

Abstract
BACKGROUND AND PURPOSE
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder with no effective treatment. Fasudil hydrochloride (fasudil), a potent rho kinase (ROCK) inhibitor, is useful for the treatment of ischaemic diseases. In previous reports, fasudil improved pathology in mouse models of Alzheimer's disease and spinal muscular atrophy, but there is no evidence in that it can affect ALS. We therefore investigated its effects on experimental models of ALS.
EXPERIMENTAL APPROACH
In mice motor neuron (NSC34) cells, the neuroprotective effect of hydroxyfasudil (M3), an active metabolite of fasudil, and its mechanism were evaluated. Moreover, the effects of fasudil, 30 and 100 mg·kg−1, administered via drinking water to mutant superoxide dismutase 1 (SOD1G93A) mice were tested by measuring motor performance, survival time and histological changes, and its mechanism investigated.
KEY RESULTS
M3 prevented motor neuron cell death induced by SOD1G93A. Furthermore, M3 suppressed both the increase in ROCK activity and phosphorylated phosphatase and tensin homologue deleted on chromosome 10 (PTEN), and the reduction in phosphorylated Akt induced by SOD1G93A. These effects of M3 were attenuated by treatment with a PI3K inhibitor (LY294002). Moreover, fasudil slowed disease progression, increased survival time and reduced motor neuron loss, in SOD1G93A mice. Fasudil also attenuated the increase in ROCK activity and PTEN, and the reduction in Akt in SOD1G93A mice.
CONCLUSIONS AND IMPLICATIONS
These findings indicate that fasudil may be effective at suppressing motor neuron degeneration and symptom progression in ALS. Hence, fasudil may have potential as a therapeutic agent for ALS treatment.
Keywords: amyotrophic lateral sclerosis, fasudil hydrochloride, motor neuron degeneration, rho kinase (ROCK), PTEN/Akt pathway
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal and incurable neurodegenerative disease characterized by an age-related progressive degeneration of both upper and lower motor neurons (Leigh and Ray-Chaudhuri, 1994). Approximately 90% of ALS cases are sporadic, while the remaining 10% are familial (fALS; Gurney, 1997). The sporadic and familial forms of the disease have some features in common, and a common pathogenesis has been identified (Boillee et al., 2006). Mutations of the gene coding for the enzyme copper–zinc superoxide dismutase 1 (SOD1) account for approximately 20% of the cases of fALS and represent a major known cause of the disease (Rosen et al., 1993). Moreover, several ALS-linked genes have been identified about 43 kDa transactivation response DNA-binding protein (TDP-43), fused in sarcoma/translocated in liposarcoma, and others (Vande Velde et al., 2011). Within the last 10 years, new models based on these genes have been established, and these have improved the mechanistic understanding of the pathogenesis of ALS (Jackson et al., 2010; Vande Velde et al., 2011). However, despite extensive investigations, the mechanism of motor neuron death in ALS is still unknown, and no effective therapies have been developed to retard or prevent the progression of ALS.
Rho kinase (ROCK), one of the serine/threonine kinases mediating the activities of rho GTPases, consists of two family members, ROCK1 and ROCK2, with redundant functions (Ishizaki et al., 1996). ROCK reportedly activates the caspase signalling cascades and promotes cellular apoptosis (Coleman et al., 2001), indicating its involvement in cell death. In a previous study, fasudil hydrochloride (fasudil), which is a potent ROCK inhibitor, was shown to improve the pathology in mouse models of brain ischaemia (Yamashita et al., 2007), Alzheimer's disease (Hou et al., 2012) and spinal muscular atrophy (SMA; Bowerman et al., 2012). These results suggest that fasudil has potential as a treatment for many neurodegenerative disorders. However, there is no data indicating whether fasudil has similar effects in ALS.
In the present study, we investigated whether fasudil might exert protective effects against motor neuron death in SOD1G93A mice (an ALS model; see Methods) and therefore whether it might be a promising agent for the treatment of ALS.
Methods
Expression plasmids, cell culture and transfection
Empty vector (mock), human wild-type (WT) SOD1 (SOD1wt) and mutant SOD1G93A vector with either enhanced GFP or Myc-tagged were used. These vectors were provided by Dr Gen Sobue (Nagoya University Graduate School of Medicine, Nagoya, Japan). Mice motor neuron (NSC34) cells, a hybrid neuroblastoma × spinal cord cell line purchased from Cosmo Bio Co. Ltd. (Tokyo, Japan), were maintained in DMEM containing 10% FBS (Valeant, Costa Mesa, CA, USA), 100 U·mL−1 penicillin (Meiji Co. Ltd., Tokyo, Japan) and 100 μg·mL−1 streptomycin (Meiji Co. Ltd.) under a humidified atmosphere of 95% air and 5% CO2 at 37°C. The cells were passaged by trypsinization every 3–4 days. Mock, SOD1wt or SOD1G93A with either enhanced GFP or Myc-tagged was transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) by following the manufacturer's instructions.
Chemicals
Fasudil hydrochloride (fasudil) was synthesized by Asahi Kasei Co. Ltd. (Tokyo, Japan). Hydroxyfasudil (M3), an active metabolite of fasudil, and Y-27632 [(R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide, a ROCK inhibitor] were purchased from Calbiochem (San Diego, CA, USA).
Cell death assay
NSC34 cells were seeded at a density of 8000 cells per well into 96-well plates with DMEM, and transfected using Lipofectamine 2000 (Invitrogen) mixed with 2 μg·mL−1 of plasmid vector in DMEM for 6 h. After 6 h, the cell culture medium was replaced with fresh DMEM and the incubation allowed to proceed for a further 42 h. Then, the cells were transferred to serum-free DMEM and immediately treated with M3 or Y-27632, each at a final concentration of 0.3, 3 or 30 nM, or else treated with M3 (30 nM) at 15, 21, 24 and 26 h after serum deprivation. Cell death was assessed based on combination staining with Hoechst 33342 and propidium iodide (PI); this was performed 27 h after M3 or Y-27632 treatment. In the PI3K/Akt inhibitor (LY294002) study, the transfected cells were placed in serum-free DMEM and immediately treated with M3 (30 nM). After 12 h of serum deprivation, the cells were treated with LY294002 at a final concentration of 0.3, 1 or 10 μM. Images were collected by means of an Olympus IX70 inverted epifluorescence microscope (OLYMPUS, Tokyo, Japan). The total number of cells was counted and the percentage of PI-positive cells calculated as a measure of dead cells, as described previously (Shimazawa et al., 2010). A total of at least 200 cells per condition were counted, in a blind manner, using image-processing software (Image-J version 1.33f; National Institutes of Health, Bethesda, MD, USA).
Immunoblot analysis
NSC34 cells or mice spinal cord were lysed using a buffer (RIPA buffer; Sigma-Aldrich, St. Louis, MO, USA) with protease (Sigma-Aldrich) and phosphatase inhibitor cocktails (Sigma-Aldrich). The protein concentration was measured by comparison with a known concentration of BSA using a bicinchoninic acid Protein Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Equal amounts of protein in sample buffer with 10% 2-mercaptoethanol were subjected to SDS-PAGE using 5–20% gradient gels (SuperSep Ace; Wako Pure Chemicals, Osaka, Japan), and the separated proteins were transferred onto a PVDF membrane (Immobilon-P; Millipore Corporation, Bedford, MA, USA). After blocking with Blocking One-P (Nacalai Tesque, Kyoto, Japan), membranes were incubated with the following primary antibodies: rabbit anti-ROCK1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-ROCK2 (Santa Cruz), rabbit anti-phospho-Akt (Cell Signaling, Beverly, MA, USA), rabbit anti-Akt (Cell Signaling), rabbit anti-phosphatase and tensin homologue deleted on chromosome 10 (PTEN; Cell Signaling), rabbit anti-PTEN (Cell Signaling), rabbit anti-phospho-adducin (Abcam, Cambridge, MA, USA), rabbit anti-α-adducin (Abcam), rabbit anti-SOD1 (Cell Signaling), mouse anti-myc (Cell Signaling) and mouse anti-β-actin (Sigma-Aldrich). Subsequently, the membrane was incubated with peroxidase goat anti-rabbit or goat anti-mouse IgG (Pierce, Rockford, IL, USA) as the secondary antibody. Band intensities were measured using an ImmunoStar LD (Wako Pure Chemicals).
ROCK activity assay
ROCK activity was assassed by use of the CycLex Rho-kinase Assay Kit (CycLex Co. Ltd., Nagano, Japan) by following manufacturer's recommendations. Briefly, NSC34 cells were plated in 10 cm dishes at 1.0 × 107 cells per dish, and transfected using Myc-tagged mock, SOD1WT or SOD1G93A for 48 h. After 27 h with or without M3 treatment, cells were immediately prepared in extraction buffer [25 mM Tris-HCl, pH 7.4, 150 mM sodium chloride, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 5% glycerol, 10 mM β-mercaptoethanol and protease (Sigma-Aldrich) and phosphatase inhibitor cocktails (Sigma-Aldrich)] at 4°C, then centrifuged at 12 000× g to obtain the lysates. The lysates were added to precoated plates with myosin-binding subunit of myosin phosphate MBS, including a threonine residue that is phosphorylated by ROCK, for 60 min at room temperature. After the plated lysates had been washed, HRP-conjugated anti-phospho-specific MBS threonine-697 specific antibody was applied to the wells and incubated for 1 h at room temperature. The products were developed by incubation with the HRP substrate, tetramethylbenzidine, at room temperature for 10 min. The reaction was stopped by adding stop solution containing 0.5 M H2SO4. The coloured products were quantified by spectrophotometry at 450 nm. Purified ROCK (CycLex Co. Ltd.) was used as a positive control.
Animals
Transgenic mice overexpressing SOD1G93A [B6SJL-Tg (SOD1-G93A) 1Gur·J−1] were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The hemizygous SOD1G93A mice were maintained by mating transgenic male mice with WT female mice. Sixty-five mice were used in the experiments. Mouse genotypes were determined by PCR analysis, as previously reported (Ito et al., 2009; Tanaka et al., 2011). WT littermates served as controls. All mice were housed at 24 ± 2 °C under a 12 h light-dark cycle and had ad libitum access to food and water. Fasudil was diluted in water and administered in drinking water to SOD1G93A mice from 5 weeks until the experimental endpoint. Vehicle-treated mice received water. To determine the doses of fasudil, we administered fasudil 100 mg·kg−1 dissolved in drinking water to 4–6-week-old WT male mice and collected their plasma as a pre-test. In liver, fasudil is metabolized into M3, which has pharmacological effects. Blood concentrations of the total amounts of fasudil and M3 were determined; the maximum (Cmax) and minimum (trough levels) concentrations of total fasudil in plasma were approximately 3 and 1 μM respectively (Table 1). Hence, in the present study, we decided to use the following two doses of fasudil hydrochloride: 30 (a low dose) and 100 mg·kg−1 (a high dose). All animal care and experimental procedures were approved and monitored by the Institutional Animal Care and Use Committee of Gifu Pharmaceutical University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Table 1.
Blood concentrations of total fasudil in WT mice
| Plasma concentration (μM) | ||||||
|---|---|---|---|---|---|---|
| No. | Age (weeks) | Weight (g) | Fasudil | M3 | Total | |
| Cmax | 1 | 6 | 21 | 0.05 | 2.40 | 2.45 |
| 2 | 5 | 22 | 0.07 | 1.67 | 1.74 | |
| 3 | 4 | 13 | 5.26 | 0.93 | 6.19 | |
| 4 | 4 | 15 | 1.99 | 1.34 | 3.33 | |
| 5 | 4 | 14 | 1.28 | 1.07 | 2.35 | |
| Trough levels | 6 | 6 | 20 | 0.02 | 1.12 | 1.14 |
| 7 | 5 | 18 | 0.06 | 0.71 | 0.77 | |
| 8 | 4 | 14 | 0.48 | 0.57 | 1.05 | |
| 9 | 4 | 11 | 0.04 | 0.09 | 0.13 | |
| 10 | 4 | 15 | 0.35 | 1.69 | 2.04 | |
To determine the doses of fasudil, 4–6-week-old male WT mice were administered fasudil 100 mg·kg−1 dissolved in their drinking water and their plasma collected, as a pre-test. In liver, fasudil is metabolized into M3, which has pharmacological effects. Blood concentrations of fasudil, M3 and their total are shown. The maximum (Cmax) and minimum (trough levels) concentrations of total fasudil in plasma were approximately 3 and 1 μM respectively. Therefore, with reference to a previous study, we decided to use doses of fasudil hydrochloride of 30 (a low dose) and 100 mg·kg−1 (a high dose) for further study.
Symptomatic analysis
Mice (vehicle, n = 15; fasudil 30 mg·kg−1, n = 13 and fasudil 100 mg·kg−1, n = 12) were tested for their ability to maintain balance on a rod rotating at 5 r.p.m. using a rotarod apparatus (Bio Medica Ltd., Osaka, Japan), as described previously (Tanaka et al., 2012). To train the mice to the apparatus, they were allowed to adjust to balancing on the rod as it rotated for 600 s each time for a week from 4 weeks old. After this period of adaptation, rotarod performance on the rod was evaluated starting at 5 weeks old. Each experimental session consisted of three trials (10 min per trial; intervals between trials 2 min), and the best performance of three trials was recorded. We performed such sessions once a week from 5 to 9 weeks old, and then twice a week until the animals became moribund. The disease onset was defined as the day when a mouse first dropped off the rotarod within 600 s, as described previously (Shimazawa et al., 2010; Tanaka et al., 2012). The age of death was taken as an animal's inability to right itself after 15 s. The date on which the animal showed signs of dying indicated the experimental endpoint, and that date was recorded.
Tissue preparation
To obtain tissues for immunohistochemical analysis, SOD1G93A (vehicle, n = 3; fasudil 30 mg·kg−1, n = 4) and WT (n = 4), mice were anaesthetized with sodium pentobarbital (Nacalai Tesque) at 80 mg·kg−1, then perfused with 4% (w v-1) paraformaldehyde solution in 0.01 M PBS at pH 7.4. Spinal cord tissues were removed after a 15 min perfusion at 4°C and immersed in the same fixative solution for 24 h, then soaked in 25% (w v-1) sucrose solution at 4°C for 1 day. Embedded tissues were immediately frozen in liquid nitrogen and stored at −80°C. Serial transverse sections were cut on a cryostat at a thickness of 14 μm and used for cresyl violet staining.
Data analysis
Data are presented as means ± SEM. Statistical comparisons were made by Dunnett's test or Student's t-test, using StatView version 5.0 (SAS Institute, Cary, NC, USA). Statistical analysis of the cumulative probability of survival was performed using the Kaplan–Meier life test. Statistical analysis of the mean age of disease onset and survival was performed, with a value of P < 0.05 being considered to indicate statistical significance.
Results
M3 protects motor neurons against the cell death resulting from SOD1G93A-induced neurotoxicity
We first examined the effects of M3, an active metabolite of fasudil, on SOD1G93A-induced motor neuron degeneration. Representative photographs of Hoechst 33342-staining and PI-staining are shown (Figure 1A). Hoechst 33342 stains all cells (live and dead), whereas PI stains only dead cells. At concentrations of 3–30 nM, M3 reduced SOD1G93A-induced cell death in a concentration-dependent manner, its effect being significant at 3 nM (P < 0.05) and 30 nM (P < 0.01; Figure 1B). Additionally, M3 had a neuroprotective effect on SOD1G93A-expressing cells at 15 h after serum deprivation (Supporting Information Figure S1). Moreover, we investigated whether M3 induced this neuroprotective effect by reducing the expression of mutant SOD1. M3 treatment had no effect on myc expression (Supporting Information Figure S2), suggesting that M3 did not induce its neuroprotective effect by altering the expression of mutant SOD1. Next we investigated the effect of Y-27632, another ROCK inhibitor, on SOD1G93A-induced motor neuron death. Representative photographs of Hoechst 33342-staining and PI-staining are shown (Supporting Information Figure S3A). As with M3, concentrations of 3–30 nM Y-27632 suppressed SOD1G93A-induced cell death in a concentration-dependent manner, its effect, too, being significant at 3 (P < 0.05) and 30 nM (P < 0.01; Supporting Information Figure S3B). Together, these data suggest that ROCK is involved in the motor neuron cell death induced by SOD1G93A, and that M3 protects against such cell death.
Figure 1.

M3 protected motor neurons against the cell death resulting from mutant SOD1G93A-induced neurotoxicity. (A) Representative fluorescence microscopic images of Hoechst 33342 (blue) and PI (red) staining, 27 h after serum deprivation, of NSC34 cells transfected with enhanced GFP-tagged (green), WT SOD1 (SOD1WT) or SOD1G93A. Scale bar represents 50 μm. (B) Quantitative analysis of Hoechst- and PI-positive NSC34 cells expressing SOD1WT or SOD1G93A after 27 h of serum deprivation. Each column represents the mean ± SEM (n = 6). $$P < 0.01 versus SOD1G93A (Student's t-test), *P < 0.05, **P < 0.01 versus vehicle (Dunnett's test).
M3 attenuates the elevated ROCK activity associated with SOD1G93A-induced neurotoxicity
Next, we determined whether SOD1G93A affects the expressions and/or activities of ROCK proteins. There was no difference among mock-, SOD1WT- and SOD1G93A-expressing NSC34 cells in the expression of either ROCK1 or ROCK2 protein at 48 or 75 h after transfection (Figure 2A and B). Moreover, 27 h treatment with M3 had no effect on the expressions of ROCK1 and ROCK2 (Figure 2A and B). On the other hand, ROCK activity at 75 h after transfection was significantly greater in the SOD1G93A-transfected cells than in those transfected with mock or SOD1WT, although no activity was detected in either group of cells at 48 h (Figure 2C). Furthermore, 27 h treatment with M3 reduced the elevated ROCK activity (Figure 2C). It has been reported that ROCK activity can be increased by activated caspase-3 and granzyme B, which are enzymes with critical roles in mammalian apoptosis (Sebbagh et al., 2001; 2005). SOD1G93A-induced motor neuron cell death was significantly increased (compared to mock and SOD1WT) at 75 h, but not at 48 h, after transfection (Supporting Information Figure S4). These results suggest that ROCK activation by SOD1G93A may play a pivotal role in SOD1G93A-induced motor neuron cell death.
Figure 2.

M3 attenuated the elevated ROCK activity associated with SOD1G93A-induced neurotoxicity. (A, B) NSC34 cells were lysed after transfection with Myc-tagged mock, SOD1WT or SOD1G93A for 48 h, then treated with PBS or M3 (30 nM) for 27 h. (A) Expressions of ROCK1 and ROCK2 were examined by immunoblotting. (B) The protein levels of ROCK1 and ROCK2 were quantified relative to the β-actin level. Each column represents the mean ± SEM (n = 4). (C) ROCK activity was measured in NSC34 cells expressing mock, SOD1WT or SOD1G93A at 48 h after transfection and after 27 h subsequent treatment with or without M3 (30 nM). Each column represents the mean ± SEM (n = 4). ##P < 0.01 versus mock (Student's t-test), *P < 0.05 versus SOD1G93A (Student's t-test). N.S., not significant.
M3 regulates the phosphorylation levels of Akt and PTEN
In a recent study, activated ROCK was shown to suppress the phosphorylation of Akt through activation of PTEN (Wu et al., 2012). We therefore examined whether M3 treatment might affect the expressions of Akt and PTEN proteins. Compared with their mock- or SOD1WT-expressing counterparts, SOD1G93A-expressing NSC34 cells exhibited an up-regulation of phosphorylated PTEN and a down-regulation of phosphorylated Akt, and these changes were attenuated by M3 treatment (Figure 3A–C). These data suggest that M3 up-regulates the expression of phosphorylated Akt via a down-regulation of the phosphorylated level of PTEN.
Figure 3.

M3 regulated the phosphorylation levels of Akt and PTEN. (A–C) NSC34 cells were lysed at 48 h after transfection with Myc-tagged mock, SOD1WT or SOD1G93A, then treated with PBS or M3 (30 nM) for 12 or 27 h. (A) Expressions of Akt and PTEN were examined by immunoblotting. The protein levels of phosphorylated Akt (B) and phosphorylated PTEN (C) were quantified relative to the total Akt and total PTEN levels respectively. Each column represents the mean ± SEM (n = 4). #P < 0.05, ##P < 0.01 versus mock + PBS (Student's t-test), *P < 0.05 versus SOD1G93A + PBS (Student's t-test).
Akt inhibition eliminates the neuroprotective effects of M3 against SOD1G93A-induced neurotoxicity
We next examined whether M3 might protect motor neurons against the cell death resulting from SOD1G93A-induced neurotoxicity by modulating the PTEN/Akt pathway. Representative photographs of Hoechst 33342- and PI- staining are shown (Figure 4A). At a concentration of 30 nM, M3 reduced SOD1G93A-induced cell death (Figure 4B). LY294002 inhibited the neuroprotective effect of M3 in a concentration-dependent manner, these effects of LY294002 were significant at both 1 and 10 μM (P < 0.01; Figure 4B). These findings suggest that the PTEN/Akt pathway is involved in the neuroprotective effects of M3.
Figure 4.

Akt inhibition eliminated the neuroprotective effects of M3 against SOD1G93A-induced neurotoxicity. (A) Representative fluorescence microscopic images of Hoechst 33342 (blue) and PI (red) staining after 27 h serum deprivation in NSC34 cells transfected for 48 h with enhanced GFP-tagged (green) SOD1G93A. Scale bar represents 50 μm. (B) Quantitative analysis of Hoechst- and PI-positive NSC34 cells expressing SOD1WT or SOD1G93A after 27 h serum deprivation. Each column represents the mean ± SEM (n = 6). $$P < 0.01 versus SOD1G93A (Student's t-test), ##P < 0.01 versus vehicle (Student's t-test), **P < 0.01 versus SOD1G93A + serum deprivation + M3 (30 nM; Dunnett's test).
Fasudil delays disease onset and prolongs survival time, and reduces the loss of motor neurons, in SOD1G93A mice
To investigate the effects of fasudil on SOD1G93A mice, we administered fasudil at 30 and 100 mg·kg−1 in drinking water. Disease onset was determined by the loss of motor function, as measured in a rotarod test. In SOD1G93A mice, fasudil at 30 or 100 mg·kg−1 delayed the mean disease onset from 108.8 ± 1.9 days (n = 15) in the vehicle group to 118.8 ± 1.8 days (30 mg·kg−1, n = 13) or 119.0 ± 3.4 days (100 mg·kg−1, n = 12; Figures 5A and Supporting Information Figure S5). These values represent almost a 10% delay in the onset of the motor deficit [9.2% at 30 mg·kg−1, (P = 0.041) and 9.4% at 100 mg·kg−1, (P = 0.033); Figure 5C].
Figure 5.
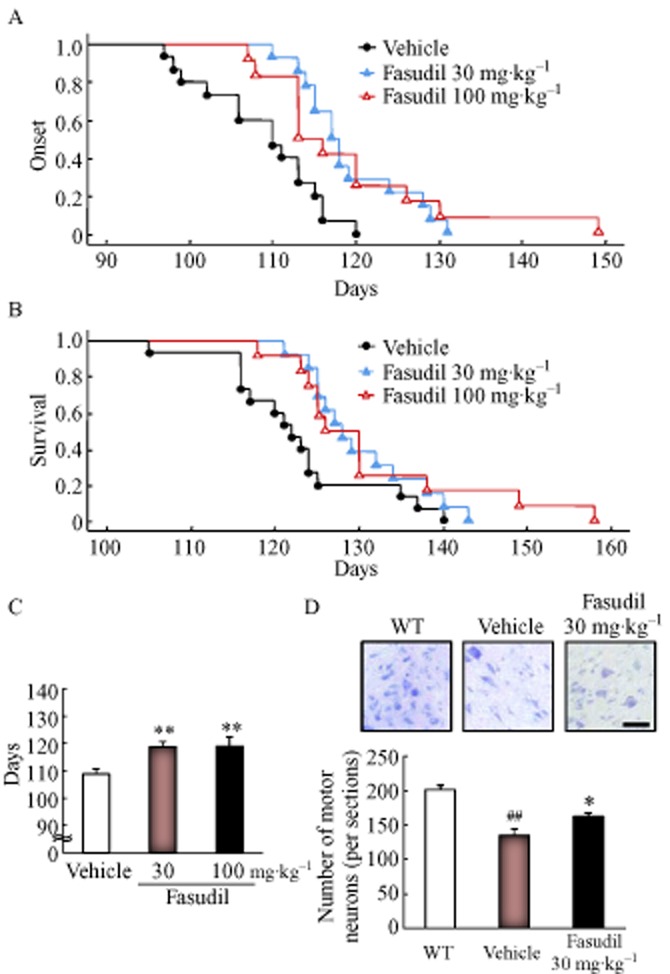
Fasudil hydrochloride (fasudil) delayed the disease onset and prolonged the survival time, and suppressed motor neuron loss, in SOD1G93A mice. The effects of fasudil at 30 and 100 mg·kg−1 were examined by evaluating (A) motor performance in the rotarod test, (B) survival time and (C) the number of days before the beginning of an observable motor deficit in vehicle-, fasudil 30 or 100 mg·kg−1-treated SOD1G93A mice. Each column represents the mean ± SEM (vehicle, n = 15; fasudil 30 mg·kg−1, n = 13; fasudil 100 mg·kg−1, n = 12). **P < 0.01 versus vehicle (Student's t-test). (D) The numbers of motor neurons in WT, vehicle- or fasudil 30 mg·kg−1-treated SOD1G93A mice were evaluated using cresyl violet staining. Scale bar represents 50 μm. Each column represents the mean ± SEM (WT, n = 4; vehicle, n = 3; fasudil 30 mg·kg−1, n = 4). ##P < 0.01 versus WT (Student's t-test), *P < 0.05 versus vehicle (Student's t-test).
Furthermore, treatment of SOD1G93A mice with fasudil at 30 and 100 mg·kg−1 extended their mean survival time by 6.0% (P = 0.002) and 7.0% (P = 0.001), respectively, the mean survival times being 130.2 ± 1.9 days (30 mg·kg−1) and 131.3 ± 3.4 days (100 mg·kg−1) respectively (Figure 5B). In contrast, no significant difference in body weight was found among the vehicle- and fasudil- (30 or 100 mg·kg−1) treated groups during disease progression (Supporting Information Figure S6). Next, we evaluated the effect of fasudil on motor neuron loss in the spinal cord in SOD1G93A mice at 15 weeks of age. The number of motor neurons in the lumbar anterior horn was decreased in SOD1G93A mice compared to their WT littermates (Figure 5D). Fasudil 30 mg·kg−1 significantly prevented the motor neuron loss in SOD1G93A mice (compared to the vehicle-treated SOD1G93A group; Figure 5D). Furthermore, the expression of mutant SOD1 was not changed between vehicle- and fasudil-treated groups (Supporting Information Figure S7). These results suggest that fasudil significantly extends survival in the present ALS model by delaying disease onset and protecting motor neurons against degeneration but not by reducing the expression of mutant SOD1.
Fasudil attenuates the elevated ROCK activity and phosphorylated PTEN, and the reduction of phosphorylated Akt in SOD1G93A mice
According to in vitro study, we investigated ROCK expression and activity, and PTEN and Akt expression in SOD1G93A mice. There was no difference among the WT, vehicle- and fasudil 30 mg·kg−1-treated mice in the expression of either ROCK1 or ROCK2 (Figure 6A–C). Next, ROCK activity was measured by determining the level of phosphorylated adducin. The expression of phosphorylated adducin in SOD1G93A mice was significantly increased compared with WT mice and this increase was attenuated by fasudil treatment (Figure 6D, E). Moreover, compared with WT mice, phosphorylation of PTEN was increased and phosphorylation of Akt was decreased in SOD1G93A mice (Figure 7A–C). Treatment with fasudil attenuated these changes (Figure 7A–C). These data suggest that fasudil up-regulates the expression of phosphorylated Akt by down-regulating the activation of ROCK and decreasing the level of phosphorylated PTEN in SOD1G93A mice.
Figure 6.

Fasudil attenuated the elevated ROCK activity in SOD1G93A mice. (A) Expressions of ROCK1 and ROCK2 were examined by immunoblotting. (B, C) The protein levels of ROCK1 and ROCK2 were quantified relative to the β-actin level. Each column represents the mean ± SEM (n = 4). (D, E) ROCK activity was measured by the phosphorylation of adducin level in WT, vehicle- or fasudil 30 mg·kg−1-treated mice. (D) Expressions of adducin were examined by immunoblotting. (E) The protein levels of phosphorylation of adducin were quantified relative to the α-adducin level. Each column represents the mean ± SEM (n = 4). ##P < 0.01 versus WT (Student's t-test), *P < 0.05 versus vehicle (Student's t-test). N.S., not significant.
Figure 7.

Fasudil regulated the phosphorylation levels of Akt and PTEN in SOD1G93A mice. (A) Expressions of Akt and PTEN were examined by immunoblotting. The protein levels of phosphorylated Akt (B) and phosphorylated PTEN (C) were quantified relative to the total Akt and total PTEN levels respectively. Each column represents the mean ± SEM (n = 4). #P < 0.05 versus WT (Student's t-test), *P < 0.05 versus vehicle (Student's t-test).
Discussion
In the present study, fasudil was found to be effective at preventing motor neuron degeneration through inhibition of ROCK activation in ALS transgenic mice and in in vitro studies. The activation of ROCK by SOD1G93A down-regulates phosphorylated Akt by enhancing the phosphorylation of PTEN, and this results in neuronal cell death. Here, we obtained evidence suggesting that fasudil suppresses this activation of ROCK and thereby reduces the phosphorylation of PTEN, resulting in neuronal protection via increasing phosphorylated Akt (Figure 8).
Figure 8.

The putative mechanism underlying the protective effect of fasudil on ALS. ROCK activity is increased by the neurotoxic action of SOD1G93A. The activated ROCK suppresses phosphorylation of Akt through phosphorylation of PTEN. Fasudil attenuates the elevated ROCK activity and thereby attenuates the increased phosphorylation of PTEN, resulting in survival of neuronal cells via an increase in phosphorylated Akt.
Our initial results indicated that ROCK is associated with neuronal cell death in experimental in vitro and in vivo models of ALS. We found that fasudil and M3 protected motor neuron cells against SOD1G93A-induced neurotoxicity (Figures 1 and 5) and modulated the PTEN/Akt pathway (Figures 3 and 7) via inhibition of ROCK activation by SOD1G93A (Figures 2C and 6D, E). Previous research has shown that the ROCK/PTEN/Akt pathway regulates cell death in neurons (Yang and Kim, 2012). Moreover, the PTEN/Akt pathway is linked to motor neuron survival in human SOD1-related ALS (Kirby et al., 2011); indeed, up-regulation of phosphorylated Akt via the inactivation of phosphorylated PTEN leads to motor neuron survival in ALS patients and in transgenic mouse models. In addition, the level of phosphorylated Akt is decreased in motor neuron cells in patients with sporadic or fALS, and also in mutant SOD1 mice, in the early stages of the disease before the onset of clinical signs or symptoms (Dewil et al., 2007). This suggests that Akt may play a pivotal role in protecting neurons from apoptosis. A recent study showed that fasudil prevented neuronal apoptosis by regulating ROCK activity and the PTEN/Akt pathway in cerebral ischaemia in ratz (Wu et al., 2012). These findings suggest ROCK may play an important role in neuronal cell death, and that ROCK may have potential as a new treatment target in ALS.
In our ALS model mice, fasudil slowed the disease progression and prolonged the survival time (Figure 5A–C). In a mouse model of a similar neurodegenerative disease, SMA, oral administration of fasudil (30 mg·kg−1) has been found to be efficacious and safe (Bowerman et al., 2012). It is supportive and potentially very significant that fasudil had neuroprotective effects in our ALS model at doses with a confirmed safety profile.
Despite extensive research, the mechanism underlying ALS remains unclear, and no effective treatments for ALS have so far been developed. Moreover, new potentially therapeutic compounds have been tested in animal models of ALS, and some of these agents have gone forward to clinical trials. Of necessity, safety and efficacy assessments in humans take a long time. However, fasudil is already well known as a vasodilator agent with confirmed safety and efficacy in humans (Zhao et al., 2011). Indeed, its safety for clinical use is well established (Shibuya et al., 1992) and fasudil has been confirmed as effective for the treatment of diseases such as ischaemic disease (including cerebral vasospasm after subarachnoid haemorrhage) and pulmonary hypertension, based on its inhibitory action on cell contractility (Shimokawa et al., 2002; Shibuya et al., 2005; Vicari et al., 2005). This could progress fasudil, an existing medication, to being considered for clinical use as a treatment for ALS.
In conclusion, we have demonstrated here that fasudil inhibits SOD1G93A-induced motor neuron cell death by inactivating ROCK, suggesting that ROCK plays a critical role in motor neuron degeneration. The present findings indicate that fasudil warrants serious consideration as a candidate for a potential therapeutic agent for ALS.
Acknowledgments
We thank Dr Gen Sobue (Department of Neurology, Nagoya University Graduate School of Medicine, Aichi, Japan) for providing several SOD1 plasmid vectors.
Glossary
- ALS
amyotrophic lateral sclerosis
- Fasudil
fasudil hydrochloride
- NSC34
mice motor neuron
- PI
propidium iodide
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- ROCK
rho kinase
- SMA
spinal muscular atrophy
- SOD1
superoxide dismutase 1
- WT
wild-type
Conflict of interest
This work was supported in a research grant by Asahi Kasei Pharma. KT, KK and MS are employees of Asahi Kasei Pharma.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 M3 protected motor neuron cells expressing SOD1G93A after serum deprivation. (A) Representative fluorescence microscopic images of Hoechst 33342 (blue) and PI (red) staining after 27 h serum deprivation in NSC34 cells transfected with enhanced GFP-tagged (green) SOD1G93A. Scale bar represents 50 μm. (B) Quantitative analysis of Hoechst- and PI-positive NSC34 cells expressing SOD1G93A after 27 h serum deprivation. Each column represents the mean ± SEM (n = 6). $$P < 0.01 versus SOD1G93A (Student's t-test), **P < 0.01 versus vehicle (Dunnett's test).
Figure S2 M3 treatment did not affect the expression of SOD1G93A. (A, B) NSC34 cells were lysed after transfection with Myc-tagged mock, SOD1WT or SOD1G93A for 48 h, then treated with PBS or M3 (30 nM) for 27 h. (A) Expression of myc was examined by immunoblotting. (B) The protein levels of myc were quantified relative to the β-actin level. Each column represents the mean ± SEM (n = 4).
Figure S3 Y-27632, a ROCK inhibitor, prevented motor neuron cell death induced by SOD1G93A. (A) Representative fluorescence microscopic images of Hoechst 33342 (blue) and PI (red) staining after 27 h serum deprivation in NSC34 cells transfected with enhanced GFP-tagged (green) SOD1G93A. Scale bar represents 50 μm. (B) Quantitative analysis of Hoechst- and PI-positive NSC34 cells expressing SOD1G93A after 27 h serum deprivation. Each column represents the mean ± SEM (n = 6). $$P < 0.01 versus SOD1G93A (Student's t-test), **P < 0.01 versus vehicle (Dunnett's test).
Figure S4 Increased motor neuron cell death was detected at 75 h, but not at 48 h, after transfection with SOD1G93A. Quantitative analysis of Hoechst- and PI-positive NSC34 cells expressing mock, SOD1WT or SOD1G93A at 48 or 75 h after transfection. Each column represents the mean ± SEM (n = 6), **P < 0.01 versus SOD1WT 75 h (Student's t-test).
Figure S5 The disease onset was defined as the day when a mouse first dropped off the rotarod within (A and B) 180 s and (C and D) 300 s. (A and C) The motor performance in the rotarod test and (B and D) the number of days before the beginning of an observable motor deficit in vehicle-, fasudil 30 or 100 mg·kg−1-treated SOD1G93A transgenic mice. Each column represents the mean ± SEM (vehicle, n = 15; fasudil 30 mg·kg−1, n = 13; fasudil 100 mg·kg−1, n = 12). **P < 0.01 versus vehicle (Student's t-test).
Figure S6 Body weight data for WT, vehicle- or fasudil (30 or 100 mg·kg−1)-treated SOD1G93A mice.
Figure S7 Fasudil did not affect the expression of mutant SOD1 in SOD1G93A mice. (A) Expression of SOD1 was examined by immunoblotting. (B) The protein level of SOD1 was quantified relative to the β-actin level. Each column represents the mean ± SEM (n = 4).
References
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bowerman M, Murray LM, Boyer JG, Anderson CL, Kothary R. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. 2012;10:24. doi: 10.1186/1741-7015-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- Dewil M, Lambrechts D, Sciot R, Shaw PJ, Ince PG, Robberecht W, et al. Vascular endothelial growth factor counteracts the loss of phospho-Akt preceding motor neurone degeneration in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2007;33:499–509. doi: 10.1111/j.1365-2990.2007.00850.x. [DOI] [PubMed] [Google Scholar]
- Gurney ME. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci. 1997;152(Suppl. 1):S67–S73. doi: 10.1016/s0022-510x(97)00247-5. [DOI] [PubMed] [Google Scholar]
- Hou Y, Zhou L, Yang QD, Du XP, Li M, Yuan M, et al. Changes in hippocampal synapses and learning-memory abilities in a streptozotocin-treated rat model and intervention by using fasudil hydrochloride. Neuroscience. 2012;200:120–129. doi: 10.1016/j.neuroscience.2011.10.030. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996;15:1885–1893. [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Yamada M, Tanaka H, Aida K, Tsuruma K, Shimazawa M, et al. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol Dis. 2009;36:470–476. doi: 10.1016/j.nbd.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Jackson M, Ganel R, Rothstein JD. Models of amyotrophic lateral sclerosis. 2002. Curr Protoc Neurosci, Chapter 9, Unit 9.13. [DOI] [PubMed]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby J, Ning K, Ferraiuolo L, Heath PR, Ismail A, Kuo SW, et al. Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134:506–517. doi: 10.1093/brain/awq345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh PN, Ray-Chaudhuri K. Motor neuron disease. J Neurol Neurosurg Psychiatry. 1994;57:886–896. doi: 10.1136/jnnp.57.8.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–352. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- Sebbagh M, Hamelin J, Bertoglio J, Solary E, Breard J. Direct cleavage of ROCK II by granzyme B induces target cell membrane blebbing in a caspase-independent manner. J Exp Med. 2005;201:465–471. doi: 10.1084/jem.20031877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, et al. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Results of a prospective placebo-controlled double-blind trial. J Neurosurg. 1992;76:571–577. doi: 10.3171/jns.1992.76.4.0571. [DOI] [PubMed] [Google Scholar]
- Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E. Effects of fasudil in acute ischemic stroke: results of a prospective placebo-controlled double-blind trial. J Neurol Sci. 2005;238:31–39. doi: 10.1016/j.jns.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Tanaka H, Ito Y, Morimoto N, Tsuruma K, Kadokura M, et al. An inducer of VGF protects cells against ER stress-induced cell death and prolongs survival in the mutant SOD1 animal models of familial ALS. PLoS ONE. 2010;5:e15307. doi: 10.1371/journal.pone.0015307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H, Hiramori K, Iinuma H, Hosoda S, Kishida H, Osada H, et al. Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in patients with stable effort angina: a multicenter study. J Cardiovasc Pharmacol. 2002;40:751–761. doi: 10.1097/00005344-200211000-00013. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shimazaki H, Kimura M, Izuta H, Tsuruma K, Shimazawa M, et al. Apoptosis-inducing factor and cyclophilin A cotranslocate to the motor neuronal nuclei in amyotrophic lateral sclerosis model mice. CNS Neurosci Ther. 2011;17:294–304. doi: 10.1111/j.1755-5949.2010.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Shimazawa M, Kimura M, Takata M, Tsuruma K, Yamada M, et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci Rep. 2012;2:573. doi: 10.1038/srep00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, Dion PA, Rouleau GA. Amyotrophic lateral sclerosis: new genes, new models, and new mechanisms. F1000 Biol Rep. 2011;3:18. doi: 10.3410/B3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, et al. Efficacy and safety of fasudil in patients with stable angina: a double-blind, placebo-controlled, phase 2 trial. J Am Coll Cardiol. 2005;46:1803–1811. doi: 10.1016/j.jacc.2005.07.047. [DOI] [PubMed] [Google Scholar]
- Wu J, Li J, Hu H, Liu P, Fang Y, Wu D. Rho-kinase inhibitor, fasudil, prevents neuronal apoptosis via the Akt activation and PTEN inactivation in the ischemic penumbra of rat brain. Cell Mol Neurobiol. 2012;32:1187–1197. doi: 10.1007/s10571-012-9845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita K, Kotani Y, Nakajima Y, Shimazawa M, Yoshimura S, Nakashima S, et al. Fasudil, a Rho kinase (ROCK) inhibitor, protects against ischemic neuronal damage in vitro and in vivo by acting directly on neurons. Brain Res. 2007;1154:215–224. doi: 10.1016/j.brainres.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Yang S, Kim HM. The RhoA-ROCK-PTEN pathway as a molecular switch for anchorage dependent cell behavior. Biomaterials. 2012;33:2902–2915. doi: 10.1016/j.biomaterials.2011.12.051. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhou D, Guo J, Ren Z, Zhou L, Wang S, et al. Efficacy and safety of fasudil in patients with subarachnoid hemorrhage: final results of a randomized trial of fasudil versus nimodipine. Neurol Med Chir (Tokyo) 2011;51:679–683. doi: 10.2176/nmc.51.679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.