

Abstract
BACKGROUND AND PURPOSE
The contribution of endothelin-1 (ET-1) in a B2KO mouse model of a high salt-induced arterial hypertension was investigated.
EXPERIMENTAL APPROACH
Wild-type (WT) or B2KO mice receiving a normal diet (ND) or a high-salt diet (HSD) were monitored by radiotelemetry up to a maximum of 18 weeks. At the 12th week of diet, subgroups under ND or HSD received by gavage the ETA antagonist A127722 during 5 days. In addition, blood samples were collected and, following euthanasia, the lungs, heart and kidneys were extracted, homogenized and assayed for ET-1 by RIA. In a separate series of experiments, the ETA antagonist, BQ123 was tested against the pressor responses to a NOS inhibitor L-NG-nitroarginine methyl ester (L-NAME) in anaesthetized WT and B2KO mice.
KEY RESULTS
In B2KO, but not WT mice, 12 weeks of HSD triggered a maximal increase of the mean arterial pressure (MAP) of 19.1 ± 2.8 mmHg, which was corrected by A127722 to MAP levels found in B2KO mice under ND. Significant increases in immunoreactive ET-1 were detected only in the lungs of B2KO mice under HSD. On the other hand, metabolic studies showed that sodium urinary excretion was markedly reduced in B2KO compared with WT mice under ND. Finally, BQ123 (2 mg·kg−1) reduced by 50% the pressor response to L-NAME (2 mg·kg−1) in B2KO, but not WT mice under anaesthesia.
CONCLUSIONS AND IMPLICATIONS
Our results support the concept that functional B2 receptors oppose high salt-induced increments in MAP, which are corrected by an ETA receptor antagonist in this mouse model of experimental hypertension.
Keywords: high-salt diet, A127722, radiotelemetry, RIA, ET-1, knockout mice, bradykinin
Introduction
Katori and Majima (2006) suggested that multiple factors have been linked with high-salt hypertension, albeit none provides a full explanation for this clinical situation. Dietary sodium restrictions, however, lower the incidence of cardiovascular complications in groups of hypertensive patients, as shown by Cook et al. (2007). African-American hypertensive patients are particularly sensitive to salt intake even in conditions of aggressive treatment with ACE inhibitors (ACEI; Weir et al., 1995; Grim and Robinson, 1996; Sanders, 2009a). These latter patients, some of whom show functional polymorphism for the B2 receptor (Gainer et al., 2000), also have a marked elevation in plasma levels of endothelin-1 (Ergul et al., 1998; Ergul, 2000).
Endothelin-1 (ET-1), a potent endothelial derived pressor peptide discovered by Yanagisawa and colleagues in 1988 (Yanagisawa et al., 1988), has been suggested to play a significant contribution in the aetiology of vascular diseases such as primary pulmonary hypertension (Giaid et al., 1993), scleroderma (Dhaun et al., 2007), renal hypertension (Takeda et al., 1997) and ACEI-resistant systemic hypertension (Weber et al., 2009). It is noticeable that antagonists of ETA and ETB receptors for ET-1, such as bosentan and macitentan have, up until now, only been proven clinically useful in primary pulmonary hypertension (Channick et al., 2001; Bolli et al., 2012) as well as in the prevention of digital ulcer in systemic sclerosis (Matucci-Cerinic et al., 2011).
Pharmacological cross-talk between ET-1 and bradykinin (BK), on the other hand, has recently been identified, for the first time, in Chagas's disease. Indeed, Andrade and colleagues have demonstrated a significant cooperation between endothelin receptor activation and the toll-like receptor 2/C-X-C motif receptor-2/B2 kinin receptor (TLR2/CXCR2/B2) complex in Chagas's-induced inflammation (Andrade et al., 2012). Whether such cooperation between the ET-1 and the BK pathways can be reproduced in cardiovascular diseases remained to be determined. On a more clinical point of view, Elijovich et al. (2001) have suggested the importance of testing an endothelin antagonist in salt-sensitive hypertensive patients, by showing an increase in endothelin levels in salt depleted salt-sensitive patients. The usefulness of endothelin antagonists in these particular patients remains, however, to be investigated.
Based on these observations mentioned earlier, it was of keen interest for our group to further explore the putative cross-talk between B2 receptors for kinins and ET-1, in experimental settings of high salt-induced hypertension.
The advent of mice knocked out for the BK B2 receptor (Borkowski and Hess, 1995) has allowed the identification of several roles for this particular receptor type in inflammation, immune reactions and pain (Boyce et al., 1996; Seabrook et al., 1997; Samadfam et al., 2000) as well as in the onset on hypertensive states (Alfie et al., 1996; Madeddu et al., 1997). Madeddu et al. (1997), for example, have shown by tail plethysmography that a high-salt diet triggered a significant increase in mean arterial pressure in B2KO mice, but not in wild-type (WT) counterparts. Alterations in the BK-dependent release of endothelial-derived NO have been documented as well in B2KO mice (Berthiaume et al., 1997; Schanstra et al., 2003). Loiola et al. (2011) also showed decreased NO bioavailability in blood vessels derived from B2KO mice. Finally, our group has reported the role of B2 and ETB receptors in the prostacyclin producing properties of BK and ET-1, respectively (Labonte et al., 2001).
In the present study, we hypothesized that the endothelin pathway is significantly involved in a high-salt diet (HSD) induced hypertensive murine model knocked out for the B2 receptor of kinins (B2KO mice). Thus, the principal aim of the present study was to assess whether an orally available ETA antagonist is able to reverse the salt-induced hypertensive state found in B2KO mice.
Our results show that the high salt-induced hypertension, which develops in the B2KO mouse, is corrected by the orally available ETA receptor antagonist A127722 (Opgenorth et al., 1996). These observations support the hypothesis that BK, via its B2 receptors, represents a physiological antagonist to the ET–1-induced blood pressure elevation in the murine model receiving a high-salt diet.
Material and methods
Animals
C57BL/6 WT mice or knocked out for BK B2 receptor (–/–; B2KO) on the same genetic background (Borkowski and Hess, 1995), weighing 25–35 g and of either sex, were used in these experiments. From breeding pairs originally donated by Dr Fred Hess, B2KO mice are currently bred and housed in our local colony where they are kept at constant room temperature (±23°C) and humidity (∼78%) under a controlled light–dark cycle (0600–1800 h).
Experimental design
WT and B2KO mice were each divided in two groups and fed ad libitum with normal diet (ND; 0.49% NaCl with normal tap water) or HSD (8% NaCl with tap water at 1% of NaCl) during a period of 8–18 weeks before euthanasia by cervical dislocation.
Anaesthesia was performed with the intra-muscular administration of ketamine/zylazine (87/13 mg kg−1), with maintenance doses of 29/4 mg·kg−1 every 30 min when needed. Anaesthesia was monitored by reflex measurement and breathing rhythm.
Animal care and experimentations were approved by the Ethics Committee on Animal Research of the Université de Sherbrooke, according to the guidelines of the Canadian Council on Animal Care. This manuscript follows the ARRIVE guidelines for reporting in vivo experiments (Kilkenny et al., 2010; McGrath et al., 2010).
Metabolic studies
WT or B2KO mice priorly subjected to 10 weeks of ND or HSD were placed in metabolic cages with free access to water and food for 24 h. Urine samples were collected at the end of experiments and their content in sodium and potassium were analysed by an AVL electrolyte analyser 9140 (Roche Diagnostic, Laval, QC, Canada).
Experimental design in chronic radiotelemetry-implanted mice
Telemetry probe implantation was achieved in accordance to Carlson and Wyss (2000) and Butz and Davisson (2001) and as described in the supplementary methods.
Mice were monitored by radiotelemetry, for a continuous 24 h, once a week, for a total of 18 weeks. In a second series of experiments, WT and B2KO mice under ND or HSD for 12 weeks were treated for 4 days by oral gavage with the ETA antagonist A127722 (5 mg kg−1; Opgenorth et al., 1996) or the ETB antagonist A192621 (30 mg·kg−1; von Geldern et al., 1999; graciously supplied by Abbott Laboratories, Abbott Park, IL, USA) twice a day (0800 and 1800 h). Doses for each antagonist used in the present study were selected in accordance to our previous study in radiotelemetry-instrumented hamsters (Honore et al., 2005). Those mice were continuously monitored for 2 days prior to treatment, during the ET receptor antagonist treatment and for a 4-day washout period.
Reverse transcription and quantitative real-time PCR (RT-PCR)
Tissues (heart, lung, renal cortex and renal medulla) were homogenized in Trizol reagent (Invitrogen, Burlington, ON, Canada) by using a tissue homogenizer (polytron; ultra-turrax T8, IKA, Wilmington, NC, USA). The extraction of total RNA was performed in a phenol/chloroform solution, precipitation in isopropanol and washing in 75% ethanol. The RNA was dried and re-dissolved in diethyl-pyrocarbonate-treated water. A deoxyribonuclease treatment (DNase I, Invitrogen) was made on 1 μg of total RNA. Finally, a reverse transcription was performed using 25 units of avian myeloblastosis virus reverse transcriptase (Roche Diagnostics). The reaction was performed at 42°C for 60 min.
Each PCR reactions contained 2 μL (∼100 ng) cDNA, 300 nM of primers in 25 μL of reactive mixture with 12.5 μL of SYBR Green master mix (Stratagene, La Jolla, CA, USA).
Quantitative PCR for actin, ETA and ETB receptors of endothelin were performed by monitoring in real time the fluorescence increase of SYBR Green using the MX3000P Multiplex Quantitative PCR System (Stratagene). Actin served as internal control (supplemental methods, section on real-time PCR).
Monitoring of immunoreactive ET-1
Mice were anaesthetized and a polyethylene catheter was inserted into the right carotid artery for blood sampling and plasma was obtained by a centrifugation of 1 min at 20 000 g. The mice were then euthanized and the organs (heart, kidney, aorta and lungs) were rapidly removed, weighted and frozen on dry ice. The tissues were homogenized in 1.4 mL of chloroform : methanol (2:1) at 4°C. 350 μL of water was added to the homogenates and a centrifugation at 3000 g for 25 min (at 4°C) was performed. The supernatant of tissue homogenate and the plasma were acidified with 1 volume of trifluoroacetic acid (TFA) 0.2%, before being passed trough an Amprep Octadecyl C18 minicolumn 100 mg (RPN 1900, Amersham Biosciences, Baie D'Urfé, QC, Canada).
The column was beforehand activated with 1 mL of methanol, washed with 4 mL of TFA 0.1%, the sample was applied, the column washed with 9 mL of TFA 0.1% and finally the column was eluted with 1.5 mL of acetonitrile 60%/TFA 0.1%. Eluates were evaporated to dryness in polypropylene tubes using a vacuum concentrator. The desiccated residues were re-dissolved in RIA buffer of the immuno-reactive endothelin (IR-ET) double-antibodies assay kit (RPA 555, Amersham Biosciences; modified from Gratton et al., 1997). Results are expressed in fmol·g−1 of tissue.
Acute monitoring of blood pressure in response to NOS inhibition
Eight-week-old mice on ND were anaesthetized and cannulated in the carotid artery as described earlier. Another catheter was inserted in the jugular vein for i.v. drug administration. After surgery completion, the carotid artery catheter was connected to a Blood Pressure Analyzer 200A (Micro-Med, Tustin, CA, USA) for arterial pressure measurements. A 15 min stabilization period was allowed before i.v. administration of either vehicle or BQ123 (2 mg·kg−1). Another 20 min period was allowed before the i.v. administration of the NOS inhibitor L-NG-nitroarginine methyl ester (L-NAME; 2.5 mg·kg−1). Data were then recorded for 20 min before the mice were euthanized.
Chemicals
A127722 and A192621 were dissolved into 2 M equivalents of sodium hydroxide and the volume was completed with saline 0.9%. BQ123 was dissolved in PBS containing 10% dimethyl sulfoxide (DMSO). L-NAME (Sigma-Aldrich, Oakville, ON, Canada) was dissolved in PBS. Vehicles were tested and found to be inactive in WT and B2KO mice subjected to ND or HSD.
Nomenclature
All drug and molecular target nomenclature are in concordance with the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2011).
Statistical analysis
Data are expressed as mean ± SEM of n experiments. Statistical analysis were performed by unpaired Student's t-test or by an ANOVA test followed by a Tukey post-test whenever applicable. *P < 0.05; **P < 0.01; ***P < 0.001. Blood pressure variations within each group for A127722 treatment were also evaluated with a paired Student's t-test.
Results
Development of hypertension during a HSD in non-anaesthetized B2KO mice
B2KO mice were subjected to either ND or HSD and monitored by radiotelemetry for 18 weeks (Figure 1).
Figure 1.
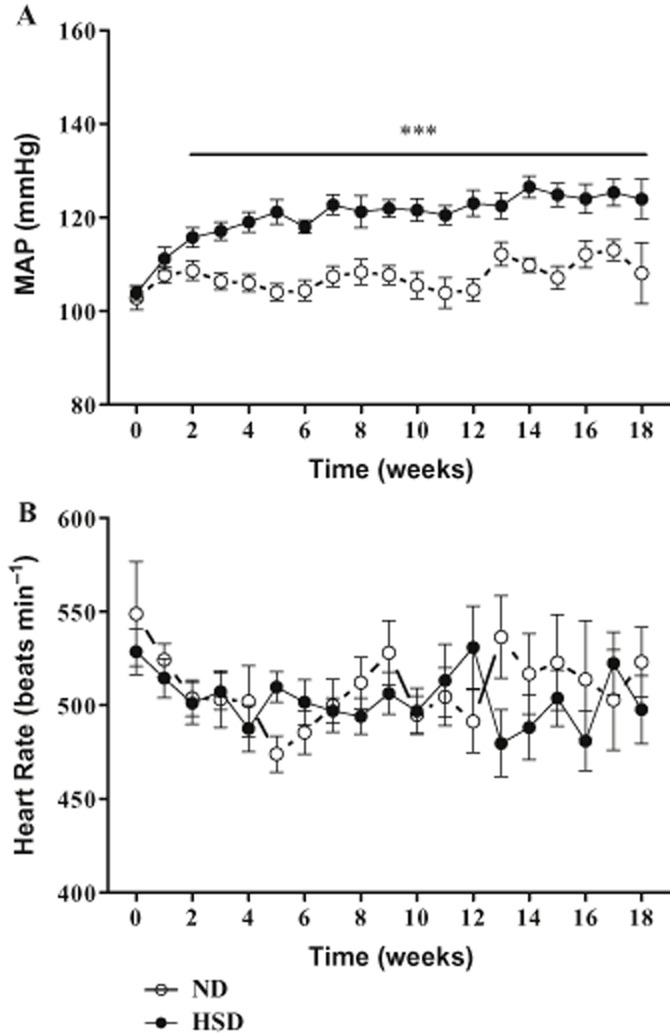
Weekly profile of MAP (A) and heart rate (HR; B) of B2KO mice submitted to ND or HSD. Data were collected by radiotelemetry. Each point represents the mean ± SEM of 7–23 experiments. Seven (ND) and 13 mice (HSD) completed the whole 18-week protocol; the other mice completed 12 weeks and underwent ETA or ETB antagonist treatments. ***P < 0.001.
Figure 1A illustrates that the onset of mean arterial pressure increase in B2KO mice developed after 1 week of HSD and reached a maximal increase of 19.1 ± 2.8 and 20.4 ± 4.3 mmHg after the 12th and 18th week of treatment respectively (Figure 1A and Table 1). On the other hand, B2KO mice on ND maintained a stable mean arterial blood pressure (MAP; week 0: 105.7 ± 2.6; 18th week: 110.6 ± 6.0 mmHg). Table 1 shows a similar pattern of slightly, but non-significantly increased MAP (5–7 mmHg) between the 0- and 18-week period in WT mice on either diet. Furthermore, Figure 1B shows no difference in terms of heart rate between ND and the HSD groups during the entire treatment period.
Table 1.
Blood pressure parameters of mice either on ND or HSD for 18 weeks
| WT (ND) n = 6 | WT (HSD) n = 5 | B2KO (ND) n = 7 | B2KO (HSD) n = 13 | |||||
|---|---|---|---|---|---|---|---|---|
| Week 0 | Week 18 | Week 0 | Week 18 | Week 0 | Week 18 | Week 0 | Week 18 | |
| MAP (mmHg) | 95.6 ± 3.5 | 101.7 ± 0.6 | 103.2 ± 0.6 | 110.7 ± 4.3 | 105.7 ± 2.6 | 110.6 ± 6.0 | 103.6 ± 2.5 | 124.0 ± 4.3*** |
| Systolic (mmHg) | 108.4 ± 3.4 | 115.3 ± 1.5 | 116.8 ± 1.6 | 126.0 ± 6.4 | 116.8 ± 4.4 | 125.4 ± 6.3 | 118.2 ± 4.7 | 141.0 ± 8.1* |
| Diastolic (mmHg) | 80.2 ± 4.3 | 87.5 ± 1.4 | 88.9 ± 2.8 | 95.4 ± 5.2 | 87.5 ± 4.1 | 96.5 ± 6.3 | 88.4 ± 2.9 | 99.8 ± 3.5* |
| Pulse pressure (mmHg) | 27.3 ± 2.4 | 27.8 ± 2.8 | 27.8 ± 4.3 | 30.7 ± 7.8 | 30.4 ± 1.7 | 28.6 ± 4.2 | 33.1 ± 2.9 | 41.3 ± 5.3 |
| Heart rate (beats·min−1) | 536.19 ± 12.0 | 555.1 ± 19 | 591.0 ± 9.3 | 483.0 ± 11.4* | 531.1 ± 21.0 | 525.1 ± 19.8 | 510.0 ± 32.3 | 490.9 ± 29.2 |
Data expressed as mean ± SEM. Significant changes between week 0 and week 18 of treatment: *P ≤ 0.05; ***P ≤ 0.001.
Renal excretory functions impaired in B2KO mice
To confirm the contribution of B2 receptor in the maintenance of electrolyte homeostasis, sodium and potassium were measured in the urine of WT and B2KO mice submitted to the different diets. Supporting Information Table S1 shows that, under ND, B2KO mice excrete significantly less sodium and potassium than WT mice. Under HSD, B2KO mice show no difference in sodium excretion compared with WT while potassium excretion remains significantly lower in the KO congeners.
ET receptors expression is not modulated at the transcriptional level by HSD in B2KO mice
The mRNA levels for the ETA and ETB receptors were analysed by RT-PCR. Supporting Information Figure S3 shows that none of the mRNAs levels mentioned earlier were modified in lungs or heart homogenates derived from WT or B2KO mice subjected to 18 weeks of HSD when compared with animals under ND. Earlier experiments using the conventional RT-PCR approach did not show any enhancement of cardiac and pulmonary- pre–pro-ET-1, angiotensin receptor type-1a (AT1a) or angiotensinogen mRNA levels in B2KO mice subjected to a HSD (results not shown).
Interestingly, ETB receptor mRNA levels are expressed in significantly higher levels (P < 0.05) than the ETA in both the renal cortex and medulla of WT or B2KO mice, with no significant alterations however for the former receptor type in both strains subjected to HSD (Supporting Information Figure S2).
As a final control, albeit an up-regulation of the B1 receptor mRNA was observed in the heart as well as in the renal medulla and cortex obtained from B2KO mice when compared with WT congeners, HSD did not influence the expression of either receptor in all the other organs studied (Supporting Information Figure S4).
HSD increases ET-1 pulmonary concentration in B2KO mice
In another series of experiments, tissue and plasma immunoreactive ET-1 concentrations were measured by RIA. Overall tissue and plasma levels of immunoreactive ET-1 were significantly lower in B2KO mice when compared with the same tissues derived from WT congeners with the notable exception of the lungs (Figure 2C). Basal tissue levels of immunoreactive ET-1 were found to be 10-fold higher in the lungs (Figure 2C) than in the heart or kidneys of both WT mice and B2KO mice (Figure 2A). Worthy of notice is that the levels of the peptide were significantly increased by 45% only in pulmonary tissues of B2KO but not in the WT mice tissue when subjected to HSD. Finally albeit no significant variations in cardiac, renal or plasma ET-1 levels were found in WT or B2KO mice subjected to HSD (Figure 2), the same regimen triggered a marked increase in aortic ET-1 levels in WT but not in B2KO mice.
Figure 2.
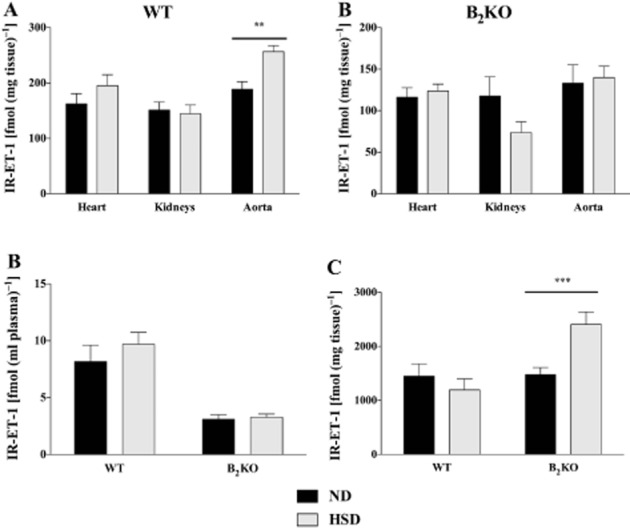
Immunoreactive levels of ET(IR-ET-1) [fmol·(g of tissue)-1] in the heart, kidney and aorta (A), plasma (B) [fmol·(ml of plasma)-1] or the lungs (C) [fmol·(g of tissue)-1] of WT and B2KO mice submitted to 18 weeks of ND or HSD. **P < 0.01, ***P < 0.001 (n = 8–10).
Effect of ETA or ETB receptor blockade on blood pressure and heart rate in the hypertensive condition of B2KO mice on HSD
WT or B2KO mice under ND or HSD were treated twice daily with the selective ETA or ETB antagonist administered orally, A127722 (5 mg·kg−1; Figures 3 and 4) or A192621 (30 mg·kg−1; Figure 5) respectively. Interestingly, after 2 days of treatment, A127722 significantly decreased MAP in all groups (reduction of 9–11 mmHg); however, with a more pronounced hypotensive response in B2KO mice on HSD (reduction of 18 mmHg; P < 0.01 when compared with each of the other groups). On the fourth day of treatment with A127722, systolic, diastolic and MAP of B2KO mice under HSD were reduced back to baseline levels found in WT or B2KO mice subjected to ND (Figures 3 and 4). Washout of the ETA antagonist promoted the return to the elevation of blood pressure in the HSD mice whereas the same ETA antagonist did not modify the heart rate in WT or B2KO mice under ND or HSD (Figure 3 and 4).
Figure 3.
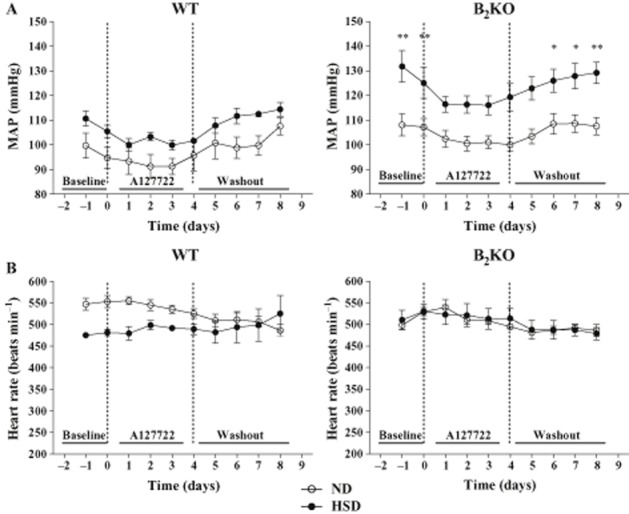
Effects of chronic A127722 treatment (5 mg·kg−1, per os) administered twice a day for 4 days, on MAP (A) and heart rate (HR; B) of WT and B2KO mice submitted to ND or high salt diet (HSD). Left and right dotted lines represent the starting point with the ET antagonist and the initiation of washout period, respectively. Each point represents the mean ± SEM of three to eight experiments. *P < 0.05, **P < 0.01 of HSD versus Day 0 of ND.
Figure 4.
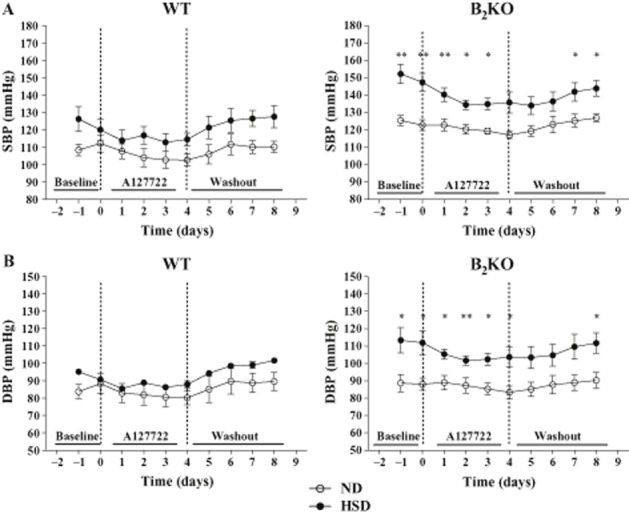
Effects of chronic A127722 treatment (5 mg·kg−1, per os) administered twice a day for 4 days, on SBP (A) and DBP (B). Left and right dotted lines represent the starting point with the ET antagonist and the initiation of washout period respectively. Each point represents the mean ± SEM of 3–8 experiments. *P < 0.05, **P < 0.01 of HSD versus Day 0 of ND.
Figure 5.
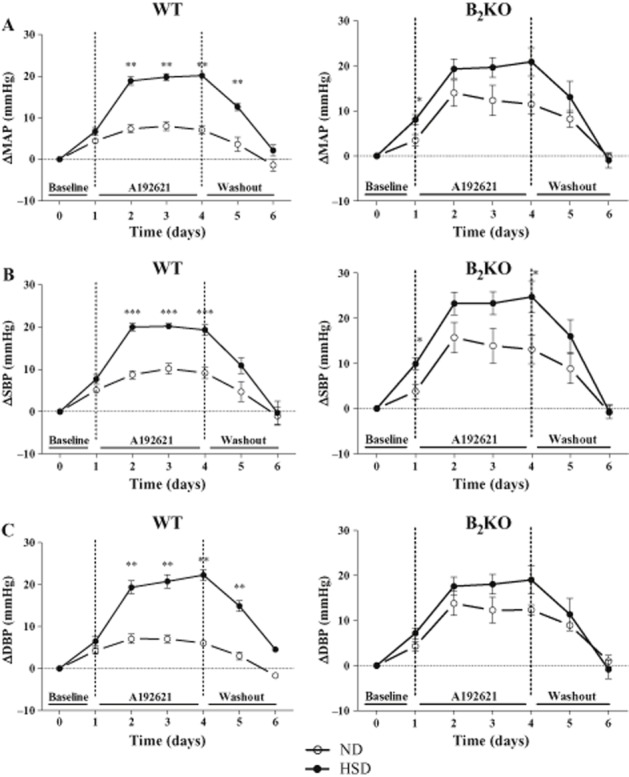
Effect of chronic A192621 treatment (30 mg·kg−1, per os) administered twice a day for 4 days (Days 1–4), on the maximal delta MAP (A), the maximal delta systolic blood pressure (SBP; B) and the maximal delta diastolic blood pressure (DBP; C) of WT and B2KO mice submitted to normal (ND) or high salt diet (HSD). Day 0 corresponds to the baseline and days 5–6 to the washout period. Each point represents the mean ± SEM of at least three different experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Finally, a 4-day treatment with the ETB antagonist A192621 was associated with a significant increase in MAP, systolic blood pressure (SBP) and diastolic blood pressure (DBP) after the second day of administration in WT mice under ND and HSD, albeit to a lesser extent in the former salt diet (Figure 5). Meanwhile, A192621 increased MAP and DBP similarly in B2KO mice receiving the ND or HSD (Figure 5A and C).
An ETA receptor antagonist reduces the blood pressure response to a NO synthase inhibitor in B2KO mice, but not in WT mice
In a final series of experiments, WT or B2KO mice, aged 8 weeks under ND, were pretreated with vehicle or the ETA selective antagonist BQ123 (2 mg·kg−1) before the administration of the non-selective NOS inhibitor L-NAME (2.5 mg·kg−1; Figure 6). The BQ123 treatment failed to reduce the blood pressure increase in response to L-NAME in WT mice, but reduced the response to L-NAME by close to 40% in B2KO mice.
Figure 6.
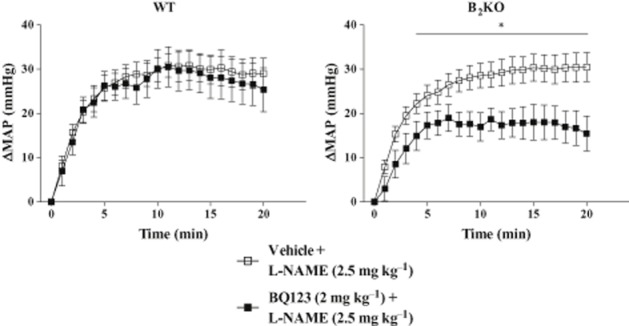
Effect of pretreatment with BQ123 (2 mg·kg−1, i.v.) on the blood pressure response to L-NAME (2.5 mg·kg−1, i.v.) over 20 min in WT and B2KO mice on ND. Each point represents the mean ± SEM of five to six experiments. *P < 0.05.
Discussion
We show in the present study, by radiotelemetry, that HSD induces an increase in systolic, diastolic and MAP in B2KO, but not in WT mice, thus confirming the initial observations of Madeddu et al. (1997) where blood pressure was measured by tail plethysmography in the same murine strains. Cervenka et al. (1999) and Carlson and Wyss (2000) have previously reported, by tail cuff and radiotelemetry respectively, that long term HSD does not increase MAP in WT mice thus confirming the present data in these animals.
Interestingly in the present study, the selective ETA antagonist A127722 was able to reduce the hypertensive state induced by 12 consecutive weeks of HSD in B2KO mice to MAP levels found in the same strain subjected to normal salt diet. The dynamic role of the ET system was further demonstrated following washout of A127722 as haemodynamic parameters reverted to a hypertensive state 2 days following withdrawal of the antagonist. In contrast, a selective ETB antagonist, A192621 (von Geldern et al., 1999), was associated with a marked increase in blood pressure highlighting the physiological antagonism afforded by the ETB receptor type in the systemic circulation as previously reported (Berthiaume et al., 2000). Notably, the hypertensive properties of A192621 are further enhanced in B2KO mice subjected to the normal salt diet.
We had previously reported that the lack of B2 receptors abolished the production of endothelial-derived NO afforded by exogenous BK in isolated mesenteric circuits of the mouse (Berthiaume et al., 1997). Schanstra et al. (2003) have also shown much lower excreted nitrites levels in B2KO mice than their WT congeners. It is also well established that stimulated or basal NO levels are both negative modulators of the ET-1 synthesis and release (Boulanger and Glick, 2000; Gratton et al., 2000). The studies mentioned earlier thus support the concept that the high salt-induced hypertensive condition is linked to a reduced capacity of the B2KO mice to activate BK-induced endogenous NO-dependent inhibition of the ET-1 pathway. Because HSD did not influence mRNA levels for either the ETA nor the ETB receptor in all organs examined in B2KO mice, we suggest that this enhanced hypertensive response to A192621 is caused by a loss of NO-dependent negative regulation of vascular tone (Sanders, 2009b), albeit this remains to be validated in our experimental model.
Our data also show that tissue and plasma ET-1 levels measured were similar to previous reports with the largest concentrations of the peptide localized in the lungs (Okumura et al., 1993; 1995; Ashizawa et al., 1994; Carr et al., 1998). This state of events is suggested to be due to the high-density location of endothelial cells involved in ET-1 clearance in this particular organ when compared with the heart or the kidney for example (D'Orleans-Juste et al., 2002). It was thus interesting that a 45% increase in pulmonary content of ET-1 occurs in B2KO mice subjected to HSD when compared with same animal strain treated with a normal salt diet. Such increases in pulmonary ET-1 levels were not observed in WT congeners under ND or HSD. Therefore, we suggest that the repression of B2 receptor functions may be partially responsible for the increased ET-1 level observed in the pulmonary tissue. Importantly, mRNA levels of prepro-ET-1 in several organs including the lungs were not modified by the HSD in B2KO mice (results not shown) suggesting that transcriptional mechanisms are not implicated in the increased pulmonary ET-1 levels found in B2KO mice subjected to HSD. It is also noticeable that immunoreactive ET-1 was not elevated in the aortae of B2KO mice under HSD. Thus, albeit tissue ET-1 content in resistance vessels were not monitored in the present study, we suggest that the increase in pulmonary content of the same peptide plays no role in the A127722-sensitive hypertensive state reported here in B2KO mice under HSD.
Finally, considering that antagonism of ETA receptors was efficient in reversing the effect of HSD in B2KO mice and that ETB antagonism was more efficacious as a pressor agent in WT than in B2KO mice, our results supports the postulate that arterial hypertension found in these B2KO mice is due to ET-1 overproduction as well as reduced clearance of the same peptide in organs such as the lungs (Sirvio et al., 1990), because no high salt-induced alterations in ETB receptor mRNA nor significant increases in tissue or plasma ET-1 levels were found in B2KO mice. Importantly, angiotensinogen and AT1 receptor mRNA levels are not enhanced by HSD in B2KO mice (Cervenka et al., 1999). In addition, as shown in the present study, a second ETA antagonist, BQ123 (Ihara et al., 1992) was able to significantly reduce the hypertensive response afforded by a NO-synthase inhibitor, L-NAME (Rees et al., 1990), in B2KO, but not in WT congeners.
Based on the experimental evidences mentioned earlier as well as on the concept of ET-1 interacting with the (TLR2/CXCR2/B2) complex in Chagas's disease put forward by Andrade et al. (2012), we suggest that BK (via B2 receptors) and ET-1 (via ETB receptors) may cooperate as well in releasing NO and prostacyclin (Labonte et al., 2001). We suggest that this BK/ET-1 cooperation is hampered in B2KO mice, thus explaining why the ETB antagonist is less efficient as a pressor agent in B2KO mice than in WT counterparts. We further suggest that ET-1 then binds to the underlying vascular smooth muscle cells and activates vasoconstrictor ETA receptors unopposed by kinin-dependent NO and PGI2 release.
We also confirm in the present study an impairment of sodium and potassium excretion in B2KO mice as previously reported by Alfie et al. (1996). However, under HSD conditions used in our study, potassium, but not sodium excretion remained significantly lower than in WT mice subjected to the same regime. Interestingly, Katori and Majima (2006) reported that high potassium intake reduces the sensitivity of blood pressure to HSD. This state of event would suggest a compensatory reduction of potassium excretion in B2KO mice under HSD. Some study has already been performed in that regard with clinical primary aldosteronism (PA), in which a functional polymorphism of the B2 promoter was associated with increased blood pressure in PA patients (Mulatero et al., 2002). However, no differences were accounted for between B2 genotypes in plasma K+ levels in those PA patients (Mulatero et al., 2002).
Duka et al. (2001) and Tan et al. (2007), on the other hand, previously reported a compensatory role from an up-regulated B1 renal receptor population in B2KO mice; a phenomenon significantly enhanced in one kidney clipped or in bi-nephrectomized mice given a HSD (Duka et al., 2001). Kakoki et al. (2007) have also shown that mRNA levels of preproET-1 in kidney homogenates between WT and B2KO mice are not significantly different unless the animals are prior subjected to ischemic insults. In accordance with these data, HSD did not trigger a significant increase in B1 receptor expression in all organs derived from WT or B2KO mice in our hands, thus suggesting the lack of contribution of up-regulated B1 receptors, in HSD-induced hypertension in our KO model.
Finally, it is noteworthy that high densities of both ETB receptors and B1 receptor mRNAs were found in the renal medulla when compared with the cortex, the later mRNA being further enhanced by repression of the B2 receptor. Albeit endogenous ET-1 and both ETA and ETB receptors have been shown to be fundamental in the regulation of sodium and water excretion (Kohan, 2011; Nakano and Pollock, 2012) and in the control of vascular tone (Amiri et al., 2010) and that ETB receptors in the kidney may be protective against high salt-induced hypertension (Gariepy et al., 2000), less is known on the role of BK in these physiological mechanisms. BK and ET-1 are probably two factors among many that are involved in the fine-tuning of Na+ and water metabolism in the distal nephron (Kohan, 2011; Zaika et al., 2011; Mamenko et al., 2012). Both BK (Zaika et al., 2011) and ET-1 (Kohan, 2011) can inhibit epithelial sodium channel (ENaC). In a salt-loading model, we suggest that both peptides play a protective role by increasing Na+ excretion, predominantly through B2 and ETB receptors. In our 4-day A192621 experiment, impaired B2-dependent ENaC inhibition could well be one mechanism of increased blood pressure on top of the blockade of ETB-dependent endothelium derived relaxing factor release. In fact, collecting-duct specific ETB receptor deletion seems to mimic the blood pressure variations we see in our B2KO mouse model (Ge et al., 2006).
Our study did not show any changes in ETB mRNA, either between WT and B2KO mice or between ND and HSD. However, WT mice on HSD were significantly more sensitive to ETB antagonism than B2KO mice. This suggests a possible change in ETB protein levels. An alternate explanation would be an increase in ET-1 levels in the distal nephron caused by HSD. We did not investigate this aspect in the present study however. It is possible that any alterations specific to the collecting duct-derived ET-1 levels may be masked by the whole organ measurements performed in the present study.
Another possible interaction between BK and ET-1 is through NO production, which by itself can inhibit ENaC and other sodium reabsorption mechanisms (Helms et al., 2005). Considering that B2 and ETB receptors prompt NO synthesis upon activation, it remains to be determined whether NO formation is required for BK and/or ET-1 inhibition of ENaC.
In conclusion, high salt-induced hypertension afforded by non-anaesthetized mice genetically repressed for the B2 receptor can be reversed by an ETA selective antagonist. The present study also provides evidences towards a protective role for ETB receptors in this particular animal model of hypertension. Whether the same paradigm can be translated to ACEI resistant-hypertensive patients in which functional B2 receptor polymorphism has been identified remains to be investigated.
Acknowledgments
The authors gratefully acknowledge Mrs Angèle Tremblay and Cécile Monpays for technical assistance, as well as Abbott Laboratories (Abbott Park, IL, USA) for graciously providing the antagonists A127722 and A192621. This project is financially supported by the Canadian Institutes for Health Research as well as by the Etienne Lebel Clinical Reseach Center of the Centre Hospitalier Universitaire de Sherbrooke.
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Weekly profile of the systolic blood pressure (SBP) of B2KO mice subjected to normal (ND) or high salt diet (HSD). Data were collected by radiotelemetry. Each point represents the mean ± SEM of seven to eight experiments. ***P < 0.001.
Figure S2 Monitoring of the relative mRNA expression of ETA and ETB receptors by real-time PCR in the kidney of WT and B2KO mice submitted to both diets. The values are normalized against actin and expressed as 2–ΔΔCt. Each value represents mean ± SEM of five to eight experiments.
Figure S3 Monitoring of the relative mRNA expression of ETA and ETB receptors by real-time PCR in the lung (A) and the heart (B) of WT and B2KO mice submitted to both diets. The values are normalized against actin and expressed as 2–ΔΔCt. Each value represents mean ± SEM of five to eight experiments.
Figure S4 Monitoring of the relative mRNA expression of B1 receptors by real-time PCR in the lung (A), the heart (B) and the kidney of WT and B2KO mice submitted to both diets. The values are normalized against actin and expressed as 2–ΔΔCt. Each value represents mean ± SEM of four to five experiments.
Figure S5 Maximal increase of mean arterial pressure (MAP) in anaesthetized WT and B2KO mice in response to ET-1 (0.1 mg·kg−1, i.v.), following pretreatment with either vehicle (4% DMSO in PBS) or BQ123 (2 mg·kg−1, i.v.). n = 4–6. **P < 0.01, ***P < 0.001 versus vehicle.
Table S1 Urinary volume, urinary excretion and plasmatic concentration of Na+ and K+ from mice either on ND or HSD for 18 weeks.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfie ME, Yang XP, Hess F, Carretero OA. Salt-sensitive hypertension in bradykinin B2 receptor knockout mice. Biochem Biophys Res Commun. 1996;224:625–630. doi: 10.1006/bbrc.1996.1076. [DOI] [PubMed] [Google Scholar]
- Amiri F, Ko EA, Javeshghani D, Reudelhuber TL, Schiffrin EL. Deleterious combined effects of salt-loading and endothelial cell restricted endothelin-1 overexpression on blood pressure and vascular function in mice. J Hypertens. 2010;28:1243–1251. doi: 10.1097/HJH.0b013e328338bb8b. [DOI] [PubMed] [Google Scholar]
- Andrade D, Serra R, Svensjo E, Lima AP, Ramos ES, Jr, Fortes FS, et al. Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: a converging pathway leading to chagasic vasculopathy. Br J Pharmacol. 2012;165:1333–1347. doi: 10.1111/j.1476-5381.2011.01609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashizawa N, Okumura H, Matsuura A, Kobayashi F. Diltiazem facilitates endothelin clearance from the blood stream to reduce toxic elevation of plasma endothelin level in rodents. J Pharm Pharmacol. 1994;46:200–204. doi: 10.1111/j.2042-7158.1994.tb03778.x. [DOI] [PubMed] [Google Scholar]
- Berthiaume N, Hess F, Chen A, Regoli D, D'Orleans-Juste P. Pharmacology of kinins in the arterial and venous mesenteric bed of normal and B2 knockout transgenic mice. Eur J Pharmacol. 1997;333:55–61. doi: 10.1016/s0014-2999(97)01096-0. [DOI] [PubMed] [Google Scholar]
- Berthiaume N, Yanagisawa M, Labonte J, D'Orleans-Juste P. Heterozygous knock-out of ETB receptors induces BQ-123-sensitive hypertension in the mouse. Hypertension. 2000;36:1002–1007. doi: 10.1161/01.hyp.36.6.1002. [DOI] [PubMed] [Google Scholar]
- Bolli MH, Boss C, Binkert C, Buchmann S, Bur D, Hess P, et al. The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N'-p ropylsulfamide (Macitentan), an orally active, potent dual endothelin receptor antagonist. J Med Chem. 2012;55:7849–7861. doi: 10.1021/jm3009103. [DOI] [PubMed] [Google Scholar]
- Borkowski JA, Hess JF. Targeted disruption of the mouse B2 bradykinin receptor in embryonic stem cells. Can J Physiol Pharmacol. 1995;73:773–779. doi: 10.1139/y95-104. [DOI] [PubMed] [Google Scholar]
- Boulanger SC, Glick PL. Positive end expiratory pressure and response to inhaled nitric oxide therapy. Surgery. 2000;128:871–872. doi: 10.1067/msy.2000.110854. [DOI] [PubMed] [Google Scholar]
- Boyce S, Rupniak NM, Carlson EJ, Webb J, Borkowski JA, Hess JF, et al. Nociception and inflammatory hyperalgesia in B2 bradykinin receptor knockout mice. Immunopharmacology. 1996;33:333–335. doi: 10.1016/0162-3109(96)00101-4. [DOI] [PubMed] [Google Scholar]
- Butz GM, Davisson RL. Long-term telemetric measurement of cardiovascular parameters in awake mice: a physiological genomics tool. Physiol Genomics. 2001;5:89–97. doi: 10.1152/physiolgenomics.2001.5.2.89. [DOI] [PubMed] [Google Scholar]
- Carlson SH, Wyss JM. Long-term telemetric recording of arterial pressure and heart rate in mice fed basal and high NaCl diets. Hypertension. 2000;35:E1–E5. doi: 10.1161/01.hyp.35.2.e1. [DOI] [PubMed] [Google Scholar]
- Carr MJ, Spalding LJ, Goldie RG, Henry PJ. Distribution of immunoreactive endothelin in the lungs of mice during respiratory viral infection. Eur Respir J. 1998;11:79–85. doi: 10.1183/09031936.98.11010079. [DOI] [PubMed] [Google Scholar]
- Cervenka L, Harrison-Bernard LM, Dipp S, Primrose G, Imig JD, El-Dahr SS. Early onset salt-sensitive hypertension in bradykinin B(2) receptor null mice. Hypertension. 1999;34:176–180. doi: 10.1161/01.hyp.34.2.176. [DOI] [PubMed] [Google Scholar]
- Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119–1123. doi: 10.1016/S0140-6736(01)06250-X. [DOI] [PubMed] [Google Scholar]
- Cook NR, Cutler JA, Obarzanek E, Buring JE, Rexrode KM, Kumanyika SK, et al. Long term effects of dietary sodium reduction on cardiovascular disease outcomes: observational follow-up of the trials of hypertension prevention (TOHP) BMJ. 2007;334:885–888. doi: 10.1136/bmj.39147.604896.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Orleans-Juste P, Labonte J, Bkaily G, Choufani S, Plante M, Honore JC. Function of the endothelin(B) receptor in cardiovascular physiology and pathophysiology. Pharmacol Ther. 2002;95:221–238. doi: 10.1016/s0163-7258(02)00235-8. [DOI] [PubMed] [Google Scholar]
- Dhaun N, Pollock DM, Goddard J, Webb DJ. Selective and mixed endothelin receptor antagonism in cardiovascular disease. Trends Pharmacol Sci. 2007;28:573–579. doi: 10.1016/j.tips.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Duka I, Kintsurashvili E, Gavras I, Johns C, Bresnahan M, Gavras H. Vasoactive potential of the b(1) bradykinin receptor in normotension and hypertension. Circ Res. 2001;88:275–281. doi: 10.1161/01.res.88.3.275. [DOI] [PubMed] [Google Scholar]
- Elijovich F, Laffer CL, Amador E, Gavras H, Bresnahan MR, Schiffrin EL. Regulation of plasma endothelin by salt in salt-sensitive hypertension. Circulation. 2001;103:263–268. doi: 10.1161/01.cir.103.2.263. [DOI] [PubMed] [Google Scholar]
- Ergul A. Hypertension in black patients: an emerging role of the endothelin system in salt-sensitive hypertension. Hypertension. 2000;36:62–67. doi: 10.1161/01.hyp.36.1.62. [DOI] [PubMed] [Google Scholar]
- Ergul S, Ergul A, Hudson JA, Puett D, Wieman BM, Durham MD, et al. The effect of regulation of high blood pressure on plasma endothelin-1 levels in blacks with hypertension. Am J Hypertens. 1998;11(11 Pt 1):1381–1385. doi: 10.1016/s0895-7061(98)00150-2. [DOI] [PubMed] [Google Scholar]
- Gainer JV, Brown NJ, Bachvarova M, Bastien L, Maltais I, Marceau F, et al. Altered frequency of a promoter polymorphism of the kinin B2 receptor gene in hypertensive African-Americans. Am J Hypertens. 2000;13:1268–1273. doi: 10.1016/s0895-7061(00)01215-2. [DOI] [PubMed] [Google Scholar]
- Gariepy CE, Ohuchi T, Williams SC, Richardson JA, Yanagisawa M. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. J Clin Invest. 2000;105:925–933. doi: 10.1172/JCI8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Bagnall A, Stricklett PK, Strait K, Webb DJ, Kotelevtsev Y, et al. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. Am J Physiol Renal Physiol. 2006;291:F1274–F1280. doi: 10.1152/ajprenal.00190.2006. [DOI] [PubMed] [Google Scholar]
- von Geldern TW, Tasker AS, Sorensen BK, Winn M, Szczepankiewicz BG, Dixon DB, et al. Pyrrolidine-3-carboxylic acids as endothelin antagonists. 4. Side chain conformational restriction leads to ET(B) selectivity. J Med Chem. 1999;42:3668–3678. doi: 10.1021/jm990170q. [DOI] [PubMed] [Google Scholar]
- Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–1739. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- Gratton J, Rae G, Bkaily G, D'Orléans-Juste P. ET(B) receptor blockade potentiates the pressor response to big endothelin-1 but not big endothelin-2 in the anesthetized rabbit. Hypertension. 2000;35:726–731. doi: 10.1161/01.hyp.35.3.726. [DOI] [PubMed] [Google Scholar]
- Gratton JP, Cournoyer G, Loffler BM, Sirois P, D'Orleans-Juste P. ET(B) receptor and nitric oxide synthase blockade induce BQ-123-sensitive pressor effects in the rabbit. Hypertension. 1997;30:1204–1209. doi: 10.1161/01.hyp.30.5.1204. [DOI] [PubMed] [Google Scholar]
- Grim CE, Robinson M. Blood pressure variation in blacks: genetic factors. Semin Nephrol. 1996;16:83–93. [PubMed] [Google Scholar]
- Helms MN, Yu L, Malik B, Kleinhenz DJ, Hart CM, Eaton DC. Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. Am J Physiol Cell Physiol. 2005;289:C717–C726. doi: 10.1152/ajpcell.00006.2005. [DOI] [PubMed] [Google Scholar]
- Honore JC, Fecteau MH, Brochu I, Labonte J, Bkaily G, D'Orleans-Juste P. Concomitant antagonism of endothelial and vascular smooth muscle cell ETB receptors for endothelin induces hypertension in the hamster. Am J Physiol Heart Circ Physiol. 2005;289:H1258–H1264. doi: 10.1152/ajpheart.00352.2005. [DOI] [PubMed] [Google Scholar]
- Ihara M, Noguchi K, Saeki T, Fukuroda T, Tsuchida S, Kimura S, et al. Biological profiles of highly potent novel endothelin antagonists selective for the ETA receptor. Life Sci. 1992;50:247–255. doi: 10.1016/0024-3205(92)90331-i. [DOI] [PubMed] [Google Scholar]
- Kakoki M, McGarrah RW, Kim HS, Smithies O. Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc Natl Acad Sci U S A. 2007;104:7576–7581. doi: 10.1073/pnas.0701617104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katori M, Majima M. A missing link between a high salt intake and blood pressure increase. J Pharmacol Sci. 2006;100:370–390. doi: 10.1254/jphs.crj06003x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG Group NCRRGW. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE. Endothelin and collecting duct sodium and water transport. Contrib Nephrol. 2011;172:94–106. doi: 10.1159/000328687. [DOI] [PubMed] [Google Scholar]
- Labonte J, Brochu I, Honore JC, D'Orleans-Juste P. Role of ETB and B2 receptors in the ex vivo platelet inhibitory properties of endothelin and bradykinin in the mouse. Br J Pharmacol. 2001;132:934–940. doi: 10.1038/sj.bjp.0703880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiola RA, Reis FC, Kawamoto EM, Scavone C, Abdalla DS, Fernandes L, et al. Role of vascular Kinin B1 and B2 receptors in endothelial nitric oxide metabolism. Peptides. 2011;32:1700–1705. doi: 10.1016/j.peptides.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Madeddu P, Varoni MV, Palomba D, Emanueli C, Demontis MP, Glorioso N, et al. Cardiovascular phenotype of a mouse strain with disruption of bradykinin B2-receptor gene. Circulation. 1997;96:3570–3578. doi: 10.1161/01.cir.96.10.3570. [DOI] [PubMed] [Google Scholar]
- Mamenko M, Zaika O, Doris PA, Pochynyuk O. Salt-dependent inhibition of epithelial Na+ channel-mediated sodium reabsorption in the aldosterone-sensitive distal nephron by bradykinin. Hypertension. 2012;60:1234–1241. doi: 10.1161/HYPERTENSIONAHA.112.200469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matucci-Cerinic M, Denton CP, Furst DE, Mayes MD, Hsu VM, Carpentier P, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2011;70:32–38. doi: 10.1136/ard.2010.130658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulatero P, Williams TA, Milan A, Paglieri C, Rabbia F, Fallo F, et al. Blood pressure in patients with primary aldosteronism is influenced by bradykinin B(2) receptor and alpha-adducin gene polymorphisms. J Clin Endocrinol Metab. 2002;87:3337–3343. doi: 10.1210/jcem.87.7.8666. [DOI] [PubMed] [Google Scholar]
- Nakano D, Pollock D. New concepts in endothelin control of sodium balance. Clin Exp Pharmacol Physiol. 2012;39:104–110. doi: 10.1111/j.1440-1681.2011.05517.x. [DOI] [PubMed] [Google Scholar]
- Okumura H, Ashizawa N, Kobayashi F, Arai K, Asakura R, Ashikawa N, et al. Comparison of haemoconcentration induced by big endothelin-1 and endothelin-1 in mice. Br J Pharmacol. 1993;110:1395–1400. doi: 10.1111/j.1476-5381.1993.tb13975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura H, Ashizawa N, Asakura R, Aotsuka T, Kobayashi F, Matsuura A. Possible involvement of different mechanisms in sudden death induced by endothelin-1 and big endothelin-1. Biol Pharm Bull. 1995;18:18–23. doi: 10.1248/bpb.18.18. [DOI] [PubMed] [Google Scholar]
- Opgenorth TJ, Adler AL, Calzadilla SV, Chiou WJ, Dayton BD, Dixon DB, et al. Pharmacological characterization of A-127722: an orally active and highly potent ETA-selective receptor antagonist. J Pharmacol Exp Ther. 1996;276:473–481. [PubMed] [Google Scholar]
- Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadfam R, Teixeira C, Bkaily G, Sirois P, de Brum-Fernandes A, D'Orleans-Juste P. Contribution of B(2) receptors for bradykinin in arthus reaction-induced plasma extravasation in wild-type or B(2) transgenic knockout mice. Br J Pharmacol. 2000;129:1732–1738. doi: 10.1038/sj.bjp.0703225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders PW. Dietary salt intake, salt sensitivity, and cardiovascular health. Hypertension. 2009a;53:442–445. doi: 10.1161/HYPERTENSIONAHA.108.120303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders PW. Vascular consequences of dietary salt intake. Am J Physiol Renal Physiol. 2009b;297:F237–F243. doi: 10.1152/ajprenal.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanstra JP, Duchene J, Praddaude F, Bruneval P, Tack I, Chevalier J, et al. Decreased renal NO excretion and reduced glomerular tuft area in mice lacking the bradykinin B2 receptor. Am J Physiol Heart Circ Physiol. 2003;284:H1904–H1908. doi: 10.1152/ajpheart.01150.2002. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Bowery BJ, Heavens R, Brown N, Ford H, Sirinathsinghi DJ, et al. Expression of B1 and B2 bradykinin receptor mRNA and their functional roles in sympathetic ganglia and sensory dorsal root ganglia neurones from wild-type and B2 receptor knockout mice. Neuropharmacology. 1997;36:1009–1017. doi: 10.1016/s0028-3908(97)00065-8. [DOI] [PubMed] [Google Scholar]
- Sirvio ML, Metsarinne K, Saijonmaa O, Fyhrquist F. Tissue distribution and half-life of 125I-endothelin in the rat: importance of pulmonary clearance. Biochem Biophys Res Commun. 1990;167:1191–1195. doi: 10.1016/0006-291x(90)90649-8. [DOI] [PubMed] [Google Scholar]
- Takeda M, Saito K, Tsutsui T, Mizusawa T, Obara K, Tomita Y, et al. Plasma endothelin-1 level in patients with renovascular hypertension – does the kidney with stenosis of the renal artery upregulate production of endthelin-1? Eur J Med Res. 1997;2:315–320. [PubMed] [Google Scholar]
- Tan Y, Keum JS, Wang B, McHenry MB, Lipsitz SR, Jaffa AA. Targeted deletion of B2-kinin receptors protects against the development of diabetic nephropathy. Am J Physiol Renal Physiol. 2007;293:F1026–F1035. doi: 10.1152/ajprenal.00203.2007. [DOI] [PubMed] [Google Scholar]
- Weber MA, Black H, Bakris G, Krum H, Linas S, Weiss R, et al. A selective endothelin-receptor antagonist to reduce blood pressure in patients with treatment-resistant hypertension: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1423–1431. doi: 10.1016/S0140-6736(09)61500-2. [DOI] [PubMed] [Google Scholar]
- Weir MR, Gray JM, Paster R, Saunders E. Differing mechanisms of action of angiotensin-converting enzyme inhibition in black and white hypertensive patients. The Trandolapril Multicenter Study Group. Hypertension. 1995;26:124–130. doi: 10.1161/01.hyp.26.1.124. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- Zaika O, Mamenko M, O'Neil RG, Pochynyuk O. Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. Am J Physiol Renal Physiol. 2011;300:F1105–F1115. doi: 10.1152/ajprenal.00606.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.