

Abstract
Purpose
The synthetic compound 3-(5′-hydroxymethyl-2’-furyl)-1-benzylindazole (YC-1) reduces the protein stability of hypoxia-inducible factor (HIF)-1α and can serve as a potential anticancer agent. Our previous study elucidated that YC-1 decreased the protein level of HIF-1α and inhibited cell proliferation under normoxic conditions. In the present study, we explored the inhibitory effect of YC-1 on the regulation of HIF-1α and cell survival under hypoxia.
Methods
Chemical and physical hypoxia using cobalt chloride and an anaerobic incubator, respectively, was induced in the photoreceptor cell line 661W. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay and morphological observation were used to analyze cell survival. Flow cytometry with a LIVE/DEAD cell viability assay and annexin V was used to determine the number of live and dead cells or cell apoptosis, respectively. Cell proliferation was analyzed with high-content screening of MKI67 (Ki-67) immunofluorescent staining. Immunoblotting and a quantitative reverse-transcription PCR were used to assess the protein and mRNA levels, respectively.
Results
Our results showed that 661W cells exposed to YC-1 decreased cell survival through the induction of cell apoptosis and cell-cycle arrest under hypoxia. We also found that YC-1 reduced the HIF-1α protein level after 2 h of hypoxia, but the mRNA level of HIF-1α was not affected. In addition, YC-1 significantly increased levels of p53, the proapoptotic gene BCL2-associated X protein (Bax), and cell proliferation-related gene, cyclin-dependent kinase inhibitor 1A (p21) mRNAs under hypoxia.
Conclusions
Unlike normoxia, YC-1 not only inhibited cell proliferation but also induced cell death under hypoxia. We also found that YC-1 inhibited hypoxia-induced HIF-1α and partially affected hypoxia-regulated gene expression.
Introduction
The lack of oxygen induces many adaptive responses and stimulates many hypoxia-responsive transcription factors [1]. Among them, hypoxia-inducible factor (HIF)-1, a heterodimeric transcription factor, is the major hypoxic signaling protein that allows cells to adapt to low-oxygen conditions [2,3]. Under hypoxia, the HIF-1α subunit containing an oxygen-dependent degradation domain interacts with the HIF-1β subunit to form the HIF-1 dimer [4,5]. Consequently, HIF-1 is involved in many pathophysiological processes, such as angiogenesis, metabolism, apoptosis, and cell proliferation [6], through transcriptional factors, such as nuclear factor (NF)-κB [7,8] and protein 53 (p53) [9], in response to hypoxia. Many target genes, such as vascular endothelial growth factor (VEGF), erythropoietin (EPO), inducible nitric oxide synthase (iNOS), carbonic anhydrase 9 (Car9), and glucose transporter 1 (GLUT1), can be activated by HIF-1 under hypoxia [2,3]. Under cell stress, p53 can transactivate proapoptotic genes, such as BCL2-associated X protein (Bax) and apoptotic peptidase activating factor 1 (Apaf1), or transrepress antiapoptotic genes, such as B-cell CLL/lymphoma 2 (Bcl2), subsequently inducing cell apoptosis [10]. P53 can also induce cyclin-dependent kinase inhibitor 1A (p21) gene expression, which results in cell-cycle arrest [11]. For the relationship between p53 and HIF-1 during hypoxia, a previous study noted that mutation of p53 in tumor cells can lead to an accumulation of HIF-1α and an increase in HIF-1-dependent transcriptional activation of VEGF [12]. Therefore, it is important to know the relationship between p53 and HIF-1 under cell stress and hypoxia.
Hypoxia can be the cause of many central nervous diseases and ocular diseases, such as diabetic retinopathy and glaucoma [13-15], that exhibit extensive neuroretinal cell apoptosis [16-18] because the neuronal cells and neuroretinal cells are particularly vulnerable to transient, mild, systemic hypoxia in the human [19] and animal neuroretina [20]. Here, we used a photoreceptor cell line 661W to study the possible effects of hypoxia on the neuronal cells in the eye. We also applied 3-(5′-hydroxymethyl-2’-furyl)-1-benzylindazole (YC-1), a potential anticancer agent that suppresses HIF-1 and VEGF expression in cancer cells [21], to this cell line under hypoxia. We hypothesized that YC-1 might inhibit hypoxia-induced HIF-1α and subsequently affect HIF-1-regulated cell apoptosis and proliferation in 661W cells under hypoxia.
Briefly, we analyzed the cell viability, proliferation, and death and apoptosis of 661W cells in response to YC-1 under cobalt chloride (CoCl 2)-mediated chemical hypoxia. The protein and mRNA levels of HIF-1α and other hypoxia-related gene expression were also estimated. Finally, we used physical hypoxia with a low-oxygen supply to confirm our findings.
Methods
Cell culture
Our cell line was purchased from the American Type Culture Collection (Manassas, VA) and recharacterized as a murine photoreceptor cell line 661W. 661W cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen Life Technologies, Carlsbad, CA) containing 100 U/ml penicillin/streptomycin (Invitrogen), 0.125 mg/L amphotericin B (Invitrogen), and 5% heat-inactivated fetal calf serum (Invitrogen) at 37 °C in a humidified incubator with 5% CO2. TrypLE (Invitrogen) was used for cell passages and was removed by centrifugation at 112 ×g for 3 min.
Chemical and physical hypoxia
The hypoxia-mimicking agent CoCl2 (Sigma-Aldrich, St. Louis, MO) was dissolved in sterile distilled water and used to induce chemical hypoxia as previously described [22]. Physical hypoxia was induced using a humidified anaerobic workstation INVIVO2 200 (Ruskinn Technology, Pencoed, UK) at 37 °C with 0.5% O2, 5% CO2, and 94.5% N2. Cultured 661W cells were incubated with different concentrations of YC-1 (Sigma-Aldrich) in dimethyl sulfoxide (DMSO; Sigma-Aldrich) for 5 min before induction of both chemical and physical hypoxia.
MTT assay
We dissolved MTT powder (Sigma-Aldrich) in distilled H2O (5 mg/ml) and sterilized this mixture through a 0.22-µm filter before use. Cultured 661W cells were seeded in a 96-well plate (5,000 cells/well, total 100 µl) overnight and treated until 80% confluent in our experiments. After treatment, 10 µl of the MTT stock solution was added to each well and incubated at 37 °C at least 1 h during normoxia. After removing the medium, the formazan product was dissolved in 200 µl DMSO in each well. Absorbance was measured at 570 nm with a μQuant microplate reader (BioTek Instruments, Winooski, VT). The test was performed in four wells and repeated three times.
Morphological imaging and fluorescent staining
Photographic images were captured by a Zeiss Axiovert 35 Inverted Fluorescence Microscope (Carl Zeiss, Oberkochen, Germany) equipped with an ImagingSource DBK 41AU02.AS digital camera (Stuttgart, Germany). Fluorescence microscopy used a Leica DM 2500 stereomicroscope (Wetzlar, Germany) equipped with a Leica DFC490 digital camera. For fluorescence staining, cultured 661W cells were incubated with LIVE/DEAD® cell viability assay dyes (calcein acetoxymethyl ester [AM]/ethidium homodimer-1 [EthD-1]; Molecular Probes, Eugene, OR) and Hoechst 33,342 (1:1,000; Invitrogen) at room temperature for 30 min.
Flow cytometry
After subculturing and filtrating through a 0.45-µm filter, the 661W cells were counted and diluted to 1 ml (5×106 cell/ml) in each assay tube. The LIVE/DEAD® cell viability assay dyes and Hoechst were added to the assay tubes and allowed to sit at room temperature for 30 min. The staining of annexin V conjugated to allophycocyanin (Invitrogen) was used to detect apoptotic cells and was processed according to the manufacturer's instructions (Invitrogen). Propidium iodide (PI; 1:1,000; Sigma-Aldrich) was added to the assay tubes for 5 min at room temperature before use. The stained cells were analyzed by BD LSR II or FACSCalibur flow cytometry (Becton Dickinson, San Jose, CA).
High-content screening and immunofluorescence staining of antigen identified by the Ki-67 monoclonal antibody
661W cells were seeded in a Costar® 96-well black solid plate (5,000 cells/well, total 100 µl; Bio-Rad Laboratories, Hercules, CA), using a MicroFill Microplate Dispenser (BioTek). After a brief wash with phosphate-buffered saline (PBS; pH 7.4, 1.06 mM KH2PO4, 155.17 mM NaCl and 2.97 mM Na2HPO4.7H2O; Invitrogen), treated cells were fixed with 4% paraformaldehyde in PBS and subsequently permeabilized with 0.2% Triton-X in PBS for 15 min. Then, cells were blocked with 5% BSA in PBS for 1 h, followed by incubation with a monoclonal anti-MKI67 (Ki-67) antibody NCL-L-Ki67-MM1 (1:200; Novocastra Lab, Newcastle upon Tyne, UK) for 1.5 h and washing three times with PBS. Finally, cells were stained with the secondary antibody, Alexa Fluor® 488 goat anti-mouse immunoglobulin G (1:200, Invitrogen) for 1 h and washed three times with PBS. After nuclear staining with Hoechst 33,342 (1:1,000; Invitrogen), the sample was analyzed by a Thermo Scientific Cellomics® ArrayScan® VTI HCS Reader (Thermo Fisher Scientific, Pittsburgh, PA) and Columbus™ Image Data Storage and Analysis System software (PerkinElmer, Columbus, OH).
Quantitative reverse-transcription PCR
Total RNA of the 661W cells was extracted using a GeneJETTM RNA Purification Kit (Fermentas International, Burlington, ON, Canada) and reverse-transcribed using a RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas). cDNA was mixed with Maxima® SYBR Green/ROX qPCR Master Mix (2X; Fermentas) and 400 nM of specific forward and reverse primers and analyzed with a 7900 HT real-time PCR system (2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C; Applied Biosystems, Foster City, CA). All primers in this study were designed using the online software Primer-Blast (National Center for Biotechnology Information, Bethesda, MD; available at Primer-Blast) and are listed in Table 1. Multiples of changes in expression were derived by the comparative threshold cycle method. The internal control used β-actin mRNA to compare with others for relative quantification.
Table 1. Primers used in the qRT-PCR.
| Primer | Sequence (5′→3′) |
|---|---|
|
β-actin |
F: CTGTCGAGTCGCGTCCACCC |
|
β-actin |
R: ACATGCCGGAGCCGTTGTCG |
|
HIF-1α |
F: CCCACCCCGCCTCTGGACTT |
|
HIF-1α |
R: TCGGTGCCCGCGTTGTCTTC |
|
VEGF-A |
F: ACTCGGATGCCGACACGGGA |
|
VEGF-A |
R: AAATGTGCCCCCGTGCCCTG |
|
Car9 |
F: GGTGCACCTCAGTACTGCTT |
|
Car9 |
R: ATGGGACAGCAACTGTTCGT |
|
GLUT1 |
F: ACCATCTTGGAGCTGTTCCG |
|
GLUT1 |
R: GCCTTCTCGAAGATGCTCGT |
|
NF-κB |
F: GGCCTTCCGAATTTGGCGTCCT |
|
NF-κB |
R: GAGTGCAGCTGAGCGAGCGA |
|
p53 |
F: GTGGGTCAGCGCCACACCTC |
|
p53 |
R: GGGGAGGAGCCAGGCCATCA |
|
Bax |
F: GTGAGGGCCGCACGTCCAC |
|
Bax |
R: CTCCCCGGACCCGTCCATCA |
|
Bcl2 |
F: CTGCCTTTTTGCCCCGCTGC |
|
Bcl2 |
R: GGGGTCTGGGGGCTAGGTGG |
|
Apaf1 |
F: ATGAAGATGAGGTGCTCTGCTGC |
|
Apaf1 |
R: GCTTCCCAGTCGCAGAATCCCA |
|
p21 |
F: TCCAGGAGGCCCGAGAACGG |
| p21 | R: CCTCCCAGGTCGTCACGGCT |
Immunoblotting
Total protein of the 661W cells was extracted using ice-cold lysis buffer composed of 10% radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) and a protease inhibitor cocktail (Sigma-Aldrich), and the concentration was determined using an R-250 Protein Assay kit (Bio-Rad Laboratories, Philadelphia, PA). Protein samples were mixed with 5X sample buffer (312.5 mM Tris-base, 50% glycerol, 12.5% β-mercaptoethanol, 10% sodium dodecylsulfate [SDS], and 0.01% bromophenol blue) and dry heated to 95 °C for 5 min. After SDS–PAGE with an 8% acrylamide gel, separated proteins were transferred onto polyvinylidene difluoride membranes and blocked with PBS containing 5% nonfat milk overnight. The membrane was incubated with a rabbit anti-HIF-1α antibody (1:1,000; Epitomics, Burlingame, CA) or a mouse anti-β-actin antibody (1:10,000; Sigma-Aldrich) at room temperature for 2 h. Then, the membrane was washed with tris-buffered saline with Tween-20 (TBST; 25 mM Tris, 150 mM NaCl, 2 mM KCl, and 0.1% Tween-20; pH 7.4) for 5 min 3 times each before being incubated with an anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody (1:10,000, Santa Cruz Biotechnology, Santa Cruz, CA) at room temperature for 1 h. Finally, the membrane was washed with TBST three times and visualized using an enhanced chemiluminescence detection kit (Millipore, Bedford, MA) and film. Image analysis was performed using the software ImageJ (National Institutes of Health, Bethesda, MD).
Statistical analysis
Calculations in this study were performed with the software Microsoft Excel 2003 (Microsoft, Redmond, WA). All data are presented as the mean±standard deviation. The statistical significance of differences between groups was analyzed using the Student t test. A p value of <0.05 indicated a statistically significant difference.
Results
YC-1 decreased cell survival during chemical hypoxia
To evaluate cell survival under hypoxia, we treated 661W cells with different concentrations of CoCl2, a chemical hypoxia-mimicking agent. The MTT assay was used to measure changes in the number of live cells of 661W. Our results revealed that the absorbance at 570 nm in the MTT assay displayed a dose-dependent effect in response to CoCl2 (Figure 1A). The MTT survival of 661W cells exposed to 200 μM CoCl2 for 24 h decreased to 50% of that under normoxia. This concentration was adopted for our in vitro chemical hypoxia model.
Figure 1.

3-(5′-hydroxymethyl-2’-furyl)-1-benzylindazole (YC-1) decreased cell survival in a dose- and time-related manner under chemical hypoxia. A: 661W cells were treated with 25, 50, 100, 200, and 400 μM CoCl2 for 24 h, and cell viability was measured with an MTT assay (n=4). B: The viability of 661W cells in the presence of 0.066% DMSO or 20 μM YC-1 for 24 h during chemically induced hypoxia (by 200 μM CoCl2) was evaluated using an MTT assay (n=4). C: Morphological changes in cell density were observed with light microscopy. The black arrows indicate the cell debris. D: Concentration-dependent reduction in cell survival by YC-1 during chemical hypoxia. 661W cells were exposed to 5, 10, 20, and 40 μM YC-1 for 5 min, followed by 200 μM CoCl2 for 24 h (n=4). E: Time-dependent curves of cell survival in response to YC-1 in hypoxia. 661W cells were incubated with 200 μM CoCl2 in the absence or presence of 20 μM YC-1 for 1, 4, 12, and 24 h (n=4). * Indicates p<0.05 compared to the control group or the DMSO vehicle group; # indicates p<0.05 compared to the chemically induced hypoxic group.
To clarify the effect of YC-1 on cell survival during hypoxia, 661W cells were pretreated with 20 μM YC-1 for 5 min, followed by co-treatment with 200 μM CoCl2 for 24 h. We found that the MTT survival of cells treated with 20 μM YC-1 decreased to 30% of that of the DMSO vehicle group during hypoxia (Figure 1B). YC-1 also decreased cell density and increased cell debris under chemical hypoxia (Figure 1C). Moreover, exposure to 20 μM YC-1 showed a maximal effect compared to treatments at 5 and 10 μM YC-1 (Figure 1D). Furthermore, 20 μM YC-1 had no inhibitory effects on MTT survival until 12 h of treatment (Figure 1E). These findings revealed that YC-1 reduced survival of 661W cells under chemical hypoxia and suggest that YC-1 could reduce hypoxic cell viability/proliferation and induce cell death.
YC-1 decreased live and increased dead cells under chemical hypoxia
Because decreased MTT survival may have been due to induction of cytotoxicity or inhibition of cell proliferation in the MTT assay, we used a LIVE/DEAD® cell viability assay, which indicates live and dead cells using the fluorescent dyes, calcein AM and EthD-1, respectively. Our findings demonstrated that YC-1 significantly decreased the density of live cells and increased the number of dead cells compared to those of the control group under chemical hypoxia (Figure 2A), as was also shown by flow cytometry (Figure 2B,C). These results suggest that YC-1 could decrease live cell number through inhibition of cell proliferation and increase dead cell number through induction of cell death under hypoxia.
Figure 2.

LIVE/DEAD cell viability assay of YC-1-treated cells under chemical hypoxia. A: After treatment with 200 μM CoCl2 in the absence or presence of 20 μM YC-1 for 24 h, 661W cells were stained with calcein AM (green for live cells), EthD-1 (red for dead cells), and Hoechst 33,342 (blue for nuclei). Representative fluorescence staining shows the cell density and composition. B: 661W cells were exposed to 0.066% DMSO, 20 μM YC-1, 200 μM CoCl2, or both YC-1 and CoCl2 for 24 h. After fluorescence staining, cells was measured and analyzed with flow cytometry. C: Quantitative data of flow cytometric analysis are from three independent experiments (n=3). * Indicates p<0.05 compared to the control group or DMSO vehicle group; # indicates p<0.05 compared to the chemically induced hypoxic group.
To further confirm the effect of YC-1 on cell proliferation and cytotoxicity under chemical hypoxia, we counted the number of proliferative and apoptotic cells. The Ki-67 is a proliferation marker that is located in nuclei except in cells in the resting stage [23]. To measure cell proliferation in response to YC-1 under hypoxia, HCS, an efficient technique that quantitatively measures cellular imaging [24], was used to detect nuclear Ki-67 expression in 661W cells. Morphological results showed that YC-1 markedly reduced the intensity of fluorescence-labeled Ki-67 compared to that of the DMSO vehicle group under hypoxia (Figure 3A). Quantitative results revealed that YC-1 significantly decreased the number (~550 cell/well) and ratio (~9%) of Ki-67-expressing nuclei compared to that of the DMSO vehicle group (Figure 3B,C), indicating that YC-1 may reduce 661W cellular proliferation during hypoxia.
Figure 3.

High-content screening (HCS) of YC-1-treated cells under chemical hypoxia. 661W cells were exposed to 0.066% DMSO, 20 μM YC-1, 200 μM CoCl2, or both YC-1 and CoCl2 for 24 h. After fixation and immunofluorescence staining, cells were detected and analyzed using a Cellomics ArrayScan VTI HCS Reader. A: Representative fluorescent photographs show the distribution of Ki-67 (green) and Hoechst-labeled nuclei (blue) using the Columbus Image Data Storage and Analysis System. Panels (B) and (C) show quantified HCS results for the number and ratio of Ki-67-stained nuclei (n=4). * Indicates p<0.05 compared to the control group or the DMSO vehicle group; # indicates p<0.05 compared to the chemically induced hypoxic group.
To clarify YC-1-induced cytotoxicity under hypoxia, we used flow cytometry with the annexin V assay and PI to detect apoptotic and necrotic 661W cells, respectively. The data showed that YC-1 (20 μM for 24 h) could increase the percentage of annexin V-labeled apoptotic cells under hypoxia (Figure 4A,B), and an increase of PI-labeled necrotic cells was also observed (Figure 4A). In summary, we demonstrated that YC-1 could inhibit cell proliferation, induce cell apoptosis and death, and lead to reduced cell survival under hypoxia.
Figure 4.

YC-1 increased cell apoptosis under chemical hypoxia. A: After treatment with 0.066% DMSO or 200 µM CoCl2 in the absence or presence of 20 µM YC-1 for 24 h, 661W cells were stained with annexin V (for apoptotic cells) and propidium iodide (for necrotic cells) and measured and analyzed by flow cytometry. B: The frequency of apoptosis was quantified from three independent experiments (n=3). * Indicates p<0.05 compared to DMSO vehicle group; # indicates p<0.05 compared to the chemically induced hypoxic group.
YC-1 reduced HIF-1α protein but not mRNA expression during chemical hypoxia
Our previous study demonstrated that YC-1 decreased the HIF-1α protein and consequently reduced cell viability under normoxia [25]. Therefore, we hypothesized that YC-1 might inhibit the level of HIF-1α protein and subsequently reduce cell viability under hypoxia. To clarify the role of HIF-1α in cell survival under hypoxia, immunoblotting and quantitative reverse-transcription PCR (qRT-PCR) were used to determine changes in the protein and mRNA levels, respectively, of HIF-1α. Immunoblotting results presented in Figure 5A show that the HIF-1α protein increased by about 3.67-fold (lane 3) over that of the DMSO vehicle group (lane 1) in cells exposed to hypoxia for 2 h, and pretreatment with 20 μM YC-1 for 5 min (lane 4) suppressed HIF-1α expression (0.26-fold lower than that of the chemical hypoxia group). Interestingly, we also found that a longer treatment (24 h) with CoCl2 downregulated rather than upregulated the expression of HIF-1α (Figure 5B). These findings suggest that YC-1 possibly inhibits HIF-1α protein expression and further reduces cell survival during hypoxia. On the other hand, the qRT-PCR results revealed that YC-1 had no effect on the transcription of HIF-1α except with short-term incubation (2 h) under normoxia (Figure 6A), suggesting that YC-1 might directly affect the level of HIF-1α protein during hypoxia.
Figure 5.

Effect of YC-1 on HIF-1α protein expression under chemical hypoxia. Total proteins were extracted from 661W cells incubated with 0.066% DMSO, 20 µM YC-1, 200 µM CoCl2, or both YC-1 and CoCl2 for 2 h (A) and 24 h (B). Expressions of HIF-1α and β-actin (which served as the internal control) were determined with immunoblotting.
Figure 6.

Quantitative reverse-transcription PCR (qRT-PCR) analysis of related genes in response to YC-1 under chemical hypoxia. 661W cells were incubated with 0.066% DMSO, 20 μM YC-1, 200 μM CoCl2, or both YC-1 and CoCl2 for 2 h or 24 h. After mRNA was extracted and reverse transcription was performed, relative HIF-1α (A), VEGF (B), glucose transporter 1 (GLUT1; C), carbonic anhydrase 9 (Car9; D), NF-κB (E), p53 (F), Bax (G), Bcl2 (H), Apaf1 (I), and cyclin-dependent kinase inhibitor 1A (p21; J) mRNAs were measured and compared to β-actin mRNA with qRT-PCR (n=3). * Indicates p<0.05 compared to the control group or the DMSO vehicle group; # indicates p<0.05 compared to the chemically induced hypoxic group; + indicates p<0.05 compared to the YC-1-treated group. D, DMSO; Y, YC-1; C, CoCl2; Y+C, both YC-1 and CoCl2.
Furthermore, we wanted to identify whether reduction of hypoxia-induced HIF-1α resulted in decrease of HIF-1-dependent target gene transcription. Our immunoblotting results showed that YC-1 reduced hypoxia-induced HIF-1α at 2 h (Figure 5A), so we estimated the mRNA levels of HIF-1 downstream target genes (VEGF, Car9, and GLUT1) to assess the HIF-1-regulated transcription at 2 and 24 h. The qRT-PCR results revealed that there was no significant effect of YC-1 on inhibition of VEGF (VEGF-A) and GLUT1 mRNA expression under hypoxia (Figure 6B,C), but long-term incubation (24 h) of YC-1 could significantly inhibit hypoxia-induced Car9 (Figure 6D). These results suggest that YC-1 could reduce hypoxia-induced HIF-1α and affect the transcriptional activity of HIF-1.
YC-1 regulates HIF-1α-related genes involved in cell apoptosis and proliferation under chemical hypoxia
NF-κB and p53 are HIF-1α-related transcription factors that are involved in cell apoptosis and the cell cycle [9,26]. We hypothesized that YC-1 might affect the expression of NF-κB or p53 in 661W cells under hypoxia. qRT-PCR data revealed that the level of NF-κB mRNA increased during hypoxia but that YC-1 had no additive effect on its expression (Figure 6E). In contrast, we found that YC-1 significantly increased the level of p53 mRNA compared to that of the DMSO vehicle group during hypoxia (Figure 6F). These findings suggest the possibility that YC-1 could affect not only HIF-1α but also hypoxia-related genes that are involved in cell apoptosis and proliferation under hypoxia.
To confirm the impact of YC-1 on the p53-regulated pathway under hypoxia, we further analyzed downstream genes of p53 involved in cell apoptosis and proliferation. qRT-PCR results showed that YC-1 significantly upregulated proapoptotic gene Bax and the cell proliferation-related gene p21 mRNA compared to that of the DMSO vehicle group (Figure 6G,J). Similarly, YC-1 was found to affect mRNA levels of the apoptosis-related genes Bcl2 (Figure 6H) and Apaf1 (Figure 6I). These findings suggest that YC-1-reduced cell survival was probably correlated to p53-mediated apoptosis and cell-cycle arrest under hypoxia.
YC-1 reduced cell survival during physical hypoxia
Finally, to confirm the inhibitory effect of YC-1 on 661W cell survival during physical hypoxia, we treated 661W cells with 20 μM YC-1 for 24 h in a physical hypoxia model with a low-oxygen supply (0.5% O2, 5% CO2). Based on morphological analysis, our results revealed that YC-1 still decreased the cell density of 661W cells during physical hypoxia (Figure 7A); MTT survival was reduced to 30% of that in the DMSO vehicle group (Figure 7B). Therefore, we confirmed that YC-1 induced dose-dependent inhibition of MTT survival during physical hypoxia (Figure 7C).
Figure 7.

YC-1 reduced cell survival under physical hypoxia. A: 661W cells were exposed to 0.066% DMSO or 20 μM YC-1 for 24 h under normoxia or hypoxia (0.5% O2, 5% CO2). Morphological changes in cell density were observed with light microscopy. Panel (B) is the quantified cell viability determined with an MTT assay (n=4). Panel (C) shows a concentration-dependent cell survival curve of YC-1 during hypoxia (n=4). 661W cells were incubated with 5, 10, 20, and 40 μM YC-1 for 5 min, followed by exposure to physical hypoxia for 24 h. * Indicates p<0.05 compared to the control group or the DMSO vehicle group; # indicates p<0.05 compared to the hypoxic group.
Discussion
We demonstrated that YC-1 reduced the viability of 661W cells in CoCl2-induced (chemical) and low-oxygen (physical) hypoxia (as summarized in Figure 8). Our results further showed that YC-1 decreased the protein expression of HIF-1α and subsequently induced cell death and apoptosis and cell-cycle arrest during chemical hypoxia. We also demonstrated that YC-1 significantly increased hypoxia-induced p53 mRNA levels without changes in NF-κB. The p53-regulated genes Bax and p21 were also observed to significantly increase in response to YC-1 during hypoxia.
Figure 8.
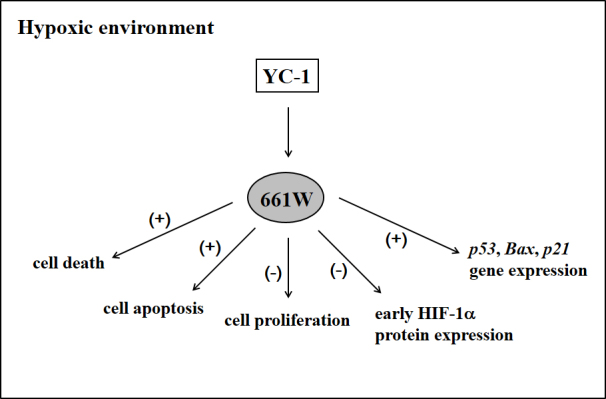
Summary of the effects of YC-1 on 661W cell viability under hypoxia.
We showed that YC-1-reduced cell survival resulting from the decrease in live cells and the increase in dead cells under chemical hypoxia. We also found that YC-1 decreased cell proliferation and decreased the number of live cells under hypoxia, but the effect was less than that of the decrease in cell viability. A possible explanation for this might be perturbation of mitochondrial function. YC-1 has been reported to induce or enhance apoptosis through the mitochondrial dysfunction pathway in several cancer cell lines [27-29]. Our results were compatible with these studies that YC-1 can induce mitochondrial dysfunction (decrease in MTT absorbance) and increase cell apoptosis under hypoxia. Furthermore, our results demonstrated that YC-1 had no effect on apoptosis and cell death under normoxia [25]. Thus, we suggest that YC-1 might not be cytotoxic for cells under normoxia but could increase cell apoptosis and death under hypoxia.
We further investigated the role of HIF-1α in YC-1-reduced cell survival under hypoxia. YC-1 was reported to be able to directly or indirectly interact with the domain of HIF-1α and result in promoting HIF-1α degradation [30,31]. Our data revealed that YC-1 had no significant effect on the level of HIF-1α mRNA under normoxia or chemical hypoxia but reduced the expression of the HIF-1α protein level under hypoxia after 2-h treatment and subsequently decreased cell survival under hypoxia 12 h and 24 h later. These findings are consistent with our hypothesis that YC-1 might affect HIF-1α expression and cell survival under hypoxia.
Under hypoxia, HIF-1α stabilizes, translocates from the cytoplasm to the nucleus, and forms activated HIF-1 [2,3]. Furthermore, YC-1 was reported to decrease the protein level of HIF-1α- and HIF-1-inducible genes [32]. These findings suggest that YC-1 could affect HIF-1-regulated gene expression via inhibiting HIF-1α. However, our results showed that YC-1 inhibited only the HIF-1 target gene Car9 (not VEGF and GLUT1) under hypoxia. Our results also showed that YC-1 did not inhibit HIF-1α expression in western blots, and further future studies using proteasomal inhibitors should determine the effect of YC-1 on HIF-1 target genes under hypoxia.
We tried to investigate HIF-1α-related genes involved in cell apoptosis and cell cycle in response to YC-1 under hypoxia. NF-κB responds to many extrinsic stresses, such as cytokine activation and infectious diseases; however, p53 was reported to respond to many intrinsic stresses, such as DNA damage, hypoxia, and even oncogene activation [33]. Our data showed that YC-1 significantly increased the level of hypoxia-induced p53 mRNA, but had no effect on the level of NF-κB mRNA under hypoxia. We reasoned that YC-1-induced p53 transcription may have resulted from disruption of the balance between HIF-1α and p53 under hypoxia because hypoxia-induced wild-type p53 expression was shown to result from an increase in p53 stabilization mediated by HIF-1α [34]. Our results also revealed that an early decrease in the HIF-1α protein level in response to YC-1 also significantly affected mRNA expression of the p53 downstream target genes Bax and p21 during hypoxia. Therefore, we speculate that YC-1 affects not only the protein degradation of HIF-1α but also the transcriptional regulation of p53 through regulating the stability of HIF-1α.
YC-1 was demonstrated to affect many types of cell survival via the p53-regulated pathway. Under hypoxia, treatment with YC-1 induced cell apoptosis [35,36], cell-cycle arrest [37-40], or both [41,42]. HIF-1α knockdown also enhanced hypoxia-induced cell apoptosis and inhibited cell proliferation [36]. In human esophageal squamous cell carcinoma Eca109 cells, YC-1-mediated cell apoptosis corresponded to induction of p53 and Bax and suppression of HIF-1α and Bcl2 under hypoxia [35]. In human, pulmonary artery, smooth muscle cells, YC-1 inhibited hypoxia-induced cell proliferation via induction of p53 and p21 [39]. Interestingly, we found that YC-1 downregulated the protein expression of HIF-1α, then affected the expression of Bcl2 and Apaf1, and significantly increased p53, Bax, and p21 mRNAs under hypoxia. Our results are consistent with previous studies and suggest that YC-1 may induce apoptosis and cell-cycle arrest through a p53-related mechanism regulated by the inhibition of HIF-1α.
We used two hypoxia models in this study. Our data showed that chemical hypoxia (CoCl2) was more potent than physical hypoxia (low oxygen) in reducing MTT survival. Although both hypoxic conditions increased HIF-1α expression, their transcriptional regulations are different [43]. Another mechanism should be involved in CoCl2-induced cell apoptosis other than HIF-1α [44]. Because of the limitation of chemical hypoxia, we used physical hypoxia to confirm our findings on cell viability. We suggest that YC-1-enhanced 661W cell loss may be due to the damage from different hypoxic conditions, and this may be why YC-1-reduced MTT survival under physical hypoxia was weaker than that under chemical hypoxia. However, CoCl2 is widely used to induce chemical hypoxia through the direct attenuation of HIF-1α degradation [45]. A previous study indicated that CoCl2 can lead to the accumulation of HIF-1α protein with 1 h of hypoxia [46]. In our study, we observed that CoCl2 upregulated the HIF-1α protein with 2 h of treatment but downregulated the protein after 24 h of treatment. We also found that 24 h of treatment with CoCl2 resulted in a decreased mRNA level of HIF-1α. Our findings suggest that CoCl2 can induce early protein expression of HIF-1α and subsequently lead to downregulation of HIF-1α transcription, indicating that early protein expression of HIF-1α is important in maintaining cell survival during hypoxia and may play a neuroprotective role against hypoxic damage.
A recent study reported that YC-1 inhibited not only HIF-1α but also HIF-2α and regulated hypoxia-induced genes [31]. We suggest that inhibition of HIF-1α, at least in part, may be correlated to effect of the hypoxia-regulated genes in response to YC-1, but the mechanism of YC-1-mediated cell loss under hypoxia must be further explored. In addition, we used CoCl2 to mimic the hypoxic environment and used an anaerobic incubator to confirm our findings, but more physiologic conditions should be adopted, especially in an ischemia primary culture cell or animal model.
In conclusion, we demonstrated that YC-1 can inhibit HIF-1α and affect cell viability under hypoxia. Our findings also provided new insights into drugs targeting HIF-1α, especially in neuronal cells of eye.
Acknowledgments
The study was supported in part by Taiwan National Science Council grants NSC97–2627-B-002–007, NSC98–2627-B-002–004, NSC98–3112-B002–040, NSC99–3112-B002–029, NSC99–2314-B002–039-MY3, NSC99–2314-B002–040-MY3, and NSC100–2314-B002–061-MY3, and Department of Health, Executive Yuan, R.O.C. (TAIWAN), DOH100-TD-PB-111-TM005, and National Taiwan University Hospital NTUH.98-S1105, NTUH.99-MSN 01, NTUH.100–001637, NTUH.VN101–06.We especially appreciate the assistance of the National RNAi Core Facility (Academia Sinica, Taipei, R.O.C.) with the HCS experiment. Dr. I-Jong Wang (ijong@ms8.hinet.net) and Dr. Tsorng-Harn Fong (thfong@tmu.edu.tw) are co-corresponding authors for this paper.
References
- 1.Cummins EP, Taylor CT. Hypoxia-responsive transcription factors. Pflugers Arch. 2005;450:363–71. doi: 10.1007/s00424-005-1413-7. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–82. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 3.Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006;70:1469–80. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 4.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pugh CW, O'Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. Activation of hypoxia-inducible factor-1; definition of regulatory domains within the alpha subunit. J Biol Chem. 1997;272:11205–14. doi: 10.1074/jbc.272.17.11205. [DOI] [PubMed] [Google Scholar]
- 6.Goda N, Dozier SJ, Johnson RS. HIF-1 in cell cycle regulation, apoptosis, and tumor progression. Antioxid Redox Signal. 2003;5:467–73. doi: 10.1089/152308603768295212. [DOI] [PubMed] [Google Scholar]
- 7.Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201:105–15. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor CT, Cummins EP. The role of NF-kappaB in hypoxia-induced gene expression. Ann N Y Acad Sci. 2009;1177:178–84. doi: 10.1111/j.1749-6632.2009.05024.x. [DOI] [PubMed] [Google Scholar]
- 9.Chen D, Li M, Luo J, Gu W. Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function. J Biol Chem. 2003;278:13595–8. doi: 10.1074/jbc.C200694200. [DOI] [PubMed] [Google Scholar]
- 10.Slee EA, O'Connor DJ, Lu X. To die or not to die: how does p53 decide? Oncogene. 2004;23:2809–18. doi: 10.1038/sj.onc.1207516. [DOI] [PubMed] [Google Scholar]
- 11.Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010;22:1003–12. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Valenzuela E, Gorczyca W, Darzynkiewicz Z, Sharma SC. Apoptosis in adult retinal ganglion cells after axotomy. J Neurobiol. 1994;25:431–8. doi: 10.1002/neu.480250408. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Valenzuela E, Shareef S, Walsh J, Sharma SC. Programmed cell death of retinal ganglion cells during experimental glaucoma. Exp Eye Res. 1995;61:33–44. doi: 10.1016/s0014-4835(95)80056-5. [DOI] [PubMed] [Google Scholar]
- 15.Kerrigan LA, Zack DJ, Quigley HA, Smith SD, Pease ME. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 1997;115:1031–5. doi: 10.1001/archopht.1997.01100160201010. [DOI] [PubMed] [Google Scholar]
- 16.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–91. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol. 2008;586:4401–8. doi: 10.1113/jphysiol.2008.156695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaur C, Foulds WS, Ling EA. Hypoxia-ischemia and retinal ganglion cell damage. Clin Ophthalmol. 2008;2:879–89. doi: 10.2147/opth.s3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kergoat H, Herard ME, Lemay M. RGC sensitivity to mild systemic hypoxia. Invest Ophthalmol Vis Sci. 2006;47:5423–7. doi: 10.1167/iovs.06-0602. [DOI] [PubMed] [Google Scholar]
- 20.Luo X, Lambrou GN, Sahel JA, Hicks D. Hypoglycemia induces general neuronal death, whereas hypoxia and glutamate transport blockade lead to selective retinal ganglion cell death in vitro. Invest Ophthalmol Vis Sci. 2001;42:2695–705. [PubMed] [Google Scholar]
- 21.Yeo EJ, Chun YS, Park JW. New anticancer strategies targeting HIF-1. Biochem Pharmacol. 2004;68:1061–9. doi: 10.1016/j.bcp.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 22.Maxwell P, Salnikow K. HIF-1: an oxygen and metal responsive transcription factor. Cancer Biol Ther. 2004;3:29–35. doi: 10.4161/cbt.3.1.547. [DOI] [PubMed] [Google Scholar]
- 23.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–22. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Zanella F, Lorens JB, Link W. High content screening: seeing is believing. Trends Biotechnol. 2010;28:237–45. doi: 10.1016/j.tibtech.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Tsui L, Fong TH, Wang IJ. YC-1 targeting of hypoxia-inducible factor-1alpha reduces RGC-5 cell viability and inhibits cell proliferation. Mol Vis. 2012;18:1594–603. [PMC free article] [PubMed] [Google Scholar]
- 26.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen CJ, Hsu MH, Huang LJ, Yamori T, Chung JG, Lee FY, Teng CM, Kuo SC. Anticancer mechanisms of YC-1 in human lung cancer cell line, NCI-H226. Biochem Pharmacol. 2008;75:360–8. doi: 10.1016/j.bcp.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Chung JG, Yang JS, Huang LJ, Lee FY, Teng CM, Tsai SC, Lin KL, Wang SF, Kuo SC. Proteomic approach to studying the cytotoxicity of YC-1 on U937 leukemia cells and antileukemia activity in orthotopic model of leukemia mice. Proteomics. 2007;7:3305–17. doi: 10.1002/pmic.200700200. [DOI] [PubMed] [Google Scholar]
- 29.Lee CS, Kwak SW, Kim YJ, Lee SA, Park ES, Myung SC, Kim W, Lee MS, Lee JJ. Guanylate cyclase activator YC-1 potentiates apoptotic effect of licochalcone A on human epithelial ovarian carcinoma cells via activation of death receptor and mitochondrial pathways. Eur J Pharmacol. 2012;683:54–62. doi: 10.1016/j.ejphar.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 30.Kim HL, Yeo EJ, Chun YS, Park JW. A domain responsible for HIF-1alpha degradation by YC-1, a novel anticancer agent. Int J Oncol. 2006;29:255–60. [PubMed] [Google Scholar]
- 31.Li SH, Shin DH, Chun YS, Lee MK, Kim MS, Park JW. A novel mode of action of YC-1 in HIF inhibition: stimulation of FIH-dependent p300 dissociation from HIF-1{alpha}. Mol Cancer Ther. 2008;7:3729–38. doi: 10.1158/1535-7163.MCT-08-0074. [DOI] [PubMed] [Google Scholar]
- 32.Yeo EJ, Chun YS, Cho YS, Kim J, Lee JC, Kim MS, Park JW. YC-1: a potential anticancer drug targeting hypoxia-inducible factor 1. J Natl Cancer Inst. 2003;95:516–25. doi: 10.1093/jnci/95.7.516. [DOI] [PubMed] [Google Scholar]
- 33.Ak P, Levine AJ. p53 and NF-kappaB: different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010;24:3643–52. doi: 10.1096/fj.10-160549. [DOI] [PubMed] [Google Scholar]
- 34.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392:405–8. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 35.Feng Y, Zhu H, Ling T, Hao B, Zhang G, Shi R. Effects of YC-1 targeting hypoxia-inducible factor 1 alpha in oesophageal squamous carcinoma cell line Eca109 cells. Cell Biol Int. 2011;35:491–7. doi: 10.1042/CBI20090419. [DOI] [PubMed] [Google Scholar]
- 36.Zhu H, Feng Y, Zhang J, Zhou X, Hao B, Zhang G, Shi R. Inhibition of hypoxia inducible factor 1alpha expression suppresses the progression of esophageal squamous cell carcinoma. Cancer Biol Ther. 2011;11:981–7. doi: 10.4161/cbt.11.11.15707. [DOI] [PubMed] [Google Scholar]
- 37.Cheng Y, Li W, Liu Y, Cheng HC, Ma J, Qiu L. YC-1 exerts inhibitory effects on MDA-MB-468 breast cancer cells by targeting EGFR in vitro and in vivo under normoxic condition. Chin J Cancer. 2012;31:248–56. doi: 10.5732/cjc.011.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeNiro M, Alsmadi O, Al-Mohanna F. Modulating the hypoxia-inducible factor signaling pathway as a therapeutic modality to regulate retinal angiogenesis. Exp Eye Res. 2009;89:700–17. doi: 10.1016/j.exer.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 39.Huh JW, Kim SY, Lee JH, Lee YS. YC-1 attenuates hypoxia-induced pulmonary arterial hypertension in mice. Pulm Pharmacol Ther. 2011;24:638–46. doi: 10.1016/j.pupt.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Yeo EJ, Ryu JH, Chun YS, Cho YS, Jang IJ, Cho H, Kim J, Kim MS, Park JW. YC-1 induces S cell cycle arrest and apoptosis by activating checkpoint kinases. Cancer Res. 2006;66:6345–52. doi: 10.1158/0008-5472.CAN-05-4460. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Zhao X, Tang H, Zhong Z, Zhang L, Xu R, Li S, Wang Y. Effects of YC-1 on hypoxia-inducible factor 1 alpha in hypoxic human bladder transitional carcinoma cell line T24 cells. Urol Int. 2012;88:95–101. doi: 10.1159/000331881. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Q, Du J, Gu H, Teng X, Zhang Q, Qin H, Liu N. Effects of YC-1 on hypoxia-inducible factor 1-driven transcription activity, cell proliferative vitality, and apoptosis in hypoxic human pancreatic cancer cells. Pancreas. 2007;34:242–7. doi: 10.1097/01.mpa.0000250135.95144.b6. [DOI] [PubMed] [Google Scholar]
- 43.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–20. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo M, Song LP, Jiang Y, Liu W, Yu Y, Chen GQ. Hypoxia-mimetic agents desferrioxamine and cobalt chloride induce leukemic cell apoptosis through different hypoxia-inducible factor-1alpha independent mechanisms. Apoptosis. 2006;11:67–77. doi: 10.1007/s10495-005-3085-3. [DOI] [PubMed] [Google Scholar]
- 45.Yuan Y, Hilliard G, Ferguson T, Millhorn DE. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J Biol Chem. 2003;278:15911–6. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
- 46.Whitlock NA, Agarwal N, Ma JX, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–8. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]