

Abstract
BACKGROUND AND PURPOSE
Genistein is an isoflavone phytoestrogen found in a number of plants such as soybeans and there is accumulating evidence that it has beneficial effects on the regulation of glucose homeostasis. In this study we evaluated the effect of genistein on glucose homeostasis and its underlying mechanisms in normal and insulin-resistant conditions.
EXPERIMENTAL APPROACH
To induce insulin resistance, mice or differentiated 3T3-L1 adipocytes were treated with macrophage-derived conditioned medium. A glucose tolerance test was used to investigate the effect of genistein. Insulin signalling activation, glucose transporter-4 (GLUT4) translocation and AMP-activated PK (AMPK) activation were detected by Western blot analysis or elisa.
KEY RESULTS
Genistein impaired glucose tolerance and attenuated insulin sensitivity in normal mice by inhibiting the insulin-induced phosphorylation of insulin receptor substrate-1 (IRS1) at tyrosine residues, leading to inhibition of insulin-mediated GLUT4 translocation in adipocytes. Mac-CM, an inflammatory stimulus induced glucose intolerance accompanied by impaired insulin sensitivity; genistein reversed these changes by restoring the disturbed IRS1 function, leading to an improvement in GLUT4 translocation. In addition, genistein increased AMPK activity under both normal and inflammatory conditions; this was shown to contribute to the anti-inflammatory effect of genistein, which leads to an improvement in insulin signalling and the amelioration of insulin resistance.
CONCLUSION AND IMPLICATIONS
Genistein showed opposite effects on insulin sensitivity under normal and inflammatory conditions in adipose tissue and this action was derived from its negative or positive regulation of IRS1 function. Its up-regulation of AMPK activity contributes to the inhibition of inflammation implicated in insulin resistance.
Keywords: genistein, insulin resistance, inflammation, AMP-activated PK, adipocyte
Introduction
In skeletal muscle and adipose tissue, insulin induces glucose transporter-4 (GLUT4) translocation to the cell membrane through insulin receptor substrate-1 (IRS1)/PI3K/Akt pathway. Insulin resistance is one of the great challenges in diabetes and obesity management. Many inflammatory molecules can attenuate insulin-mediated tyrosine phosphorylation of IRS1 and this impairs insulin PI3K signalling, leading to insulin resistance (Gual et al., 2005). This event establishes the link between inflammation and insulin resistance. In adipose tissue of obese patients, more macrophages are recruited that then activate the inflammatory response in neighbouring adipocytes by releasing pro-inflammatory cytokines. Consequently the inflammation is exacerbated and insulin resistance occurs (Rasouli and Kern, 2008). In addition to insulin, AMP-activated PK (AMPK) also plays an important role in the regulation of glucose homeostasis. Agents that activate AMPK, such as 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) and metformin, have been shown to increase muscle glucose transport (Sajan et al., 2010). AMPK antagonizes fatty acid-induced inflammation in macrophages (Yang et al., 2010) and reduces insulin resistance in obesity (Galic et al., 2011). Salicylate reduces insulin resistance both by inhibition of IκB kinase β (IKKβ; Yuan et al., 2001) and activation of AMPK, induced by inhibiting the dephosphorylation of p-AMPK (Hawley et al., 2012). These findings indicate that AMPK activation, by inhibiting inflammation, is involved in the regulation of insulin sensitivity.
Genistein is an isoflavone phytoestrogen found in a number of plants such as soybeans and there is accumulating evidence that it has beneficial effects on the regulation of glucose homeostasis. It has been reported that genistein significantly reduces fasting glucose and insulin in osteopaenic, postmenopausal women (Atteritano et al., 2007), ameliorates glycaemia and improves glucose tolerance in diabetic animals (Rauter et al., 2010; Fu et al., 2012) and stimulates the insulin-dependent signalling pathway in hepatocytes (Haneishi et al., 2011). In addition, it has been demonstrated to have anti-diabetic effects. However, in contrast, genistein has also been shown to inhibit insulin-stimulated glucose uptake by down-regulation of GLUT4 translocation (Bazuine et al., 2005; Nomura et al., 2008) and to counteract the antilipolytic action of insulin in adipocytes (Szkudelska et al., 2008), suggesting that it has negative effects on the regulation of insulin's action in adipocytes. These findings indicate that the effects of genistein on insulin's action in glucose homeostasis need to be clarified. Because the negative regulation of insulin action by genistein was mostly derived from studies in vitro, further studies need to be performed to elucidate its effects in vivo. In fact, genistein is a multifunctional agent and has been demonstrated to inhibit inflammation (Geng et al., 1993) and stimulate AMPK activation (Arunkumar and Anuradha, 2012). Hence, we determined whether these activities contribute to its regulation of insulin sensitivity, especially in insulin-resistant conditions. In an attempt to gain further insights into the regulation of glucose homeostasis by genistein in vivo, we observed the effects of genistein on glucose tolerance in mice under normal and insulin-resistant conditions and investigated the underlying mechanism by focusing on the regulation of IRS1 in adipose tissue. We further showed that genistein affects AMPK activity in adipose tissue, and this action contributes to the amelioration of insulin resistance through inhibition of inflammation. A better understanding of the effects of genistein on glucose homeostasis could be beneficial for indicating whether it has potential as a drug for controlling obesity and diabetes associated with insulin resistance.
Methods
Animals
Male ICR mice (6–8 weeks of age) were supplied by the Laboratory Animal Center of Nanjing Qinglongshan; the total number of mice used was 360. The care and treatment of these mice were in accordance with the Provisions and General Recommendation of Chinese Experimental Animals Administration Legislation. The animal care and experimental procedures were approved by the Administrative Committee of Experimental Animal Care and Use of China's Pharmaceutical University. Animals were housed in a room with a constant temperature (22 ± 1°C) and a 12-h light-dark cycle, and allowed free access to a standard diet (the Laboratory Animal Center of Nanjing Qinglongshan) and water ad libitum. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Preparation of macrophages-derived conditioned medium
Peritoneal macrophages were collected from mice and activated with LPS (Escherichia coli serotype 055:B5, LPS) as described previously (Liu et al., 2011). Briefly, peritoneal macrophages were seeded in 6-well plates (2 × 106 per well) for 2 h. After being washed twice with PBS, adherent macrophages were cultured in serum-free DMEM or KRH (Krebs–Ringer phosphate–HEPES buffer, containing 118-mmol·L−1 NaCl, 5-mmol·L−1 KCl, 1.3-mmol·L−1 CaCl2, 1.2-mmol·L−1 MgSO4, 1.2-mmol·L−1 KH2PO4, 30 mmol·L−1 HEPES and 0.2 % BSA, pH 7.4, to test for AMPK activity in adipocytes) and stimulated with LPS (5 μg·mL−1) for 24 h. The supernatant, which contained high levels of TNF-α and IL-6 (quantified with elisa kits; R&D, Shanghai, China) was collected as conditioned medium (Mac–CM) and stored at −70°C.
Oral glucose load test and insulin sensitivity assay in mice
Mice deprived of food overnight (fasted mice; 10 mice per group) were given genistein (10, 25 and 50 mg·kg−1) or metformin (200 mg·kg−1) by oral gavage. After 30 min, the mice were administered glucose (2 g·kg−1, p.o.). In the case of Mac–CM challenge, mice were injected with Mac–CM i.p. (0.1 mL 10 g−1, diluted with saline, 1:1, v v−1) 30 min before glucose load. Blood was collected from the orbital sinus at regular intervals after glucose load. Serum glucose was determined with a commercial kit based on the glucose oxidase peroxidase (GOD-POD) method and blood glucose AUC (AUC-G) was calculated as follows: 0.5 × (Bg0 + Bg30) / 2 + 0.5 × (Bg30 + Bg60) / 2 + 1 × (Bg60 + Bg120) / 2 (Bg0, Bg30, Bg60 and Bg120 refers to the blood glucose concentration at 0, 30, 60 and 120 min after glucose load). To determine insulin sensitivity, blood was collected at 30 min after glucose load for the assessment of insulin (with ELISA kit) and glucose. Insulin sensitivity index (ISI) was calculated according to the formula: ISI = 1 / [blood glucose (mmol·L−1) × blood insulin (pmol·L−1)].
Assay of plasma concentration of genistein
Overnight-fasted male mice (three to four mice per group) were given 50 mg·kg−1 genistein by oral gavage. Blood samples were collected at 30, 60, 90 and 120 min after genistein administration and anticoagulated with 3.8% buffered sodium citrate (9:1, v v−1). Then, each blood sample was centrifuged at 1000× g for 10 min. 4-Hydroxybenzophenone (internal standard) and 4 mL of ethyl acetate (extraction solvent) were added to plasma samples (200 μL). Extraction was conducted by vortexing vigorously for 50 min and centrifuged at 1000× g for 15 min. The supernatant was transferred to the glass tube and evaporated to dryness under a gentle stream of nitrogen at 40°C. The residue was reconstituted with 0.1 mL of mobile phase (27:73 mixture of acetonitrile and 0.05 M ammonium acetate buffer solution). Genistein and 4-hydroxybenzophenone in plasma samples were separated on a C18 column (4.6 × 150 mm, 5 mm; Inertsil ODS-SP, Shanghai, China) maintained at 30°C. The mobile phase used was a 27:73 mixture of acetonitrile and 0.05 M ammonium acetate buffer solution. The flow rate was set at 1 mL·min−1 and detection was performed at 262 nm. The accuracy and precision of the HPLC method were evaluated according to the method used by the Kwon SH group (Kwon et al., 2007).
Cell culture and differentiation
3T3-L1 cells (from the cell bank of the Chinese Academy of Sciences, Shanghai, China) were cultured in 6-well plates (1 × 106 cells per well) in DMEM (25 mM glucose), supplemented with 10% FBS, 100 u·mL−1 penicillin, and 100 μg·mL−1 streptomycin, until the cells grew to confluence. Postconfluent cells were differentiated by incubation in DMEM containing 0.5 mM IBMX, 1 mM dexamethasone (Dex), 10 μg·mL−1 insulin for 48 h, then for 2 days in DMEM (10% FBS) containing 10 μg·mL−1 insulin alone. Thereafter, cells were maintained in DMEM (10% FBS) for 8–12 days, until over 80% of the cells exhibited the mature adipocyte phenotype.
RNA isolation, cDNA synthesis and real-time RT-PCR
Mice (three to four animals per group) were fasted overnight and then administered either genistein, metformin or salicylate, p.o., at given doses 30 min before an i.p. injection of diluted Mac–CM (0.1 mL 10 g−1). Mice were killed at 2 h post injection and the fat tissue located in the epididymis was immediately removed. Then 100 mg fat tissue was chopped into small pieces and homogenized with 1 mL Trizol on ice. Total RNA from tissues was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized with the iScript Synthesis kit from Bio-Rad (Hercules, CA, USA). Quantitative PCR was performed in 20 μL reactions using SYBR-Green Quantitative PCR Kit (Thermo, Waltham, MA, USA). PCR reagents were obtained from Thermo and used according to the instructions of the manufacturer. Amplification was performed with the ABI 7500 sequence detection system using the following protocol: 40 cycles (30 s at 95°C and 30 s at 60°C) after an initial activation step for 10 min at 95°C. Primer sequences were shown as follows: β-actin [222 base pairs (bp)], forward primer: 5′-ACGGATGTCAACGTCACACT-3′, reverse primer: 5′-CACTATTGGCAAC GAGCGGTT CC G-3′; TNF-α (111 bp), forward primer: 5′-GGGACAGTGACCTGGACTGT-3′, reverse primer: 5′- CTCCCTTTGCAGAACTCAGG −3′; IL-6 (173 bp), forward primer: 5′-AACCCAA GGGCATT T CAATC-3′, reverse primer: 5′- CACCGCATCTATCACCACAG-3′. The mRNA level of individual genes was normalized and presented as a ratio to β-actin.
AMPK and PI3K activity assay in adipocytes
To determine AMPK activity, adipocytes (1 × 106 per well) were seeded onto 6-well plates for 24 h. Cells were washed with and cultured in KRH, pretreated with genistein (10 μM), metformin (1 mM) or Compound C (25 μM) for 30 min, then stimulated with or without Mac–CM (diluted in KRH, 1:1, v v−1) for 30 min followed by the addition of glucose (11 mM) for the subsequent 30 min. For the detection of PI3K activity, adipocytes (1 × 106 per well) were incubated for 4 h in serum-free DMEM, pretreated with genistein (10 μM), Compound C (25 μM), genistein plus Compound C or AICAR (500 μM) for 30 min, then exposed to Mac–CM (diluted in DMEM, 1:1, v v−1) for another 30 min followed by 20-min treatment of insulin (100 nM). After washed with ice-cold PBS, cells were lysed in cell lysis buffer (Tris–HCl 50 mM, pH 7.2, containing 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% NP-40, NaCl 0.15 M, sodium orthovanadate 1 mM). The lysates were centrifuged at 13 000 x g for 15 min at 4°C and the supernatant was collected for AMPK activity assay or for the quantification of PIP3, the product of PI3K, with elisa Kits (Dizhao Biotech, Nanjing, China) respectively.
Western blot analysis
Mice (three to four animals per group) were treated with different drugs as mentioned earlier in the oral glucose tolerance test. Thirty minutes after oral glucose load, mice were killed by cervical dislocation and epididymal adipose tissue was separated and homogenized in ice-cold cell lysis buffer (1: 4, w v−1, g·mL−1) to extract the protein. Homogenates were centrifuged at 13 000 x g for 15 min at 4°C and supernatants were collected. The protein concentration of each sample was determined using a Bicinchoninic Acid Protein Assay kit (Biosky Biotechnology Corporation, Nanjing, China). Twenty to 40 μg of protein were mixed with sample buffer, boiled for 5 min, separated by SDS-PAGE and transferred to PVDF membranes. Following incubation with primary antibodies, the membranes were incubated with secondary antibodies conjugated to HRP. Antibody reactivity was detected by an ECL Western Blotting Detection System (Beyotime Institute of Biotechnology, Haimen, Jiangsu, China).
For Western blot analysis in adipocytes, differentiated 3T3-L1 adipocytes (1 × 106 per well) were seeded onto 6-well plates and deprived of serum for 4 h in DMEM or KRH. Cells were pretreated with genistein, wortmannin, Compound C, genistein plus Compound C or AICAR, respectively, at given concentrations for 0.5 h, followed by addition of Mac–CM or insulin. After 30 min, cells were washed with ice-cold PBS, collected and lysed in cell lysis buffer and the lysates were centrifuged at 13 000× g for 15 min at 4°C for the collection of supernatants. For the assay of membrane GLUT4, membrane protein was prepared with a plasma membrane protein extraction kit (Sangon Biotech Co., Ltd., Shanghai, China).
Statistical analysis
Results are expressed as mean ± SD. Differences were analysed by one-way anova followed by Newman–Keuls test. Values of P < 0.05 were considered statistically significant.
Materials
Genistein (98% in purity) was purchased from Nanjing Zelang Medical Technology Co., Ltd. (Nanjing, China); Metformin was from Sino-American Shanghai Squibb Pharma and sodium salicylate from Tianjin Kemiou Chemical Agent Center (Tianjin, China). For cell experiments, genistein, metformin and sodium salicylate were dissolved in DMSO (final concentration < 0.1% v v−1); For animal experiments, they were suspended in 0.5 % sodium carboxymethyl cellulose. The following items were obtained from the commercial sources cited: LPS (E. coli serotype 055:B5, LPS) and Compound-C, Sigma (St. Louis, MO, USA); insulin, Wanbang Biochemical Pharmaceutical Company (Xuzhou, Jiangsu, China); anti-IRS1 (R301; BS1408), anti-phospho-IRS1 (Ser307; BS4725), anti-Akt (A444; BS1810), anti-Phospho-Akt (T308; BS4008), anti-IKKβ (F182; BS1407), anti-Phospho-IKKβ (Y199; BS4320), anti-Na+/K+-ATPase α-1 (BS1436), GAPDH (AP0063), HRP-conjugated anti-rabbit and anti-mouse IgG antibodies, Bioworld Technology (St. Paul, MN, USA); PY99 (sc-7020), Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); anti-AMPKα, anti-phospho-AMPKα (T172) and anti-GLUT4, Cell Signaling Technology, Inc. (Beverly, MA, USA); AICAR and wortmannin, Beyotime Institute of Biotechnology (Shanghai, China).
Results
Genistein impaired oral glucose tolerance and attenuated insulin sensitivity in normal mice
We first examined the effect of genistein on oral glucose tolerance in mice. Glucose load led to an increase in blood glucose, with its Cmax reached at 30 min and then it gradually returned to the basal level. Administration of genistein p.o. at doses ranging from 10 to 50 mg·kg−1 increased blood glucose at 30 and 60 min by delaying glucose disposal, as indicated by the increase in AUC-G (Figure 1A and B). Insulin sensitivity was also attenuated by genistein, as indicated by a 58.4% decrease (50 mg·kg−1) in ISI (Figure 1C). As a positive control, the anti-diabetic agent metformin promoted glucose disposal and enhanced insulin sensitivity in mice.
Figure 1.
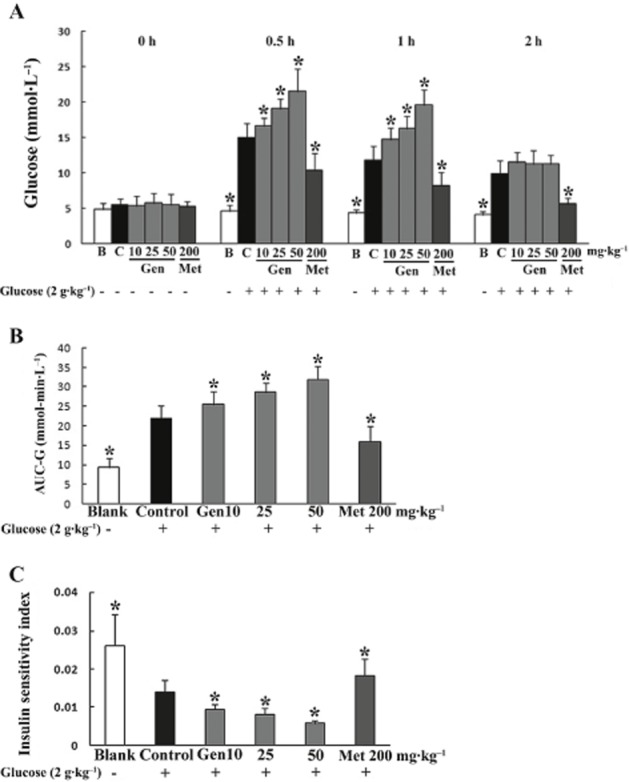
Genistein impaired glucose tolerance and attenuated insulin sensitivity in normal mice. Fasted mice were pretreated with genistein or metformin by gavage 30 min before p.o. administration of 2 g·kg−1 glucose. Blood was collected from the orbital sinus at regular intervals for glucose detection (A) and the AUC-G calculated (B). ISI (C) was calculated as the product of blood glucose and blood insulin at 30 min after glucose load. The results are expressed as the mean ± SD (n = 10). *P < 0.05 versus control. (B, blank group only treated with vehicle; C, control group treated only with glucose.)
Genistein reversed glucose intolerance and ameliorated insulin sensitivity in Mac–CM treated mice
We also determined the influence of genistein on glucose tolerance under inflammatory conditions. As an inflammatory challenge, Mac–CM treatment impaired glucose tolerance in mice, as demonstrated by the higher serum glucose level compared with the vehicle-treated group. This change was reversed by genistein in a dose-dependent manner (Figure 2A). The increase in total glucose was also reduced by genistein, as shown by the decreased AUC-G (Figure 2B). In addition, the changes in insulin sensitivity induced by the Mac–CM insult were effectively restored by genistein, as evidenced by a 55.1% increase (50 mg·kg−1 genistein) in ISI compared with the control (Figure 2C). Similar to genistein, metformin significantly reversed glucose intolerance and restored insulin sensitivity.
Figure 2.
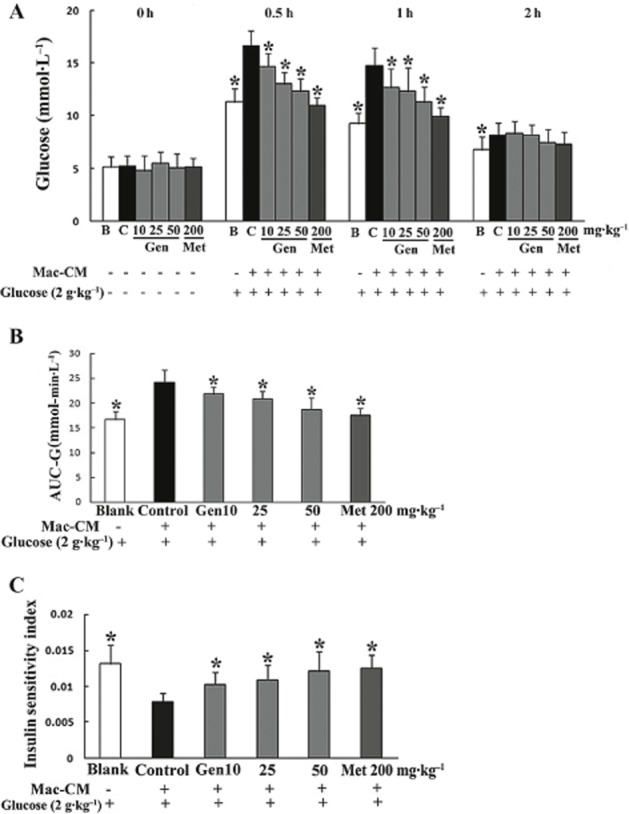
Genistein ameliorated glucose intolerance and improved insulin sensitivity in Mac–CM treated mice. Fasted mice were given genistein or metformin, p.o. After 30 min, mice were treated with Mac–CM by i.p. injection 30 min before the administration of 2 g·kg−1 glucose, p.o. Blood was collected at regular intervals for glucose or insulin detection. (A) Glucose concentrations within 2 h after glucose load. (B) AUC-G. (C) ISI. The results are expressed as the mean ± SD (n = 10). *P < 0.05 versus control. (B, blank group treated with vehicle; C, control group treated with Mac–CM.)
Blood concentration after administration of genistein p.o
After administration of 50 mg·kg−1 genistein p.o., the plasma concentration was assayed by HPLC. As shown in Figure 3, the retention times of genistein and 4-hydroxybenzophenone (internal standard) were about 24 min and 26 min respectively. The average concentration of genistein at 30, 60, 90 and 120 min after gavage was 115, 263, 424 and 153 ng·mL−1 respectively (Table 1).
Figure 3.
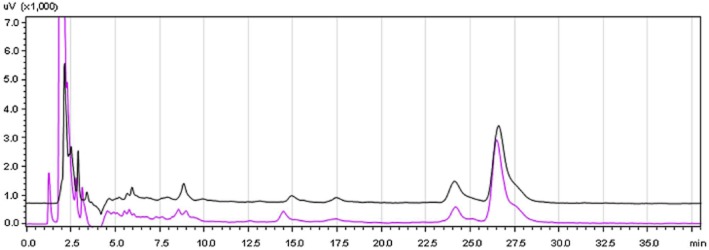
Retention time of genistein measured by HPLC. Overnight-fasted male mice (three to four mice per group) were given 50 mg·kg−1 genistein or distilled water by gavage, p.o. Blood samples were collected at 90 min after oral administration and anticoagulated with sodium citrate. The plasma concentration of genistein was measured by HPLC. Red curve: plasma sample (from mice exposed to water) containing standard genistein (500 ng·mL−1) and 4-hydroxybenzophenone (internal standard, 1 μg·mL−1); Black curve: plasma sample from mice given 50 mg·kg−1 genistein, p.o.
Table 1.
The plasma concentration of genistein assayed by HPLC after its administration
| Time (min) | 30 | 60 | 90 | 120 |
|---|---|---|---|---|
| Plasma concentration (ng·mL−1) | 115 ± 29 | 263 ± 78 | 424 ± 99 | 153 ± 34 |
Overnight-fasted male mice were given 50 mg·kg−1 genistein by gavage, p.o. Blood samples were collected at 30, 60, 90 and 120 min after its administration and anticoagulated with sodium citrate. Plasma concentrations of genistein were measured by HPLC. The results are expressed as the mean ± SD (n = 3–4).
Genistein inhibited the phosphorylation of IRS1 at tyrosine residues and Akt phosphorylation after glucose load in adipose tissue
In normal mice, glucose load induced both phosphorylation of IRS1 at tyrosine residues and the subsequent phosphorylation of Akt in adipose tissue. This enhanced phosphorylation was attenuated by administration of genistein at doses of 25 and 50 mg·kg−1 respectively (Figure 4A and B), indicating an inhibitory effect of genistein on insulin IRS1/Akt signalling.
Figure 4.
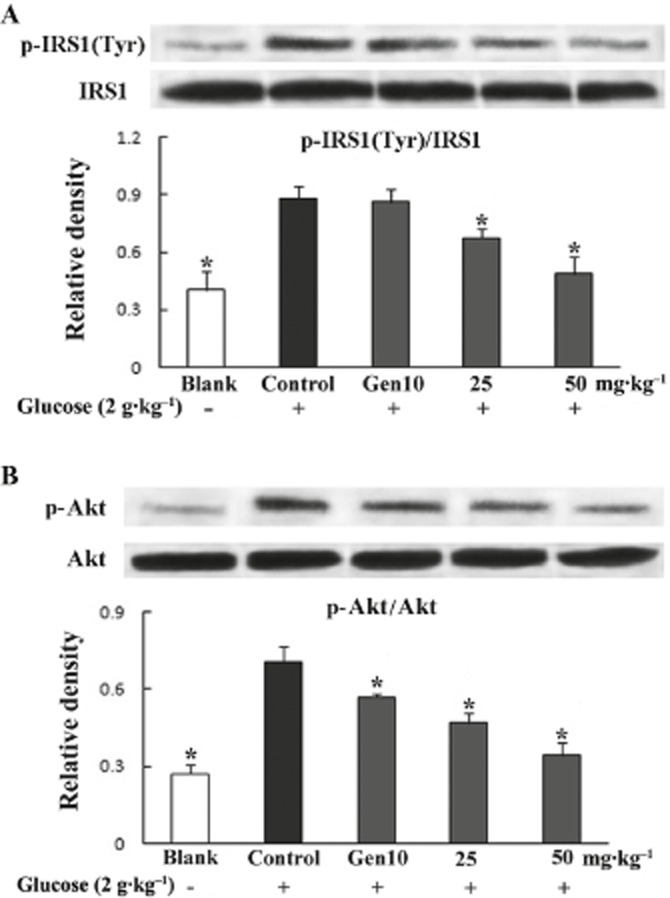
Genistein inhibited glucose-load induced phosphorylation of IRS1 (at tyrosine residues; A) and Akt (B) in adipose tissue from normal mice. Fasted mice were exposed to genistein and glucose as described in the glucose tolerance test. Epididymal adipose tissue was separated and homogenized at 30 min after glucose load. Phosphorylation of IRS1 and Akt were assayed by Western blot analysis. Data were derived from at least three mice. *P < 0.05 versus control.
Genistein inhibited IKKβ phosphorylation and reduced mRNA expressions for TNF-α and IL-6 in adipose tissue
An i.p. injection of Mac–CM induced the phosphorylation of IKKβ in epididymal adipose tissue from control mice. In line with this alteration, mRNA expressions for TNF-α and IL-6 in fat were increased to a higher level compared with that of the blank group. Genistein, as well as metformin and salicylate, reversed these Mac-CM-induced changes in inflammatory mediators dramatically (Figure 5A–C).
Figure 5.
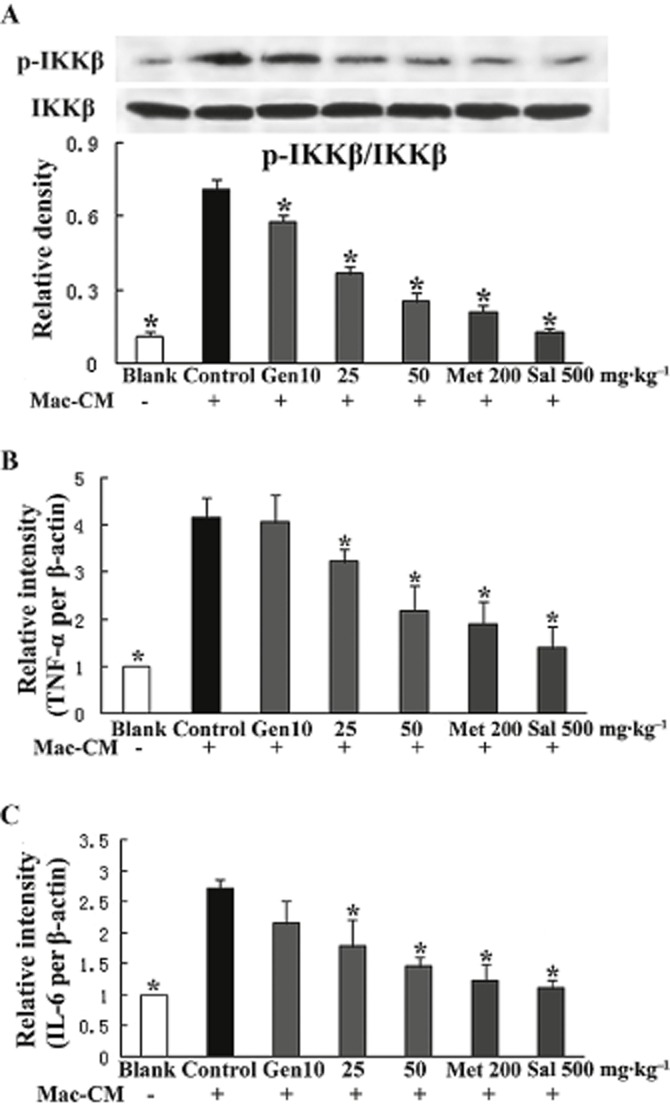
Genistein suppressed the inflammatory response in adipose tissue from mice treated with Mac–CM. Fasted mice were exposed to genistein, metformin or salicylate, respectively, 30 min before the treatment with Mac–CM. For the detection of p-IKKβ by Western blot (A), epididymal adipose tissue was separated and homogenized at 30 min after Mac–CM injection. For the measurement of gene expression of TNF-α and IL-6 by qPCR (B and C), adipose tissue homogenates were prepared at 2 h after Mac–CM injection. Data were derived from at least three mice. *P < 0.05 versus control.
Genistein modulated serine/tyrosine phosphorylation at IRS1 and restored Akt phosphorylation in mice exposed to Mac–CM
In mice exposed to Mac–CM, the effect of genistein on the insulin signal was different from that in normal mice. Mac–CM injection led to an increased serine phosphorylation of IRS1 and a lower level of IRS1 tyrosine phosphorylation after glucose load, and these changes in phosphorylation of serine/tyrosine residues were reversed by genistein (25 and 50 mg·kg−1), metformin or salicylate respectively (Figure 6A and B). As expected, genistein, as well as metformin and salicylate, restored the impaired phosphorylation of Akt in adipose tissue (Figure 6C).
Figure 6.
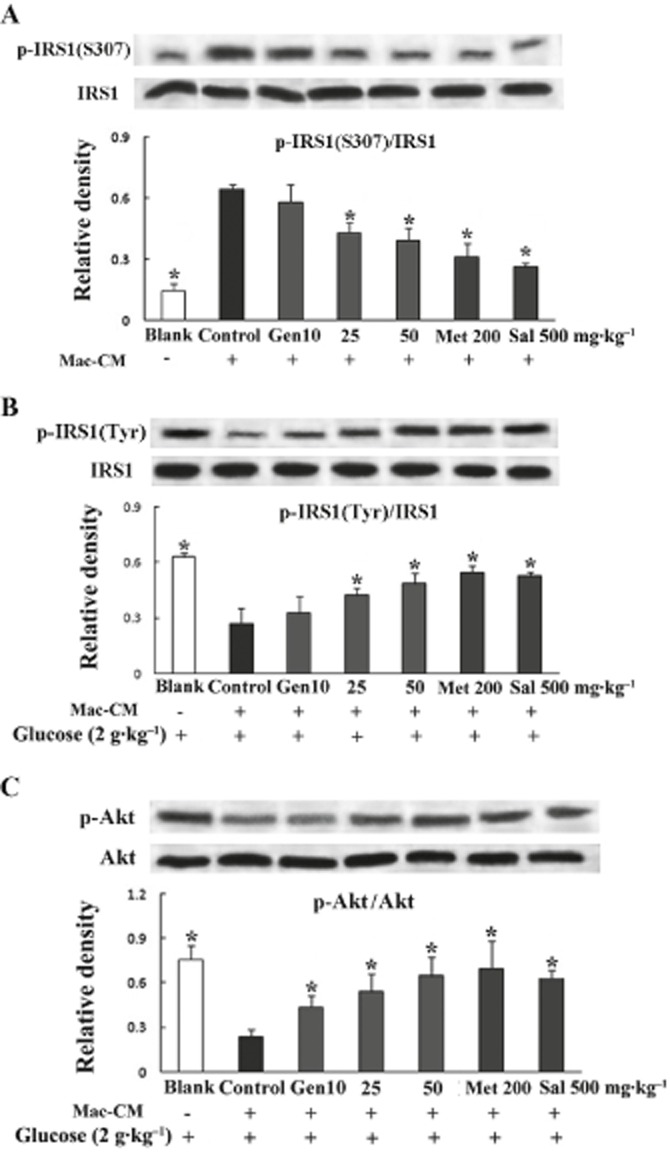
Genistein improved glucose-load induced insulin signalling in adipose tissue from mice treated with Mac–CM. Fasted mice were exposed to genistein, metformin or salicylate, respectively, then treated with Mac–CM and subsequent glucose load as described in the glucose tolerance test. For the detection of p-IRS1 at S307 (A), epididymal adipose tissue was separated and homogenized at 30 min after Mac–CM injection; for the assay of p-IRS1 at tyrosine residues (B) and p-Akt at T308 (C), adipose tissue homogenates were prepared at 30 min after glucose load. Data were derived from at least three mice. *P < 0.05 versus control.
Genistein affects insulin-mediated GLUT4 translocation in adipocytes
In 3T3-L1 adipocytes, insulin stimulation led to the translocation of GLUT4 to cell membranes, whereas genistein treatment markedly reduced the GLUT4 content in the plasma membrane fraction. Wortmannin, a specific inhibitor of PI3K, also inhibited insulin-mediated GLUT4 translocation (Figure 7A). In adipocytes exposed to Mac–CM, insulin-stimulated GLUT4 translocation to the membrane was decreased, and this change was inhibited by genistein (Figure 7B). Interestingly, we observed that the beneficial effect of genistein on GLUT4 translocation in the presence of Mac–CM was abolished by co-treatment with the AMPK inhibitor Compound C, suggesting that AMPK is involved in this effect of genistein.
Figure 7.
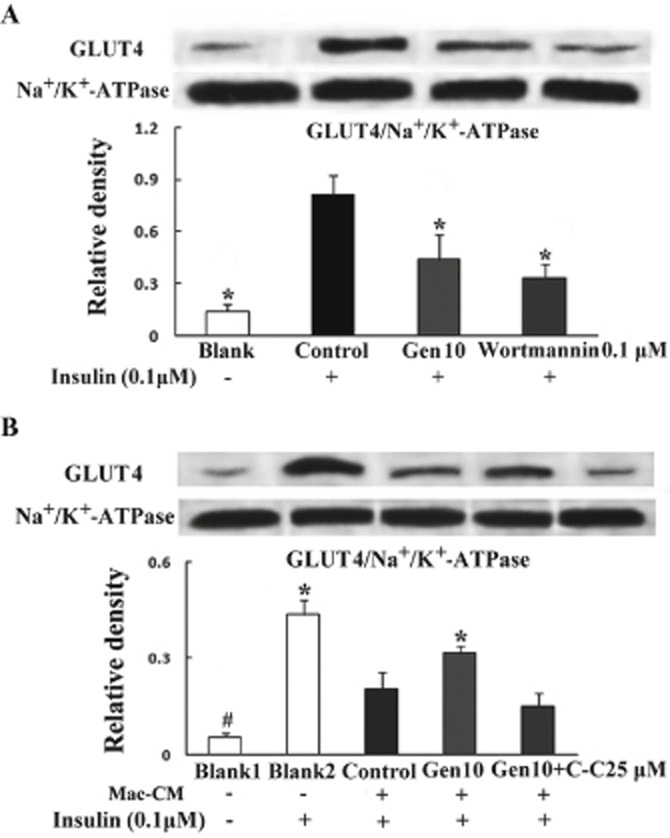
Genistein differently regulated insulin-mediated GLUT4 translocation under normal (A) and inflammatory conditions (B). Differentiated adipocytes were pretreated with genistein, wortmannin or genistein plus Compound C, respectively, for 30 min followed by stimulation with or without Mac–CM for another 30 min. Following exposure to insulin (100 nM) for 20 min, membrane protein was extracted and GLUT4 content was determined by Western blot analysis. Data were derived from three independent experiments. *P < 0.05 versus control, #P < 0.05 versus Blank2.
Genistein affects AMPK activation in adipocytes and fat
Because the AMPK inhibitor Compound C abolished the ability of genistein to restore insulin-mediated GLUT4 translocation, we investigated the potential effect of genistein on AMPK. In adipocytes, results from the elisa assay show that genistein (10 μM) increased AMPK activity under both normal and inflammatory conditions (Figure 8A and B). The Western blot analysis also demonstrated that genistein enhanced basal AMPK phosphorylation and significantly restored AMPK phosphorylation when adipocytes were exposed to Mac–CM (Figure 8C).
Figure 8.
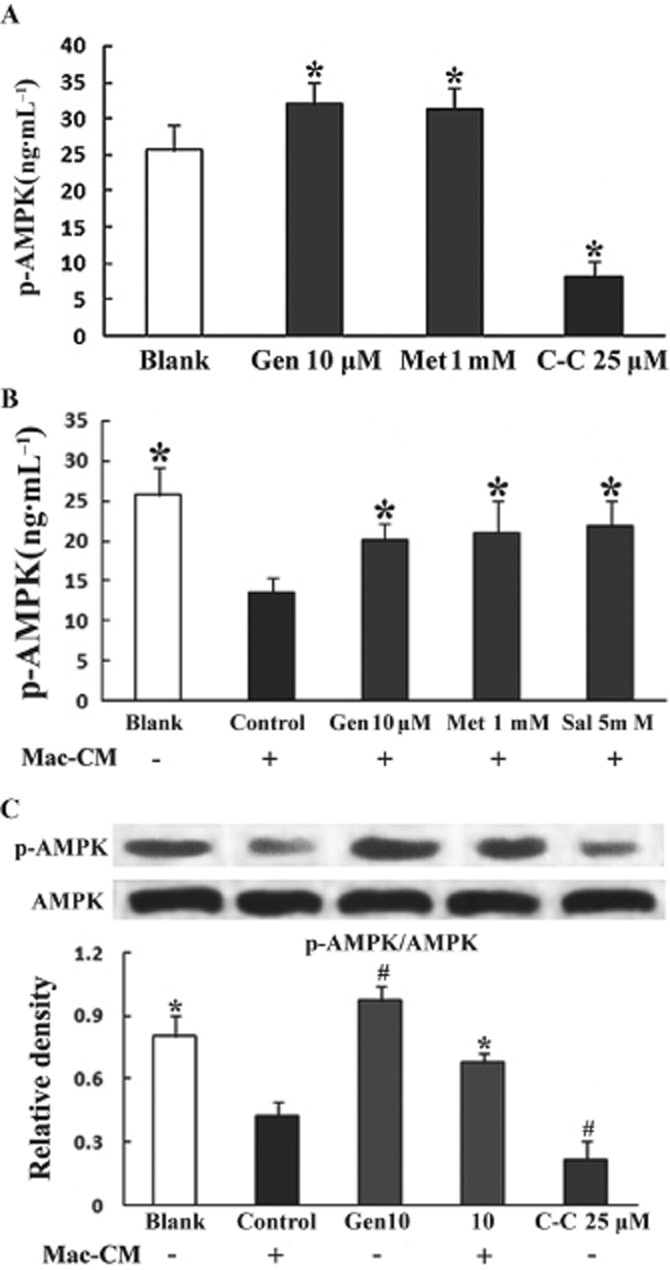
Genistein promoted AMPK activation in adipocytes. Differentiated adipocytes were serum-starved for 4 h in KRH before pretreatment with genistein, metformin or Compound C, respectively, for 30 min, then they were either untreated (A) or treated with (B) Mac–CM for 30 min followed by incubation in the presence of glucose (11 mM) for 30 min. The results are expressed as the mean ± SD (five to six independent experiments). *P < 0.05 versus Blank (A) or control (B). As for the detection of p-AMPK, adipocytes were pretreated with genistein or Compound C for 30 min followed by a 30 min stimulation of Mac–CM, then incubated in the presence of glucose (11 mM) for 30 min. p-AMPK (C) was detected by Western blot analysis. Data were derived from three independent experiments. *P < 0.05 versus control, #P < 0.05 versus Blank.
We also investigated the effect of genistein on AMPK in vivo. Treating mice with genistein enhanced basal AMPK phosphorylation in a dose-dependent manner in adipose tissue (Figure 9A). Ma–CM attenuated AMPK phosphorylation in vivo and this alteration was effectively reversed by genistein treatment (Figure 9B). These results demonstrate that genistein has a positive effect on AMPK activity in vivo.
Figure 9.
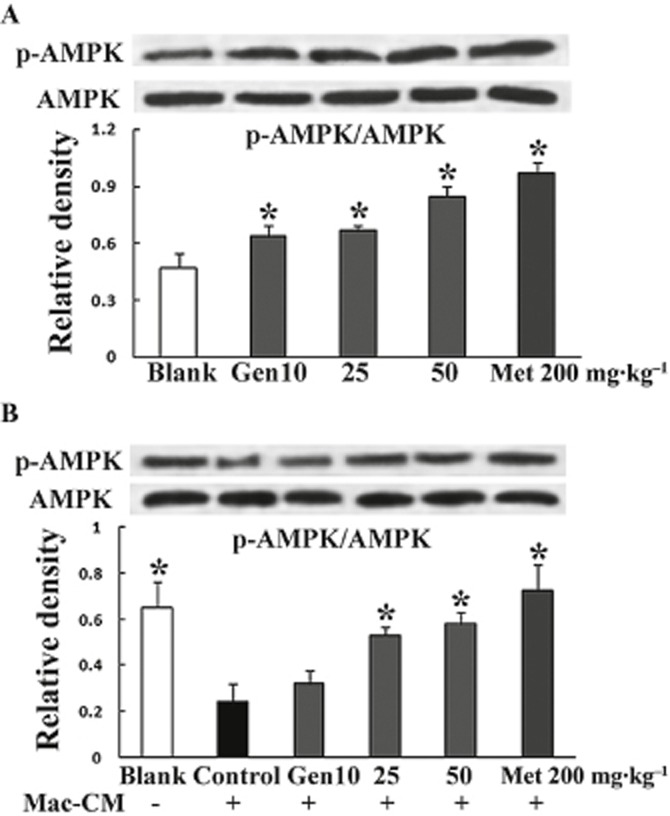
Genistein regulated AMPK phosphorylation in mouse fat. (A) Mice were administered genistein or metformin p.o. (B) Mice were administered genistein or metformin p.o. followed by an i.p. injection of Mac–CM. Two hours after the last treatment, epididymal fat was isolated for determination of AMPK phosphorylation by Western blot. Data were derived from three independent experiments. *P < 0.05 versus control.
Genistein restored insulin PI3K signalling by inhibiting inflammation in adipocytes
Mac–CM stimulation of adipocytes induced IKKβ phosphorylation, and this change was reversed by AICAR (Figure 10A), indicating that AMPK activation is involved in the suppression of inflammatory responses. Genistein decreased IKKβ phosphorylation and restored the reduced PI3K activity induced by the inflammatory insult. These effects of genistein were abolished by co-culture with Compound C (Figure 10A & B), suggesting that AMPK activation contributes to the anti-inflammatory effect of genistein and this leads to an improvement in insulin signalling.
Figure 10.
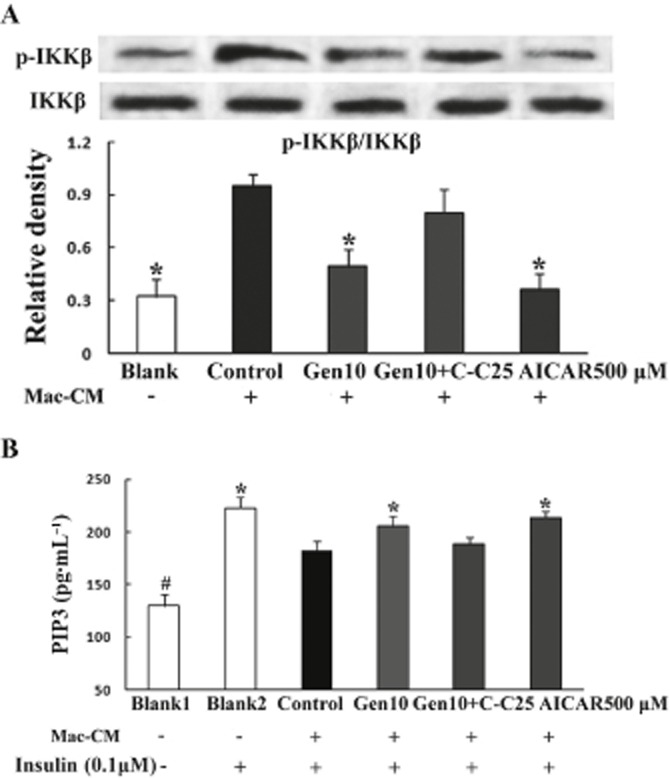
AMPK activation by genistein contributed to its anti-inflammatory effect implicated in the improvement in insulin PI3K signalling. (A) Adipocytes were pretreated with genistein, genistein plus Compound C or AICAR, respectively, for 30 min followed by stimulation with Mac–CM for another 30 min. IKKβ phosphorylation was determined by Western blot. Data were derived from three independent experiments. *P < 0.05 versus control. (B) Adipocytes were pretreated with genistein, genistein plus Compound C or AICAR for 30 min, then stimulated with Mac–CM for another 30 min followed by 20 min exposure to insulin (100 nM). The product of PIP3 was determined with an elisa kit for PI3K activity. The results are expressed as the mean ± SD (n = 5). *P < 0.05 versus control, #P < 0.05 versus Blank2.
Discussion and conclusions
Insulin regulates glucose homeostasis in skeletal muscle and adipose tissue by promoting glucose uptake through GLUT4 translocation. In the present study, we observed that genistein induced glucose intolerance in normal mice and inhibited insulin-stimulated GLUT4 in adipocytes. In contrast, genistein reversed the impaired glucose tolerance and inhibition of insulin-stimulated GLUT4 translocation induced in mice challenged with an inflammatory mediator. These results demonstrate that genistein can have opposite effects on the actions of insulin in adipocytes.
To clarify the effects of genistein on the actions of insulin in vivo, we first observed the effects of genistein on glucose tolerance in mice. Glucose load stimulates insulin secretion from pancreatic islets, and insulin in turn promotes glucose disposal. Administration of genistein induced glucose intolerance and reduced ISI in normal mice. These results suggest that genistein induced glucose intolerance by reducing insulin sensitivity. It has been shown previously that genistein counteracts the effects of insulin in adipocytes (Nomura et al., 2008; Szkudelska et al., 2008), and our findings further confirm this negative effect of genistein on insulin's actions in vivo.
According to previously reported data, the bioavailability of genistein (administered p.o.) is not good. To further confirm that the effects on glucose tolerance and ISI were due to genistein, we determined the plasma levels of genistein in mice by HPLC. We observed that after a single p.o. dose, the plasma concentration of genistein increased gradually, reached 263 ng·mL−1 at 60 min and then peaked at about 90 min, to 424 ng·mL−1; it then decreased to 153 ng·mL−1 at 120 min. From these measurements it was deduced that the higher levels of plasma genistein corresponded to the more marked effects of genistein on the blood glucose level in glucose tolerance tests. These changes in plasma levels of genistein in mice were similar to those reported previously in rats (Uehara et al., 2001; Kwon et al., 2007), although it seems that the bioavailability in mice is less than that in rats.
Insulin regulates glucose uptake in adipocytes through the insulin receptor/IRS1/PI3K/Akt pathways, along which tyrosine phosphorylation of IRS1 is a key control mechanism linking insulin receptor activation with downstream signalling. Genistein is a potent inhibitor of tyrosine PK (TPK), and we observed that IRS1 tyrosine phosphorylation in response to a glucose load was inhibited in adipose tissue from genistein-treated mice. This suggests that TPK inhibition is involved in the effect of genistein on IRS1. This inhibitory effect of genistein led to a decrease in the downstream PI3K signalling, as indicated by attenuated Akt phosphorylation. This finding is consistent with the observation that PP2 (4-amino-5-(4-chlorophenyl)-7 (t-butyl) pyrazolo [3,4-d] pyridine) and herbimycin (two tyrosine kinase inhibitors) inhibit insulin-stimulated muscle glucose transport (Wright et al., 2006). However, in contrast to these findings, Nomura et al. (2008) showed that genistein inhibits insulin-mediated glucose uptake without affecting Akt activity. These contradictory results may be due to differences in the assaying techniques. Nomura et al. directly determined Akt activity by using a specific peptide as the Akt substrate, but we measured the Akt phosphorylation level and the attenuated Akt phosphorylation we observed should be secondary to the inhibition of IRS1 tyrosine phosphorylation. In the present study, impaired insulin signalling is thought to be responsible for the glucose intolerance induced by genistein in normal mice. This finding is very interesting. For many years, the lower frequency of insulin resistance-associated diseases in Asian countries has been linked to the higher consumption of isoflavones (such as genistein)-containing soy-based products; a hypothesis that appears to contradict the findings of the present study. However, the results presented here were derived from an acute experiment that only focused on some specific effects of genistein. Whereas in Asian countries the genistein is consumed over a long period, therefore, other effects of genistein, such as activation of AMPK and its oestrogenic properties, as well as the cross-talk between these effects and its TPK-inhibiting abilities, might affect glucose homeostasis (Alonso et al., 2010; Arunkumar and Anuradha, 2012). In addition, it is possible that other factors such as species and gender differences in rodents and humans might play a role (Penza et al., 2006). Hence, further studies, in particular chronic experiments in different genders of rodents and humans, are needed to obtain a better understanding of the effect of genistein on glucose homeostasis.
Inflammation is closely associated with insulin resistance. Quite a few studies have demonstrated that the anti-inflammatory activity of genistein is involved in its anti-diabetic actions (Elmarakby et al., 2011; Valsecchi et al., 2011; Babu et al., 2012). In the present study, Mac–CM challenge induced insulin resistance in mice, as seen by the impaired glucose disposal and attenuated ISI. These effects of Mac–CM were counteracted by genistein treatment, demonstrating it has a positive effect on glucose homeostasis in mice under inflammatory conditions and also suggest that these effects of genistein are due to its anti-inflammatory properties. As expected, we observed significant phosphorylation of IKKβ in the adipose tissue from mice exposed to Mac–CM challenge, and this was accompanied by a significant mRNA expression of TNF-α and IL-6; all these changes were reversed by genistein as well as metformin and salicylate. These results confirm that genistein acts as an anti-inflammatory in adipose tissue and are in line with its influence on insulin signalling. IRS1 serine phosphorylation is a key event linking inflammation and insulin resistance (Kanety et al., 1995; Gual et al., 2005). In the present study, we observed that Mac–CM stimulation induced serine phosphorylation (S307) of IRS1, attenuated IRS1 tyrosine phosphorylation and Akt phosphorylation, while genistein reversed these changes, leading to an improvement in downstream PI3K signalling. This positive effect of genistein on insulin signalling is thought to be responsible for its beneficial effect on glucose tolerance in mice under inflammatory conditions.
The anti-diabetic agent metformin ameliorates insulin resistance by activation of AMPK and its anti-inflammatory activity has been studied both in vitro and in vivo (Hattori et al., 2006; Isoda et al., 2006; Tsoyi et al., 2011). Genistein is also an AMPK activator (Hsu et al., 2011; Arunkumar and Anuradha, 2012). Hence, we investigated whether there is a link between its AMPK-activating effect and its anti-inflammatory property associated with the regulation of insulin signalling. In 3T3-L1 adipocytes, we observed the different regulation of insulin-mediated GLUT4 translocation by genistein, which is in agreement with the in vivo data about its influence on insulin IRS1/Akt signalling. More interestingly, under inflammatory conditions, the positive effect of genistein on GLUT4 translocation was abolished by the AMPK inhibitor Compound C, indicating that the activation of AMPK is involved in this effect of genistein. We then determined the effect of genistein on the activity and phosphorylation of AMPK in adipocytes, and the results confirmed its ability to activate AMPK. The in vivo data also support this finding. IKKβ activation is a key event linking inflammation and insulin resistance because it inhibits the response of IRS1 to insulin (Feinstein et al., 1993; Cai et al., 2005). We observed that genistein inhibits IKKβ phosphorylation induced by Mac–CM stimulation and restores insulin-stimulated activation of PI3K in adipocytes, similar to its positive regulation of IRS1 function under inflammatory conditions in vivo. More importantly, Compound C attenuated the beneficial effects of genistein on both IKKβ phosphorylation and PI3K activity, strongly suggesting that the AMPK-activating property of genistein contributes to its ability to inhibit inflammation. These results demonstrate that genistein regulates glucose homeostasis under insulin-resistant conditions in an AMPK-dependent fashion.
In conclusion, our study shows that genistein has opposite effects on insulin IRS1/Akt signalling in adipose tissue under normal and inflammatory conditions, its negative effect on TPK and positive regulation of AMPK play a key role in these actions respectively (Figure 11). Importantly, genistein is a multifunctional agent that has other properties that may also be involved in its effects on glucose homeostasis. Therefore, more studies need to be done to determine the long-term effects of genistein on glucose homeostasis.
Figure 11.
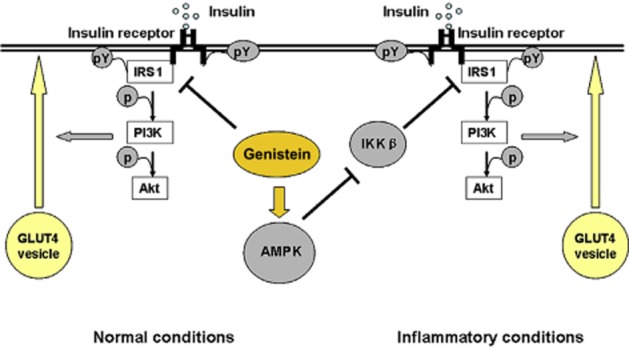
Proposed action pathway of genistein to regulate insulin IRS1/Akt signalling in adipose tissue. Under normal conditions, genistein suppressed insulin tyrosine phosphorylation of IRS1, leading to a suppression of insulin-mediated GLUT4 translocation. In contrast, under inflammatory conditions, it inhibited IKKβ phosphorylation via activation of AMPK and this resulted in the restoration of GLUT4 translocation mediated by the insulin IRS1/Akt pathway.
Acknowledgments
This work is funded by the National Natural Science Foundation of China (Grant no. 81072976) and the Fundamental Research Funds for the Central Universities (Program No. JKZ2011018). The work in our department was supported by 2011’ Program for Excellent Scientific and Technological Innovation Team of Jiangsu Higher Education.
Glossary
- AMPK
AMP-activated PK
- GLUT4
glucose transporter-4
- IKKβ
IκB kinase β
- IRS1
insulin receptor substrate-1
- ISI
inuslin sensitivity index
- Mac–CM
macrophage-derived conditioned medium
- TPK
tyrosine PK
Conflict of interest
None.
References
- Alonso A, González-Pardo H, Garrido P, Conejo NM, Llaneza P, Díaz F, et al. Acute effects of 17 β-estradiol and genistein on insulin sensitivity and spatial memory in aged ovariectomized female rats. Age (Dordr) 2010;32:421–434. doi: 10.1007/s11357-010-9148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunkumar E, Anuradha CV. Genistein promotes insulin action through adenosine monophosphate-activated protein kinase activation and p70 ribosomal protein S6 kinase 1 inhibition in the skeletal muscle of mice fed a high energy diet. Nutr Res. 2012;32:617–625. doi: 10.1016/j.nutres.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Atteritano M, Marini H, Minutoli L, Polito F, Bitto A, Altavilla D, et al. Effects of the phytoestrogen genistein on some predictors of cardiovascular risk in osteopenic, postmenopausal women: a two-year randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. 2007;92:3068–3075. doi: 10.1210/jc.2006-2295. [DOI] [PubMed] [Google Scholar]
- Babu PV, Si H, Fu Z, Zhen W, Liu D. Genistein prevents hyperglycemia-induced monocyte adhesion to human aortic endothelial cells through preservation of the cAMP signaling pathway and ameliorates vascular inflammation in obese diabetic mice. J Nutr. 2012;142:724–730. doi: 10.3945/jn.111.152322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazuine M, van den Broek PJ, Maassen JA. Genistein directly inhibits GLUT4-mediated glucose uptake in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2005;326:511–514. doi: 10.1016/j.bbrc.2004.11.055. [DOI] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKKβ and NF-κB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmarakby AA, Ibrahim AS, Faulkner J, Mozaffari MS, Liou GI, Abdelsayed R. Tyrosine kinase inhibitor, genistein, reduces renal inflammation and injury in streptozotocin- induced diabetic mice. Vascul Pharmacol. 2011;55:149–156. doi: 10.1016/j.vph.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A. Tumor necrosis factor-α suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem. 1993;268:26055–26058. [PubMed] [Google Scholar]
- Fu Z, Gilbert ER, Pfeiffer L, Zhang Y, Fu Y, Liu D. Genistein ameliorates hyperglycemia in a mouse model of nongenetic type 2 diabetes. Appl Physiol Nutr Metab. 2012;37:480–488. doi: 10.1139/h2012-005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. 2011;121:4903–4915. doi: 10.1172/JCI58577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Zhang B, Lotz M. Protein tyrosine kinase activation is required for lipopolysaccharide induction of cytokines in human blood monocytes. J Immunol. 1993;151:6692–6700. [PubMed] [Google Scholar]
- Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Haneishi A, Takagi K, Asano K, Nakamura S, Kagawa N, Yamada K. Genistein stimulates the insulin-dependent signaling pathway. Front Biosci (Elite Ed) 2011;3:1534–1540. doi: 10.2741/e354. [DOI] [PubMed] [Google Scholar]
- Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–1188. doi: 10.1161/01.HYP.0000221429.94591.72. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu MH, Savas U, Lasker JM, Johnson EF. Genistein, resveratrol, and 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside induce cytochrome P450 4F2 expression through an AMP-activated protein kinase-dependent pathway. J Pharmacol Exp Ther. 2011;337:125–136. doi: 10.1124/jpet.110.175851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26:611–617. doi: 10.1161/01.ATV.0000201938.78044.75. [DOI] [PubMed] [Google Scholar]
- Kanety H, Feinstein R, Papa MZ, Hemi R, Karasik A. Tumor necrosis factor alpha- induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin stimulated tyrosine phosphorylation of IRS-1. J Biol Chem. 1995;270:23780–23784. doi: 10.1074/jbc.270.40.23780. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SH, Kang MJ, Huh JS, Ha KW, Lee JR, Lee SK, et al. Comparison of oral bioavailability of genistein and genistein in rats. Int J Pharm. 2007;337:148–154. doi: 10.1016/j.ijpharm.2006.12.046. [DOI] [PubMed] [Google Scholar]
- Liu K, Luo T, Zhang Z, Wang T, Kou J, Liu B, et al. Modified Si-Miao-San extract inhibits inflammatory response and modulates insulin sensitivity in hepatocytes through an IKKβ/IRS-1/Akt-dependent pathway. J Ethnopharmacol. 2011;136:473–479. doi: 10.1016/j.jep.2011.01.051. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Takahashi T, Nagata N, Tsutsumi K, Kobayashi S, Akiba T, et al. Inhibitory mechanisms of flavonoids on insulin-stimulated glucose uptake in MC3T3-G2/PA6 adipose cells. Biol Pharm Bull. 2008;31:1403–1409. doi: 10.1248/bpb.31.1403. [DOI] [PubMed] [Google Scholar]
- Penza M, Montani C, Romani A, Vignolini P, Pampaloni B, Tanini A, et al. Genistein affects adipose tissue deposition in a dose-dependent and gender-specific manner. Endocrinology. 2006;147:5740–5751. doi: 10.1210/en.2006-0365. [DOI] [PubMed] [Google Scholar]
- Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocr Metab. 2008;93:S64–S73. doi: 10.1210/jc.2008-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauter AP, Martins A, Borges C, Mota-Filipe H, Pinto R, Sepodes B, et al. Antihyperglycaemic and protective effects of flavonoids on streptozotocin-induced diabetic rats. Phytother Res. 2010;24:S133–S138. doi: 10.1002/ptr.3017. [DOI] [PubMed] [Google Scholar]
- Sajan MP, Bandyopadhyay G, Miura A, Standaert ML, Nimal S, Longnus SL, et al. AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC. Am J Physiol Endocrinol Metab. 2010;298:E179–E192. doi: 10.1152/ajpendo.00392.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkudelska K, Nogowski L, Szkudelski T. Genistein, a plant-derived isoflavone, counteracts the antilipolytic action of insulin in isolated rat adipocytes. J Steroid Biochem Mol Biol. 2008;109:108–114. doi: 10.1016/j.jsbmb.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Tsoyi K, Jang HJ, Nizamutdinova IT, Kim YM, Lee YS, Kim HJ, et al. Metformin inhibits HMGB1 release in LPS-treated RAW 264.7 cells and increases survival rate of endotoxaemic mice. Br J Pharmacol. 2011;162:1498–1508. doi: 10.1111/j.1476-5381.2010.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara M, Ohta A, Sakai K, Suzuki K, Watanabe S, Adlercreutz H. Dietary fructooligosaccharides modify intestinal bioavailability of a single dose of genistein and daidzein and affect their urinary excretion and kinetics in blood of rats. J Nutr. 2001;131:787–795. doi: 10.1093/jn/131.3.787. [DOI] [PubMed] [Google Scholar]
- Valsecchi AE, Franchi S, Panerai AE, Rossi A, Sacerdote P, Colleoni M. The soy isoflavone genistein reverses oxidative and inflammatory state, neuropathic pain, neurotrophic and vasculature deficits in diabetes mouse model. Eur J Pharmacol. 2011;650:694–702. doi: 10.1016/j.ejphar.2010.10.060. [DOI] [PubMed] [Google Scholar]
- Wright DC, Geiger PC, Han DH, Holloszy JO. Are tyrosine kinases involved in mediating contraction-stimulated muscle glucose transport? Am J Physiol Endocrinol Metab. 2006;290:E123–E128. doi: 10.1152/ajpendo.00280.2005. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem. 2010;285:19051–19059. doi: 10.1074/jbc.M110.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]