzed as in Fig. 1. The data are presented as Means ± SD (*, P<0.05; Student’s t-test, n=9).
STK24 and CCM3 interact with UNC13
To understand how STK24 and CCM3 regulate exocytosis, we used the TAP (Tandem Affinity Purification) technique, followed by mass spectrometry, to identify proteins that may bind to STK24 in HEK293. CCM3 was the top hit. Among the other hits, there was UNC13B (Fig. S4A). UNC13B (also known as Munc13-2) is a member of the UNC13 family of exocytic regulators (Koch et al., 2000; Feldmann et al., 2003; Li and Chin, 2003). While Unc13b is expressed at a very low level in neutrophils, its family member Unc13d is expressed at a higher level in neutrophils (Fig. S4B). In addition, UNC13D (also known as Munc13-4) has been shown to play a role in neutrophil degranulation (Pivot-Pajot et al., 2008; Johnson et al., 2011). Thus, we tested its interaction with STK24. UNC13D co-immunoprecipitated with WT or kinase-dead STK24, when expressed in HEK293 cells (Fig. 4A). We also found that CCM3 and UNC13D interacted by co-immunoprecipitation (Fig. 4A). To determine if these interactions are direct, recombinant proteins were used in a pull-down assay. UNC13D interacted with CCM3 as well as WT (Fig. 4B) and kinase-dead (data not shown) STK24.
Figure 4. STK24 and CCM3 interact with UNC13D.

A. Detection of the interactions by co-immunoprecipitation. HEK293 cells were cotransfected with plasmids as indicated in the figure, and immunoprecipitation was performed 24 hours later. Proteins were detected by Western analysis. See also Figure S4.
B. Detection of the direct interactions by GST pulldown using recombinant proteins isolated from E. coli. Proteins were detected by Western analysis.
C–E. STK24 and CCM3 do not interfere with the interaction of UNC13D with RAB27, SYNTAXIN7, or DOC2α. HEK293 cells were cotransfected with plasmids as indicated, followed by immunoprecipitation and Western analysis. STX7, SYNTAXIN7.
The UNC13 family of proteins interacts with several exocytosis-related proteins, including RAB27, DOC2α, and syntaxins (STXs) (Neeft et al., 2005; Higashio et al., 2008; Boswell et al., 2012). Given that STK24 regulates degranulation independently of its kinase activity (Fig. 1J), a possible mechanism for STK24 to inhibit exocytosis may be to disrupt one of these interactions with UNC13D. However, expression of STK24 and/or CCM3 did not attenuate the interaction of UNC13D with STX7, DOC2α, or RAB27 (Fig. 4C–E).
STK24 inhibits UNC13D binding to liposomes
UNC13D also binds with membrane lipids in a Ca2+-sensitive manner and has an important role in vesicle docking and SNARE-dependent vesicle fusion (Boswell et al., 2012). We examined effects of STK24 and CCM3 on the binding of UNC13D to lipids using buoyant density flotation of phosphatidylcholine (PC)/phosphatidylserine (PS) liposomes as previously described (Boswell et al., 2012). While CCM3 had little effect on the binding of UNC13D to liposomes regardless of the presence of Ca2+, STK24 strongly inhibited the binding both in the presence and absence of Ca2+ (Fig. 5A) and abrogated most of the stimulatory effect of Ca2+ on the binding (Fig. 5B). Boswell et al. have showed that UNC13D bound to liposomes via its C2B domain in a Ca2+-sensitive manner (Boswell et al., 2012). We found that mutating the two Ca2+-binding Asp residues (941 and 947) into Asn in the C2B domain not only abrogated Ca2+-stimulated binding of UNC13D to liposomes as Boswell et al showed (Boswell et al., 2012)(Fig. S5A), but also attenuated the interaction of UNC13D with STK24 (Fig. 5C). This result suggests that these Asp residues may be involved in Ca2+-mediated regulation of UNC13D binding to both lipids and STK24. Consistent with this conclusion, STK24, via its kinase domain, interacted with the C2B domain in a co-immunoprecipitation assay (Fig. S5B–D) and pull-down assay using recombinant proteins (Fig. 5D & S5E). In addition, STK24 without this kinase domain, unlike WT STK24, was not able to revert STK24-deficiency-induced increase in MPO release (Fig. S5F), underscoring the importance of STK24-UNC13D C2B interaction in STK24-mediated suppression of exocytosis. Therefore, putting these results together, we conclude that STK24 may compete with lipids for binding to the C2B domain.
Figure 5. STK24 inhibits UNC13D binding to liposomes.

A & B. STK24, but not CCM3, inhibits UNC13D binding to liposomes. Recombinant UNC13D (33 nM) and STK24 (66 nM) or CCM3 (66 nM) were incubated with liposomes. After ultracentrifugation, liposome layers and inputs were analyzed by Western blotting (A). The ratios of UNC13D contents in the liposome layers in the presence of Ca2+ to those in the presence of EGTA (-Ca2+) were obtained from quantification of the blots from three independent experiments (B) and are shown as Means ± SD (*, P<0.01; Student’s t-test).
C. Ca2+-binding site mutations in UNC13D C2B domain disrupt the interaction with STK24. GST pull-down was carried out using recombinant protein as indicated in the presence of Ca2+ or EGTA (-Ca2+). Proteins were detected by Western analysis. C2B*, UNC13D C2B mutant (D941N, D947N). See also Figure S5A.
D. Direct interaction between SKT24 and UNC13D C2B. A pull-down assay was carried out using recombinant proteins (40 nM C2B and 40 nM STK24), which was detected by Coomassie Blue staining. See also Figure S5B–F.
CCM3 counteracts STK24 in UNC13 binding to liposomes
While CCM3 alone had little effect on UNC13D-liposome binding (Fig. 5A, B), it unexpectedly attenuated the inhibition of UNC13D-liposome binding by STK24 (Fig. 6A). Moreover, this attenuation was much greater in the presence of Ca2+ than in its absence, because there was significantly greater Ca2+-induced liposome binding of UNC13D in the presence of CCM3 than that in the presence of STK24 alone (Fig. 6B). In other words, CCM3 appears to have a role in restoring Ca2+-sensitivity suppressed by STK24. Given that chemoattractants increase intracellular Ca2+ concentrations, this finding may underlie a mechanism by which degranulation of the reserved granule pool is regulated by chemoattractants.
Figure 6. Ca2+ stimulates CCM3-UNC13D interaction to relieve the inhibition by STK24.

A,B. CCM3 counteracts STK24-mediated inhibition of UNC13D binding to liposomes. Recombinant UNC13D (33 nM), STK24 (66 nM), and CCM3 (66 nM) were incubated with liposomes. After ultracentrifugation, liposome layers and inputs were analyzed by Western blotting (A). The ratios of UNC13D contents in the liposome layers in the presence of Ca2+ to those in the presence of EGTA (-Ca2+) were obtained from quantification of the blots from three independent experiments (B) and are shown as Means ± SD (*, P<0.01; Student’s t-test). See also Figure S6A.
C. Ca2+ stimulates the formation of UNC13D, CCM3 and STK24 complex. GST pulldown was carried out using recombinant proteins as indicated in the presence of Ca2+ or EGTA (-Ca2+). Proteins were detected by Western analysis.
D. Ca2+-binding site mutations in UNC13D C2A domain enhance the interaction with CCM3. GST pulldown was carried out using recombinant proteins as indicated in the presence of Ca2+ or EGTA (-Ca2+). C2A* stands for the UNC13D mutant carrying the D127N and D133N mutations. Proteins were detected by Western analysis.
E. Ca2+-binding site mutations in UNC13D C2A domain enhance liposome binding. Recombinant proteins were incubated with liposomes in the presence of Ca2+ or EGTA (-Ca2+). After ultracentrifugation, liposome layers and inputs were analyzed by Western blotting. Blots from three independent experiments were quantified, and the value for WT binding alone (Lane 6) was taken as 1. The data are shown as Means ± SD. (AU, arbitrary unit; *, P<0.05, Student’s t-test).
F. A model depicting the regulation of degranulation of the reserved granule pool by STK24 and CCM3 in response to Ca2+. At the basal state, STK24 binds to the C2B domain of UNC13D and inhibits its binding to the plasma membrane lipid. When cells are stimulated, the intracellular Ca2+ concentrations increase. Ca2+ stimulates the interaction of UNC13D C2A domain with CCM3, which presumably leads to a reduction in STK24 binding to UNC13D at the C2B domain and hence STK24-mediated inhibition of UNC13D binding to lipid and granule docking/priming. F, FATH domain; NT, N terminal domain; CT, C-terminal domain; K, kinase domain. See also Figure S6B.
G. Effect of Ca2+ on the formation of the ternary complex of UNC13D C2A domain (C2A), CCM3 and STK24. GST pulldown was carried out using recombinant proteins (20 nM C2A, 40 nM CCM3, and 40 nM STK24) in the presence or of Ca2+ or EGTA (-Ca2+) and detected by Coomassie staining.
One possible way for CCM3 to counteract STK24-mediated inhibition is to disrupt the interaction between STK24 and UNC13D. Contrary to this possibility, the presence of CCM3 did not disrupt the STK24-UNC13D interaction (compare Lane 2 and 4 of Fig. 6C), but rather markedly enhanced the interaction when Ca2+ was present (compare Lanes 3 and 5 of Fig. 6C). Because CCM3 interacts with UNC13D N-terminal (NT) region that contains the C2A domain (Fig. S5B and S6A), we mutated two of the Ca2+-binding Asp residues (127 and 133) into Asn in this domain. While these mutations did not affect Ca2+-dependent liposome binding (Fig. S5A) as previously reported (Boswell et al., 2012), they, unlike those in the C2B domain that disrupt the interaction with STK24 (Fig. 5C), unexpectedly augmented the interaction with CCM3 (Fig. 6D). We do not understand the effect of these C2A mutations on the interaction. One simple explanation is that the mutated domain may mimic the Ca2+-bound state, given that Ca2+ also enhanced the interaction of UNC13D with CCM3 (Fig. 6D). Nevertheless, this UNC13D mutant may help us to understand how CCM3 and its interaction with UNC13D are involved in counteracting STK24’s effect. Thus, we tested the effect of these C2A mutations on CCM3-mediated reversal of the inhibition by STK24. UNC13D carrying these C2A mutations appeared to bind to liposomes as strongly in the absence of Ca2+ as the WT UNC13D did in the presence of Ca2+ in the liposome-binding assay in which both STK24 and CCM3 were present (compare lanes 1 and 4 of Fig. 6E). In addition, the interaction of this mutant, unlike the WT UNC13D, with liposomes was largely insensitive to Ca2+ (compare the difference between lanes 3 and 4 with that between lanes 1 and 2 in Fig. 6E). These results indicate a correlation between greater interaction of CCM3 with UNC13D, which can be the result of either increased Ca2+ concentration or the mutations in the C2A domain, and stronger reversal of STK24-mediated inhibition of UNC13D binding to liposomes. This correlation can be further interpreted to suggest that the enhanced interaction of CCM3 with UNC13D, which is mediated by the C2A domain, might be an important step in Ca2+-stimulated CCM3-mediated relief of STK24 inhibition of UNC13D-liposome binding. Together with the knowledge of the interactions between CCM3’s NT domain and STK24’s CT domain (Ceccarelli et al., 2011) and between CCM3 FATH domain and UNC13D N-terminus (Fig. S6A), we propose a model to explain how ligand/Ca2+ may regulate exocytosis through the CCM3 and STK24 complex (Fig. 6F). In this model, STK24 binds to UNC13D C2B domain to inhibit UNC13D binding to liposomes at the resting state. When Ca2+ concentration arises upon ligand stimulation, Ca2+ binds to UNC13D C2B domain and stimulates the binding of the C2B domain to liposomes on one hand. On the other hand, Ca2+ binds to the C2A domain and enhances the interaction of the C2A domain with CCM3. The binding of CCM3 to the C2A domain may recruit STK24 away from its binding to the C2B domain, thus relieving STK24-mediated inhibition of liposome binding. This model is supported by the findings that Ca2+ augmented the formation of a ternary complex of UNC13D C2A domain with CCM3 and STK24 (Fig. 6G) and that the CCM3 mutant that cannot interact with STK24 failed to reverse STK24-mediated inhibition of lipid binding (Fig. S6B).
STK24 or CCM3-deficiency exacerbates tissue injury in an ischemia-reperfusion model
We next examined the in vivo significance of enhanced degranulation associated with the loss of STK24 or CCM3. A renal ischemia-reperfusion injury model was adopted, because neutrophils are important for the reperfusion-induced renal tissue damage (Heinzelmann et al., 1999). Reperfusion after renal arterial ischemia resulted in infiltration of neutrophils (Fig. S7A, B). Importantly, the reperfusion induced significantly greater increases in serum creatinine contents (Fig. 7A, B) and the number of coagulative necrotic renal tubules (Fig. 7C–F) in both Stk24h/h and Ccm3m/m mice than their wildtype controls. These results indicated that the lack of STK24 or CCM3 was associated with increased renal tissue damages after ischemia-reperfusion. Given that STK24 or CCM3-deficiency did not significantly affect the numbers of neutrophils infiltrated into the kidneys (Fig. S7C, D), augmented tissue damage occurred in these mutant mice should be at least in part attributed to the enhanced degranulation. In support of this idea, we found that there were significantly higher levels of plasma MPO in the STK24 or CCM3 mutant mice than their controls (Fig. 7G, H).
Figure 7. Loss of STK24 or CCM3 exacerbates tissue damages in a renal ischemia-reperfusion model.
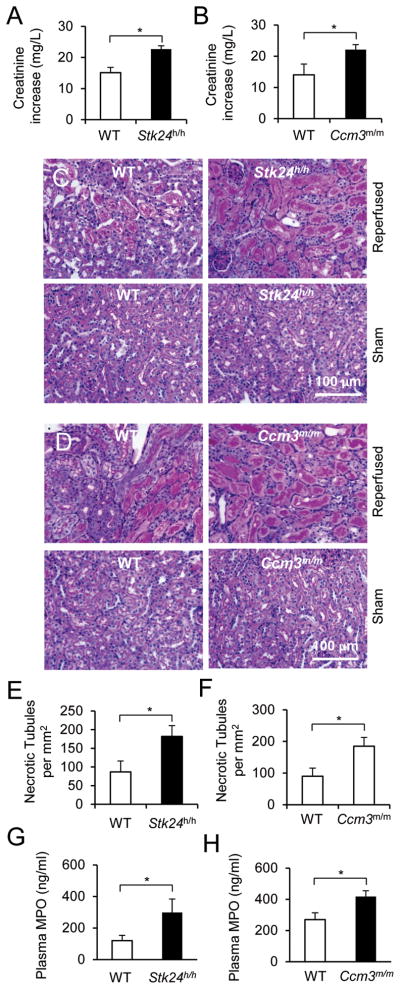
A, B. Blood creatinine contents from mice that were subjected to the renal ischemia-reperfusion procedure. Data are presented as Means ± SEM (*, p<0.05, Student’s t-test, n=4).
C–F. Histological examination of renal tissue injury. Renal histological sections from mice that were subjected to the renal ischemia-reperfusion procedure were stained with Periodic acid-Schiff (PAS) dye, and the representative sections are shown (C, D). The numbers of coagulative necrotic tubules were counted from 6 random PAS-stained sections per kidney and are presented as Means ± SEM (*, p<0.05, Student’s t-test, n=4).
G, H. Blood MPO contents from mice that were subjected to the renal ischemia-reperfusion procedure. Data are presented as Means ± SEM (*, p<0.05, Student’s t-test, n=4). See also Figure S7.
DISCUSSION
In this study, we have uncovered an important cellular function for the protein complex of CCM3 and one of the GCK III sub-family members, STK24. We present solid evidence to demonstrate that the protein complex plays significant roles in ligand-stimulated exocytosis in neutrophils. Importantly, we have characterized the biochemical mechanism by which CCM3 and STK24 regulate this cellular function.
Most of the work on ligand-stimulated exocytosis has been done with neurons, endocrine cells, platelets, and cytotoxic T cells. Not much is known about the mechanisms for ligand-regulated exocytosis or degranulation in neutrophils, other than the involvement of UNC13D and RAB27 (Munafo et al., 2007; Brzezinska et al., 2008; Herrero-Turrion et al., 2008; Pivot-Pajot et al., 2008; Elstak et al., 2011). In this study, we found that MPO secretion had at least two phases. The first phase probably corresponds to the release of the readily releasable pool, which is rapidly released upon ligand stimulation, whereas the second phase may be from the reserved pool, which may not be docked prior to the initial stimulation. The STK24-CCM3 complex seems to primarily regulate the release of the reserved pool. This conclusion is consistent with our mechanistic results, which show that STK24 appears to regulate steps upstream of vesicle fusion, because its deficiency doesn’t affect vesicle fusion kinetics. In further support of this conclusion, STK24 inhibits the binding of UNC13D to lipids, a step that is important for vesicle docking (Pivot-Pajot et al., 2008; Boswell et al., 2012). The involvement of CCM3 in exocytosis regulation seems more complicated than STK24. It appears to play dual roles. On one hand, it stabilizes the STK24 protein. This may explain the similar degranulation phenotypes of its deficiency to those of STK24-deficiency. On the other hand, CCM3 is required for restoration of Ca2+-stimulated binding of UNC13D to lipids that is strongly inhibited by STK24. This restoration provides a means for ligands that can increase intracellular Ca2+ concentrations to regulate the second phase of degranulation from the reserved pool, as depicted in the model (Fig. 6F). In this model, Ca2+ stimulates the interaction of CCM3 with UNC13D C2A domain, which recruits STK24 away from the C2B domain to allow the binding of the C2B domain to lipids. We, however, do not know exactly why the lack of either STK24 or CCM3 did not significantly impact the first phase of exocytosis immediately after ligand stimulation. A simple explanation is that certain equilibrium between STK24 and membrane lipids for binding to UNC13D C2B domain may have been reached at the resting state.
Our renal ischemia-reperfusion model study of STK24-deficient and particularly myeloid-specific CCM3-deficient mice have demonstrated the in vivo significance of this degranulation regulation imposed by the STK24 and CCM3 complex. Given the broad tissue expression patterns of CCM3 and GCKIII sub-family members, this model may also be applicable to the regulation of exocytosis in other cell types. It may also be of particular significance in the regulation of the second phase of exocytosis from the reserved pool, because the mechanism for this regulation remains largely elusive (Sugita, 2008; Pores-Fernando and Zweifach, 2009; Seino et al., 2011). Another potential impact of this study beyond neutrophils may be on the investigation of the pathogenic basis by which CCM3 mutations cause the CCM disease. Our preliminary evidence suggests that CCM3 and STK24 are also involved in the regulation of endothelial cell exocytosis. Knockdown of CCM3 or STK24 resulted in an increase in the level of cell surface P-selectin upon TNFα stimulation without altering the mRNA levels of P-selectin (data not shown). P-selectin is a cell surface adhesion molecule and stored in endothelial granules called Weibel-Palade Bodies (WPBs). It is transported to cell surfaces through exocytosis in response to stimulation by pro-inflammatory ligands including TNFα (Valentijn et al., 2011). This preliminary observation not only indicates the importance of this protein complex in exocytic regulation beyond neutrophils, but also provides a basis for a plausible hypothesis to explain the contribution of CCM3 inactivation to the CCM pathogenesis. We hypothesize that enhanced exocytosis in response to certain stimulations in the brain may contribute to vascular defects observed in CCM disease. It is not difficult to postulate how enhanced exocytosis, through which cell surface and secreted proteins and molecules are transported to their functional sites, causes the vasculature defects. For instance, enhanced release of angiopoietin-2, which is also stored from WPBs, may cause capillary destabilization (Augustin et al., 2009), whereas increased levels of cell adhesion or junction proteins may result in defects in the formation and maintenance of vessel tubular morphology. This hypothesis is also compatible for the anatomic restriction of the CCM lesions, which may be the result of the unique nature of the stimulations and/or effect of hyper-exocytic activity on the blood vessels in the brain. Therefore, more studies are warranted to further characterize the role of CCM3 and STK24 as well as other members of the GCKIII subfamily in endothelial cell exocytosis and determine if altered exocytosis contributes to the CCM phenotypes. Of note, our hypothesis is consistent with the findings from a recent fly genetic study. This fly work shows that the loss-of-function mutant of fly CCM3 or GCKIII kinase ortholog have dilated tracheal tubes, which resembles dilated vessels found in CCM patients. This tube dilation phenotype can be suppressed by the reduction in the expression of N-ethylmaleimide sensitive factor 2(NSF2) (Song et al., 2013). Given the important role of NSF2 in SNARE recycling and the secondary phase of exocytosis (Zhao et al., 2012), these fly results are in agreement with our observation that the STK24-CCM3 complex regulates exocytosis of the reserved pool of granules and support our hypothesis that enhanced exocytosis may contribute to defective vessel tubular morphology observed in the CCM disease.
Despite the shared heart development phenotypes in zebrafish (Zheng et al., 2010) and exocytosis in cultured endothelial cells by CCM3 and STK24 knockdown, we have not observed overt cardiac or consistent vascular defects in the Stk24h/h mice. The lack of the obvious cardio-vascular phenotypes may be attributed to greater molecular redundancy of the GCKIII sub-family members in mice, which is consistent with the weaker effect of STK24 knockdown on P-selectin cell surface expression than CCM3 knockdown (data not shown). Alternatively, exocytosis may only be one of many cellular activities regulated by CCM3, as CCM3 was found to regulate Golgi assembly with STK25 (Fidalgo et al., 2010). These questions need to be resolved in future investigation.
EXPERIMENTAL PROCEDURES
Reagents and constructs
CCM3 antibody was generated as described before (He et al., 2010). Other antibodies were acquired commercially: STK24 antibodies (rabbit, 3723, Cell Signaling), GST antibody (2624, Cell Signaling), His antibody (2366, Cell Signaling), MST4 antibody (3822, Cell Signaling), HA antibody (MMS-101R, Covance), Myc antibody (MMS-150R, Covance), GFP antibody (sc-9996, Santa Cruz Biotechnology), Gβ2 antibody (sc-380, Santa Cruz Biotechnology), and MPO antibody (HM1051BT, HyCult Biotech). Protein A/G-PLUS-agarose beads were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Formyl-Met-Leu-Phe (fMLP) and Cytochalasin B (CB) were purchased from Sigma (St. Louis, MO). Human UNC13D cDNA, and mouse STK24 cDNA were acquired from Open Biosystems (Lafayette, CO).
Mice
STK24h/h mice were generated using a gene-trap ESC line AM0826 (Wellcome Trust Sanger Institute) from International Gene Trap Consortium at the Mutant Mouse Regional Resource Center (UC Davis) using the standard method. Mice heterozygous for the mutation were initially generated on a mixed 129/Ola and C57BL/6 background and were subsequently backcrossed to the C57BL/6 background for more than 5 generations. Myeloid-specific CCM3 knockout mice were generated by crossing floxed CCM3 mice (He et al., 2010) with the B6.129-Lyzstm1(cre)Ifo/J mice from Jackson Lab. All animal studies were approved by the institutional animal care and use committees of Yale University.
White blood cell counts
A Hemavet system (Drew Scientific, Ramsey, MN), which is a multiparameter, automated hematology analyzer designed for in vitro diagnostic use (Li et al., 2009), was used to count the leukocyte numbers in circulating blood. To quantify neutrophil numbers in spleens, livers and kidneys, cells were dissociated from the tissues and total cell numbers were counted using a Guava EasyCyte System (Millipore, Billerica, Massachusetts). The cells were also stained with APC-CD11b antibody (17-0112, eBioscience) and Percp-Ly-6G (560602, BD) antibody and analyzed by a LSR II FACS analyzer. Neutrophils were defined as Ly-6G+CD11b+ cells.
Neutrophil preparation and transfection
Murine neutrophils were purified from bone marrows as previously described (Zhang et al., 2010). Briefly, bone marrow cells collected from mice were treated with the ACK buffer (155 mM NH4Cl, 10 mM KHCO3 and 127 μM EDTA) for red blood cell lysis, followed by a discontinuous Percoll density gradient centrifugation. Neutrophils were collected from the band located between 81% and 62% of Percoll. For transient transfection of primary mouse neutrophils, three million neutrophils were electroporated with 1.6 μg endotoxin-free plasmids using the human monocyte nucleofection kit (Lonza, Switzerland) with an Amaxa electroporation system. The cells were then cultured for overnight in the medium supplied with the kit containing 10% FBS and 25 ng/ml recombinant GM-CSF (PeproTech, Rocky Hill, NJ). GFP+ Cells were sorted by a FACS Aria sorter (BD, San Jose, CA).
Immunostaining of neutrophils
Neutrophils were placed onto the coverslips for 15 minutes before stimulation. The cells were then fixed with 4% Para-formaldehyde and permeabilized with 0.01% saponin and blocked with 2% BSA in PBS (Brzezinska et al., 2008). Neutrophils were stained with a STK24 antibody (3723, Cell Signaling, 1:100) and/or biotinylated MPO antibody (HM1051BT, Hycult Biotech, 1:100), followed by Alexa-488-Goat-anti-Rabbit (Invitrogen, 1:300 for the STK antibody) and APC-streptavidin (BD, 1:300 for the MPO antibody) staining using a previously described protocol (Xu et al., 2010). Mounted slides were observed using a Leica SP5 confocal microscopy.
TIRF microscopy
TIRF imaging was performed using an IX-70 inverted microscope (Olympus), equipped with argon (488 nm) and argon/krypton (568 nm) laser lines (Melles Griot), a 60× 1.45 NA oil immersion objective lens (Plan-ApoN; Olympus), and a custom TIRFM condenser. Cells were imaged in one channel at 5–10 Hz or two channels by sequential excitation at 3–5 Hz without binning and detected with a back-illuminated iXon887 EMCCD camera (512 × 512, 0.18 μm per pixel, 16 bits; Andor Technologies) with a 1.6× magnification lens. The system was controlled by the Andor iQ software (Xu et al., 2011). For live-cell imaging, neutrophils were resuspended in 2 ml Hank’s buffer with 0.25% BSA and seeded into 35 mm petri dish with a No. 1.5 coverglass at the bottom (MatTek, Ashland, MA). For cell stimulation, an Eppendorf Femotip was loaded with 50 μM fMLP and 50 μM CB, and ligands were loaded by applying air pressure to the micropipette using a Picospritzer II microinjection system (Parker Hannifin, Cleveland, OH). Each puff lasts 1 second, and the volume for each puff is about 5 nanoliters. Time-lapsed images were acquired at 200 mill-second intervals at 37°C. For fixed cell imaging, neutrophils were stimulated with 1 μM fMLP and 10 μM CB, fixed, and stained with anti-MPO antibody (HyCult Biotech) before imaging.
MPO and MMP detection
One million neutrophils were incubated with 10 μM CB for 5 min at 37°C prior to stimulation with fMLP (500 nM) or MIP2 (10 nM) for another 10 min. The reaction was stopped by being placed on ice, and the suspension was centrifuged at 500xg for 5 min at 4°C. Supernatants were assayed for MPO and MMP contents using the EnzChek Myeloperoxidase Activity Assay Kit and EnzChek Gelatinase/Collagenase Assay kit (Life Technologies, Grand Island, NY), respectively (Lee et al., 2007; Li et al., 2009).
Immunoprecipitation
HEK293T cells were maintained in Dulbecco’s modifies Eagle’s medium (DMEM) with 4.5 g/liter glucose supplemented with 10% fetal bovine serum (FBS). Transient transfection was carried out using Lipofectamine Plus (Life Technologies, Grand Island, NY), and samples were collected 24 hours after transfection. Cells were lysed in a cell lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 5 mM EDTA, Roche’s protease inhibitor cocktail, Roche’s phosphatase inhibitor cocktail [Indianapolis, IN]). After removing insoluble materials by centrifugation, immunoprecipitation was carried out by adding various antibodies and Protein A/G-PLUS-agarose beads into supernatants. Immunocomplexes were washed three times with lysis buffer before the SDS sample buffer was added for SDS-PAGE analysis.
Phagocytosis, chemotaxis and superoxide production assays
For testing phagocytosis, neutrophils (0.5 million) were incubated with 20 μl pHrodo™ E. coli BioParticles Conjugate (Invitrogen) at 37 °C. After fixation, cells were analyzed by flow cytometry using a BD LSRII FACS analyzer (Hsieh et al., 2009).
The chemotaxis assay was carried out using a Dunn chamber as previously described (Zicha et al., 1997) with some modifications as detailed in (Xu et al., 2010; Zhang et al., 2010).
For superoxide production assay, neutrophils were suspended in Hank’s buffer at 1 × 106/ml on ice. Fifty microliters of cells were mixed with 20 μl HRP (100 U/ml, Sigma), 1.5 μl luminol (10 mM, Sigma), and 28.5 μl Hank’s buffer in a well of a 96-well plate. Chemiluminescence was detected by a Wallac plate reader (Dong et al., 2005).
Tandem Affinity Purification (TAP) and Mass Spectrometry Analyses
TAP were performed with the InterPlay Mammalian TAP System (Agilent, Clara, CA). The purified proteins were separated on SDS-PAGE, and the protein bands were excised from the gel for MS analyses. An LTQ-Orbitrap was used for identification of CCM3 and STK24-associated proteins at W.M. Keck MS and Proteomics Resource of Yale University. MS/MS spectra were searched against Homo sapiens subset of the NCBI protein sequence database (Release 20110610) using Mascot Daemon software.
Protein preparation and pull-down
Recombinant proteins were expressed in Escherichia coli BL21(DE3) and purified by affinity chromatography using glutathione agarose beads for GST-fusion proteins or Ni-NTA agarose beads (GE Healthcare) for His-tagged proteins. Proteins were dialyzed to a buffer containing 20 mM HEPES, pH 7.5, 100 mM KCl, 0.1% Triton X-100, 0.2 mM EGTA, 10% glycerol and 1 mM DTT. For the pull-down assays, His- and GST-tagged proteins were incubated with either glutathione-agarose or Ni-NTA agarose beads as indicated in the figures at 4°C for 5 hours. After extensive washes, proteins on the beads were resolved by SDS/PAGE and detected by Western Blot or Coomassie blue staining.
Buoyant density flotation of liposome assay
The assay was performed as previously described (Boswell et al., 2012). Liposomes were consisted of 85% PC and 15% PS which were purchased from Avanti Polar Lipids and were incubated with UNC13D and indicated proteins (BSA or STK24/CCM3) for 30 min on ice in 75 μl of 25 mM HEPES, pH 7.4, 100 mM KCl, 10% glycerol, and 1 mM DTT (reconstitution buffer). Liposomes binding reactions were conducted in the absence of Ca2+ (0.2 mM EGTA) or presence of 100 μM of free Ca2+. 75 μl of 80% Accudenz was added to the binding reaction to yield a final concentration of 40% Accudenz. 30% Accudenz and reconstitution buffers were then layered on top and centrifuged for 4 h in a TLA100 rotor at 32,000 rpm. Either the floated fraction or inputs were collected and run on SDS-PAGE and analyzed by Western blotting.
Murine kidney ischemia/reperfusion models
The left and right renal arteries of mutant and their littermate control mice (9–10 weeks old) were clamped for 30 min using vascular clamps (Fine Science Tools, CA) through a midline abdominal incision under general anesthesia. After ischemia, the clamps were removed, the wounds were sutured, and the animals were allowed to recover for 24 h before euthanization. Blood was collected by cardiac puncture in heparin-containing tube, and creatinine levels were determined using the DetectX serum creatinine kit (Arbor Assays, Ann Arbor, MI). Kidneys were dissected from mice, fixed in 4% paraformaldehyde, embedded in paraffin, and stained for the Periodic acid-Schiff (PAS) dye and a Ly6B antibody. Plasma MPO levels were determined using an ELISA kit (HyCult Biotechnology).
Supplementary Material
Highlights.
STK24 inhibits the release of the reserve pool of neutrophil granules
STK24 binds to UNC13D and inhibits UNC13D-lipid binding and granule docking
CCM3 stabilizes STK24, and its deficiency phenocopies STK24-deficiency
Upon stimulation, CCM3 plays a role in reversal of the inhibition imposed by STK24
Acknowledgments
We thank Michelle Orsulak for technical assistance, Abdul Basit for discussion and the Microsurgery Core of Yale Cardiovascular Research Center for performing renal ischemic procedures. The work is supported by NIH grants to D.W. (HL108430, HL070694, AI094525), W.M. (HL115148, HL109420), T.B. (GM102262), D.T. (NIH DP2-OD002980), H.Z. (AHA SDG 13SDG17110045), Yale/NIDA Neuroproteomics Center (DA018343), and W.P. (National Thousand Talents Program for Distinguished Young Scholars).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- Boswell KL, James DJ, Esquibel JM, Bruinsma S, Shirakawa R, Horiuchi H, Martin TF. Munc13-4 reconstitutes calcium-dependent SNARE-mediated membrane fusion. J Cell Biol. 2012;197:301–312. doi: 10.1083/jcb.201109132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezinska AA, Johnson JL, Munafo DB, Crozat K, Beutler B, Kiosses WB, Ellis BA, Catz SD. The Rab27a effectors JFC1/Slp1 and Munc13-4 regulate exocytosis of neutrophil granules. Traffic. 2008;9:2151–2164. doi: 10.1111/j.1600-0854.2008.00838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalcanti DD, Kalani MY, Martirosyan NL, Eales J, Spetzler RF, Preul MC. Cerebral cavernous malformations: from genes to proteins to disease. J Neurosurg. 2012;116:122–132. doi: 10.3171/2011.8.JNS101241. [DOI] [PubMed] [Google Scholar]
- Ceccarelli DF, Laister RC, Mulligan VK, Kean MJ, Goudreault M, Scott IC, Derry WB, Chakrabartty A, Gingras AC, Sicheri F. CCM3/PDCD10 heterodimerizes with germinal center kinase III (GCKIII) proteins using a mechanism analogous to CCM3 homodimerization. J Biol Chem. 2011;286:25056–25064. doi: 10.1074/jbc.M110.213777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat K, Hoebe K, Ugolini S, Hong NA, Janssen E, Rutschmann S, Mudd S, Sovath S, Vivier E, Beutler B. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med. 2007;204:853–863. doi: 10.1084/jem.20062447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10:568–579. doi: 10.1038/nri2803. [DOI] [PubMed] [Google Scholar]
- Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol. 2005;15:1874–1879. doi: 10.1016/j.cub.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Elstak ED, Neeft M, Nehme NT, Voortman J, Cheung M, Goodarzifard M, Gerritsen HC, van Bergen En Henegouwen PM, Callebaut I, de Saint Basile G, van der Sluijs P. The munc13-4-rab27 complex is specifically required for tethering secretory lysosomes at the plasma membrane. Blood. 2011;118:1570–1578. doi: 10.1182/blood-2011-02-339523. [DOI] [PubMed] [Google Scholar]
- Faurobert E, Albiges-Rizo C. Recent insights into cerebral cavernous malformations: a complex jigsaw puzzle under construction. FEBS J. 2010;277:1084–1096. doi: 10.1111/j.1742-4658.2009.07537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachee-Chardin M, Chedeville G, Tamary H, Minard-Colin V, Vilmer E, Blanche S, Le Deist F, Fischer A, de Saint Basile G. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–473. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- Fidalgo M, Fraile M, Pires A, Force T, Pombo C, Zalvide J. CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J Cell Sci. 2010;123:1274–1284. doi: 10.1242/jcs.061341. [DOI] [PubMed] [Google Scholar]
- Fukuda M. Versatile role of Rab27 in membrane trafficking: focus on the Rab27 effector families. Journal of biochemistry. 2005;137:9–16. doi: 10.1093/jb/mvi002. [DOI] [PubMed] [Google Scholar]
- He B, Guo W. The exocyst complex in polarized exocytosis. Cur Opin in Cell Biol. 2009;21:537–542. doi: 10.1016/j.ceb.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhang H, Yu L, Gunel M, Boggon TJ, Chen H, Min W. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci Signal. 2010;3:ra26. doi: 10.1126/scisignal.2000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzelmann M, Mercer-Jones MA, Passmore JC. Neutrophils and renal failure. Am J Kidney Dis. 1999;34:384–399. doi: 10.1016/s0272-6386(99)70375-6. [DOI] [PubMed] [Google Scholar]
- Herrero-Turrion MJ, Calafat J, Janssen H, Fukuda M, Mollinedo F. Rab27a regulates exocytosis of tertiary and specific granules in human neutrophils. J Immunol. 2008;181:3793–3803. doi: 10.4049/jimmunol.181.6.3793. [DOI] [PubMed] [Google Scholar]
- Higashio H, Nishimura N, Ishizaki H, Miyoshi J, Orita S, Sakane A, Sasaki T. Doc2 alpha and Munc13-4 regulate Ca(2+) -dependent secretory lysosome exocytosis in mast cells. J Immunol. 2008;180:4774–4784. doi: 10.4049/jimmunol.180.7.4774. [DOI] [PubMed] [Google Scholar]
- Hsieh CH, Nickel EA, Chen J, Schwacha MG, Choudhry MA, Bland KI, Chaudry IH. Mechanism of the salutary effects of estrogen on kupffer cell phagocytic capacity following trauma-hemorrhage: pivotal role of Akt activation. J Immunol. 2009;182:4406–4414. doi: 10.4049/jimmunol.0803423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Wu YM, Hsu CY, Lee WS, Lai MD, Lu TJ, Huang CL, Leu TH, Shih HM, Fang HI, Robinson DR, Kung HJ, Yuan CJ. Caspase activation of mammalian sterile 20-like kinase 3 (Mst3). Nuclear translocation and induction of apoptosis. J Biol Chem. 2002;277:34367–34374. doi: 10.1074/jbc.M202468200. [DOI] [PubMed] [Google Scholar]
- Irwin N, Li YM, O’Toole JE, Benowitz LI. Mst3b, a purine-sensitive Ste20-like protein kinase, regulates axon outgrowth. Proc Natl Acad Sci U S A. 2006;103:18320–18325. doi: 10.1073/pnas.0605135103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski A, Kim JH, Collins RF, Daneman R, Walton P, Grinstein S. In situ measurements of the pH of mammalian peroxisomes using the fluorescent protein pHluorin. J Biol Chem. 2001;276:48748–48753. doi: 10.1074/jbc.M109003200. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Hong H, Monfregola J, Kiosses WB, Catz SD. Munc13-4 restricts motility of Rab27a-expressing vesicles to facilitate lipopolysaccharide-induced priming of exocytosis in neutrophils. J Biol Chem. 2011;286:5647–5656. doi: 10.1074/jbc.M110.184762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kean MJ, Ceccarelli DF, Goudreault M, Sanches M, Tate S, Larsen B, Gibson LC, Derry WB, Scott IC, Pelletier L, Baillie GS, Sicheri F, Gingras AC. Structure-function analysis of core STRIPAK Proteins: a signaling complex implicated in Golgi polarization. J Biol Chem. 2011;286:25065–25075. doi: 10.1074/jbc.M110.214486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch H, Hofmann K, Brose N. Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem J. 2000;349:247–253. doi: 10.1042/0264-6021:3490247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy P, Eitzen G. Control of granule exocytosis in neutrophils. Front Biosci. 2008;13:5559–5570. doi: 10.2741/3099. [DOI] [PubMed] [Google Scholar]
- Lee CW, Lin CC, Lin WN, Liang KC, Luo SF, Wu CB, Wang SW, Yang CM. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-kappaB/p300 binding in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L799–812. doi: 10.1152/ajplung.00311.2006. [DOI] [PubMed] [Google Scholar]
- Li L, Chin LS. The molecular machinery of synaptic vesicle exocytosis. Cell Mol Life Sci. 2003;60:942–960. doi: 10.1007/s00018-003-2240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jia Y, Pichavant M, Loison F, Sarraj B, Kasorn A, You J, Robson BE, Umetsu DT, Mizgerd JP, Ye K, Luo HR. Targeted deletion of tumor suppressor PTEN augments neutrophil function and enhances host defense in neutropenia-associated pneumonia. Blood. 2009;113:4930–4941. doi: 10.1182/blood-2008-06-161414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber B, Howe ML, Benowitz LI, Irwin N. Mst3b, an Ste20-like kinase, regulates axon regeneration in mature CNS and PNS pathways. Nat Neurosci. 2009;12:1407–1414. doi: 10.1038/nn.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TJ, Lai WY, Huang CY, Hsieh WJ, Yu JS, Hsieh YJ, Chang WT, Leu TH, Chang WC, Chuang WJ, Tang MJ, Chen TY, Lu TL, Lai MD. Inhibition of cell migration by autophosphorylated mammalian sterile 20-like kinase 3 (MST3) involves paxillin and protein-tyrosine phosphatase-PEST. J Biol Chem. 2006;281:38405–38417. doi: 10.1074/jbc.M605035200. [DOI] [PubMed] [Google Scholar]
- Menager MM, Menasche G, Romao M, Knapnougel P, Ho CH, Garfa M, Raposo G, Feldmann J, Fischer A, de Saint Basile G. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat Immunol. 2007;8:257–267. doi: 10.1038/ni1431. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Munafo DB, Johnson JL, Ellis BA, Rutschmann S, Beutler B, Catz SD. Rab27a is a key component of the secretory machinery of azurophilic granules in granulocytes. Biochem J. 2007;402:229–239. doi: 10.1042/BJ20060950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeft M, Wieffer M, de Jong AS, Negroiu G, Metz CH, van Loon A, Griffith J, Krijgsveld J, Wulffraat N, Koch H, Heck AJ, Brose N, Kleijmeer M, van der Sluijs P. Munc13-4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. Mol Biol Cell. 2005;16:731–741. doi: 10.1091/mbc.E04-10-0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Sudhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Decoding cytosolic Ca2+ oscillations. Trends Biochem Sci. 2011;36:78–87. doi: 10.1016/j.tibs.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Payton JE, Grieselhuber NR, Chang LW, Murakami M, Geiss GK, Link DC, Nagarajan R, Watson MA, Ley TJ. High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J Clin Invest. 2009;119:1714–1726. doi: 10.1172/JCI38248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivot-Pajot C, Varoqueaux F, de Saint Basile G, Bourgoin SG. Munc13-4 regulates granule secretion in human neutrophils. J Immunol. 2008;180:6786–6797. doi: 10.4049/jimmunol.180.10.6786. [DOI] [PubMed] [Google Scholar]
- Pombo CM, Force T, Kyriakis J, Nogueira E, Fidalgo M, Zalvide J. The GCK II and III subfamilies of the STE20 group kinases. Front Biosci. 2007;12:850–859. doi: 10.2741/2107. [DOI] [PubMed] [Google Scholar]
- Pores-Fernando AT, Zweifach A. Calcium influx and signaling in cytotoxic T-lymphocyte lytic granule exocytosis. Immunol Rev. 2009;231:160–173. doi: 10.1111/j.1600-065X.2009.00809.x. [DOI] [PubMed] [Google Scholar]
- Ren Q, Wimmer C, Chicka MC, Ye S, Ren Y, Hughson FM, Whiteheart SW. Munc13-4 is a limiting factor in the pathway required for platelet granule release and hemostasis. Blood. 2010;116:869–877. doi: 10.1182/blood-2010-02-270934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci. 2001;4:129–136. doi: 10.1038/83949. [DOI] [PubMed] [Google Scholar]
- Schinkmann K, Blenis J. Cloning and Characterization of a Human STE20-like Protein Kinase with Unusual Cofactor Requirements. J Biol Chem. 1997;272:28695–28703. doi: 10.1074/jbc.272.45.28695. [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest. 2011;121:2118–2125. doi: 10.1172/JCI45680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Eng M, Ghabrial AS. Focal Defects in Single-Celled Tubes Mutant for Cerebral Cavernous Malformation 3, GCKIII, or NSF2. Dev Cell. 2013;25:507–519. doi: 10.1016/j.devcel.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rizo J. Synaptic vesicle exocytosis. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S. Mechanisms of exocytosis. Acta Physiol (Oxf) 2008;192:185–193. doi: 10.1111/j.1748-1716.2007.01803.x. [DOI] [PubMed] [Google Scholar]
- Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. 2011;117:5033–5043. doi: 10.1182/blood-2010-09-267492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss K, Stahl S, Hogan BM, Reinders J, Schleider E, Schulte-Merker S, Felbor U. Functional analyses of human and zebrafish 18-amino acid in-frame deletion pave the way for domain mapping of the cerebral cavernous malformation 3 protein. Hum Mutat. 2009;30:1003–1011. doi: 10.1002/humu.20996. [DOI] [PubMed] [Google Scholar]
- Wu HY, Lin CY, Chen TC, Pan ST, Yuan CJ. Mammalian Ste20-like protein kinase 3 plays a role in hypoxia-induced apoptosis of trophoblast cell line 3A-sub-E. Int J Biochem Cell Biol. 2011;43:742–750. doi: 10.1016/j.biocel.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Wu HY, Lin CY, Lin TY, Chen TC, Yuan CJ. Mammalian Ste20-like protein kinase 3 mediates trophoblast apoptosis in spontaneous delivery. Apoptosis. 2008;13:283–294. doi: 10.1007/s10495-007-0161-x. [DOI] [PubMed] [Google Scholar]
- Xu W, Wang P, Petri B, Zhang Y, Tang W, Sun L, Kress H, Mann T, Shi Y, Kubes P, Wu D. Integrin-induced PIP5K1C kinase polarization regulates neutrophil polarization, directionality, and in vivo infiltration. Immunity. 2010;33:340–350. doi: 10.1016/j.immuni.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Rubin BR, Orme CM, Karpikov A, Yu C, Bogan JS, Toomre DK. Dual-mode of insulin action controls GLUT4 vesicle exocytosis. J Cell Biol. 2011;193:643–653. doi: 10.1083/jcb.201008135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T. Ca2+-dependent regulation of synaptic vesicle endocytosis. Neurosci Res. 2012;73:1–7. doi: 10.1016/j.neures.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Yoruk B, Gillers BS, Chi NC, Scott IC. Ccm3 functions in a manner distinct from Ccm1 and Ccm2 in a zebrafish model of CCM vascular disease. Dev Biol. 2012;362:121–131. doi: 10.1016/j.ydbio.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tang W, Jones MC, Xu W, Halene S, Wu D. Different roles of G protein subunits beta1 and beta2 in neutrophil function revealed by gene expression silencing in primary mouse neutrophils. J Biol Chem. 2010;285:24805–24814. doi: 10.1074/jbc.M110.142885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Smith EC, Whiteheart SW. Requirements for the catalytic cycle of the N-ethylmaleimide-Sensitive Factor (NSF) Biochim Biophys Acta. 2012;1823:159–171. doi: 10.1016/j.bbamcr.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Xu C, Di Lorenzo A, Kleaveland B, Zou Z, Seiler C, Chen M, Cheng L, Xiao J, He J, Pack MA, Sessa WC, Kahn ML. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J Clin Invest. 2010;120:2795–2804. doi: 10.1172/JCI39679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zicha D, Dunn G, Jones G. Analyzing chemotaxis using the Dunn direct-viewing chamber. Methods Mol Biol. 1997;75:449–457. doi: 10.1385/0-89603-441-0:449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.