

Abstract
Genomic instability underlies the transformation of host cells toward malignancy, promotes development of invasion and metastasis and shapes the response of established cancer to treatment. In this review, we discuss recent advances in our understanding of genomic stability in squamous cell carcinoma of the head and neck (HNSCC), with an emphasis on DNA repair pathways. HNSCC is characterized by distinct profiles in genome stability between similarly staged cancers that are reflected in risk, treatment response and outcomes. Defective DNA repair generates chromosomal derangement that can cause subsequent alterations in gene expression, and is a hallmark of progression toward carcinoma. Variable functionality of an increasing spectrum of repair gene polymorphisms is associated with increased cancer risk, while aetiological factors such as human papillomavirus, tobacco and alcohol induce significantly different behaviour in induced malignancy, underpinned by differences in genomic stability. Targeted inhibition of signalling receptors has proven to be a clinically-validated therapy, and protein expression of other DNA repair and signalling molecules associated with cancer behaviour could potentially provide a more refined clinical model for prognosis and treatment prediction. Development and expansion of current genomic stability models is furthering our understanding of HNSCC pathophysiology and uncovering new, promising treatment strategies.
1. Introduction
Carcinogenesis and evolution of the cancer genome are driven by genomic instability. We review here advances in our understanding of the pathways that preserve genome integrity that have improved insight into cancer behaviour, prediction, prognosis, and personalised therapy. Traditional anticancer therapy has exploited the inherent genomic instability of malignancy; however, this mutagenic pressure also promotes the emergence of treatment resistance, invasion, and metastasis (Figure 1). Squamous cell carcinoma of the head and neck (HNSCC) is the sixth most-common cancer in the developed world [1, 2] and represents a therapeutically-challenging, behaviourally-heterogenous category of disease. Genomic instability is a defining characteristic of HNSCC [3]. Subregional differences in patterns in risk, treatment response, and prognosis in HNSCC are underpinned by aetiological factors that affect genomic stability in different ways. In HNSCC, the principal subsites of the upper aerodigestive tract are oral cavity (including tongue, floor of mouth, and buccal surface), nasopharynx, oropharynx (including tonsil and base of tongue), and larynx. An important emergent epidemiological change in HNSCC has been the increasing prevalent of human papillomavirus- (HPV-) associated cancer.
Figure 1.
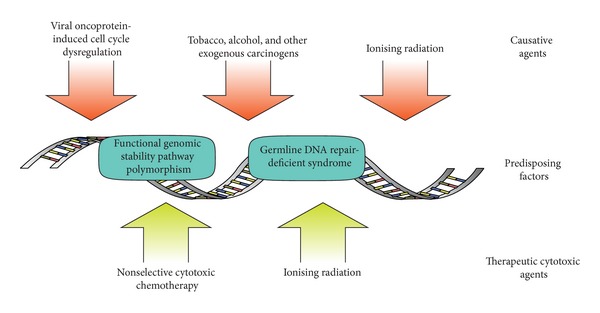
Common genomic stressors in carcinogenesis and therapy.
Chemotherapy regimens in HNSCC are based on platinum compounds, prototypically cisplatin, with or without 5-fluorouracil [4], with greatest effect when given concurrently with radiotherapy [5, 6]. The absolute 5-year survival benefit conferred by concurrent cisplatin/5-fluorouracil chemotherapy is a modest 4.5% [5, 7] and is associated with significantly increased treatment morbidity and mortality [8]. Newer chemotherapeutic agents include taxols and epidermal growth factor receptor- (EGFR-) inhibitors [9]. Addition of docetaxel, a mitotic spindle stabilizer, to PF induction chemotherapy has been shown to confer an additional survival benefit [10], although heterogeneity between trial treatment arms and high loss-to-followup make absolute benefit difficult to interpret. EGFR inhibitor therapy has emerged as an effective adjuvant in HNSCC and is discussed in detail below.
Chromosomal mutation is one of the most readily observable features of cancer and has been known to underpin malignancy for over a hundred years [11]. The mechanism behind most common chromosomal lesions is genomic instability, which is influenced by the interdependent triad of accumulating DNA damage, defective DNA damage repair, and replication stress. In HNSCC, DNA damage is increased by exposure to carcinogens (tobacco, alcohol, and various regionally specific botanicals). Ineffective repair by a number of germline variations in the DNA repair pathways, long recognised in congenital repair syndromes such as Fanconi anemia, have been increasingly implicated as factors in HNSCC, particularly in phenotypically silent individuals with single-nucleotide polymorphisms (SNPs) of DNA repair genes. The DNA damage response is a highly conserved pathway, that is, activated on the basis of a “threshold” of DNA damage events and functions to inhibit cell cycle progression via checkpoint signalling for either repair or apoptosis as well as directly in the repair of the lesions. These pathways are known to be constitutively active in dysplastic lesions [12–14]. This genetically unstable environment drives mutagenic stress, with selective pressure for inactivation of growth restricting/apoptotic processes [13, 15]. Replication stress drives the accumulation of DNA lesions in HNSCC, and is promoted by genetic loss of cell-cycle checkpoint control through mutation and epigenetic loss via oncoviral coinfection with high-risk human papillomavirus (HPV).
Head and neck squamous-cell carcinoma has a male predominance and is associated with tobacco, alcohol, and use of a number of alkaloid-rich regional stimulants (such as betel nut). A subset of HNSCC is strongly associated with “high-risk” HPV subtypes 16 and 18 infection [16] that are present in up to 60–80% of nonoral HNSCC and 30% of oral cavity SCC in a Western population [17–20]. This group of disease that affects younger patients is proportionally associated with number of sexual partners and has a distinctly improved prognosis and different treatment profile from HPV-unrelated oropharyngeal HNSCC [21–27] as well as a distinct pattern of chromosomal mutation [28, 29]. Prognostic significance in oral cavity SCC is less clear (reviewed in [19]). The association of HPV with oral squamous-cell carcinoma was observed 30 years ago [30]; however, the epidemiological changes became more evident over the following decades as decreased tobacco use and increased HPV prevalence highlighted this subgroup [16]. Viral oncoproteins E6 and E7 have been implicated as the causative agents in HPV-associated HNSCC. E6-induced ubiquitin-mediated proteosomal degradation of tumour suppressor p53 causes cell-cycle dysregulation and increased replication stress [31–33]. Oncoprotein E7 binds to and disrupts pRb-mediated degradation of transcription activator E2F, which promotes genes responsible for cell-cycle progression [34–36]. Tumour suppressor protein p16, a CDK4A inhibitor, is characteristically elevated in E7-induced pRb suppression, to the extent that p16 levels are typically used as a surrogate marker for HPV-related SCC [37–39]. E7 has also been shown to induce aberrant centrosome number and mitotic spindle formation [40, 41]. These proteins function to allow viral replication in the normally postmitotic upper differentiated epithelial layers [42]. Whole-exome sequencing has shown that HPV-related HNSCC is associated with less instability, with a 2-fold lower mutation rate [43]. Interestingly, while suppression of the upstream DNA damage response ataxia-telangectasia mutated protein (ATM) and ataxia-telangectasia and Rad3-related protein (ATR) is deleterious for HPV episome stability [44]; cell line studies have shown that the radiosensitivity of HPV-16 related SCC is attributable in part to a repair defect with accumulation of unrepaired DNA double strand breaks and resultant cell-cycle arrest [45]. This study was conducted using a limited number of cell lines rather than fresh tissue; however, the relative contribution of HR and NHEJ in HPV-related SCC has yet to be explored.
While p16 is characteristically elevated in HPV-associated oropharyngeal cancer, up to 56–63% of OSCC and 59% of premalignant leukoplakia lesions show specific downregulation of p16, as expected of the tumour-suppressor role it has [46–49]. HPV-associated oral cavity SCC shows a lower incidence of p16 positivity (65% high risk HPV c.f. 44% low-risk/HPV negative cancers) [50], as does hypopharyngeal cancer (11% high-risk HPV c.f. 0% low-risk/HPV negative cancers) [51]. CDK2NA (encoding p16) is commonly inhibited via promoter hypermethylation, and this is associated with a worse prognosis [52, 53].
2. Chromosomal Mutations in HNSCC
One of the most morphologically apparent features of neoplasia is chromosomal mutations and rearrangements, and these have been well described in HNSCC. Flow cytometric analysis of both dysplastic and malignant lesions shows the expected derangement of chromosomal ploidy. Clinically precancerous lesions show rates of 46% diploidy, 37% tetraploidy, and 17% aneuploidy, while malignant lesions show 10% diploidy and 90% aneuploidy [54]. In addition to deletions and duplications (copy number variation, CNV), a critical component of carcinogenesis is allelic loss or loss-of-heterozygosity (LOH).
The first chromosomal aberrations in premalignant lesions are losses of 3p, 9p, 5p, and 17p [55–57]. Importantly, CDKN2A encoding p16 is localised to the most commonly affected 9p21-22 locus [58, 59], which shows LOH in 46–71% of premalignant lesions and 72% of carcinoma [59, 60] and is strongly associated with progression to cancer and metastasis [60, 61]. Lydiatt et al. have suggested that this deletion is overrepresented in HNSCC cell lines [62], although this study found much lower rates of primary tumour deletions. While germline p16 mutations have been described in familial HNSCC syndromes, these are rare [63, 64].
Allelic loss of 3p is present in 74–81% HNSCC [56, 65, 66], with PCR and hybrid clone studies suggesting three discrete areas at 3p13-p14 3p21.2-p21.3, and 3p25 [65, 67] are positively associated with tobacco-related disease and nodal status [68]. The FHIT gene encoding a common epithelial tumour suppressor is found at locus 3p14.2. Expression of FHIT is suppressed in 65% of HNSCC and is associated with worse overall survival [69]. Locus 3p21.31 contains a number of tumour-suppressor genes (LIMD1, LTF, CDC25A, SCOTIN, RASSF1a, and CACNA2D2) of which alterations to LTF and RASSF1A were associated with significantly worse outcome in oral cavity disease in an Indian cohort [70]. 3p25 contains the gene locus for hOGG1 and von Hippel-Lindau tumour suppressor gene. hOGG1 is an important component of the base excision DNA repair pathway and shows LOH in 55–61% of HNSCC and is underexpressed in 49% [71, 72]. As discussed below, this mechanism is important in the repair of ionizing radiation and common oxidative and tobacco-related DNA lesions [73]. Unrepaired tobacco-induced benzo[a]pyrene lesions commonly result in G:T transversion mutations [74, 75]. Repair of the oxidative lesion 8-oxoG also relies on this pathway, which can result in a stable G:C to 8oxoG:A missense substitution when encountered by the DNA polymerase [76]. Together these substitutions comprise 54% of the most common inactivating p53 point mutations in HNSCC [77]. This mutation pattern, which has also been strongly linked to smoking-induced lung cancer [78], is correlated with smoking exposure-related HNSCC [43].
Chromosomal aberrations associated with late disease and metastasis are less well characterised. It has been found that metastatic deposits mostly retain clonality with their primary cancers [79]. Gains of 3q, 11q13, 7q11.2, and 1q21-q22 and losses of 8p, 11p14, 10p12, 10q, and 14q were reported in association with metastasis by Bockmühl et al. [79, 80], who also found a high rate of 45% deletions in the 8p23 region correlated with poor prognosis [81]. This region corresponds to putative tumour suppressor gene CUB and SUSHU multiple domain protein 1 (CSMD1) which is associated with a high risk, although higher-resolution deletion mapping has shown inconsistent targeting of this gene [82]. Many more chromosomal mutations have been described in later diseases [61, 83–89]. These rearrangements occur at common fragile sites, late-replicating regions which can preferentially stall replication forks under conditions of replication stress [90]. Inappropriate recovery of these forks can cause double-strand DNA breaks or inversions [91], and ineffective repair can lead to translocations and deletions [92, 93]. For more detail on other chromosomal rearrangements see [94, 95] for a comprehensive review.
3. The DNA Damage Response
Highly conserved across all domains of life is a robust DNA damage recognition and repair system. Genomic material, both DNA and RNA, is under constant degradation from intrinsic and extrinsic factors. These include oxidative stress, ultraviolet radiation, ionising radiation, and other chemical alterations such as alkylation [96, 97]. Cells under active division, such as deregulated tumour cells, are particularly sensitive. This differential sensitivity is exploited by chemotherapy and external-beam radiation. There are many different types of DNA lesions, which are broadly dealt with via interacting pathways (Figure 2):
mismatch repair (MMR),
nucleotide excision repair (NER),
base excision repair (BER),
single-strand break repair (SSB-R)*,
double-strand break repair (DSB-R).
Figure 2.
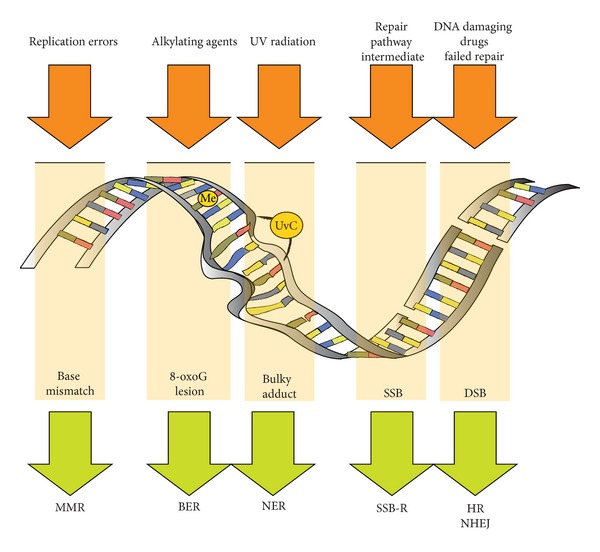
Examples of DNA lesions and repair pathway choice.
Single-strand break repair is not usually considered a distinct repair pathway and is integrated into BER, NER, and DSB-R. The most severe form of DNA damage is the double-strand break [98]. Base excision repair and nucleotide excision repair pathways involve single-strand break intermediates, which unrepaired become double-strand breaks when encountered by a replication fork [12]. Double-strand break repair occurs via two pathways nonhomologous end-joining (NHEJ) and homologous recombination (HR). The primary pathway in nonreplicating mammalian cells is NHEJ [12].
ATM is an upstream phosphatidylinositol 3 kinase-like kinase (PIKK), that is, recruited to double-strand break sites and coordinates repair. Limited studies in ATM have shown a strong increased cancer risk with a specific SNP in an East Asian population [99]. Promoter hypermethylation of ATM has been linked with early age of onset and poor prognosis in one mixed subsite study [100]; however, this has not been reproduced in a large oral cavity cancer series [101]. The mutation most common in the ataxia-telangectasia phenotype does not seem to be a significant contributor to the development of HNSCC [102].
4. Mismatch Repair Pathway
Microsatellite instability is a distinct, measurable form of genomic damage, that is, linked to the mismatch repair pathway, principally involving mut-S homologin 2 (MSH2) and mut-L Homologin 1 (MLH1). The archetypical germline defect in this pathway is the hereditary nonpolyposis colorectal carcinoma syndrome [103]; however, the involvement of MSI in HNSCC has been less clearly elucidated. Early studies showed that direct mutation or deletion of hMLH1/hMSH2 was uncommon [104, 105], and reported frequency of MSI in HNSCC has ranged from 7% to 100%, varying with marker choice, tumour site, and patient demographic [104–109]. MSI has been found to be more prevalent in younger patients [104], and Ha et al. have shown that it becomes more frequent as dysplastic lesions progress to malignancy (14% and 55%, resp.) [110].
The dominant moderator of mismatch repair in HNSCC is promoter hypermethylation rather than direct mutation [111]. MLH1 promoter hypermethylation has been found in 76% of one OSCC series, associated with early disease, where 38% of tumours exhibited protein underexpression [112]. There is however wide variation in the reported prevalence of promoter methylation, from 8% to 69% [113–115], and normal tissue demonstrates promoter methylation of up to 45% [115]. Most studies show no correlation with tobacco exposure; however, there may be an association with metachronous primary cancers [114]. A more recent study of MLH1, MSH2, and MGMT in mobile tongue SCC showed increased expression of the mismatch repair proteins (55.1% MSH2, 36.73% MLH1), which was associated paradoxically with less muscle invasion, less perineural invasion but lower overall survival, higher rates of lymphatic metastasis, and more aggressive morphology, implying that this may be a reactive rather than causative finding [116].
5. Base Excision Repair Pathway
hOGG1 and NEIL1 and 2 are DNA glycosylases that are involved in the first step of damage recognition. They demonstrate significant functional cross-over with each other [117] and show affinity for lesions from 5-fluorouracil (5-FU), UV damage, and 8-oxoguanine (a highly mutagenic by-product of ROS). The hOGG1 locus commonly undergoes LOH as discussed above, with up to 49% of HNSCC demonstrating underexpression [72, 118]. Interestingly, there is some evidence that this defect is present systemically and may be due to a germline rather than sporadic mutation [119], although haplotype studies have been inconclusive as shown above. APEX1, a processing enzyme immediately downstream of hOGG1, has been shown to be overexpressed in HNSCC in a Pakistani population and is associated with nodal positivity and later stage [120]. NEIL1/2 form a redundant BER system, substituting AP endonuclease (APEX1/2) for polynucleotide kinase (PNK) [121], and the g.4102971CC polymorphism has been linked to increased risk of HNSCC [122].
XRCC1, as discussed above under SNPs, has been extensively investigated for its role in HSNCC. It functions in DNA single-strand break repair downstream of APEX1/PNK to scaffold and coordinate nucleotide synthesis [123]. Early studies of mRNA expression in head and neck cell lines implied that XRCC1 was not a significant contributor to cancer radiosensitivity [124]; however, a more recent protein expression series in primary laryngeal cancer looking at outcomes following definitive radiotherapy has contradicted this [125]. Overexpression of XRCC1, in a more recent series, has been linked to poor outcome in chemoradiation-treated HNSCC but not other modalities [126], and other studies have reported a predominant pattern of underexpression in an Indian population [118].
Poly- (ADP-Ribose) polymerase (PARP) 1 and 2 are DNA repair proteins that have direct affinity to many DNA lesions and once bound autocatalyze the generation of a PAR polymer which induces chromatin decondensation and recruits many other repair molecules, including ATM [127]. Although it has no direct repair role, it is required for efficient base excision repair, nucleotide excision repair, HR, and alternative-NHEJ [127–131], with the most well-understood role of single-strand break binding to facilitate polymerisation and ligation. Of particular importance in malignancy, PARP-1 is required for safe traversal of damaged DNA by the replication machinery and subsequent HR-mediated repair [128], which is significantly exacerbated under replication stress. Importantly, PARP inhibition has been found to be synthetically lethal in BRCA- (HR-) deficient breast cancer [132, 133]. As discussed above, EGFR-inhibition induces an HR and NHEJ deficiency in addition to antiproliferative effects. There is evidence that combined therapy can replicate an HR “block” in a similar fashion [134] that may be linked to Nbs1 suppression [135, 136]. Indeed, cosuppression of many proteins involved in HR induces in vitro PARP susceptibility [137]. Blocking upstream signalling by PI3K also induces HR defects and PARP-sensitive synthetic lethality in breast cancer cell lines [138]. There is also limited data in HNSCC demonstrating a link between PARP1 overexpression and cisplatin resistance [139], suggesting a possible role for chemoresistant tumours.
6. Nucleotide Excision Repair Pathway
The nucleotide excision pathway (NER) corrects “bulky” DNA adducts and lesions such as UV photoproducts, platinum lesions, and tobacco-linked lesions [140]. Germline defects in this pathway are responsible for the xeroderma pigmentosum phenotype, for which many key proteins are eponymous. There are two principal monitoring pathways, global genomic NER and transcription-coupled NER.
Global genomic NER is initiated by direct recognition of DNA lesions irrespective of transcription [141, 142], and polymorphism of several key proteins have been implicated in HNSCC as shown above. Transcription-coupled NER differs in that detection and recruitment of repair molecules is initiated by stalled RNA polymerase II (RNAP II), with repair mediated by UVSSA-USP7 and BRCA1 [143, 144]. The lack of a clear association with a cancer syndrome, in vitro or de novo cancers make the transcription-coupled pathway a less promising factor in cancer genome stability [145, 146].
ERCC1-XPF has been extensively studied in lung cancer, breast cancer, and HNSCC. High ERCC1 expression in HNSCC has been associated with cisplatin resistance and poor survival in a betel chewing endemic area [147]. Two larger retrospective trials showed prognostic benefit of low ERCC1 expression in HNSCC (HR 0.42, CI95% 0.20–0.90, P 0.03 and HR 0.12, P 0.043) [148, 149]. In vivo ERCC1 has been associated with enhanced cisplatin resistance [150] and is involved in alternative NHEJ [151]. Metastasis has also been associated with increased expression of XPF [152].
SNPs in all eight major NER genes have been implicated in increasing background risk of oral leukoplakia [153], and altered PBL expression of ERCC1, ERCC5, ERCC6/CSB [154], and ERCC4 [155] have been found to be higher in patients with oral cancer. Similarly to peripheral blood lymphocyte assays done in hOGG1 (discussed above), there is limited evidence that there may be a systemic NER deficient phenotype in a small cohort of patients with HNSCC [156]. Some SNPs have been implicated as protective or hazardous in single SNP studies (refer Table 1), while multiple (5–7) risk NER genotypes have been associated with a 2.4-fold increased relative risk of second primary HNSCC [157].
Table 1.
| Reference | Subsite | Population | Study size | Gene SNP | Odds ratio (P value) |
|---|---|---|---|---|---|
| Sliwinski et al. [240] | Laryngeal | Polish | 641 (288 cases, 353 controls) | Rad51 G135C XRCC3 C722T |
2.92 (<0.0001) 4.03 (<0.0001) |
|
| |||||
| Romanowicz-Makowska et al. [241] | Laryngeal |
Polish | 506 (253 cases, 253 controls) | Rad51 G135C XRCC2 G31479A XRCC2 C41657T |
NS [3.43 (0.073)] NS [3.81 (0.061)] NS [2.14 (0.061)] |
|
| |||||
| Werbrouck et al. [242] | All | Belgium | 169 cases, 352 controls | XRCC3 C722T Rad51 G-3429C Lig4 C26T Ku70 C1310G Ku80 c.2110-2408 G>A |
1.96 (0.02) 0.43 (0.05) 0.43 (0.01) NS NS |
|
| |||||
| Cui et al. [243] | North American | 443 cases (including oesophageal), 912 controls | XPG c.1104 | 0.47 (<0.05) | |
|
| |||||
| dos Reis et al. [244] | Oral cavity | Brazil | 300 (150 cases, 150 controls) | XRCC1 Arg194Trp, Arg399Gln XRCC3 Thr241Met |
NS NS |
|
| |||||
| Pawlowska et al. [245] | Laryngeal | Polish | 506 (256 cases, 256 controls) | hOGG1 Ser326Cys | 2.96 (0.012) |
|
| |||||
| Kietthubthew et al. [246] | Oral cavity | Thai (betel quid endemic) | 106 cases, 164 controls | XRCC1 Arg194Trp, exon 6 | NS |
| XRCC3 Thr241Met | 3.3 (0.01) | ||||
|
| |||||
| XRCC3 rs3212057 | 6.0 (<0.01) | ||||
| Gresner et al. [247] | All | Polish | 81 cases, 111 controls | Rad51 rs5030789 AA |
0.33 (<0.05) |
| Rad51 rs1801321 TT |
0.1 (<0.05) | ||||
|
| |||||
| Zhang et al. [248] | All | North American | 706 cases, 1196 controls | hOGG1 Ser326Cys | NS |
|
| |||||
| Elahi et al. [249] | All | North American | 169 cases, 338 controls | hOGG1 Ser326Cys Cys326Cys |
1.6 (<0.05) 4.1 (<0.05) |
|
| |||||
| Majumder et al. [225] | All | Indian | 180 cases, 150 controls | XRCC1 c.194, c.399 | NS |
|
| |||||
| Mahjabeen et al. [120] | Not specified | Pakistani | 300 cases, 300 controls | APEX1 13T<GG Ser129Arg Val131Gly |
4.97* (0.0001) 11.74* (0.0001) 5.12* (0.0001) |
|
| |||||
| Ramachandran et al. [250] | Oral cavity | Indian | 110 cases, 110 controls | XRCC1 c.194 c.280 c.399 XPD c.751 |
3.09 (<0.0001) NS 2.37 (<0.0001) 2.10 (0.003) |
|
| |||||
| Tae et al. [251] | All | Korean | 147 cases, 168 controls | XRCC1 R194W R280H, R399G |
2.61 (<0.05) NS |
|
| |||||
| Kumar et al. [118] | Not specified | North Indian | 75 cases, 75 controls | XRCC1 Arg399Gln XPD Lys 751Gln hOGG1 Ser326Cys |
NS |
|
| |||||
| Kumar et al. [228] | Not specified | North Indian | 278 cases, 278 controls | XRCC1 Arg194Trp Arg280His Arg399Gln XPD Lys751Gln |
0.72 (0.03) NS 0.64 (0.003) 1.75 (0.002) |
|
| |||||
| Kowalski et al. [252] | All | Polish | 92 cases, 124 controls | XRCC1 Arg194Trp Arg399Gln |
NS |
|
| |||||
| Yen et al. [253] | Oral cavity | Taiwan | 103 cases, 98 controls | XRCC1 Arg194Trp XRCC2 (5′ locus) XRCC3 Thr241Met XRCC4 T139G |
NS |
| “pseudo-haplotype” multiple concurrent SNPs | 2–2.45 (0.03) 3–5.03 (0.013) 4–10.1 (0.036) |
||||
|
| |||||
| Tseng et al. [254] | Oral cavity | Taiwan | 318 cases, 318 controls(1) | XRCC4 c247 G1394T rs28360317 rs1805377 |
2.04 (<0.05) NS NS NS |
|
| |||||
| Chiu et al. [255] | Oral cavity | Taiwan | 318 cases, 318 controls(1) | XRCC4 G1394T rs28360071 rs28360217 rs1805377 |
NS 1.55 (<0.05) NS NS |
|
| |||||
| Chiu et al. [256] | Oral cavity | Taiwan | 292 cases, 290 controls | ERCC6 c.399 (A/A) c.399 (G/G, G/A) c.399 (Any A) c.1097 c.1413 |
1.82 (<0.05) NS 1.43 (<0.05) NS NS |
|
| |||||
| Hsu et al. [257] | Oral cavity | Taiwan | 600 cases, 600 controls(1) | Ku80 G-1401T C-319T Intron19 |
1.603 (<0.05)(2)
NS NS |
|
| |||||
| Bau et al. [258] | Oral cavity | Taiwan | 318 cases, 318 controls(1) | Ku70 T991C G-57C A-31G Intron 3 |
2.15 (<0.05) NS NS NS |
|
| |||||
| Bau et al. [99] | Oral cavity | Taiwan | 620 cases, 620 controls | ATM rs189037 rs600931 rs652311 rs228589 rs227092 rs227060 |
1.61 (<0.05) NS NS NS NS NS |
|
| |||||
| Flores-Obando et al. [259] | Meta-analysis | Multiple | XPA A23G XPD C22541A/A XPD A35931C XPD Asp312Asn XPD C23047G XPD C23051G XPC PAT XPC Lys939Gln XPC Ala499Val XPF T2063A ERCC1 C8092A XRCC1 exon 6 T/T XRCC1 exon 10 XRCC1 exon 9 XRCC3 Thr241Met |
NS 0.74 (<0.05) NS 1.14 (0.05) NS NS NS NS 1.56 (<0.05) NS NS 1.69 (<0.05) NS NS NS |
|
|
| |||||
| Zhai et al. [122] | All | North America | 872 cases, 1044 controls | NEIL1 g.46434077 NEIL1 g.46438282 NEIL2 g.4102971CC |
NS NS 1.30 (<0.05) |
*Crude odds ratio.
(1) Same group/cohort.
(2)Only in Areca-nut exposed subgroup.
7. Nonhomologous End Joining Pathway
The Ku heterodimer is comprised of Ku70 and Ku80 functions as a sensor, binding to free dsDNA ends, an end-processing 5′-dRP/AP lyase, and a recruiter of DNA-PKcs [158] and telomere protection [159]. Ku80 overexpression has been linked to poor prognosis, local failure, and recurrence following radiation [160]. While this study did not include tongue cancer specimens, the predictive value was greatest in HPV negative cancers (RR 9, P < 0.01). In contrast, two similarly-sized mixed studies found Ku70 mRNA levels and protein overexpression improved recurrence-free survival in locally advanced HNSCC [161] and tonsillar cancer [162]; however, these did not stratify for p16 status and had unusual treatment regimens. In support of these findings are in vitro studies showing that Ku70 loss improved radioresistance in late G2/S, possibly by allowing the more error-free HR [163].
8. Homologous Recombination Pathway
The Fanconi anemia pathway encompasses eleven proteins involved in the HR-mediated repair of interstrand crosslink (ICL) lesions [164]. Fanconi anemia homozygous patients have a much greater rate of head & neck solid tumours [165], and underexpression of many Fanconi proteins is common in primary HNSCC [166], particularly in young patients [167], although the underlying mechanism is unclear. Tobacco exposure has been shown to selectively suppress FANCD2 expression in a respiratory epithelial cell line [168]. Promoter hypermethylation of FANCF has been found in 15% of HNSCC [169], while 42% of OSCC show FANCC methylation in both malignant and premalignant lesions in an Indian population [170]. In contrast, another series demonstrated 31% PHM of FANCB, and found other Fanconi genes to be rarely methylated [171]. While cisplatin commonly induces interstrand crosslinks, specific upregulation of the Fanconi pathway has not been implicated in cisplatin-resistant HNSCC cell lines [172].
Hyperphosphorylation of replication protein A (RPA), a single-strand DNA binding protein, that is, integral to HR, has been implicated as a mechanism for cisplatin resistance in HNSCC cell lines [173]. RPA signalling activity is heavily modulated by phosphorylation status, and this posttranslational resistance mechanism promotes inappropriate cell-cycle progression.
BRCA1 and 2 are essential for efficient HR, and germline mutation is causally linked to breast and ovarian cancers [174, 175]. Low BRCA expression has been correlated with better cisplatin response and survival in lung cancer [176]. BRCA-deficient breast cancers have shown survival benefits with synthetically lethal PARP inhibition in Phase II trials [177]. An interesting pattern in tongue SCC pathogenesis shows BRCA1 overexpression in leukoplakia followed by subsequent underexpression in carcinoma [178]. Sporadic mutation of BRCA2 is rare [179] as is promoter hypermethylation [169].
9. The Epidermal Growth Factor Receptor in DNA Repair
Epidermal growth factor receptor (EGFR) is a transmembrane signalling protein belonging to the receptor tyrosine kinase family, which has significant implications in HNSCC. EGFR is physiologically activated via ligand binding (transforming growth factor-α, TGF-α), and active EGFR promotes cell proliferation and enhances radioprotective mechanisms through the mitogen-activated protein kinase (MAPK) pathway and the extracellular signal-regulated kinases (ERK1/2). [180, 181]. The strongest effect of EGFR activation on DNA repair is via upregulation of the phosphoinositol-3 kinase (PI3K)/AKT signalling pathway (Figure 3). PI3K mutations are common in HNSCC and linked with higher genomic instability [182]. PI3K promotes transcription of the nonhomologous end-joining complex DNA-PK [183, 184], mediates Nbs1 binding for DNA damage detection [185], and promotes effective DNA double strand break repair [186], while AKT1 promotes transcription of the DNA-PKcs subunit [187] and negatively regulates BRCA1/Rad51 nuclear transport [188]. Particularly relevant to DNA repair, inhibition of the EGFR receptor induces a NHEJ and HR defect. Erlotinib has been shown to decrease BRCA1-dependent HR by twofold in breast cancer cell lines [189] and attenuate radiation-induced Rad51 expression (a key HR protein) [190]. Gefitinib delays Nbs1 recruitment to DNA double strand break sites in lung cancer cell lines and significantly reduces DNA double strand break repair [136]. Downstream inhibition of PI3K has also been shown to induce a BRCA1/2 deficiency in breast cancer cell lines [138]. The functionality of the BRCA/HR pathway is of particular importance for the utility of PARP inhibitors (discussed below).
Figure 3.
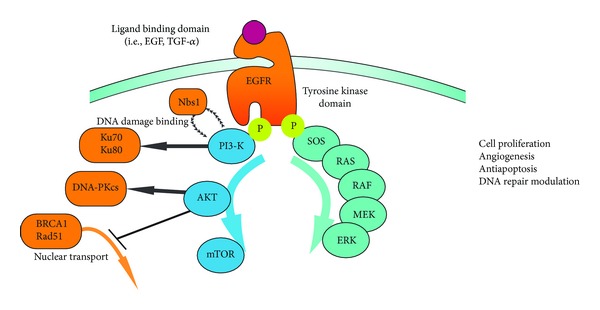
Downstream effects of EGFR on DNA repair proteins.
Constitutively-active inhibitor-sensitive EGFR mutants are an important mechanism in 10% of nonsmall cell lung cancer (NSCLC) cases, conferring a significant survival benefit when cancer cells dependent on this proliferative signal are deprived via EGFR-inhibition [191, 192]. Such kinase domain mutations are rare however in HNSCC, with mutation rates of 7–16% in Asian and 0–4% in Japanese and Caucasian populations and are not present in HPV-positive tumours [193–197]. This suggests that they do not have a significant role in oncogenesis and may instead reflect underlying instability. In striking contrast, an EGFR inhibitor-resistant truncation mutant EGFRvIII has been found in up to 42% of HNSCC [198]. This mutant has a deletion of exons 2 to 7, involving the extracellular binding domain rather than the kinase domain [199]. This results in a protein, that is, constitutively active, has altered kinase dynamics that favour PI3K signalling, and that promotes ligand-independent invasion and migration [200, 201].
Oncogenic mutations in other components of this pathway are less common. PI3KCA somatic mutations have been described in 0–11% of HNSCC tumour samples and are overrepresented in cell lines [202–204], while PTEN and Akt1 mutations are rare [205].
Overexpression of EGFR is very common in HNSCC, with protein immunohistochemical studies showing high expression in 43–68% of HNSCC [197, 206–208] and Areca quid-associated OSCC demonstrating a lower rate of 23% [209]. High EGFR protein levels are strongly linked to poor cause-specific survival [207, 208], nodal stage and dedifferentiation [197], and emergence of stem-cell like characteristics [210]. Altered EGFR copy numbers have been found in 24% of HNSCC cases in one study (17% increased, 7% decreased) and both are linked to a reduced cause-specific survival and disease-free survival [211]. EGFR amplifications have been demonstrated in 24% (10% deletions) of laryngeal cancers, although survival significance was only demonstrated when combined with chromosome 7 copy-number status [212]. There does not appear to be any difference between HPV-associated and HPV-negative cancers with regard to EGFR expression [50].
Cetuximab, an IgG1 monoclonal antibody against the receptor domain of EGFR, has been shown to add an 8% absolute 2-year survival advantage as a monotherapy adjunct to radiotherapy in HNSCC [213] and has become incorporated into the standard management of late-stage HNSCC [214]. The tyrosine kinase inhibitor gefitinib has also been shown to sensitize HNSCC cells to cisplatin via destabilization of Rad51 [215]; however, Phase III trials have shown no advantage as monotherapy in recurrent HNSCC compared with methotrexate [216]. Cancers with high EGFR expression demonstrate additional benefit from hyperfractionated radiotherapy regimens [217].
Radioprotection in EGFR-competent cells is improved via enhanced immediate DNA repair, protection from damage-induced apoptosis, and enhanced proliferation of the EGFR-competent subpopulation during repopulation [218], providing a selective advantage. Cetuximab resistance has been linked to EGFR translocation from the cytoplasm to the nucleus and endogenous ligand production [219]. Transport dysregulation and HER2-dependent transactivation has also been found to contribute to resistance in lung cancer cell lines [220]. Emergent resistance is also very common in colorectal cancer, where it has been found to be mediated by endogenous upregulation of K-Ras [221].
10. Single Nucleotide Polymorphisms in DNA Repair Pathways
While some areas of genomic DNA are highly conserved, the dynamic variability of large domains encompassing coding and noncoding areas is becoming increasingly important to our understanding of individual disease risks. The most common examples are single nucleotide polymorphisms (SNPs), of which 3.1 million have been identified encompassing most proteins involved in DNA repair [222]. The two principal types of SNPs are synchronous (or “silent”) and nonsynchronous, where a base substitution results in a corresponding protein substitution. The protein product may be a “functional variant,” or has some degree of impaired function. Analogous to hereditary DNA repair syndromes, much attention has been paid to SNP prevalence and the effect on HNSCC risk. Listed below are a subset of SNP studies specific for DNA repair proteins involved in the pathways discussed above.
In addition to the above studies, a series in a Pakistani population found a remarkably high prevalence of novel hOGG1 polymorphism is association with HNSCC, although this has not been subsequently validated [223]. Interestingly, both this and a Japanese study found a statistically significant link between hOGG1 Ser326Cys status and tobacco use [224], complicating statistical analysis. Likewise, HPV-16 positivity may alter the significance of XRCC1 polymorphism in HNSCC risk, possibly explaining the inconsistent results between study populations [225, 226], although these findings are in contrast to cervical SCC and HPV-16 status where no relationship has been found [227]. Complex gene-gene interactions between XRCC1 and XPD haplotypes have also been described [228].
Carles et al. [229] examined nine nucleotide excision repair gene SNPs with regard to response to definitive radiotherapy. This study found XPA (5′UTR), XPF/ERCC1 C259C, and XPG/ERCC5 G1104C/T46C SNPs resulted in a significantly worse overall survival following treatment in a cohort of 104 patients over 10 years. In a Brazilian population, ERCC1 T19007C was not found to affect response to treatment or overall survival in a modest cohort [230].
11. Discussion
Genomic instability underpins the development of dysplasia, malignancy, invasion, and metastasis in cancers. Many of our cancer therapeutic drugs exploit these genetic stability pathways by adding extra pressure onto already primed cancerous cells. The formation of these cancers is also clearly linked to genetic instability with the high risk factors such as HPV, tobacco, areca nut, betel quid, and alcohol having direct mutagenic effects on the cellular genome. There is also an emerging spectrum of DNA deficient high risk phenotypes that are improving our understanding of background genetic risk. These aetiological differences in oncogenesis translate into distinctly different disease profiles. Profiling of the DNA repair pathways in established cancer may allow the development of robust biomarker-based cancer phenotyping for both improved prognosis and personalized therapeutic selection. The success of the PARP-1 inhibitor olaparib in BRCA1-deficient breast cancer and the early translational work in HNSCC underpin the importance of future targeted therapy in exploiting synthetic lethality in DNA repair.
The inherited DNA repair deficient syndromes provide the prototype for insight into DNA repair in cancer, highlighting the importance of the double-strand repair pathways, and in particular homologous recombination. Ataxia telangiectasia presents with a significant increased all-cause cancer risk [231] consistent with its critical repair role and inherited HR protein BRCA1 and BRCA2 mutants strongly predispose toward breast cancer, prostate, and colon cancer [175]. Homologous recombination is the preferred repair pathway in cells undergoing active replication and is constitutively active in cancer. Defective HR is responsible for many chromosomal rearrangements [91, 232], and emerging evidence shows a robust redundant pathway, that is, more complex than previously thought. Complementing our knowledge of this pathway is ongoing discovery of HR-essential proteins such as hSSB1/2 [233, 234], RNF8 [235], and Exo1 [236], which coordinate the more well-understood core repair complexes such as Rad51 and MRN [237]. Further exploration of the oncogenic role of these core proteins is needed.
Head and neck cancer is a diverse category of disease, with outcomes influenced largely by primary site and p16 status. Existing studies on more well-known DNA repair biomarkers are often difficult to interpret due to conflation of anatomical subsites or limited p16 control. Focused, well-powered studies controlled for anatomical subsite are likely to be of greater clinical benefit. Similarly, the abundance of the literature on genetic polymorphism and cancer risk is tempered by conflicting reproducibility, conflation of anatomical sites, and population differences. Localisation of SNP studies is important given the significantly unique risk factor profiles in East Asian and Chinese populations; however, it makes generalisation of these highly prognostic studies to Western populations difficult.
Current gaps in the literature of cancer genomic instability include the nature of interpathway and intrapathway redundancy. Major protein complexes can be less efficiently replaced by alternate, multipurpose proteins to otherwise allow DNA repair [235, 238], an important mechanism to understand in designing synthetically lethal repair blockade. SNPs of backup 8oxoG pathways such as PNK/NEIL1 have been linked to HNSCC risk [122] and contributes to poor survival in non-HNSCC cancer [239]; however, the role of this pathway in relation to hOGG1 deficiencies has yet to be determined in HNSCC.
Improved understanding of the nature of genomic instability in head and neck cancer has helped clarify the interplay between mutagenic stresses and DNA repair efficiency. Future characterization of signature instability patterns will guide further therapeutic development targeted at the critical foundation underlying malignancy.
References
- 1.Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International Journal of Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: A Cancer Journal for Clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Maeda T, Jikko A, Hiranuma H, Fuchihata H. Analysis of genomic instability in squamous cell carcinoma of the head and neck using the random amplified polymorphic DNA method. Cancer Letters. 1999;138(1-2):183–188. doi: 10.1016/s0304-3835(99)00004-x. [DOI] [PubMed] [Google Scholar]
- 4.Merlano M, Vitale V, Rosso R, et al. Treatment of advanced squamous-cell carcinoma of the head and neck with alternating chemotherapy and radiotherapy. The New England Journal of Medicine. 1992;327(16):1115–1121. doi: 10.1056/NEJM199210153271602. [DOI] [PubMed] [Google Scholar]
- 5.Pignon J-P, Maître AL, Maillard E, Bourhis J. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): an update on 93 randomised trials and 17,346 patients. Radiotherapy and Oncology. 2009;92(1):4–14. doi: 10.1016/j.radonc.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Furness S, Glenny A-M, Worthington HV, et al. Interventions for the treatment of oral cavity and oropharyngeal cancer: chemotherapy. Cochrane Database of Systematic Reviews. 2011;4 doi: 10.1002/14651858.CD006386.pub3.CD006386 [DOI] [PubMed] [Google Scholar]
- 7.Pignon JP, Bourhis J, Domenge C, Designé L. Chemotherapy added to locoregional treatment for head and neck squamous-cell carcinoma: three meta-analyses of updated individual data. The Lancet. 2000;355(9208):949–955. [PubMed] [Google Scholar]
- 8.Logan RM. Advances in understanding of toxicities of treatment for head and neck cancer. Oral Oncology. 2009;45(10):844–848. doi: 10.1016/j.oraloncology.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 9.Murdoch D. Standard, and novel cytotoxic and molecular-targeted, therapies for HNSCC: an evidence-based review. Current Opinion in Oncology. 2007;19(3):216–221. doi: 10.1097/01.cco.0000264952.98166.99. [DOI] [PubMed] [Google Scholar]
- 10.Posner MR, Hershock DM, Blajman CR, et al. Cisplatin and fluorouracil alone or with docetaxel in head and neck cancer. The New England Journal of Medicine. 2007;357(17):1705–1715. doi: 10.1056/NEJMoa070956. [DOI] [PubMed] [Google Scholar]
- 11.Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. Journal of Cell Science. 2008;121(supplement 1):1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- 12.Hakem R. DNA-damage repair; the good, the bad, and the ugly. The EMBO Journal. 2008;27(4):589–605. doi: 10.1038/emboj.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorgoulis VG, Vassiliou L-VF, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434(7035):907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 14.DiTullio RA, Jr., Mochan TA, Venere M, et al. 53BP1 functions in a ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nature Cell Biology. 2002;4(12):998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 15.Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26(56):7773–7779. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 16.Sturgis EM, Cinciripini PM. Trends in head and neck cancer incidence in relation to smoking prevalence: an emerging epidemic of human papillomavirus-associated cancers? Cancer. 2007;110(7):1429–1435. doi: 10.1002/cncr.22963. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann M, Kahn T, Mahnke CG, Goeroegh T, Lippert BM, Werner JA. Prevalence of human papillomavirus in squamous cell carcinoma of the head and neck determined by polymerase chain reaction and Southern blot hybridization: proposal for optimized diagnostic requirements. Acta Oto-Laryngologica. 1998;118(1):138–144. doi: 10.1080/00016489850155279. [DOI] [PubMed] [Google Scholar]
- 18.Termine N, Panzarella V, Falaschini S, et al. HPV in oral squamous cell carcinoma vs head and neck squamous cell carcinoma biopsies: a meta-analysis (1988–2007) Annals of Oncology. 2008;19(10):1681–1690. doi: 10.1093/annonc/mdn372. [DOI] [PubMed] [Google Scholar]
- 19.Kansy K, Thiele O, Freier K. The role of human papillomavirus in oral squamous cell carcinoma: myth and reality. Oral and Maxillofacial Surgery. 2012 doi: 10.1007/s10006-012-0383-0. [DOI] [PubMed] [Google Scholar]
- 20.Marur S, D’Souza G, Westra WH, Forastiere AA. HPV-associated head and neck cancer: a virus-related cancer epidemic. The Lancet Oncology. 2010;11(8):781–789. doi: 10.1016/S1470-2045(10)70017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dayyani F, Etzel CJ, Liu M, Ho C-H, Lippman SM, Tsao AS. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC) Head and Neck Oncology. 2010;2(1, article 15) doi: 10.1186/1758-3284-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England Journal of Medicine. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. The New England Journal of Medicine. 2007;356(19):1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 24.D’Souza G, Agrawal Y, Halpern J, Bodison S, Gillison ML. Oral sexual behaviors associated with prevalent oral human papillomavirus infection. Journal of Infectious Diseases. 2009;199(9):1263–1269. doi: 10.1086/597755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz SM, Daling JR, Doody DR, et al. Oral cancer risk in relation to sexual history and evidence of human papillomavirus infection. Journal of the National Cancer Institute. 1998;90(21):1626–1636. doi: 10.1093/jnci/90.21.1626. [DOI] [PubMed] [Google Scholar]
- 26.Lassen P, Eriksen JG, Hamilton-Dutoit S, Tramm T, Alsner J, Overgaard J. Effect of HPV-associated p16INK4A expression on response to radiotherapy and survival in squamous cell carcinoma of the head and neck. Journal of Clinical Oncology. 2009;27(12):1992–1998. doi: 10.1200/JCO.2008.20.2853. [DOI] [PubMed] [Google Scholar]
- 27.Gillison ML, D’Souza G, Westra W, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. Journal of the National Cancer Institute. 2008;100(6):407–420. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 28.Smeets SJ, Braakhuis BJM, Abbas S, et al. Genome-wide DNA copy number alterations in head and neck squamous cell carcinomas with or without oncogene-expressing human papillomavirus. Oncogene. 2006;25(17):2558–2564. doi: 10.1038/sj.onc.1209275. [DOI] [PubMed] [Google Scholar]
- 29.Wilting SM, Smeets SJ, Snijders PJ, et al. Genomic profiling identifies common HPV-associated chromosomal alterations in squamous cell carcinomas of cervix and head and neck. BMC Medical Genomics. 2009;2(1, article 32) doi: 10.1186/1755-8794-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Syrjanen K, Syrjanen S, Lamberg M. Morphological and immunohistochemical evidence suggesting human papillomavirus (HPV) involvement in oral squamous cell carcinogenesis. International Journal of Oral Surgery. 1983;12(6):418–424. doi: 10.1016/s0300-9785(83)80033-7. [DOI] [PubMed] [Google Scholar]
- 31.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75(3):495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 32.Thomas M, Pim D, Banks L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene. 1999;18(53):7690–7700. doi: 10.1038/sj.onc.1202953. [DOI] [PubMed] [Google Scholar]
- 33.Mietz JA, Unger T, Huibregtse JM, Howley PM. The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. The EMBO Journal. 1992;11(13):5013–5020. doi: 10.1002/j.1460-2075.1992.tb05608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Current Opinion in Cell Biology. 2002;14(6):684–691. doi: 10.1016/s0955-0674(02)00388-5. [DOI] [PubMed] [Google Scholar]
- 35.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes and Development. 2000;14(19):2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Clements A, Zhao K, Marmorstein R. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. Journal of Biological Chemistry. 2006;281(1):578–586. doi: 10.1074/jbc.M508455200. [DOI] [PubMed] [Google Scholar]
- 37.Lewis JS, Jr., Thorstad WL, Chernock RD, et al. P16 positive oropharyngeal squamous cell carcinoma: an entity with a favorable prognosis regardless of tumor HPV status. American Journal of Surgical Pathology. 2010;34(8):1088–1096. doi: 10.1097/PAS.0b013e3181e84652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindquist D, Romanitan M, Hammarstedt L, et al. Human papillomavirus is a favourable prognostic factor in tonsillar cancer and its oncogenic role is supported by the expression of E6 and E7. Molecular Oncology. 2007;1(3):350–355. doi: 10.1016/j.molonc.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlecht NF, Brandwein-Gensler M, Nuovo GJ, et al. A comparison of clinically utilized human papillomavirus detection methods in head and neck cancer. Modern Pathology. 2011;24(10):1295–1305. doi: 10.1038/modpathol.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duensing S, Münger K. Human papillomaviruses and centrosome duplication errors: modeling the origins of genomic instability. Oncogene. 2002;21(40):6241–6248. doi: 10.1038/sj.onc.1205709. [DOI] [PubMed] [Google Scholar]
- 41.Duensing S, Lee LY, Duensing A, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(18):10002–10007. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duensing S, Münger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. International Journal of Cancer. 2004;109(2):157–162. doi: 10.1002/ijc.11691. [DOI] [PubMed] [Google Scholar]
- 43.Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edwards TG, Helmus MJ, Koeller K, Bashkin JK, Fisher C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. Journal of Virology. 2013;87(7):3979–3989. doi: 10.1128/JVI.03473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rieckmann T, Tribius S, Grob TJ, et al. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiotherapy and Oncology. 2013;107(2):242–246. doi: 10.1016/j.radonc.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 46.Yakushiji T, Uzawa K, Shibahara T, Noma H, Tanzawa H. Over-expression of DNA methyltransferases and CDKN2A gene methylation status in squamous cell carcinoma of the oral cavity. International Journal of Oncology. 2003;22(6):1201–1207. [PubMed] [Google Scholar]
- 47.Reed AL, Califano J, Cairns P, et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Research. 1996;56(16):3630–3633. [PubMed] [Google Scholar]
- 48.Tsai C-H, Yang C-C, Chou LS-S, Chou M-Y. The correlation between alteration of p16 gene and clinical status in oral squamous cell carcinoma. Journal of Oral Pathology and Medicine. 2001;30(9):527–531. doi: 10.1034/j.1600-0714.2001.300903.x. [DOI] [PubMed] [Google Scholar]
- 49.Pande P, Mathur M, Shukla NK, Ralhan R. pRB and p16 protein alterations in human oral tumorigenesis. Oral Oncology. 1998;34(5):396–403. doi: 10.1016/s1368-8375(98)00024-4. [DOI] [PubMed] [Google Scholar]
- 50.Duray A, Descamps G, Decaestecker C, et al. Human papillomavirus DNA strongly correlates with a poorer prognosis in oral cavity carcinoma. Laryngoscope. 2012;122(7):1558–1565. doi: 10.1002/lary.23298. [DOI] [PubMed] [Google Scholar]
- 51.Ernoux-Neufcoeur P, Arafa M, Decaestecker C, et al. Combined analysis of HPV DNA, p16, p21 and p53 to predict prognosis in patients with stage IV hypopharyngeal carcinoma. Journal of Cancer Research and Clinical Oncology. 2011;137(1):173–181. doi: 10.1007/s00432-010-0871-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marsit CJ, Christensen BC, Houseman EA, et al. Epigenetic profiling reveals etiologically distinct patterns of DNA methylation in head and neck squamous cell carcinoma. Carcinogenesis. 2009;30(3):416–422. doi: 10.1093/carcin/bgp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Merlo A, Herman JG, Mao L, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nature Medicine. 1995;1(7):686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 54.Abou-Elhamd K-EA, Habib TN. The flow cytometric analysis of premalignant and malignant lesions in head and neck squamous cell carcinoma. Oral Oncology. 2007;43(4):366–372. doi: 10.1016/j.oraloncology.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 55.Califano J, van der Riet P, Westra W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Research. 1996;56(11):2488–2492. [PubMed] [Google Scholar]
- 56.Bockmühl U, Wolf G, Schmidt S, et al. Genomic alterations associated with malignancy in head and neck cancer. Head & Neck. 1998;20(2):145–151. doi: 10.1002/(sici)1097-0347(199803)20:2<145::aid-hed8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 57.Kiaris H, Spanakis N, Ergazaki M, Sourvinos G, Spandidos DA. Loss of heterozygosity at 9p and 17q in human laryngeal tumours. Cancer Letters. 1995;97(1):129–134. doi: 10.1016/0304-3835(95)03963-w. [DOI] [PubMed] [Google Scholar]
- 58.Cairns P, Polascik TJ, Eby Y, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nature Genetics. 1995;11(2):210–212. doi: 10.1038/ng1095-210. [DOI] [PubMed] [Google Scholar]
- 59.van der Riet P, Nawroz H, Hruban RH, et al. Frequent loss of chromosome 9p21-22 early in head and neck cancer progression. Cancer Research. 1994;54(5):1156–1158. [PubMed] [Google Scholar]
- 60.Rosin MP, Cheng X, Poh C, et al. Use of allelic loss to predict malignant risk for low-grade oral epithelial dysplasia. Clinical Cancer Research. 2000;6(2):357–362. [PubMed] [Google Scholar]
- 61.Soder AI, Hopman AHN, Ramaekers FCS, Conradt C, Bosch FX. Distinct nonrandom patterns of chromosomal aberrations in the progression of squamous cell carcinomas of the head and neck. Cancer Research. 1995;55(21):5030–5037. [PubMed] [Google Scholar]
- 62.Lydiatt WM, Murty VVVS, Davidson BJ, et al. Homozygous deletions and loss of expression of the CDKN2 gene occur frequently in head and neck squamous cell carcinoma cell lines but infrequently in primary tumors. Genes, Chromosomes and Cancer. 1995;13(2):94–98. doi: 10.1002/gcc.2870130204. [DOI] [PubMed] [Google Scholar]
- 63.Yu KK, Zanation AM, Moss JR, Yarbrough WG. Familial head and neck cancer: molecular analysis of a new clinical entity. Laryngoscope. 2002;112(9):1587–1593. doi: 10.1097/00005537-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 64.Cabanillas R, Astudillo A, Valle M, et al. Novel germline CDKN2A mutation associated with head and neck squamous cell carcinomas and melanomas. Head and Neck. 2013;35(3):E80–E84. doi: 10.1002/hed.21911. [DOI] [PubMed] [Google Scholar]
- 65.Maestro R, Gasparotto D, Vukosavljevic T, Barzan L, Sulfaro S, Boiocchi M. Three discrete regions of deletion at 3p in head and neck cancers. Cancer Research. 1993;53(23):5775–5779. [PubMed] [Google Scholar]
- 66.Hogg RP, Honorio S, Martinez A, et al. Frequent 3p allele loss and epigenetic inactivation of the RASSF1A tumour suppressor gene from region 3p21.3 in head and neck squamous cell carcinoma. European Journal of Cancer. 2002;38(12):1585–1592. doi: 10.1016/s0959-8049(01)00422-1. [DOI] [PubMed] [Google Scholar]
- 67.Uzawa N, Yoshida MA, Hosoe S, Oshimura M, Amagasa T, Ikeuchi T. Functional evidence for involvement of multiple putative tumor suppressor genes on the short arm of chromosome 3 in human oral squamous cell carcinogenesis. Cancer Genetics and Cytogenetics. 1998;107(2):125–131. doi: 10.1016/s0165-4608(98)00097-1. [DOI] [PubMed] [Google Scholar]
- 68.Dasgupta S, Mukherjee N, Roy S, et al. Mapping of the candidate tumor suppressor genes’ loci on human chromosome 3 in head and neck squamous cell carcinoma of an Indian patient population. Oral Oncology. 2002;38(1):6–15. doi: 10.1016/s1368-8375(00)00131-7. [DOI] [PubMed] [Google Scholar]
- 69.Tai S-K, Lee JI, Ang KK, et al. Loss of Fhit expression in head and neck squamous cell carcinoma and its potential clinical implication. Clinical Cancer Research. 2004;10(16):5554–5557. doi: 10.1158/1078-0432.CCR-04-0208. [DOI] [PubMed] [Google Scholar]
- 70.Ghosh S, Ghosh A, Maiti GP, et al. Alterations of 3p21.31 tumor suppressor genes in head and neck squamous cell carcinoma: correlation with progression and prognosis. International Journal of Cancer. 2008;123(11):2594–2604. doi: 10.1002/ijc.23834. [DOI] [PubMed] [Google Scholar]
- 71.Blons H, Radicella JP, Laccourreye O, et al. Frequent allelic loss at chromosome 3p distinct from genetic alterations of the 8-oxoguanine DNA glycosylase 1 gene in head and neck cancer. Molecular Carcinogenesis. 1999;26(4):254–260. [PubMed] [Google Scholar]
- 72.Fan C-Y, Liu KL, Huang HY, et al. Frequent allelic imbalance and loss of protein expression of the DNA repair gene hOGG1 in head and neck squamous cell carcinoma. Laboratory Investigation. 2001;81(10):1429–1438. doi: 10.1038/labinvest.3780356. [DOI] [PubMed] [Google Scholar]
- 73.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447(7147):941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eisenstadt E, Warren AJ, Porter J, Atkins D, Miller JH. Carcinogenic epoxides of benzo[a]pyrene and cyclopenta[cd]pyrene induce base substitutions via specific transversions. Proceedings of the National Academy of Sciences of the United States of America. 1982;79(6):1945–1949. doi: 10.1073/pnas.79.6.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bernelot-Moens C, Glickman BW, Gordon AJE. Induction of specific frameshift and base substitution events by benzo[a]pyrene diol expoxide in excision-repair-deficient Escherichia coli. Carcinogenesis. 1990;11(5):781–785. doi: 10.1093/carcin/11.5.781. [DOI] [PubMed] [Google Scholar]
- 76.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349(6308):431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 77.Blons H, Laurent-Puig P. TP53 and head and neck neoplasms. Human Mutation. 2003;21(3):252–257. doi: 10.1002/humu.10171. [DOI] [PubMed] [Google Scholar]
- 78.Yoon J-H, Smith LE, Feng Z, Tang M-S, Lee C-S, Pfeifer GP. Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Research. 2001;61(19):7110–7117. [PubMed] [Google Scholar]
- 79.Bockmühl U, Petersen S, Schmidt S, et al. Patterns of chromosomal alterations in metastasizing and nonmetastasizing primary head and neck carcinomas. Cancer Research. 1997;57(23):5213–5216. [PubMed] [Google Scholar]
- 80.Bockmühl U, Schlüns K, Schmidt S, Matthias S, Petersen I. Chromosomal alterations during metastasis formation of head and neck squamous cell carcinoma. Genes, Chromosomes and Cancer. 2002;33(1):29–35. [PubMed] [Google Scholar]
- 81.Bockmühl U, Ishwad CS, Ferrell RE, Gollin SM. Association of 8p23 deletions with poor survival in head and neck cancer. Otolaryngology—Head and Neck Surgery. 2001;124(4):451–455. doi: 10.1067/mhn.2001.114794. [DOI] [PubMed] [Google Scholar]
- 82.Toomes C, Jackson A, Maguire K, et al. The presence of multiple regions of homozygous deletion at the CSMDI locus in oral squamous cell carcinoma question the role of CSMDI in head and neck carcinogenesis. Genes, Chromosomes and Cancer. 2003;37(2):132–140. doi: 10.1002/gcc.10191. [DOI] [PubMed] [Google Scholar]
- 83.Papadimitrakopoulou VA, Oh Y, El-Naggar A, Izzo J, Clayman G, Mao L. Presence of multiple incontiguous deleted regions at the long arm of chromosome 18 in head and neck cancer. Clinical Cancer Research. 1998;4(3):539–544. [PubMed] [Google Scholar]
- 84.Nawroz H, van der Riet P, Hruban RH, Koch W, Ruppert JM, Sidransky D. Allelotype of head and neck squamous cell carcinoma. Cancer Research. 1994;54(5):1152–1155. [PubMed] [Google Scholar]
- 85.van Dyke DL, Worsham MJ, Benninger MS, et al. Recurrent cytogenetic abnormalities in squamous cell carcinomas of the head and neck region. Genes, Chromosomes and Cancer. 1994;9(3):192–206. doi: 10.1002/gcc.2870090308. [DOI] [PubMed] [Google Scholar]
- 86.Mertens F, Johansson B, Höglund M, Mitelman F. Chromosomal imbalance maps of malignant solid tumors: a cytogenetic survey of 3185 neoplasms. Cancer Research. 1997;57(13):2765–2780. [PubMed] [Google Scholar]
- 87.Pershouse MA, El-Naggar AK, Hurr K, Lin H, Yung WKA, Steck PA. Deletion mapping of chromosome 4 in head and neck squamous cell carcinoma. Oncogene. 1997;14(3):369–373. doi: 10.1038/sj.onc.1200836. [DOI] [PubMed] [Google Scholar]
- 88.Yoo GH, Xu H-J, Brennan JA, et al. Infrequent inactivation of the retinoblastoma gene despite frequent loss of chromosome 13q in head and neck squamous cell carcinoma. Cancer Research. 1994;54(17):4603–4606. [PubMed] [Google Scholar]
- 89.Feenstra M, Veltkamp M, Van Kuik J, et al. HLA class 1 expression and chromosomal deletions at 6p and 15q in head and neck squamous cell carcinomas. Tissue Antigens. 1999;54(3):235–245. doi: 10.1034/j.1399-0039.1999.540304.x. [DOI] [PubMed] [Google Scholar]
- 90.Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA Repair. 2006;5(9-10):1126–1135. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 91.Mizuno K, Miyabe I, Schalbetter S, Carr AM, Murray JM. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature. 2013;493(7431):246–249. doi: 10.1038/nature11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lieber MR. NHEJ and its backup pathways in chromosomal translocations. Nature Structural and Molecular Biology. 2010;17(4):393–395. doi: 10.1038/nsmb0410-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ferguson DO, Alt FW. DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene. 2001;20(40):5572–5579. doi: 10.1038/sj.onc.1204767. [DOI] [PubMed] [Google Scholar]
- 94.Scully C, Field JK, Tanzawa H. Genetic aberrations in oral or head and neck squamous cell carcinoma. 2: chromosomal aberrations. Oral Oncology. 2000;36(4):311–327. doi: 10.1016/s1368-8375(00)00021-x. [DOI] [PubMed] [Google Scholar]
- 95.Patmore HS, Cawkwell L, Stafford ND, Greenman J. Unraveling the chromosomal aberrations of head and neck squamous cell carcinoma: a review. Annals of Surgical Oncology. 2005;12(10):831–842. doi: 10.1245/ASO.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 96.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harbor Symposia on Quantitative Biology. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 97.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(22):12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature. 2000;407(6805):777–783. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- 99.Bau D-T, Chang C-H, Tsai M-H, et al. Association between DNA repair gene ATM polymorphisms and oral cancer susceptibility. Laryngoscope. 2010;120(12):2417–2422. doi: 10.1002/lary.21009. [DOI] [PubMed] [Google Scholar]
- 100.Ai L, Vo QN, Zuo C, et al. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiology Biomarkers and Prevention. 2004;13(1):150–156. doi: 10.1158/1055-9965.epi-082-3. [DOI] [PubMed] [Google Scholar]
- 101.Rigi-Ladiz MA, Kordi-Tamandani DM, Torkamanzehi A. Analysis of hypermethylation and expression profiles of APC and ATM genes in patients with oral squamous cell carcinoma. Clinical Epigenetics. 2011;3(1, article 6) doi: 10.1186/1868-7083-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Spyridonidou S, Yapijakis C, Nkenke E, et al. A common 9 bp deletion in the ataxia-telangiectasia-mutated gene is not associated with oral cancer. Anticancer Research. 2009;29(8):3191–3193. [PubMed] [Google Scholar]
- 103.Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal (Lynch syndrome): an updated review. Cancer. 1996;78(6):1149–1167. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1149::AID-CNCR1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 104.Wang Y, Irish J, MacMillan C, et al. High frequency of microsatellite instability in young patients with head-and-neck squamous-cell carcinoma: lack of involvement of the mismatch repair genes hMLH1 and hMSH2. International Journal of Cancer. 2001;93(3):353–360. doi: 10.1002/ijc.1337. [DOI] [PubMed] [Google Scholar]
- 105.Piccinin S, Gasparotto D, Vukosavljevic T, et al. Microsatellite instability in squamous cell carcinomas of the head and neck related to field cancerization phenomena. British Journal of Cancer. 1998;78(9):1147–1151. doi: 10.1038/bjc.1998.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blons H, Cabelguenne A, Carnot F, et al. Microsatellite analysis and response to chemotherapy in head-and-neck squamous-cell carcinoma. International Journal of Cancer. 1999;84(4):410–415. doi: 10.1002/(sici)1097-0215(19990820)84:4<410::aid-ijc14>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 107.Arzimanoglou II, Gilbert F, Barber HRK. Microsatellite instability in human solid tumors. Cancer. 1998;82(10):1808–1820. doi: 10.1002/(sici)1097-0142(19980515)82:10<1808::aid-cncr2>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 108.El-Naggar AK, Hurr K, Huff V, Clayman GL, Luna MA, Batsakis JG. Microsatellite instability in preinvasive and invasive head and neck squamous carcinoma. The American Journal of Pathology. 1996;148(6):2067–2072. [PMC free article] [PubMed] [Google Scholar]
- 109.Ishwad CS, Ferrell RE, Rossie KM, et al. Microsatellite instability in oral cancer. International Journal of Cancer. 1995;64(5):332–335. doi: 10.1002/ijc.2910640509. [DOI] [PubMed] [Google Scholar]
- 110.Ha PK, Pilkington TA, Westra WH, Sciubba J, Sidransky D, Califano JA. Progression of microsatellite instability from premalignant lesions to tumors of the head and neck. International Journal of Cancer. 2002;102(6):615–617. doi: 10.1002/ijc.10748. [DOI] [PubMed] [Google Scholar]
- 111.Zuo C, Zhang H, Spencer HJ, et al. Increased microsatellite instability and epigenetic inactivation of the hMLH1 gene in head and neck squamous cell carcinoma. Otolaryngology—Head and Neck Surgery. 2009;141(4):484–490. doi: 10.1016/j.otohns.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 112.González-Ramírez I, Ramírez-Amador V, Irigoyen-Camacho ME, et al. hMLH1 promoter methylation is an early event in oral cancer. Oral Oncology. 2011;47(1):22–26. doi: 10.1016/j.oraloncology.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 113.Viswanathan M, Tsuchida N, Shanmugam G. Promoter hypermethylation profile of tumor-associated genes p16, p15, hMLH1, MGMT and E-cadherin in oral squamous cell carcinoma. International Journal of Cancer. 2003;105(1):41–46. doi: 10.1002/ijc.11028. [DOI] [PubMed] [Google Scholar]
- 114.Czerninski R, Krichevsky S, Ashhab Y, Gazit D, Patel V, Ben-Yehuda D. Promoter hypermethylation of mismatch repair genes, hMLH1 and hMSH2 in oral squamous cell carcinoma. Oral Diseases. 2009;15(3):206–213. doi: 10.1111/j.1601-0825.2008.01510.x. [DOI] [PubMed] [Google Scholar]
- 115.Steinmann K, Sandner A, Schagdarsurengin U, Dammann RH, Dammann RH. Frequent promoter hypermethylation of tumor-related genes in head and neck squamous cell carcinoma. Oncology Reports. 2009;22(6):1519–1526. doi: 10.3892/or_00000596. [DOI] [PubMed] [Google Scholar]
- 116.Theocharis S, Klijanienko J, Giaginis C, et al. Expression of DNA repair proteins, MSH2, MLH1 and MGMT in mobile tongue squamous cell carcinoma: associations with clinicopathological parameters and patients’ survival. Journal of Oral Pathology and Medicine. 2011;40(3):218–226. doi: 10.1111/j.1600-0714.2010.00945.x. [DOI] [PubMed] [Google Scholar]
- 117.Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. Journal of Biological Chemistry. 2003;278(50):49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 118.Kumar A, Pant MC, Singh HS, Khandelwal S. Reduced expression of DNA repair genes (XRCC1, XPD, and OGG1) in squamous cell carcinoma of head and neck in North India. Tumor Biology. 2012;33(1):111–119. doi: 10.1007/s13277-011-0253-7. [DOI] [PubMed] [Google Scholar]
- 119.Paz-Elizur T, Ben-Yosef R, Elinger D, et al. Reduced repair of the oxidative 8-oxoguanine DNA damage and risk of head and neck cancer. Cancer Research. 2006;66(24):11683–11689. doi: 10.1158/0008-5472.CAN-06-2294. [DOI] [PubMed] [Google Scholar]
- 120.Mahjabeen I, Baig RM, Sabir M, Kayani MA. Genetic and expressional variations of APEX1 are associated with increased risk of head and neck cancer. Mutagenesis. 2013;28(2):213–218. doi: 10.1093/mutage/ges074. [DOI] [PubMed] [Google Scholar]
- 121.Das A, Wiederhold L, Leppard JB, et al. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: evidence for a repair complex in human cells. DNA Repair. 2006;5(12):1439–1448. doi: 10.1016/j.dnarep.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhai X, Zhao H, Liu Z, et al. Functional variants of the NEIL1 and NEIL2 genes and risk and progression of squamous cell carcinoma of the oral cavity and oropharynx. Clinical Cancer Research. 2008;14(13):4345–4352. doi: 10.1158/1078-0432.CCR-07-5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair. 2003;2(9):955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 124.Dunphy EJ, Beckett MA, Thompson LH, Weichselbaum RR. Expression of the polymorphic human DNA repair gene XRCC1 does not correlate with radiosensitivity in the cells of human head and neck tumor cell lines. Radiation Research. 1992;130(2):166–170. [PubMed] [Google Scholar]
- 125.Nix P, Greenman J, Stafford N, Cawkwell L. Expression of XRCC1 and ERCC1 proteins in radioresistant and radiosensitive laryngeal cancer. Cancer Therapy. 2004;2:47–53. [Google Scholar]
- 126.Ang M-K, Patel MR, Yin X-Y, et al. High XRCC1 protein expression is associated with poorer survival in patients with head and neck squamous cell carcinoma. Clinical Cancer Research. 2011;17(20):6542–6552. doi: 10.1158/1078-0432.CCR-10-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sousa FG, Matuo R, Soares D, et al. PARPs and the DNA damage response. Carcinogenesis. 2012;33(8):1433–1440. doi: 10.1093/carcin/bgs132. [DOI] [PubMed] [Google Scholar]
- 128.Sugimura K, Takebayashi S-I, Taguchi H, Takeda S, Okumura K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. Journal of Cell Biology. 2008;183(7):1203–1212. doi: 10.1083/jcb.200806068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schultz N, Lopez E, Saleh-Gohari N, Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Research. 2003;31(17):4959–4964. doi: 10.1093/nar/gkg703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.de Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochemical Pharmacology. 2012;84(2):137–146. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 131.Schreiber V, Amé J-C, Dollé P, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. Journal of Biological Chemistry. 2002;277(25):23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 132.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 133.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England Journal of Medicine. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 134.Nowsheen S, Bonner JA, LoBuglio AF, et al. Cetuximab augments cytotoxicity with poly (ADP-Ribose) polymerase inhibition in head and neck cancer. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0024148.e24148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Abuzeid WM, Khan K, Jiang X, Cao X, O'Malley BW, Jr., Li D. R013: disruption of DNA damage signaling: a novel therapy for HNSCC. Otolaryngology—Head and Neck Surgery. 2007;137(2, supplement):P152–P153. [Google Scholar]
- 136.Tanaka T, Munshi A, Brooks C, Liu J, Hobbs ML, Meyn RE. Gefitinib radiosensitizes non-small cell lung cancer cells by suppressing cellular DNA repair capacity. Clinical Cancer Research. 2008;14(4):1266–1273. doi: 10.1158/1078-0432.CCR-07-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Research. 2006;66(16):8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 138.Ibrahim YH, García-García C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discovery. 2012;2(11):1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yamashita T, Miyamoto S, O'Malley B, Li D. The role of PARP 1 for cisplatin-based chemoresistance. Otolaryngology—Head and Neck Surgery. 2010;143(2, supplement):p. P54. [Google Scholar]
- 140.Wang L-E, Sturgis EM, Eicher SA, Spitz MR, Hong WK, Wei Q. Mutagen sensitivity to Benzo(a)pyrene diol epoxide and the risk of squamous cell carcinoma of the head and neck. Clinical Cancer Research. 1998;4(7):1773–1778. [PubMed] [Google Scholar]
- 141.Robu M, Shah RG, Petitclerc N, Brind'Amour J, Kandan-Kulangara F, Shah GM. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(5):1658–1663. doi: 10.1073/pnas.1209507110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Research. 2008;18(1):64–72. doi: 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cleaver JE. Photosensitivity syndrome brings to light a new transcription-coupled DNA repair cofactor. Nature Genetics. 2012;44(5):477–478. doi: 10.1038/ng.2255. [DOI] [PubMed] [Google Scholar]
- 144.Wei L, Lan L, Yasui A, et al. BRCA1 contributes to transcription-coupled repair of DNA damage through polyubiquitination and degradation of Cockayne syndrome B protein. Cancer Science. 2011;102(10):1840–1847. doi: 10.1111/j.1349-7006.2011.02037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kamenisch Y, Berneburg M. Progeroid syndromes and UV-induced oxidative DNA damage. Journal of Investigative Dermatology Symposium Proceedings. 2009;14(1):8–14. doi: 10.1038/jidsymp.2009.6. [DOI] [PubMed] [Google Scholar]
- 146.Wijnhoven SWP, Kool HJM, Van Oostrom CTM, et al. The relationship between benzo[a]pyrene-induce mutagenesis and carcinogenesis in repair-deficient Cockayne syndrome group B mice. Cancer Research. 2000;60(20):5681–5687. [PubMed] [Google Scholar]
- 147.Chiu T-J, Chen C-H, Chien C-Y, Li S-H, Tsai H-T, Chen Y-J. High ERCC1 expression predicts cisplatin-based chemotherapy resistance and poor outcome in unresectable squamous cell carcinoma of head and neck in a betel-chewing area. Journal of Translational Medicine. 2011;9, article 31 doi: 10.1186/1479-5876-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Handra-Luca A, Hernandez J, Mountzios G, et al. Excision repair cross complementation group 1 immunohistochemical expression predicts objective response and cancer-specific survival in patients treated by cisplatin-based induction chemotherapy for locally advanced head and neck squamous cell carcinoma. Clinical Cancer Research. 2007;13(13):3855–3859. doi: 10.1158/1078-0432.CCR-07-0252. [DOI] [PubMed] [Google Scholar]
- 149.Jun HJ, Ahn MJ, Kim HS, et al. ERCC1 expression as a predictive marker of squamous cell carcinoma of the head and neck treated with cisplatin-based concurrent chemoradiation. British Journal of Cancer. 2008;99(1):167–172. doi: 10.1038/sj.bjc.6604464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tsai LL, Yu C, Lo J-F, et al. Enhanced cisplatin resistance in oral-cancer stem-like cells is correlated with upregulation of excision-repair cross-complementation group 1. Journal of Dental Sciences. 2012;7(2):111–117. [Google Scholar]
- 151.Ahmad A, Robinson AR, Duensing A, et al. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Molecular and Cellular Biology. 2008;28(16):5082–5092. doi: 10.1128/MCB.00293-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Köberle B, Ditz C, Kausch I, Wollenberg B, Ferris RL, Albers AE. Metastases of squamous cell carcinoma of the head and neck show increased levels of nucleotide excision repair protein XPF in vivo that correlate with increased chemoresistance ex vivo. International Journal of Oncology. 2010;36(5):1277–1284. doi: 10.3892/ijo_00000612. [DOI] [PubMed] [Google Scholar]
- 153.Wang Y, Spitz MR, Lee JJ, Huang M, Lippman SM, Wu X. Nucleotide excision repair pathway genes and oral premalignant lesions. Clinical Cancer Research. 2007;13(12):3753–3758. doi: 10.1158/1078-0432.CCR-06-1911. [DOI] [PubMed] [Google Scholar]
- 154.Cheng L, Sturgis EM, Eicher SA, Spitz MR, Wei Q. Expression of nucleotide excision repair genes and dhe risk for squamous cell carcinoma of the head and neck. Cancer. 2002;94(2):393–397. doi: 10.1002/cncr.10231. [DOI] [PubMed] [Google Scholar]
- 155.Yu H, Liu Z, Huang YJ, Yin M, Wang LE, Wei Q. Association between single nucleotide polymorphisms in ERCC4 and risk of squamous cell carcinoma of the head and neck. PLoS ONE. 2012;7(7) doi: 10.1371/journal.pone.0041853.e41853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sliwinski T, Markiewicz L, Rusin P, et al. Impaired nucleotide excision repair pathway as a possible factor in pathogenesis of head and neck cancer. Mutation Research—Fundamental and Molecular Mechanisms of Mutagenesis. 2011;716(1-2):51–58. doi: 10.1016/j.mrfmmm.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 157.Zafereo ME, Sturgis E, Wei Q, Li G, Liu Z, Wang L. Nucleotide excision repair and risk of second primary cancer. Otolaryngology—Head and Neck Surgery. 2009;141(3, supplement 1):P55–P56. [Google Scholar]
- 158.Roberts SA, Strande N, Burkhalter MD, et al. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature. 2010;464(7292):1214–1217. doi: 10.1038/nature08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Indiviglio SM, Bertuch AA. Ku’s essential role in keeping telomeres intact. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(30):12217–12218. doi: 10.1073/pnas.0906427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Moeller BJ, Yordy JS, Williams MD, et al. DNA repair biomarker profiling of head and neck cancer: Ku80 expression predicts locoregional failure and death following radiotherapy. Clinical Cancer Research. 2011;17(7):2035–2043. doi: 10.1158/1078-0432.CCR-10-2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pavón MA, Parreño M, León X, et al. Ku70 predicts response and primary tumor recurrence after therapy in locally advanced head and neck cancer. International Journal of Cancer. 2008;123(5):1068–1079. doi: 10.1002/ijc.23635. [DOI] [PubMed] [Google Scholar]
- 162.Friesland S, Kanter-Lewensohn L, Tell R, Munck-Wikland E, Lewensohn R, Nilsson A. Expression of Ku86 confers favorable outcome of tonsillar carcinoma treated with radiotherapy. Head and Neck. 2003;25(4):313–321. doi: 10.1002/hed.10199. [DOI] [PubMed] [Google Scholar]
- 163.Sonoda E, Hochegger H, Saberi A, Taniguchi Y, Takeda S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair. 2006;5(9-10):1021–1029. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 164.D’Andrea AD, Grompe M. The fanconi anaemia/BRCA pathway. Nature Reviews Cancer. 2003;3(1):23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 165.Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003;101(3):822–826. doi: 10.1182/blood-2002-05-1498. [DOI] [PubMed] [Google Scholar]
- 166.Wreesmann VB, Estilo C, Eisele DW, Singh B, Wang SJ. Downregulation of Fanconi anemia genes in sporadic head and neck squamous cell carcinoma. Journal for Oto-Rhino-Laryngology and Its Related Specialties. 2007;69(4):218–225. doi: 10.1159/000101542. [DOI] [PubMed] [Google Scholar]
- 167.Tremblay S, dos Reis PP, Bradley G, et al. Young patients with oral squamous cell carcinoma: study of the involvement of GSTP1 and deregulation of the Fanconi anemia genes. Archives of Otolaryngology—Head and Neck Surgery. 2006;132(9):958–966. doi: 10.1001/archotol.132.9.958. [DOI] [PubMed] [Google Scholar]
- 168.Hays LE, Zodrow DM, Yates JE, et al. Cigarette smoke induces genetic instability in airway epithelial cells by suppressing FANCD2 expression. British Journal of Cancer. 2008;98(10):1653–1661. doi: 10.1038/sj.bjc.6604362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT. Inactivation of the Fanconi anemia/BRCA pathway in lung and oral cancers: implications for treatment and survival. Oncogene. 2004;23(4):1000–1004. doi: 10.1038/sj.onc.1207256. [DOI] [PubMed] [Google Scholar]
- 170.Ghosh A, Ghosh S, Maiti GP, et al. Association of FANCC and PTCH1 with the development of early dysplastic lesions of the head and neck. Annals of Surgical Oncology. 2012;19(supplement 3):S528–S538. doi: 10.1245/s10434-011-1991-x. [DOI] [PubMed] [Google Scholar]
- 171.Smith IM, Mithani SK, Mydlarz WK, Chang SS, Califano JA. Inactivation of the tumor suppressor genes causing the hereditary syndromes predisposing to head and neck cancer via promoter hypermethylation in sporadic head and neck cancers. Journal for Oto-Rhino-Laryngology and Its Related Specialties. 2010;72(1):44–50. doi: 10.1159/000292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Snyder ER, Ricker JL, Chen Z, Waes CV. Variation in cisplatinum sensitivity is not associated with Fanconi Anemia/BRCA pathway inactivation in head and neck squamous cell carcinoma cell lines. Cancer Letters. 2007;245(1-2):75–80. doi: 10.1016/j.canlet.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 173.Manthey KC, Glanzer JG, Dimitrova DD, Oakley GG. Hyperphosphorylation of replication protein A in cisplatin-resistant and -sensitive head and neck squamous cell carcinoma cell lines. Head and Neck. 2010;32(5):636–645. doi: 10.1002/hed.21234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Thompson D, Easton DF, The Breast Cancer Linkage Consortium Cancer incidence in BRCA1 mutation carriers. Journal of the National Cancer Institute. 2002;94(18):1358–1365. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 175.Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. The Lancet. 1994;343(8899):692–695. doi: 10.1016/s0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- 176.Taron M, Rosell R, Felip E, et al. BRCA1 mRNA expression levels as an indicator of chemoresistance in lung cancer. Human Molecular Genetics. 2004;13(20):2443–2449. doi: 10.1093/hmg/ddh260. [DOI] [PubMed] [Google Scholar]
- 177.Tobin LA, Robert C, Nagaria P, et al. Targeting abnormal DNA repair in therapy-resistant breast cancers. Molecular Cancer Research. 2012;10(1):96–107. doi: 10.1158/1541-7786.MCR-11-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vora HH, Shah NG, Patel DD, Trivedi TI, Choksi TJ. BRCA1 expression in leukoplakia and carcinoma of the tongue. Journal of Surgical Oncology. 2003;83(4):232–240. doi: 10.1002/jso.10213. [DOI] [PubMed] [Google Scholar]
- 179.Nawroz-Danish HM, Koch WM, Westra WH, Yoo G, Sidransky D. Lack of BRCA2 alterations in primary head and neck squamous cell carcinoma. Otolaryngology—Head and Neck Surgery. 1998;119(1):21–25. doi: 10.1016/S0194-5998(98)70168-8. [DOI] [PubMed] [Google Scholar]
- 180.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochemical Journal. 2000;351(2):289–305. [PMC free article] [PubMed] [Google Scholar]
- 181.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80(2):179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 182.Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discovery. 2013;3(7):761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Toulany M, Kasten-Pisula U, Brammer I, et al. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clinical Cancer Research. 2006;12(13):4119–4126. doi: 10.1158/1078-0432.CCR-05-2454. [DOI] [PubMed] [Google Scholar]
- 184.Huang S-M, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Research. 1999;59(8):1935–1940. [PubMed] [Google Scholar]
- 185.Kumar A, Fernadez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase β controls double-strand break DNA repair. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(16):7491–7496. doi: 10.1073/pnas.0914242107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. Journal of Biological Chemistry. 2007;282(29):21206–21212. doi: 10.1074/jbc.M703042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Toulany M, Kehlbach R, Florczak U, et al. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Molecular Cancer Therapeutics. 2008;7(7):1772–1781. doi: 10.1158/1535-7163.MCT-07-2200. [DOI] [PubMed] [Google Scholar]
- 188.Plo I, Laulier C, Gauthier L, Lebrun F, Calvo F, Lopez BS. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD5. Cancer Research. 2008;68(22):9404–9412. doi: 10.1158/0008-5472.CAN-08-0861. [DOI] [PubMed] [Google Scholar]
- 189.Li L, Wang H, Yang ES, Arteaga CL, Xia F. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Research. 2008;68(22):9141–9146. doi: 10.1158/0008-5472.CAN-08-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chinnaiyan P, Huang S, Vallabhaneni G, et al. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva) Cancer Research. 2005;65(8):3328–3335. doi: 10.1158/0008-5472.CAN-04-3547. [DOI] [PubMed] [Google Scholar]
- 191.Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clinical Cancer Research. 2008;14(10):2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 192.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England Journal of Medicine. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 193.Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, et al. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. European Journal of Cancer. 2006;42(1):109–111. doi: 10.1016/j.ejca.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 194.Lee JW, Soung YH, Kim SY, et al. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clinical Cancer Research. 2005;11(8):2879–2882. doi: 10.1158/1078-0432.CCR-04-2029. [DOI] [PubMed] [Google Scholar]
- 195.Schwentner I, Witsch-Baumgartner M, Sprinzl GM, et al. Identification of the rare EGFR mutation p.G796S as somatic and germline mutation in white patients with squamous cell carcinoma of the head and neck. Head and Neck. 2008;30(8):1040–1044. doi: 10.1002/hed.20831. [DOI] [PubMed] [Google Scholar]
- 196.Na II, Kang HJ, Cho SY, et al. EGFR mutations and human papillomavirus in squamous cell carcinoma of tongue and tonsil. European Journal of Cancer. 2007;43(3):520–526. doi: 10.1016/j.ejca.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 197.Ali MALS, Gunduz M, Nagatsuka H, et al. Expression and mutation analysis of epidermal growth factor receptor in head and neck squamous cell carcinoma. Cancer Science. 2008;99(8):1589–1594. doi: 10.1111/j.1349-7006.2008.00861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clinical Cancer Research. 2006;12(17):5064–5073. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 199.Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. Journal of Clinical Neuroscience. 2009;16(6):748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 200.Wheeler SE, Suzuki S, Thomas SM, et al. Epidermal growth factor receptor variant III mediates head and neck cancer cell invasion via STAT3 activation. Oncogene. 2010;29(37):5135–5145. doi: 10.1038/onc.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Batra SK, Castelino-Prabhu S, Wikstrand CJ, et al. Epidermal growth factor ligand-independent, unregulated, cell- transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth and Differentiation. 1995;6(10):1251–1259. [PubMed] [Google Scholar]
- 202.Murugan AK, Thi Hong N, Fukui Y, Munirajan AK, Tsuchida N. Oncogenic mutations of the PIK3CA gene in head and neck squamous cell carcinomas. International Journal of Oncology. 2008;32(1):101–111. [PubMed] [Google Scholar]
- 203.Qiu W, Schönleben F, Li X, et al. PIK3CA mutations in head and neck squamous cell carcinoma. Clinical Cancer Research. 2006;12(5):1441–1446. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Fenic I, Steger K, Gruber C, Arens C, Woenckhaus J. Analysis of PIK3CA and Akt/protein kinase B in head and neck squamous cell carcinoma. Oncology Reports. 2007;18(1):253–259. [PubMed] [Google Scholar]
- 205.Cohen Y, Goldenberg-Cohen N, Shalmon B, et al. Mutational analysis of PTEN/PIK3CA/AKT pathway in oral squamous cell carcinoma. Oral Oncology. 2011;47(10):946–950. doi: 10.1016/j.oraloncology.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 206.Ongkeko WM, Altuna X, Weisman RA, Wang-Rodriguez J. Expression of protein tyrosine kinases in head and neck squamous cell carcinomas. American Journal of Clinical Pathology. 2005;124(1):71–76. doi: 10.1309/BTLN5WTMJ3PCNRRC. [DOI] [PubMed] [Google Scholar]
- 207.Grandis JR, Melhem MF, Gooding WE, et al. Levels of TGF-α and EGFR protein in head and neck squamous cell carcinoma and patient survival. Journal of the National Cancer Institute. 1998;90(11):824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 208.Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Research. 2002;62(24):7350–7356. [PubMed] [Google Scholar]
- 209.Huang S-F, Chuang W-Y, Chen I-H, Liao C-T, Wang H-M, Hsieh L-L. EGFR protein overexpression and mutation in areca quid-associated oral cavity squamous cell carcinoma in Taiwan. Head and Neck. 2009;31(8):1068–1077. doi: 10.1002/hed.21067. [DOI] [PubMed] [Google Scholar]
- 210.Abhold EL, Kiang A, Rahimy E, et al. EGFR kinase promotes acquisition of stem cell-like properties: a potential therapeutic target in head and neck squamous cell carcinoma stem cells. PLoS ONE. 2012;7(2) doi: 10.1371/journal.pone.0032459.e32459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Temam S, Kawaguchi H, El-Naggar AK, et al. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. Journal of Clinical Oncology. 2007;25(16):2164–2170. doi: 10.1200/JCO.2006.06.6605. [DOI] [PubMed] [Google Scholar]
- 212.Morrison LE, Jacobson KK, Friedman M, Schroeder JW, Coon JS. Aberrant EGFR and chromosome 7 associate with outcome in laryngeal cancer. Laryngoscope. 2005;115(7):1212–1218. doi: 10.1097/01.MLG.0000163755.21035.8F. [DOI] [PubMed] [Google Scholar]
- 213.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. The New England Journal of Medicine. 2006;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 214.National Comprehensive Cancer Network. Head and Neck Cancers. 2012. http://www.nccn.org/professionals/physician_gls/pdf/head-and-neck.pdf. [Google Scholar]
- 215.Ko J-C, Ciou S-C, Cheng C-M, et al. Involvement of Rad51 in cytotoxicity induced by epidermal growth factor receptor inhibitor (gefitinib, Iressa R) and chemotherapeutic agents in human lung cancer cells. Carcinogenesis. 2008;29(7):1448–1458. doi: 10.1093/carcin/bgn130. [DOI] [PubMed] [Google Scholar]
- 216.Stewart JS, Cohen EE, Licitra L, et al. Phase III study of gefitinib compared with intravenous methotrexate for recurrent squamous cell carcinoma of the head and neck. Journal of Clinical Oncology. 2009;27(11):1864–1871. doi: 10.1200/JCO.2008.17.0530. [DOI] [PubMed] [Google Scholar]
- 217.Bentzen SM, Atasoy BM, Daley FM, et al. Epidermal growth factor receptor expression in pretreatment biopsies from head and neck squamous cell carcinoma as a predictive factor for a benefit from accelerated radiation therapy in a randomized controlled trial. Journal of Clinical Oncology. 2005;23(24):5560–5567. doi: 10.1200/JCO.2005.06.411. [DOI] [PubMed] [Google Scholar]
- 218.Chen DJ, Nirodi CS. The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage. Clinical Cancer Research. 2007;13(22):6555–6560. doi: 10.1158/1078-0432.CCR-07-1610. [DOI] [PubMed] [Google Scholar]
- 219.Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28(43):3801–3813. doi: 10.1038/onc.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wheeler DL, Huang S, Kruser TJ, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27(28):3944–3956. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Frazer KA, Ballinger DG, Cox DR, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449(7164):851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Mahjabeen I, Baig RM, Masood N, Sabir M, Malik FA, Kayani MA. OGG1 gene sequence variation in head and neck cancer patients in pakistan. Asian Pacific Journal of Cancer Prevention. 2011;12(10):2779–2783. [PubMed] [Google Scholar]
- 224.Hashimoto T, Uchida K, Okayama N, et al. Interaction of OGG1 Ser326Cys polymorphism with cigarette smoking in head and neck squamous cell carcinoma. Molecular Carcinogenesis. 2006;45(5):344–348. doi: 10.1002/mc.20140. [DOI] [PubMed] [Google Scholar]
- 225.Majumder M, Indra D, Roy PD, et al. Variant haplotypes at XRCC1 and risk of oral leukoplakia in HPV non-infected samples. Journal of Oral Pathology and Medicine. 2009;38(2):174–180. doi: 10.1111/j.1600-0714.2008.00690.x. [DOI] [PubMed] [Google Scholar]
- 226.Applebaum KM, McClean MD, Nelson HH, Marsit CJ, Christensen BC, Kelsey KT. Smoking modifies the relationship between XRCC1 haplotypes and HPV16-negative head and neck squamous cell carcinoma. International Journal of Cancer. 2009;124(11):2690–2696. doi: 10.1002/ijc.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Huang J, Ye F, Chen H, Lu W, Xie X. The nonsynonymous single nucleotide polymorphisms of DNA repair gene XRCC1 and susceptibility to the development of cervical carcinoma and high-risk human papillomavirus infection. International Journal of Gynecological Cancer. 2007;17(3):668–675. doi: 10.1111/j.1525-1438.2007.00840.x. [DOI] [PubMed] [Google Scholar]
- 228.Kumar A, Pant MC, Singh HS, Khandelwal S. Associated risk of XRCC1 and XPD cross talk and life style factors in progression of head and neck cancer in north Indian population. Mutation Research—Fundamental and Molecular Mechanisms of Mutagenesis. 2012;729(1-2):24–34. doi: 10.1016/j.mrfmmm.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 229.Carles J, Monzo M, Amat M, et al. Single-nucleotide polymorphisms in base excision repair, nucleotide excision repair, and double strand break genes as markers for response to radiotherapy in patients with Stage I to II head-and-neck cancer. International Journal of Radiation Oncology, Biology, Physics. 2006;66(4):1022–1030. doi: 10.1016/j.ijrobp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 230.De Castro G, Jr., Pasini FS, Siqueira SAC, et al. ERCC1 protein, mRNA expression and T19007C polymorphism as prognostic markers in head and neck squamous cell carcinoma patients treated with surgery and adjuvant cisplatin-based chemoradiation. Oncology Reports. 2011;25(3):693–699. doi: 10.3892/or.2011.1133. [DOI] [PubMed] [Google Scholar]
- 231.Swift M, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. The New England Journal of Medicine. 1991;325(26):1831–1836. doi: 10.1056/NEJM199112263252602. [DOI] [PubMed] [Google Scholar]
- 232.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nature Reviews Molecular Cell Biology. 2006;7(10):739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 233.Richard DJ, Bolderson E, Cubeddu L, et al. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature. 2008;453(7195):677–681. doi: 10.1038/nature06883. [DOI] [PubMed] [Google Scholar]
- 234.Richard DJ, Savage K, Bolderson E, et al. HSSB1 rapidly binds at the sites of DNA double-strand breaks and is required for the efficient recruitment of the MRN complex. Nucleic Acids Research. 2011;39(5):1692–1702. doi: 10.1093/nar/gkq1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Nakada S, Yonamine RM, Matsuo K. RNF8 regulates assembly of RAD51 at DNA double-strand breaks in the absence of BRCA1 and 53BP1. Cancer Research. 2012;72(19):4974–4983. doi: 10.1158/0008-5472.CAN-12-1057. [DOI] [PubMed] [Google Scholar]
- 236.Bolderson E, Tomimatsu N, Richard DJ, et al. Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Research. 2010;38(6):1821–1831. doi: 10.1093/nar/gkp1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Richard DJ, Cubeddu L, Urquhart AJ, et al. hSSB1 interacts directly with the MRN complex stimulating its recruitment to DNA double-strand breaks and its endo-nuclease activity. Nucleic Acids Research. 2011;39(9):3643–3651. doi: 10.1093/nar/gkq1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Mansour WY, Rhein T, Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Research. 2010;38(18):6065–6077. doi: 10.1093/nar/gkq387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Shinmura K, Tao H, Goto M, et al. Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer. Carcinogenesis. 2004;25(12):2311–2317. doi: 10.1093/carcin/bgh267. [DOI] [PubMed] [Google Scholar]
- 240.Sliwinski T, Walczak A, Przybylowska K, et al. Polymorphisms of the XRCC3 C722T and the RAD51 G135C genes and the risk of head and neck cancer in a Polish population. Experimental and Molecular Pathology. 2010;89(3):358–366. doi: 10.1016/j.yexmp.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 241.Romanowicz-Makowska H, Smolarz B, Gajęcka M, et al. Polymorphism of the DNA repair genes RAD51 and XRCC2 in smoking-and drinking-related laryngeal cancer in a Polish population. Archives of Medical Science. 2012;8(6):1065–1075. doi: 10.5114/aoms.2012.32417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Werbrouck J, De Ruyck K, Duprez F, et al. Single-nucleotide polymorphisms in DNA double-strand break repair genes: association with head and neck cancer and interaction with tobacco use and alcohol consumption. Mutation Research—Genetic Toxicology and Environmental Mutagenesis. 2008;656(1-2):74–81. doi: 10.1016/j.mrgentox.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 243.Cui Y, Morgenstern H, Greenland S, et al. Polymorphism of xeroderma pigmentosum group G and the risk of lung cancer and squamous cell carcinomas of the oropharynx, larynx and esophagus. International Journal of Cancer. 2006;118(3):714–720. doi: 10.1002/ijc.21413. [DOI] [PubMed] [Google Scholar]
- 244.dos Reis MB, Losi-Guembarovski R, de Souza Fonseca Ribeiro EM, et al. Allelic variants of XRCC1 and XRCC3 repair genes and susceptibility of oral cancer in Brazilian patients. Journal of Oral Pathology & Medicine. 2013;42(2):180–185. doi: 10.1111/j.1600-0714.2012.01192.x. [DOI] [PubMed] [Google Scholar]
- 245.Pawlowska E, Janik-Papis K, Rydzanicz M, et al. The Cys326 allele of the 8-oxoguanine DNA N-glycosylase 1 gene as a risk factor in smoking- and drinking-associated larynx cancer. Tohoku Journal of Experimental Medicine. 2009;219(4):269–275. doi: 10.1620/tjem.219.269. [DOI] [PubMed] [Google Scholar]
- 246.Kietthubthew S, Sriplung H, Au WW, Ishida T. Polymorphism in DNA repair genes and oral squamous cell carcinoma in Thailand. International Journal of Hygiene and Environmental Health. 2006;209(1):21–29. doi: 10.1016/j.ijheh.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 247.Gresner P, Gromadzinska J, Polanska K, Twardowska E, Jurewicz J, Wasowicz W. Genetic variability of Xrcc3 and Rad51 modulates the risk of head and neck cancer. Gene. 2012;504(2):166–174. doi: 10.1016/j.gene.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 248.Zhang Z, Shi Q, Wang L-E, et al. No association between hOGG1 Ser326Cys polymorphism and risk of squamous cell carcinoma of the head and neck. Cancer Epidemiology Biomarkers and Prevention. 2004;13(6):1081–1083. [PubMed] [Google Scholar]
- 249.Elahi A, Zheng Z, Park J, Eyring K, McCaffrey T, Lazarus P. The human OGG1 DNA repair enzyme and its association with orolaryngeal cancer risk. Carcinogenesis. 2002;23(7):1229–1234. doi: 10.1093/carcin/23.7.1229. [DOI] [PubMed] [Google Scholar]
- 250.Ramachandran S, Ramadas K, Hariharan R, Rejnish Kumar R, Radhakrishna Pillai M. Single nucleotide polymorphisms of DNA repair genes XRCC1 and XPD and its molecular mapping in Indian oral cancer. Oral Oncology. 2006;42(4):350–362. doi: 10.1016/j.oraloncology.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 251.Tae K, Lee HS, Park BJ, et al. Association of DNA repair gene XRCC1 polymorphisms with head and neck cancer in Korean population. International Journal of Cancer. 2004;111(5):805–808. doi: 10.1002/ijc.20338. [DOI] [PubMed] [Google Scholar]
- 252.Kowalski M, Przybylowska K, Rusin P, et al. Genetic polymorphisms in DNA base excision repair gene XRCC1 and the risk of squamous cell carcinoma of the head and neck. Journal of Experimental and Clinical Cancer Research. 2009;28(1, article 37) doi: 10.1186/1756-9966-28-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Yen C-Y, Liu S-Y, Chen C-H, et al. Combinational polymorphisms of four DNA repair genes XRCC1, XRCC2, XRCC3, and XRCC4 and their association with oral cancer in Taiwan. Journal of Oral Pathology and Medicine. 2008;37(5):271–277. doi: 10.1111/j.1600-0714.2007.00608.x. [DOI] [PubMed] [Google Scholar]
- 254.Tseng H-C, Tsai M-H, Chiu C-F, et al. Association of XRCC4 codon 247 polymorphism with oral cancer susceptibility in Taiwan. Anticancer Research. 2008;28(3A):1687–1691. [PubMed] [Google Scholar]
- 255.Chiu C-F, Tsai M-H, Tseng H-C, et al. A novel single nucleotide polymorphism in XRCC4 gene is associated with oral cancer susceptibility in Taiwanese patients. Oral Oncology. 2008;44(9):898–902. doi: 10.1016/j.oraloncology.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 256.Chiu C-F, Tsai M-H, Tseng H-C, et al. A novel single nucleotide polymorphism in ERCC6 gene is associated with oral cancer susceptibility in Taiwanese patients. Oral Oncology. 2008;44(6):582–586. doi: 10.1016/j.oraloncology.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 257.Hsu C-F, Tseng H-C, Chiu C-F, et al. Association between DNA double strand break gene Ku80 polymorphisms and oral cancer susceptibility. Oral Oncology. 2009;45(9):789–793. doi: 10.1016/j.oraloncology.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 258.Bau D-T, Tseng H-C, Wang C-H, et al. Oral cancer and genetic polymorphism of DNA double strand break gene Ku70 in Taiwan. Oral Oncology. 2008;44(11):1047–1051. doi: 10.1016/j.oraloncology.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 259.Flores-Obando RE, Gollin SM, Ragin CC. Polymorphisms in DNA damage response genes and head and neck cancer risk. Biomarkers. 2010;15(5):379–399. doi: 10.3109/13547501003797664. [DOI] [PMC free article] [PubMed] [Google Scholar]