

Abstract
BACKGROUND AND PURPOSE
Nitric oxide (NO) is known to activate NO-sensitive guanylyl cyclase (NO-GC) and to elicit cGMP production. However, NO has also been proposed to induce cGMP-independent effects. It is accepted practice to use specific NO-GC inhibitors, such as ODQ or NS2028, to assess cGMP-dependent NO effects. Consequently, NO-induced reactions seen in the presence of these inhibitors commonly serve as an affirmation of cGMP independence.
EXPERIMENTAL APPROACH
We evaluated the use of ODQ to discriminate between cGMP-dependent and cGMP-independent NO effects. NO-GC-expressing HEK cells, platelets and tissues from wild type (WT) and NO-GC-deficient mice (GCKO) were used.
KEY RESULTS
NO donors led to accumulation of cGMP in platelets and GC-expressing HEK cells and induced phosphorylation of the vasodilator-stimulated phosphoprotein in platelets; both effects were reduced by ODQ. High concentrations of NO donors, however, overrode the inhibitory effect of ODQ. Correspondingly, ODQ inhibited but did not fully eliminate NO-induced relaxation of aorta and fundus from WT mice. Relaxation induced by endogenously released NO was fully or partially inhibited by ODQ in fundus and aorta, respectively. In aorta and fundus of GCKO mice NO-induced relaxation was absent and served as standard for complete NO-GC inhibition.
CONCLUSIONS AND IMPLICATIONS
High NO concentrations can overcome the inhibitory effect of ODQ on NO-GC. Smooth muscle relaxation induced by NO donors/endogenously released NO in the presence of ODQ in WT was absent in GCKO animals indicating involvement of NO-GC. Accordingly, NO-induced effects in the presence of ODQ do not necessarily prove cGMP independence.
Keywords: ODQ, NO-sensitive guanylyl cyclase, knockout mice, cGMP, cGMP-independent effects, platelet, VASP, nitric oxide, signal transduction
Introduction
The nitric oxide (NO)/cGMP signalling cascade regulates a large number of physiological processes including cardiovascular and gastrointestinal smooth muscle relaxation (Waldman and Murad, 1987; Moncada and Higgs, 1995; Groneberg et al., 2010), neuronal signal transduction (Ignarro, 2002) and platelet aggregation (Walter and Gambaryan, 2004). In mammals, three different isoforms of NOS catalyze the production of NO from L-arginine: endothelial, neuronal and inducible NOS. Binding of NO to its most important receptor, NO-sensitive guanylyl cyclase (NO-GC; IUPAC nomenclature: soluble guanylyl cyclase), leads to an increase in cGMP production and, thereby, to an activation of effector molecules such as cGMP-dependent protein kinases, cGMP-regulated ion channels and phosphodiesterases (Kaupp and Seifert, 2002; Friebe and Koesling, 2003; Rybalkin et al., 2003; Hofmann, 2005). As a hemoprotein, NO-GC requires a prosthetic heme group for NO-induced activation (Ignarro, 1990). Heme depletion abolishes NO stimulation which can only be restored after reconstitution of NO-GC with heme (Ignarro et al., 1986). Several studies have shown that the activation of the enzyme by NO involves direct binding to the enzyme's prosthetic heme group which leads to an up to 200-fold activation of the enzyme (Humbert et al., 1990; Stone and Marletta, 1994).
The substance 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) has been established as an effective inhibitor of NO-GC by blocking NO-induced cGMP synthesis. However, NO-induced effects in the presence of ODQ have been shown to occur. Thus, inhibitors such as ODQ (Garthwaite et al., 1995) or its analogue 8-bromo-4H-2, 5-dioxa-3, 9b-diaza-cyclopenta[a]naphthalen-1-one (NS2028; Olesen et al., 1998) are commonly used to discriminate between cGMP-dependent and cGMP-independent effects of NO. These cGMP-independent effects have been attributed to S-nitrosylation, S-glutathionylation and tyrosine nitration reactions, yet, in some cases no conclusive mechanism has been described for the cGMP-independent action of NO (Marcondes et al., 2006; Lima et al., 2010; Martinez-Ruiz et al., 2011; Rukoyatkina et al., 2011; Hess and Stamler, 2012; Meng et al., 2012).
It has been demonstrated that ODQ binds to the heme group of NO-GC in an NO-competitive manner (Schrammel et al., 1996). In vitro, ODQ was shown to inhibit the stimulated enzyme irreversibly. Spectral analyses have shown oxidation of the heme iron as the mechanism underlying the inhibitory effect (Schrammel et al., 1996; Zhao et al., 2000). The IC50 value of ODQ for the NO-stimulated enzyme ranges from 0.2 to 0.7 μM depending on the 2-(N,N-diethylamino)-diazenolate-2-oxide.diethylammonium salt (DEA-NO) concentration used (Schrammel et al., 1996); thus, 10 μM ODQ can be expected to achieve virtually complete inhibition of NO-stimulated NO-GC. Due to a low specificity of other inhibitors, such as methylene blue (Mayer et al., 1993; Luo et al., 1995) or LY83583 (Mulsch et al., 1988), the use of ODQ and NS2028 is recommended to obtain specific GC inhibition. ODQ-mediated oxidation of the heme iron and subsequent loss of the heme group is known to enhance the stimulatory effect of heme-independent activators of NO-GC such as Bay 58–2667 (Stasch et al., 2002). In addition to the effects on NO-GC, ODQ in the micromolar range has been shown to be an unselective heme protein inhibitor through inhibition of NO synthase as well as cytochrome P450 enzymes involved in nitrate biotransformation (Feelisch et al., 1999; Zhao et al., 2000). These unspecific effects probably underly the fact that ODQ is rarely used in in vivo studies.
In the present study, we intended to validate the utility of ODQ as NO-GC inhibitor to characterize cGMP-independent effects. To this end, we used two cellular systems (HEK cells stably expressing NO-GC and human platelets) and two organ systems (aorta and fundus) from wild-type (WT) and NO-GC-deficient mice. Our results show that ODQ serves as valid inhibitor for NO-GC but, in contrast to the purified enzyme, is not able to completely inhibit NO-GC in intact cellular systems. Consequently, there should be no doubt on its use as NO-GC inhibitor to demonstrate cGMP-dependent effects. The incomplete inhibition of NO-GC, although, does not qualify ODQ to provide evidence for cGMP-independent effects.
Materials and methods
Animals
All experiments were conducted in accordance with the German legislation on protection of animals and approved by the local animal care committee. Mice (C57BL/6 background) were housed in standard mouse cages (267 × 207 × 140 mm; maximally three animals/cage) with woodchip bedding material and under conventional laboratory conditions [constant room temperature (22°C), humidity level (55%), a 12-h light : 12-h dark cycle (lights on at 0600 h) and either standard rodent diet (WT) or fibre-reduced rodent diet (guanylyl cyclase knockout; GCKO) (Altromin, Germany) and water available ad libitum]. Animals of either sex were sacrificed at an age of 8–14 weeks by isoflurane overdose and blood/tissues were removed. A total of 55 animals were used (platelet studies 22, organ bath experiments 33 animals). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Generation of GCKO mice
Mice lacking NO-GC globally were generated as previously described (Friebe et al., 2007). In every experiment, WT littermates were used as controls.
Cell culture of HEK cells
HEK 293 cells were cultured in DMEM supplemented with 5% heat-denatured FCS and 1% penicillin/streptomycin at 37°C in a humidified 5% CO2 atmosphere. HEK cells stably expressing bovine α1 and β1 subunits of NO-GC (HEK-GC) were generated as described (Mullershausen et al., 2004). These cells had no detectable PDE activity.
Isolation of human platelets
Human platelets were prepared and used as previously reported (Gambaryan et al., 2010). Shortly, blood was obtained according to our institutional guidelines and the Declaration of Helsinki from healthy volunteers (two males and two females, age 26–38 years) who had not taken any antiplatelet medication in the previous 10 days. Our studies with human platelets were approved and recently reconfirmed by the local ethics committee of the University of Würzburg (Studies no. 67/92 and 114/04). Blood was collected into 1/7 volume of acid citrate-dextrose (ACD) solution (12 mM citric acid, 15 mM sodium citrate, 25 mM d-glucose and 2 μM EGTA, final concentrations). PRP was obtained by 5 min centrifugation at 330× g. PRP was centrifuged for 10 min at 430× g, then the pelleted platelets were washed once in CGS buffer (120 mM sodium chloride, 12.9 mM trisodium citrate, 30 mM d-glucose, pH 6.5), and resuspended in HEPES buffer (150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM HEPES, pH 7.4, 10 mM d-glucose). After 30 min rest in a 37°C water bath, washed human platelets (2 × 108 platelets·mL−1) were pre-incubated with or without ODQ (10 μM) for 10 min at 37°C and then incubated for up to 10 min with the indicated DEA-NO concentrations. Reactions were stopped by addition of Laemmli buffer and boiling for 5 min.
Isolation of murine platelets and cGMP measurement
Mice were anaesthetized by isoflurane inhalation. Blood (400–800 μL) was drawn into 200 μL of ACD (85 mM trisodium citrate dehydrate, 65 mM citric acid, 2% d-glucose) and isolation of murine platelets was performed as described (Dangel et al., 2010). For each experiment platelets from two animals were pooled.
For cGMP RIA, platelet suspensions were adjusted to 3 × 108 platelets·mL−1 and were allowed to rest at RT for 60 min before starting the experiment. Aliquots were equilibrated at 37°C for 5 min, pre-incubated for 10 min with ODQ or carrier and then incubated with DEA-NO for 10 min. All samples routinely contained sildenafil (100 μM) to inhibit cGMP degradation. The incubation was stopped by addition of ice-cold ethanol (final concentration 66%). Isolation of cGMP and measurement by RIA were performed as described previously (Friebe et al., 1998).
For Western analysis, murine platelets (2 × 108 platelets·mL−1) were pre-incubated with or without ODQ (10 μM) for up to 30 min at 37°C and then incubated for either 5 min with the indicated DEA-NO concentrations or for 5 min or 15 s with 10 μM DEA-NO. Reactions were stopped by addition of Laemmli buffer and boiling for 5 min.
Western blot analysis
Platelet proteins were separated by SDS-PAGE (9%) and blotted on nitrocellulose. Blots were blocked for 30 min with Roti-Block (Roth, Germany) and incubated over night at 4°C with a polyclonal antibody against pVASP (Nanotools, Teningen, Germany). Immunoreactive bands at 46 and 50 kDa were detected by chemiluminescence using the ECL kit (Thermo Scientific, Rockford, IL, USA). VASP phosphorylation was monitored at serine 239 and serine 235 for human and murine vasodilator-stimulated phosphoprotein (VASP) respectively. Actin and PKG were used as loading controls. Blots were scanned using SilverFast software and analysed densitometrically by NIH Image J software for uncalibrated optical density.
Preparation of murine tissues and isometric force studies
Animals were killed by isoflurane inhalation. The abdomen was opened and the upper digestive tract and thoracic aorta were removed. The tissues were transferred to Krebs-Henseleit solution (118 mM NaCl, 4.7 mM, KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, pH 7.4, 7.5 mM d-glucose) and bubbled with 95% O2 and 5% CO2.
Aorta
Murine thoracic aortic rings were mounted in a myograph as described previously (Friebe et al., 2007). Each experiment was performed with 2–4 aortic rings derived from GCKO and WT mice. After equilibration (at least 60 min at 37°C) aortic rings were pre-contracted with phenylephrine (PE; 1 μM). When reaching a stable contraction, ODQ (1, 10 or 100 μM) or NS2028 (10 μM) were administered. After 20 min of incubation, the vasorelaxing effects of DEA-NO or S-nitrosoglutathione (GSNO) were determined. For the measurement of relaxation by endogenously released NO, we used the thromboxane A2 mimetic U46619 (10 nM) for contraction. After 20 min of incubation, the vasorelaxing effect of carbachol (CCh; 30 μM) was determined in the presence or absence of ODQ (10 μM; 10 and 30 min pre-incubation). 3-Isobutyl-1-methylxanthine (IBMX; 100 μM) was added at the end of each experiment to determine maximal relaxation.
Fundus
Murine fundus strips were mounted in a myograph as described previously (Groneberg et al., 2011). Each experiment was performed with 2–4 fundus strips derived from WT and GCKO mice. After equilibration (at least 60 min at 37°C) fundus strips were pre-contracted with CCh (0.1 or 10 μM). When reaching a stable contraction, ODQ (10 μM) was administered. After 20 min of incubation, the vasorelaxing effect of DEA-NO was determined. IBMX (100 μM) was added at the end of each experiment to determine maximal relaxation. To measure the relaxation by endogenously released NO under non-adrenergic non-cholinergic (NANC) conditions electric field stimulation was applied to fundus strips pre-contracted with U46619 (100 nM) in the presence of atropine and guanethidine (1 μM, respectively). Electrical field stimulation (EFS) was applied through two platinum wire electrodes (5 mm distance; maximal voltage, 0.5 ms, 0.5–4 Hz, 3 s. After a first protocol of EFS ODQ (10 μM) was added to the organ bath and pre-incubated for 20 min before a second protocol of EFS.
Individual statistical analyses
For calculation of statistical tests, GraphPad Prism version 4.00 for Windows, GraphPad Software, San Diego, CA, USA, was used. Mann–Whitney U-tests were used for the data in Figures 1B, 4C and 6B in a predefined sequence. In Figures 1A, 3B and D, 4B, 5A and B, and 6B, all groups were compared by Kruskal–Wallis test. If P was ≤0.05 for the global test, two groups were compared by Mann–Whitney U-test in a predefined sequence. If not stated otherwise, data are expressed as mean ± SEM (n = number of animals).
Figure 1.

ODQ inhibition of NO-stimulated NO-GC. (A) DEA-NO-induced cGMP production in HEK-GC cell homogenate in the presence of different concentrations of the NO-GC inhibitor ODQ (1, 10, 100 μM; 10 min pre-incubation); GSNO-induced cGMP production in HEK-GC cell homogenate in the presence of different concentrations of the NO-GC inhibitor ODQ (1, 10, 100 μM) (*P < 0.05, **P < 0.01 compared to non-stimulated control). (B) DEA-NO-induced cGMP production in murine platelets in the presence of ODQ (10 μM; 10 min pre-incubation). Data shown are mean ± SEM of n = 10 (***P = 0.0003, **P = 0.0019); GSNO-induced cGMP production in murine platelets in the presence of ODQ (10 μM; 10 min pre-incubation). Data shown are mean ± SEM of n = 8 (***P = 0.0003).
Figure 4.
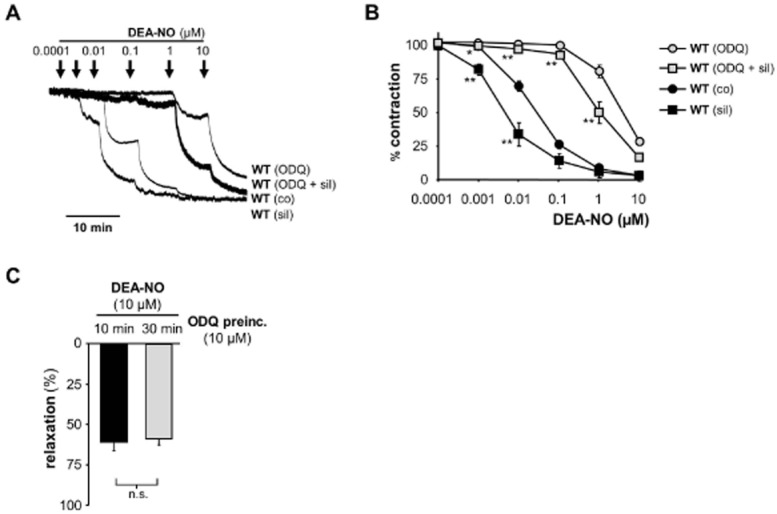
DEA-NO-induced relaxation of aortic rings. (A) Original trace of relaxation with DEA-NO from pre-contracted (1 μM PE) aortic rings of WT in the absence and presence of ODQ (10 μM), sildenafil (0.1 μM) or both. (B) Statistical analysis of DEA-NO-induced relaxation in aorta from WT in the absence and presence of 10 μM ODQ, 0.1 μM sildenafil or the combination of both drugs (*P = 0.0317, **P = 0.0079, each measurement compared to corresponding value without sil). Data shown are mean ± SEM of n = 5 WT and n = 3 GCKO animals. (C) To evaluate the time dependence of ODQ, aortic rings were pre-incubated with ODQ (10 μM) for 10 or 30 min and then relaxed by the addition of 10 μM DEA-NO. Data shown are mean ± SEM of n = 4 WT animals (n.s.: P = 0.6857).
Figure 6.

ODQ inhibition of relaxation by endogenously released NO. (A) Original traces and (B) statistical analysis of EFS-induced relaxation from pre-contracted (100 nM U46619) fundus strips from WT mice in the absence and presence of ODQ (10 μM). Prior to the addition of U46619, NANC conditions were obtained by adding atropine and guanethidine (1 μM, respectively). After the first EFS protocol ODQ was administered for 20 min with subsequent repetition of the EFS protocol. Data shown are mean ± SEM of n = 6 (**P < 0.01 compared to WT with ODQ). (C) Original traces of carbachol (CCh)-induced relaxation of aortic rings in the absence (WT and GCKO) and presence (WT) of ODQ (10 μM). Aortic rings were pre-contracted with 10 nM U46619. Approx. 30 min after addition of U46619, vehicle or ODQ were administered followed by a maximally effective concentration of CCh (30 μM). After application of ODQ, the onset of contraction was immediate and the contraction was complete after approximately 230 s indicating fast action of ODQ to block NO-GC. Note that these experiments were carried out in the absence of NOS inhibitors so that the stimulation of contraction induces counter-regulatory activation of eNOS. (D) Statistical analysis of carbachol (CCh)-induced relaxation of aortic rings in the absence (WT and GCKO) and presence (WT) of ODQ (10 μM). Data shown are mean ± SEM of n = 4 (WT) (*P < 0.005) and n = 3 (GCKO).
Figure 3.
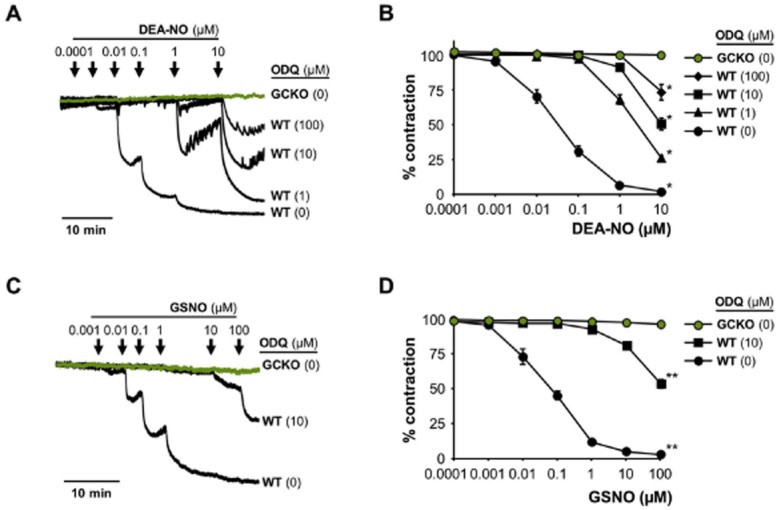
DEA-NO-induced relaxation of aortic rings. (A) Original traces of relaxation with DEA-NO from pre-contracted (1 μM PE) aortic rings of GCKO and WT in the absence and presence of ODQ (1, 10, 100 μM). ODQ had no effect on GCKO aorta. (B) Statistical analysis of DEA-NO-induced relaxation in aorta from GCKO and WT in the absence and presence of ODQ (1, 10, 100 μM) [*P < 0.05, compared to GCKO (DEA-NO 10 μM)]. (C) Original trace of relaxation with GSNO from pre-contracted (1 μM; PE) aortic rings of GCKO and WT in the absence and presence of ODQ (10 μM). (D) Statistical analysis of GSNO-induced relaxation in aorta from GCKO and WT in the absence and presence of ODQ (10 μM) [**P < 0.01, compared to GCKO (GSNO 100 μM)]. Data shown are mean ± SEM of n = 5 WT and n = 3 GCKO.
Figure 5.
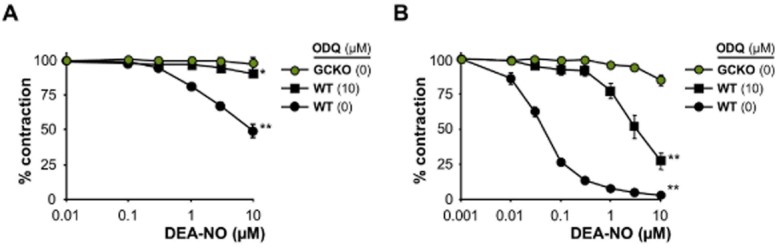
DEA-NO-induced relaxation of fundus strips. (A) DEA-NO-induced relaxation in fundus strips pre-contracted with 10 μM CCh from GCKO and WT in absence or presence of 10 μM ODQ [*P < 0.05, **P < 0.01 compared to GCKO (10 μM DEA-NO)]. (B) DEA-NO-induced relaxation in fundus strips pre-contracted with 0.1 μM CCh from GCKO and WT in absence or presence of 10 μM ODQ [**P < 0.01 compared to GCKO (10 μM DEA-NO)]. Data shown are mean ± SEM of n = 5 per genotype.
Materials
DEA-NO, GSNO, L-NAME, NS2028, U46619 and ODQ were obtained from Alexis Biochemicals (Lausen, Switzerland). CCh, Diclofenac, IBMX and PE were purchased from Sigma (Taufkirchen, Gemany). Sildenafil was a gift from Pfizer (Sandwich, UK).
Results
Measurement of cGMP in NO-GC transfected HEK cells and murine platelets
First, we used HEK cells stably expressing NO-GC (HEK-GC) to determine the inhibiting effect of ODQ. PDE activity was not detectable in these cells (Mullershausen et al., 2004). Addition of DEA-NO led to a concentration-dependent increase in intracellular cGMP concentrations (Figure 1A). Pre-incubation with ODQ reduced NO-induced cGMP increases, but even at a concentration of 100 μM ODQ the NO-induced cGMP production was not completely abrogated. This result was not dependent on the nature of the NO-donor since the non-radical GSNO showed similar results.
In order to test another cellular system, we used murine platelets (Figure 1B). Similar to the HEK-GC cells, platelets showed a pronounced cGMP increase after stimulation with DEA-NO. Incubation with 10 μM ODQ reduced but did not abolish cGMP production. When using very high DEA-NO concentration (100 μM) ODQ lost its inhibitory effect. Using GSNO, the cGMP increase in the presence of ODQ was largely diminished but still detectable at a concentration of 100 μM GSNO. Thus, ODQ only reversed, but did not fully abolish cGMP production in intact cells. This effect was more pronounced when using the NONOate DEA-NO compared to GSNO.
To prove the functionality of the cGMP signal in platelets, we measured phosphorylation of the VASP which is commonly used as an indicator of NO/cGMP signalling. Increasing concentrations of DEA-NO led to phosphorylation of VASP in human and murine platelets (Figure 2). In murine platelets, pre-incubation with ODQ (10 μM; 10 min) reduced NO-induced VASP phosphorylation, however, 10 μM DEA-NO still evoked a pronounced phosphorylation (Figure 2A, murine platelets; pSer-239). We then measured the time-dependence of ODQ pre-incubation (Figure 2B, murine platelets). VASP phosphorylation induced by a short stimulation with DEA-NO (10 μM; 15 s) could be largely blocked by a >10 min pre-incubation with 10 μM ODQ. The inhibitory effect of ODQ was, strongly reduced when stimulating platelets for 5 min with 10 μM DEA-NO. These data indicate that the inhibitory effect of ODQ can be overcome by increasing the amount of released NO.
Figure 2.

VASP phosphorylation in washed platelets. (A) Mouse platelets were pre-incubated for 10 min with or without ODQ (10 μM) and then incubated with increasing DEA-NO concentrations (0.01–10 μM; 10 min). Shown is a representative experiment and the statistical analysis of n = 6 experiments. The phosphorylation by 10 μM DEA-NO was taken as 100%. (B) Mouse platelets were pre-incubated for up to 30 min with ODQ (10 μM; co indicates absence of ODQ) and then incubated for either 5 min or 15 s with DEA-NO (10 μM). Shown is a representative experiment and the statistical analysis of n = 6 experiments. The phosphorylation in the absence of ODQ (co) was taken as 100%. (C) Human platelets were pre-incubated for 10 min with or without ODQ (10 μM) and then incubated for the indicated times with DEA-NO (10 μM; co indicates absence of DEA-NO). Shown is a representative experiment and the statistical analysis of n = 6 experiments.
To further examine the time-dependent component of DEA-NO incubation, we looked at VASP phosphorylation after different DEA-NO incubation times (Figure 2C, human platelets; pSer-239). As soon as 30 s after incubation with 10 μM DEA-NO, phosphorylation was observed showing the two characteristic bands at 46 and 50 kDa. Pre-incubation of 10 μM ODQ (10 min) inhibited VASP phosphorylation up to 2 min. However, when incubating platelets with 10 μM DEA-NO in the presence of ODQ for 3 min or longer, we saw pVASP signals indicating recovered NO/cGMP-mediated signal transduction.
NO-induced relaxation of aortic rings
Next, we tested the effect of ODQ in pre-contracted aortic rings from WT and GCKO mice using wire myography. Figure 3A shows the original traces of the myograph and Figure 3B the resulting statistics. DEA-NO led to a concentration-dependent relaxation of aorta from WT mice which was absent in GCKO mice. As can be seen in the original traces, the presence of ODQ inhibited NO-induced relaxation of WT aorta; yet, even 100 μM ODQ did not fully abolish the relaxation to DEA-NO.
Similar results were observed with the non-radical NO donor GSNO (Figures 3C and D). While GSNO caused a concentration-dependent relaxation of WT aorta, relaxation was absent in aorta from GCKO animals. As seen in Figure 3B for DEA-NO, co-incubation with 10 μM ODQ did not fully eliminate GSNO-induced relaxation. Only the use of 100 μM ODQ led to a nearly abolished relaxation (not shown) which is in line with the cGMP data obtained in platelets. Clearly, the nature of the NO donor and its rate of NO release have a major impact on the reversal of ODQ inhibition.
To confirm that the relaxation observed in the presence of ODQ was dependent on NO-GC activity (and thus cGMP), we repeated this experiment in the presence of the PDE5 inhibitor sildenafil (Figure 4A and B). Co-incubation of aorta from WT with sildenafil caused a leftward shift of the curve indicating a sensitized relaxation to DEA-NO which can be attributed to diminished cGMP degradation. Interestingly, aortic rings incubated with both ODQ and sildenafil showed a leftward shift of the curve as well. This clearly proves the involvement of cGMP in the remaining relaxation seen in the presence of ODQ.
To investigate the time dependence of ODQ inhibition in aortic tissue, we added ODQ (10 μM) to pre-contracted aortic rings. After 10 and 30 min of ODQ pre-incubation relaxation to 10 μM DEA-NO was determined (Figure 4C). There was no additional effect of prolonged ODQ pre-incubation time.
NO-induced relaxation of fundus tissue
In order to find out whether the effect seen in aortic tissue is consistent with other organs, we consequently tested the impact of ODQ in fundus tissue. Fundus strips were pre-contracted with the muscarinic agonist carbachol (CCh; 10 μM), a concentration commonly used for contraction. Under these conditions, DEA-NO-induced relaxation was basically inhibited by 10 μM ODQ matching the results from GCKO fundus (Figure 5A).
In a second attempt, the experiment was performed with a reduced pre-contraction using only 0.1 μM CCh (Figure 5B). In this case, 10 μM ODQ evoked merely a partial inhibition suggesting a cGMP-independent relaxation. However, since relaxation to DEA-NO in fundus of GCKO mice is absent, we can definitely rule out the effect to be cGMP-independent.
Inhibitory effect of ODQ on endogenously released NO
The use of pharmacological NO donors can cause artifactual results as the released NO may not be in the physiological concentration range or may act at sites that usually are not exposed to endogenous NO. In order to circumvent this problem, we tested the inhibitory effect of ODQ on endogenously released NO. First, fundus strips were pre-contracted with U46619 (100 nM) under NANC conditions and then a train of EFS was applied (Figure 6A and B). The release of NO after EFS led to a relaxation which was fully abolished by ODQ (10 μM). Higher stimulation frequencies cannot be used to increase the NO concentrations released as purinergic relaxation would confound the relaxation seen by NO. Second, we investigated the effect of ODQ on endothelial NO using aortic rings. Figure 6C shows original traces from aortic rings pre-contracted with 10 nM U46619. After stable contraction, we applied a bolus of 30 μM CCh in the absence or presence of ODQ (10 μM). The original traces show that, after application of ODQ, the onset of contraction was immediate and the contraction was complete after 203 ± 8 s indicating fast action of ODQ to block NO-GC (which becomes activated by eNOS after CCh-treatment). The statistical analysis (Figure 6D) reveals that ODQ reduced the relaxation to CCh but did not completely abolish the effect. By contrast, we were not able to detect CCh-induced relaxation mediated by endothelial NO in aorta from GCKO animals.
In sum, using GCKO mice, we corroborate ODQ to be an excellent inhibitor of NO-stimulated GC. However, our data clearly show that the use of ODQ to prove cGMP-independent mechanisms is impermissible.
Discussion
Since their implementation into laboratory use, NO-GC inhibitors such as ODQ and NS2028 have become valuable tools for the investigation of NO/cGMP signalling in biological systems (Garthwaite et al., 1995; Olesen et al., 1998). Particularly, ODQ commonly serves to prove cGMP-dependent processes but is also used to describe cGMP-independent effects. Schrammel et al. (1996) investigated the molecular mechanism of NO-GC inhibition using the purified enzyme and found that ODQ binds to NO-GC's prosthetic heme group and induces oxidation of the heme iron. In addition, binding of ODQ to the purified NO-GC was NO-competitive and led to an apparently irreversible inactivation of the enzyme.
In the present study, we looked at ODQ-mediated NO-GC inhibition in HEK-GC cells and human platelets as well as in aorta and fundus from WT mice and mice deficient in NO-GC. In order to stimulate NO-GC, we used DEA-NO as radical and GSNO as non-radical NO donor. Our results confirm that ODQ potently inhibits NO-stimulated NO-GC. Nevertheless, in all NO-GC-containing systems increasing concentrations of NO led to a reversal of ODQ-mediated inhibition contrasting the findings of Schrammel et al. (1996). Thus, we conclude that in a cellular context ODQ-induced NO-GC inhibition is not irreversible but can rather be reversed by increasing NO concentrations.
The results obtained with ODQ on murine tissues could be confirmed with another specific inhibitor of NO-GC, NS2028. In both fundus tissue (Supporting Information Figure S1A) and aortic rings (Supporting Information Figure S1B) from WT mice NS2028 inhibited NO-stimulated NO-GC which was overcome by elevated concentrations of DEA-NO. Thus, NO-mediated reversibility of NO-GC inhibition appears to be a property shared by heme-oxidizing inhibitors such as ODQ and NS2028.
Similar effects have been shown by Moro et al. (1996) in platelets and smooth muscle cells. Their study showed that a 100:1 excess of the NO donor S-nitroso-N-acetyl-D,L-penicillamine leads to partial reversal of ODQ-mediated NO-GC inhibition. In our experiments, however, ODQ-mediated NO-GC inhibition was overcome even by equimolar concentrations of NO donor and ODQ. Also, addition of 100 μM ODQ, which is far exceeding the commonly used 1 or 10 μM, did not completely block cGMP production either in HEK-GC cells or platelets. Similarly, 100 μM ODQ did not fully inhibit aortic relaxation to DEA-NO when compared to aorta from GCKO animals. Mechanistically, it is conceivable that intracellular reducing agents may convert the oxidized iron to the reduced form. Ascorbate has been proposed as has been an excess of NO (Zhao et al., 2000). In fact, our data corroborate this idea as high concentrations of NO donor or longer exposure to the NO donor overcame the inhibitory effect of ODQ on VASP phosphorylation.
ODQ has been reported to inhibit NO-GC by oxidation of the heme iron with subsequent loss of the heme group. To rule out that the reversal of ODQ inhibition was due to short pre-incubation time (routinely 10 min), we performed experiments with increased ODQ pre-incubation (up to 30 min). Neither in platelets (VASP phosphorylation) nor in aorta did we see a difference with prolonged incubation time. In fact, overall ODQ exposure to the tissues in our organ bath studies was actually up to 40 min until reaching the highest NO donor concentration (see Figure 3). Thus, we can rule out that the reversal of ODQ inhibition by NO was due to insufficient ODQ exposure.
Application of NO donors to mimic NOS-induced responses bears several disadvantages: Pharmacologically applied NO donors will elevate the general NO concentration in the tissue regardless of its physiological origin; in addition, this NO would affect cellular sites hardly reached under physiological conditions. Therefore, we evaluated the inhibitory effect of ODQ on endogenously released NO in two different organ systems, aorta and fundus. In fundus, we used EFS to release endogenous NO from the enteric nervous system in a spatially confined way. Here, ODQ fully inhibited this relaxation. In contrast, when stimulating endothelial NO release by the addition of CCh in aorta, we only saw partial inhibition by ODQ. This effect was independent of the level of pre-contraction: CCh-induced relaxation was detectable when aorta were pre-contracted with U46619+ODQ to a level similar to U46619 alone (not shown) as well as when inducing higher levels of contraction by the additive effect of U46619+ODQ as shown in Figure 6C. This could obviously be taken as proof for a cGMP-independent mechanism. Yet, under identical conditions, there was no CCh-induced relaxation of aorta from GCKO mice clearly disproving cGMP-independent mechanisms.
Why is the effectiveness of ODQ so diverging? Our findings show that reversal of ODQ-mediated NO-GC inhibition is strongly dependent on at least three experimental conditions. First, the nature of the NO donor plays an important role as seen in the difference between the radical and non-radical NO donors DEA-NO and GSNO. Using the non-radical NO donor GSNO, higher concentrations were needed to overcome the ODQ-mediated NO-GC inhibition. This indicates that the NO species released (NO. vs. NO+) influences the NO concentration required for reversal of ODQ-mediated NO-GC inhibition. Second, incubation time of ODQ and the half-life of the NO donor are important parameters for reverting ODQ-mediated NO-GC inhibition. Using Western blotting, we performed experiments on the time-dependence of ODQ-mediated NO-GC inhibition. We found that phosphorylation of VASP due to incubation with 10 μM DEA-NO was inhibited by 10 μM ODQ; however, VASP phosphorylation occurred in the presence of ODQ albeit after sustained incubation. Thus, the ODQ-mediated NO-GC inhibition can be overcome with increasing incubation time; this is of particular importance as even tiny cGMP elevations are known to lead to functional response via PKG (Mullershausen et al., 2006). Third, when using smooth muscle tissues in organ bath experiments the strength of the preceding contraction may obscure an incomplete NO-GC inhibition by ODQ and, therefore, lead to misinterpretation of the obtained results. In our experiments, after contraction with 10 μM CCh, DEA-NO induced a dose-dependent relaxation which was completely inhibited by 10 μM ODQ. With this knowledge, after contraction with a comparatively low CCh concentration (0.1 μM), relaxation by DEA-NO in the presence of 10 μM ODQ (see Figure 3B) can easily be mistaken for a cGMP-independent effect. Yet, cGMP independence can be definitely ruled out due to the lack of relaxation in GCKO tissue. Accordingly, only the comparison with GCKO allows an entirely unambiguous interpretation.
How to be sure of a cGMP-independent effect? Wanstall et al. (2005) have outlined several criteria for the proof of a cGMP-independent mechanism which include the use of a selective NO-GC inhibitor at a concentration that completely blocks NO-GC. In our study, however, we demonstrate that NO responses in the presence of ODQ (at putatively fully inhibiting concentrations of 10 and 100 μM) may not necessarily be cGMP-independent. Based on the strong influence of the varying conditions there is no concentration of ODQ which can be regarded as fully inhibitory. Consequently, the NO-induced effects in the presence of ODQ are not to be taken as evidence for cGMP-independent effects. Rather, additional controls have to be performed which depend on the cell/tissue used. Possible controls may include the addition of sildenafil (potentiation would indicate cGMP dependence) or the detection of PKG activation (phosphorylation of VASP or Rho-kinase would indicate cGMP-dependence). Measurement of cGMP itself is not suitable as in some cases smooth muscle relaxation or platelet inhibition may occur at NO concentrations which do not lead to measurable increases in cGMP (Gordge et al., 1998; Mullershausen et al., 2006).
Taken together, our experiments show that the effectiveness of NO-GC inhibition by ODQ is dependent on at least three experimental conditions: (i) the nature of the NO donor, (ii) the incubation time of ODQ and the NO donor as well as (iii) the strength of the preceding contraction when working with smooth muscle containing tissue. The choice of these parameters is of key importance and determines to what extent ODQ is able to inhibit NO-stimulated NO-GC. Without doubt, ODQ can be used as NO-GC inhibitor to reveal cGMP-dependent effects. However, we strongly advise against its use as a proof of cGMP-independent effects.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft [FR 1725/1-3] (A.F.).
The authors acknowledge the excellent technical help of Michaela Kümmel, Ulla Krabbe and Arkadius Pacha.
Glossary
- ACD
acid citrate-dextrose
- CCh
carbachol
- DEA-NO
2-(N,N-diethylamino)-diazenolate-2-oxide.diethylammonium salt
- GCKO
guanylyl cyclase knockout
- GSNO
S-nitrosoglutathione
- IBMX
3-isobutyl-1-methylxanthine
- NO-GC
nitric oxide-sensitive guanylyl cyclase
- NS2028
8-bromo-4H-2, 5-dioxa-3, 9b-diaza-cyclopenta[a]naphthalen-1-one
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- PE
phenylephrine
- VASP
vasodilator-stimulated phosphoprotein
- WT
wild type
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 (A) DEA-NO-induced relaxation of fundus strips from WT in the absence and presence of 10 μM NS2028 (**P = 0.0079). (B) DEA-NO-induced relaxation of aortic rings from WT in the absence and presence of 10 μM NS2028 (**P = 0.0079). Data shown are mean ± SEM of n = 5 animals.
References
- Dangel O, Mergia E, Karlisch K, Groneberg D, Koesling D, Friebe A. Nitric oxide-sensitive guanylyl cyclase is the only nitric oxide receptor mediating platelet inhibition. J Thromb Haemost. 2010;8:1343–1352. doi: 10.1111/j.1538-7836.2010.03806.x. [DOI] [PubMed] [Google Scholar]
- Feelisch M, Kotsonis P, Siebe J, Clement B, Schmidt HH. The soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one is a nonselective heme protein inhibitor of nitric oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol Pharmacol. 1999;56:243–253. doi: 10.1124/mol.56.2.243. [DOI] [PubMed] [Google Scholar]
- Friebe A, Koesling D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res. 2003;93:96–105. doi: 10.1161/01.RES.0000082524.34487.31. [DOI] [PubMed] [Google Scholar]
- Friebe A, Mullershausen F, Smolenski A, Walter U, Schultz G, Koesling D. YC-1 potentiates nitric oxide- and carbon monoxide-induced cyclic GMP effects in human platelets. Mol Pharmacol. 1998;54:962–967. doi: 10.1124/mol.54.6.962. [DOI] [PubMed] [Google Scholar]
- Friebe A, Mergia E, Dangel O, Lange A, Koesling D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc Natl Acad Sci U S A. 2007;104:7699–7704. doi: 10.1073/pnas.0609778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, et al. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J Biol Chem. 2010;285:18352–18363. doi: 10.1074/jbc.M109.077602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- Gordge MP, Hothersall JS, Noronha-Dutra AA. Evidence for a cyclic GMP-independent mechanism in the anti-platelet action of S-nitrosoglutathione. Br J Pharmacol. 1998;124:141–148. doi: 10.1038/sj.bjp.0701821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groneberg D, Konig P, Wirth A, Offermanns S, Koesling D, Friebe A. Smooth muscle-specific deletion of nitric oxide-sensitive guanylyl cyclase is sufficient to induce hypertension in mice. Circulation. 2010;121:401–409. doi: 10.1161/CIRCULATIONAHA.109.890962. [DOI] [PubMed] [Google Scholar]
- Groneberg D, Konig P, Koesling D, Friebe A. Nitric oxide-sensitive guanylyl cyclase is dispensable for nitrergic signaling and gut motility in mouse intestinal smooth muscle. Gastroenterology. 2011;140:1608–1617. doi: 10.1053/j.gastro.2011.01.038. [DOI] [PubMed] [Google Scholar]
- Hess DT, Stamler JS. Regulation by S-nitrosylation of protein post-translational modification. J Biol Chem. 2012;287:4411–4418. doi: 10.1074/jbc.R111.285742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F. The biology of cyclic GMP-dependent protein kinases. J Biol Chem. 2005;280:1–4. doi: 10.1074/jbc.R400035200. [DOI] [PubMed] [Google Scholar]
- Humbert P, Niroomand F, Fischer G, Mayer B, Koesling D, Hinsch KD, et al. Purification of soluble guanylyl cyclase from bovine lung by a new immunoaffinity chromatographic method. Eur J Biochem. 1990;190:273–278. doi: 10.1111/j.1432-1033.1990.tb15572.x. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ. Haem-dependent activation of guanylate cyclase and cyclic GMP formation by endogenous nitric oxide: a unique transduction mechanism for transcellular signaling. Pharmacol Toxicol. 1990;67:1–7. doi: 10.1111/j.1600-0773.1990.tb00772.x. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol. 2002;53(4 Pt 1):503–514. [PubMed] [Google Scholar]
- Ignarro LJ, Adams JB, Horwitz PM, Wood KS. Activation of soluble guanylate cyclase by NO-hemoproteins involves NO-heme exchange. Comparison of heme-containing and heme-deficient enzyme forms. J Biol Chem. 1986;261:4997–5002. [PubMed] [Google Scholar]
- Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769–824. doi: 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Das S, Vincent SR. Effects of methylene blue and LY83583 on neuronal nitric oxide synthase and NADPH-diaphorase. Eur J Pharmacol. 1995;290:247–251. doi: 10.1016/0922-4106(95)00084-4. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcondes S, Cardoso MH, Morganti RP, Thomazzi SM, Lilla S, Murad F, et al. Cyclic GMP-independent mechanisms contribute to the inhibition of platelet adhesion by nitric oxide donor: a role for alpha-actinin nitration. Proc Natl Acad Sci U S A. 2006;103:3434–3439. doi: 10.1073/pnas.0509397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic Biol Med. 2011;51:17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Mayer B, Brunner F, Schmidt K. Inhibition of nitric oxide synthesis by methylene blue. Biochem Pharmacol. 1993;45:367–374. doi: 10.1016/0006-2952(93)90072-5. [DOI] [PubMed] [Google Scholar]
- Meng E, Young JS, Cha TL, Sun GH, Yu DS, Brading AF. Neuronal-derived nitric oxide modulates the activity of mouse detrusor smooth muscle. Neurourol Urodyn. 2012;31:572–578. doi: 10.1002/nau.21247. [DOI] [PubMed] [Google Scholar]
- Moncada S, Higgs EA. Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J. 1995;9:1319–1330. [PubMed] [Google Scholar]
- Moro MA, Russel RJ, Cellek S, Lizasoain I, Su Y, Darley-Usmar VM, et al. cGMP mediates the vascular and platelet actions of nitric oxide: confirmation using an inhibitor of the soluble guanylyl cyclase. Proc Natl Acad Sci U S A. 1996;93:1480–1485. doi: 10.1073/pnas.93.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullershausen F, Russwurm M, Koesling D, Friebe A. In vivo reconstitution of the negative feedback in nitric oxide/cGMP signaling: role of phosphodiesterase type 5 phosphorylation. Mol Biol Cell. 2004;15:4023–4030. doi: 10.1091/mbc.E03-12-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullershausen F, Lange A, Mergia E, Friebe A, Koesling D. Desensitization of NO/cGMP signaling in smooth muscle: blood vessels versus airways. Mol Pharmacol. 2006;69:1969–1974. doi: 10.1124/mol.105.020909. [DOI] [PubMed] [Google Scholar]
- Mulsch A, Busse R, Liebau S, Forstermann U. LY 83583 interferes with the release of endothelium-derived relaxing factor and inhibits soluble guanylate cyclase. J Pharmacol Exp Ther. 1988;247:283–288. [PubMed] [Google Scholar]
- Olesen SP, Drejer J, Axelsson O, Moldt P, Bang L, Nielsen-Kudsk JE, et al. Characterization of NS 2028 as a specific inhibitor of soluble guanylyl cyclase. Br J Pharmacol. 1998;123:299–309. doi: 10.1038/sj.bjp.0701603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukoyatkina N, Walter U, Friebe A, Gambaryan S. Differentiation of cGMP-dependent and -independent nitric oxide effects on platelet apoptosis and reactive oxygen species production using platelets lacking soluble guanylyl cyclase. Thromb Haemost. 2011;106:922–933. doi: 10.1160/TH11-05-0319. [DOI] [PubMed] [Google Scholar]
- Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280–291. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- Stasch JP, Schmidt P, Alonso-Alija C, Apeler H, Dembowsky K, Haerter M, et al. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol. 2002;136:773–783. doi: 10.1038/sj.bjp.0704778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JR, Marletta MA. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry. 1994;33:5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- Waldman SA, Murad F. Cyclic GMP synthesis and function. Pharmacol Rev. 1987;39:163–196. [PubMed] [Google Scholar]
- Walter U, Gambaryan S. Roles of cGMP/cGMP-dependent protein kinase in platelet activation. Blood. 2004;104:2609. doi: 10.1182/blood-2004-06-2389. [DOI] [PubMed] [Google Scholar]
- Wanstall JC, Homer KL, Doggrell SA. Evidence for, and importance of, cGMP-independent mechanisms with NO and NO donors on blood vessels and platelets. Curr Vasc Pharmacol. 2005;3:41–53. doi: 10.2174/1570161052773933. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Brandish PE, DiValentin M, Schelvis JP, Babcock GT, Marletta MA. Inhibition of soluble guanylate cyclase by ODQ. Biochemistry. 2000;39:10848–10854. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.