

Abstract
BACKGROUND AND PURPOSE
Histamine H1 receptors are highly expressed in hypothalamic neurons and mediate histaminergic modulation of several brain-controlled physiological functions, such as sleep, feeding and thermoregulation. In spite of the fact that the mouse is used as an experimental model for studying histaminergic signalling, the pharmacological characteristics of mouse H1 receptors have not been studied. In particular, selective and potent H1 receptor agonists have not been identified.
EXPERIMENTAL APPROACH
Ca2+ imaging using fura-2 fluorescence signals and whole-cell patch-clamp recordings were carried out in mouse preoptic/anterior hypothalamic neurons in culture.
KEY RESULTS
The H1 receptor antagonists mepyramine and trans-triprolidine potently antagonized the activation by histamine of these receptors with IC50 values of 0.02 and 0.2 μM respectively. All H1 receptor agonists studied had relatively low potency at the H1 receptors expressed by these neurons. Methylhistaprodifen and 2-(3-trifluoromethylphenyl)histamine had full-agonist activity with potencies similar to that of histamine. In contrast, 2-pyridylethylamine and betahistine showed only partial agonist activity and lower potency than histamine. The histamine receptor agonist, 6-[2-(4-imidazolyl)ethylamino]-N-(4-trifluoromethylphenyl)heptanecarboxamide (HTMT) had no agonist activity at the H1 receptors H1 receptors expressed by mouse preoptic/anterior hypothalamic neurons but displayed antagonist activity.
CONCLUSIONS AND IMPLICATIONS
Methylhistaprodifen and 2-(3-trifluoromethylphenyl)histamine were identified as full agonists of mouse H1 receptors. These results also indicated that histamine H1 receptors in mice exhibited a pharmacological profile in terms of agonism, significantly different from those of H1 receptors expressed in other species.
Keywords: histamine, H1 receptor, H1 receptor agonist, H1 receptor signalling, methylhistaprodifen, 2-(3-trifluoromethylphenyl)histamine
Introduction
Histamine, an important biological amine, is produced by neurons of the tuberomammillary nucleus of the hypothalamus (Haas et al., 2008). Their efferent fibres project throughout the brain and are found in high density in the hypothalamus (Takada et al., 1987; Schwartz et al., 1991; Wada, 1992). Histamine modulates a variety of centrally controlled physiological functions, such as arousal, attention, sleep, feeding and thermoregulation (Haas and Panula, 2003). Recent advances in histamine receptor research have provided new drug targets in the CNS (Chazot, 2009). Thus, histamine H3 receptor antagonists have been proposed as candidates for the treatment of cognitive (Jin et al., 2009; Chazot, 2010) and sleep disorders (Guo et al., 2009). H4 receptors expressed in the cortex and hippocampus have also been proposed as targets for cognitive disorders (Connelly et al., 2009), while hypothalamic H1 receptors are considered as drug targets for preventing the weight gain induced by antipsychotics (Deng et al., 2012; receptor nomenclature follows Alexander et al., 2011). We have shown that histamine can induce hyperthermia and increase energy expenditure by activating either H1 receptors expressed by glutamatergic preoptic neurons or H3 receptors expressed by GABAergic preoptic neurons (Lundius et al., 2010; Sethi et al., 2012). In response to activation of H1 receptors, the corresponding neurons display depolarization and increased firing rates as well as Ca2+ release from intracellular stores (Lundius et al., 2010). These actions are typically observed in various preparations in response to the activation of the H1 receptors that are coupled to Gq and activate the PLC pathway (Haas et al., 2008).
While a vast amount of information about the pharmacology of human, guinea pig and rat H1 receptors is available, there is little data on the pharmacology of the mouse isoform. As the mouse is used frequently as an experimental model for studying the role of histaminergic signalling in various diseases, the pharmacology of mouse H1 receptors needs to be fully defined. As described previously, histamine activates inward currents and induces Ca2+ release from intracellular stores in a subpopulation of glutamatergic preoptic/anterior hypothalamic (PO/AH) neurons in culture or in slices (Lundius et al., 2010; Tabarean, 2012). These responses are due to activation of H1 receptors as indicated by their sensitivity to H1 receptor selective antagonists, such as trans-triprolidine, mepyramine and cetirizine, and by the presence of H1 receptor transcripts (as well as the absence of H2 and H3 receptor transcripts) in the responsive neurons (Lundius et al., 2010). Preliminary studies with the H1 receptor agonists 2-pyridylethylamine and betahistine have indicated that they can mimic the effects of histamine when applied in relatively high concentrations (Lundius et al., 2010).
In this study, I have carried out a detailed study of the pharmacology of the histamine responses in cultures of PO/AH neurons with an emphasis on the effects of H1 receptor selective agonists, as work with such compounds has been scarce. I compared the activities of three commercially available agonists (2-pyridylethylamine, 6-[2-(4-Imidazolyl)ethylamino]-N-(4-trifluoromethylphenyl)heptanecarboxamide (HTMT) and betahistine) with two other compounds, methylhistaprodifen and 2-(3-trifluoromethylphenyl)histamine (2-(3-TFMP)histamine) (Leschke et al., 1995; Elz et al., 2000) representing new classes of H1 receptor agonists. Each experiment was performed using two assays: electrophysiological measurement of the inward currents activated as well as measurement of [Ca]i changes using Fura2-AM. The results reveal two full agonists of the mouse H1 receptors, as well as differences between mouse H1 receptors and H1 receptors from other species.
Methods
Cell culture
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of the Scripps Research Institute and complied with the standards of the American Association for the Accreditation of Laboratory Animal Care (AAALAC) and the regulations in the Animal Welfare Act. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010) A total of 26 animals were used in the experiments described here. Mixed PO/AH embryonic cultures were prepared as previously described (Tabarean et al., 2005) using Swiss Webster mice. The cultures were used at 4–5 weeks after plating. A set of experiments was carried out on cultures of PO/AH embryonic tissue from C57/Bl6 mice.
Whole-cell patch-clamp recording
The extracellular solution contained (in mM): 145 NaCl, 3.5 KCl, 1.25 NaH2PO4, 10 HEPES, 2 CaCl2, 1 MgSO4 and 10 glucose; osmolarity of 315–325 mOsm (pH 7.4). A K+ pipette solution containing (in mM) 130 K-gluconate, 5 KCl, 10 HEPES, 2 MgCl2, 0.5 EGTA, 2 ATP and 1 GTP (pH 7.3) was used in all experiments. The electrode resistance after back-filling was 2–4 MΩ. All voltages were corrected for the liquid junction potential (−13 mV). Data were acquired with a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) digitized using a Digidata 1320A interface and the Pclamp9.2 software package. The sampling rate for the continuous recordings of spontaneous activity was 50 kHz. The recording chamber was constantly perfused with extracellular solution (2–3 mL·min−1). The treatments were bath-applied. The duration of agonist applications was 40 s, while antagonists were applied for 3–5 min prior to an agonist test. The washout of the bath solution lasted less than 2 min as indicated by control studies with a high K+ extracellular solution. Agonist application was separated by periods of at least 8 min. Control experiments have established that agonist applications separated by at least 8 min intervals do not induce desensitization of the H1 receptors. The temperature of the external solution was controlled with a TC-344B temperature controller and an inline heater (Warner Instruments, Hamden, CT, USA) and was maintained at 36–37°C. Extracellular solutions were bubbled with O2.
Ca2+ imaging
Fura-2 fluorescence signals were acquired with a CCD camera (Hamamatsu ORCA-ER) connected to its frame grabber operating in an 8 bit mode and driven by Slidebook software (Intelligent Imaging Innovations, Denver, CO, USA). An ultra-high-speed wavelength switcher Lambda DG-4 (Sutter Instruments, Novato, CA, USA) equipped with model 340HT15 and 380HT15 filters provided alternating excitation for ratiometric fura-2 measurements. The illumination source was a standard xenon lamp. The sampling frequency of 0.2 Hz was sufficiently fast given the relatively slow responses to histamine or H1 receptor agonists. At this excitation frequency, photobleaching and phototoxicity were minimal. Fura-2AM loading and data acquisition were carried out as described previously (Lundius et al., 2010).
Data analysis and curve fitting
All data represent mean ± SD of at least three independent experiments. Data analysis and curve fitting was carried out using the SigmaPlot software package (Systat Software, Inc., San Jose, CA, USA). One-way anova with Tukey–Kramer post hoc test (P < 0.05) was used for comparison of multiple groups. The concentration–response data of agonist actions were fitted to the Hill equation: E = Emax/{1 + (EC50/[Agonist])n}, where [Agonist] represents the agonist concentration, n is the Hill coefficient and Emax is the maximum effect as a percentage of the maximum histamine response in the same cell. The concentration–response data for an antagonist were fitted with: I = Imax/{1 + ([Antagonist]/IC50)n}, where [Antagonist] represents the antagonist concentration, n is the Hill coefficient and Imax is the maximum effect of the antagonist as a percentage of the maximum histamine response in the same cell. Statistical analysis of curve fit parameters was carried out by independently fitting the data from individual experiments and comparing the resulting curve fit values by t-test or one-way anova as appropriate.
Chemicals
Methylhistaprodifen dihydrogenoxalate and 2-(3-trifluoromethylphenyl)histamine dihydrogen maleate (2-(3-TFMP)histamine) were a kind gift from Dr. Walter Schunack (Institut für Pharmazie, Freie Universität Berlin, Germany). The synthesis of these compounds and the characterization of their actions at guinea-pig H1 receptors have been previously described (Leschke et al., 1995; Elz et al., 2000). HTMT dimaleate, 2-pyridylethylamine, mepyramine and trans-triprolidine were purchased from Tocris (Ellisville, MO, USA). Histamine, betahistine, TTX (voltage-gated sodium channel blocker), CNQX (AMPA/kainate receptor antagonist), AP-5 (NMDA receptor antagonist) and bicuculline (GABAA receptor antagonist) were purchased from Sigma (St Louis, MO, USA).
Results
Concentration–response relationships for the effects of histamine on PO/AH neurons
Experiments to determine the concentration–response relationships of histamine on cultured PO/AH neurons (Figure 1A,B,E,F) were first carried out. Figure 1A,B depicts the dose–response relationship of [Ca]i elevations by histamine. The derived Hill function had an EC50 of 39 μM and a slope factor of 1.2. Measurements of inward currents activated by the neurotransmitter yielded a very similar concentration–response curve (Figure 1E,F) and an EC50 of 36 μM and a slope factor of 1.4. The curve fit parameters were not statistically different for the two concentration–response curves (unpaired t-test, P > 0.05). The proportions of neurons affected by histamine were 22 and 20% of neurons studied with the [Ca]i and inward current assays respectively. These values are similar to those reported by us in earlier studies of PO/AH neurons and in accordance with the percentages of neurons that express H1 receptors (Lundius et al., 2010; Tabarean, 2012). All measurements were carried out in the presence of TTX (1 μM), CNQX (20 μM), AP-5 (50 μM) and bicuculline (20 μM) to eliminate synaptic activity and spontaneous action potential firing. In the presence of these blockers, we have not observed histamine effects in the opposite direction (outward currents or a decrease in [Ca]i) in any PO/AH neuron studied, confirming our previous results that inhibitory actions (due to activation of H3 receptors) are dependent on action potential firing (Lundius et al., 2010).
Figure 1.
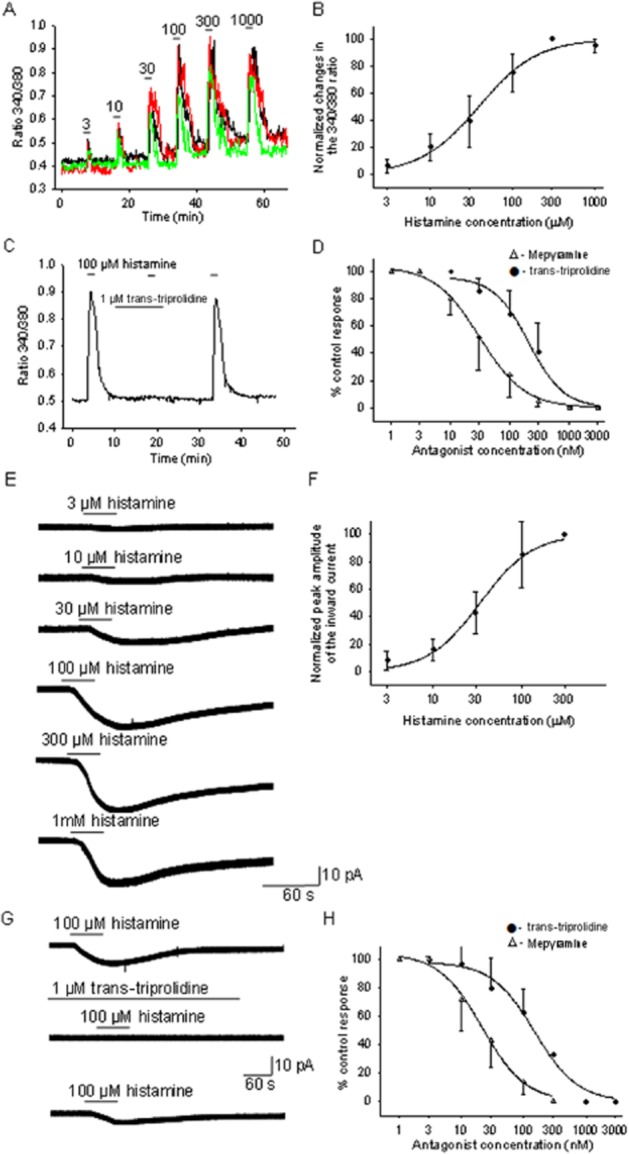
Concentration–response relationships for the histamine evoked [Ca]i increase and inward current in PO/AH neurons and for the antagonism of these actions by trans-triprolidine and mepyramine. (A) [Ca]i responses to different concentrations of histamine from 3 PO/AH neurons. (B) concentration–response curve for the histamine-induced elevations of [Ca]i. Each point represents the average of data collected from 30 different PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 39 μM. (C) [Ca]i responses to histamine (100 μM) before during and after trans-triprolidine (1 μM) incubation. The trace represents the average of the responses from five cultured PO/AH neurons. Note that trans-triprolidine (1 μM) abolished the effects of histamine. (D) Concentration–response curve for the antagonist activity of trans-triprolidine and mepyramine on the [Ca]i responses to histamine (100 μM). Each point represents the average of data collected from 30 different PO/AH neurons. The datasets were fitted with logistic functions. The fits yielded an IC50 of 0.21 and 0.32 μM for trans-triprolidine and mepyramine respectively. (E) Inward current activated by several concentrations of histamine recorded in a cultured PO/AH neurons. The holding potential was −50 mV. (F) Concentration–response curve for the histamine activation of inward currents. Each point represents the average of data collected from nine different PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 36 μM. (G) Inward currents activated by histamine (20 μM) before (upper trace), during incubation with trans-triprolidine (1 μM), and after washout of the antagonist (lower trace). The neuron was held at −50 mV. The holding potential was −50 mV. (H) Concentration–response curve for the antagonist activity of trans triprolidine and mepyramine on the [Ca]i responses to histamine (100 μM). Each point represents the average of data collected from five different PO/AH neurons. The datasets were fitted with logistic functions. The fits yielded IC50 of 0.15 and 0.03 μM for trans-triprolidine and mepyramine respectively. (A–H) TTX (1 μM), CNQX (20 μM), AP-5 (50 μM) and bicuculline (20 μM) were present in the extracellular solution.
As indicated by the standard deviation (Figure 1B,F), we have observed significant variability among neurons in terms of the concentration dependence of the histamine effects. Thus, measurements using both assays indicated that some neurons responded to concentrations as low as 3 μM, while other neurons required higher concentrations (10 μM or higher). Similarly, maximal effects were obtained at 150 μM in some neurons, while others required higher concentrations. Nevertheless, in all neurons studied, the maximal effect was obtained at 300 μM or lower concentrations.
Concentration–response relationships for the H1 receptor antagonists trans-triprolidine and mepyramine on histamine responses
Histamine responses were very sensitive to H1 receptor antagonists. Thus, incubation with 1 μM trans-triprolidine abolished the histamine effects in all the neurons studied (Figure 1C,G). The concentration–response relationship of the antagonistic action of trans-triprolidine on the effects of histamine (150 μM) on [Ca]i (Figure 1D) yielded an IC50 of 0.21 μM and a slope factor of 1.4. Similarly, the concentration–response relationship of the antagonistic action of trans-triprolidine on the inward currents activated by histamine (150 μM) indicated an IC50 of 0.15 μM and a slope factor of 1.2 (Figure 1H). The curve fit parameters were not statistically different for the two dose–response curves (unpaired t-test, P > 0.05).
The action of mepyramine, another H1 receptor selective antagonist, was also studied. Incubation with 3 μM mepyramine abolished the actions of histamine in all neurons studied. The concentration–response curve describing the antagonistic action of mepyramine on the histamine (150 μM)-induced [Ca]i elevation (Figure 1D) yielded an IC50 of 0.02 μM and a slope factor of 1.1. The concentration–response relationship of the antagonistic action of mepyramine on the inward currents activated by histamine (150 μM) yielded an IC50 of 0.03 μM and a slope factor of 1.2 (Figure 1H). The curve fit parameters were not statistically different for the two concentration–response curves (unpaired t-test, P > 0.05). The IC50 values obtained for mepyramine and trans-triprolidine were significantly different (P < 0.01, unpaired t-test).
Concentration–response relationships for the H1 receptor agonists methylhistaprodifen,2-(3-TFMP)histamine, 2-pyridylethylamine and betahistine
First, the effects of two selective H1 receptor agonists: methylhistaprodifen and 2-(3-TFMP)histamine were studied. Methylhistaprodifen was a full agonist for the H1 receptors expressed by cultured PO/AH neurons. Figure 2A,B depicts the concentration–response relationship of [Ca]i changes induced by the agonist. A Hill function with an EC50 of 28 μM and a slope factor of 1.3 fitted the data. Measurements of inward currents activated by methylhistaprodifen also yielded a very similar dose–response curve (Figure 2C,D) and an EC50 of 32 μM and a slope factor of 1.4.
Figure 2.
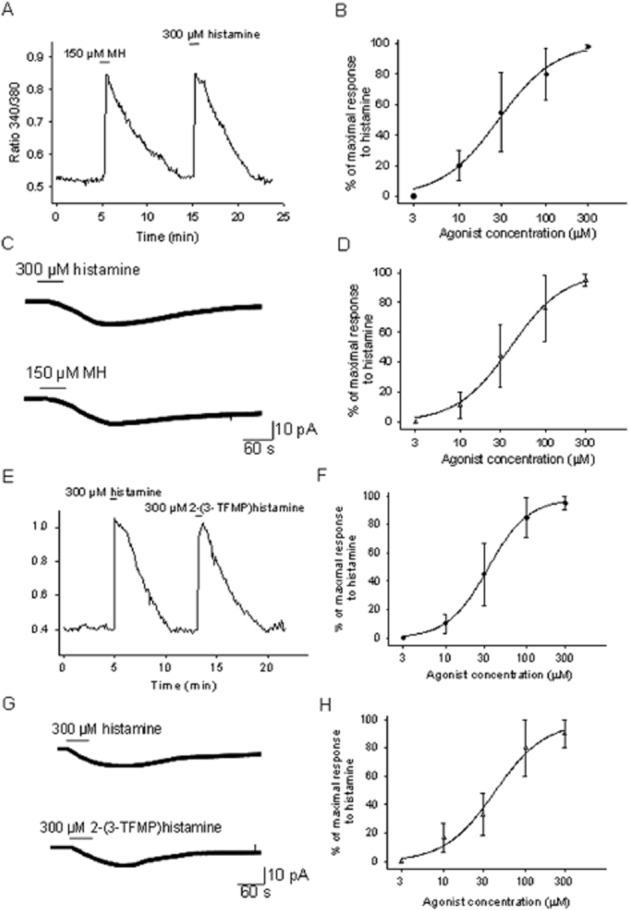
Methylhistaprodifen (MH) and 2-(3-TFMP) histamine are full agonists at the H1 receptors expressed by PO/AH neurons. (A) [Ca]i responses to histamine (300 μM) and MH (150 μM). The trace represents the average the response from six cultured PO/AH neurons. (B) Concentration–response curve for the activation of [Ca]i responses by MH normalized to the response to histamine (300 μM). Each point represents the average of data collected from 30 different PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 28 μM. (C) Inward current activated by histamine (300 μM) and by MH (150 μM) recorded in a cultured PO/AH neuron. The holding potential was −50 mV. (D) Concentration–response curve for the inward currents activated by MH. The values were normalized to the maximal inward current activated by histamine (300 μM) in the same neuron. Each point represents the average of data collected from eight different PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 32 μM. (E) [Ca]i responses to histamine (300 μM) and 2-(3-TFMP) histamine (300 μM). The trace represents the average of the responses from 5 cultured PO/AH neurons. (F) Concentration–response curve for the activation of [Ca]i responses by 2-(3-TFMP) histamine normalized to the response to histamine (300 μM). Each point represents the average of data collected from 28 PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 36 μM. (G) Inward current activated by histamine (300 μM) and by 2-(3-TFMP) histamine (300 μM) recorded in a cultured PO/AH neuron. The holding potential was −50 mV. (H) Dose–response curve for the inward currents activated by 2-(3-TFMP) histamine. The values were normalized to the maximal inward current activated by histamine (300 μM) in the same neuron. Each point represents the average of data collected from 6 different PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 41 μM. (A–H) TTX (1 μM), CNQX (20 μM), AP-5 (50 μM), and bicuculline (20 μM) were present in the extracellular solution.
Similarly, 2-(3-TFMP)histamine had full-agonist activity, measured with the [Ca]i assay, with an EC50 of 34 μM and a slope factor of 1.6 (Figure 2E,F). The concentration–response curve obtained by measurements of inward currents activated by the agonist (Figure 2F,G) yielded an EC50 of 42 μM and a slope factor of 1.4.
The activity of two commercially available compounds with agonist activity at the H1 receptor was then studied: 2-pyridylethylamine, reported to be a H1/2 receptor agonist and betahistine, a partial H1 agonist/H3 antagonist. The concentration–response studies indicated that 2-pyridylethylamine had partial agonist activity at the H1 receptors expressed in cultured PO/AH neurons. Thus, [Ca]i measurements yielded a concentration–response curve (Figure 3A,B) with an EC50 of 56 μM, a slope factor of 1.1 and a maximal effect of 76% (of the maximal effect activated by histamine). Measurements of inward currents activated by 2-pyridylethylamine produced a concentration–response curve (Figure 3C,D) and an EC50 of 61 μM, a slope factor of 1.1 and a maximal effect of 79%.
Figure 3.
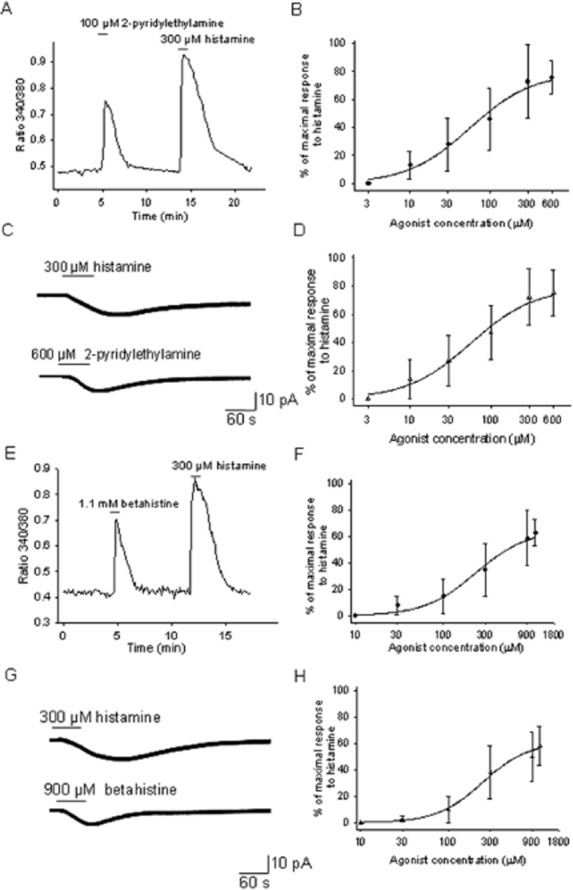
2-Pyridylethylamine and betahistine are partial agonists at the H1 receptors expressed by PO/AH neurons. (A) [Ca]i responses to 2-pyridylethylamine (100 μM) and histamine (300 μM). The trace represents the average of the responses from six cultured PO/AH neurons. (B) Concentration–response curve for the activation of [Ca]i responses by 2-pyridylethylamine normalized to the response to histamine (300 μM). Each point represents the average of data collected from 32 PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 56 μM and a maximal effect was 76%. (C) Inward current activated by histamine (300 μM) and by 2-pyridylethylamine (600 μM) recorded in a cultured PO/AH neuron. The holding potential was −50 mV. (D) Concentration–response curve for the inward currents activated by 2-pyridylethylamine. The values were normalized to the maximal inward current activated by histamine (300 μM) in the same neuron. Each point represents the average of data collected from 8 PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 61 μM and a maximal effect was 79%. (E) [Ca]i responses to betahistine (1.1 mM) and histamine (300 μM). The trace represents the average of the responses from eight cultured PO/AH neurons. (F) Concentration–response curve for the activation of [Ca]i responses by betahistine normalized to the response to histamine (300 μM). Each point represents the average of data collected from 27 PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 254 μM and a maximal effect was 64%. (G) Inward current activated by histamine (300 μM) and by betahistine (900 μM) recorded in a cultured PO/AH neuron. The holding potential was −50 mV. (H) Concentration–response curve for the inward currents activated by betahistine. The values were normalized to the maximal inward current activated by histamine (300 μM) in the same neuron. Each point represents the average of data collected from six PO/AH neurons. The data were fitted with a Hill function. The fit yielded an EC50 of 244 μM and a maximal effect was 60%. (A–H) TTX (1 μM), CNQX (20 μM), AP-5 (50 μM) and bicuculline (20 μM) were present in the extracellular solution.
Betahistine also had partial agonist activity in our experiments. Figure 3E,F represents the concentration–response relationship of [Ca]i changes induced by the agonist and the Hill function that fitted the data. The EC50 was 254 μM, the slope factor was 1.3 and the maximal effect was 65%. Measurements of inward currents activated by betahistine generated a concentration–response curve (Figure 3G,H) characterized by an EC50 of 244 μM, a slope factor of 1.6 and a maximal effect of 61%.
The EC50 values, obtained with either of the two assays, for the four agonists were significantly different [P < 0.01, anova, with the exception of the comparison between methylhistaprodifen and 2-(3-TFMP)histamine that yielded P < 0.05].
In spite of the fact that all drug treatments were of the same duration (40 s), the decay of the [Ca]i responses varied among agonists. Histamine response decay times (from peak to 20% of maximum) were 5.7 ± 1.1 min (n = 152). The decay times of 2-(3-TFMP)histamine responses averaging 5.3 ± 0.8 min (n = 49) were similar to those of histamine, while those of methylhistaprodifen were significantly longer averaging 7.1 ± 1.2 min (n = 55) (anova, P < 0.05). In contrast, the decay times of 2-pyridylethylamine and betahistine responses were significantly shorter than those of histamine averaging 2.9 ± 0.8 min (n = 41) and 2.6 ± 0.5 min (n = 32) respectively (anova, P < 0.05 for both).
All neurons responsive to histamine (using either of the two assays) responded also to the H1 receptor agonists tested (at least 20 neurons were tested for each agonist). Conversely, neurons that did not respond to histamine did not display responses to any of the H1 receptor agonists tested (at least 20 neurons were tested for each agonist). The effects on [Ca]i as well as the inward currents activated by the four H1 receptor agonists studied were abolished in the presence of trans-triprolidine (1 μM) in all neurons studied (at least 10 neurons were studied for each agonist). These observations further confirm the fact that the observed effects were due to activation of H1 receptors.
To test whether the H2 receptor might also have been involved in the histamine responses described previously, I studied the effects of two H2 receptor selective agonists dimaprit (1, 10, 100 μM) and amthamine (1, 10, 100 μM) had no effect on [Ca]i in all neurons studied (for each concentration at least 50 neurons were studied). Similarly, neither dimaprit (1, 10, 100 μM) nor amthamine (1, 10, 100 μM) activated inward currents in the neurons tested (at least 20 neurons were tested for each concentration).
Similarly, we have tested the effects of the H4 receptor agonist, 4-methylhistamine (20 μM) on PO/AH cultured neurons. The agonist had no effect on [Ca]i or on the electrophysiological characteristics of the neurons studied (n = 30 and n = 16 respectively).
The histamine receptor agonist HTMT acts as an antagonist at the H1 receptors expressed by mouse PO/AH neurons.
HTMT has been reported to be an H1/H2 receptor agonist (Khan et al., 1986). In the experiments described here, this compound did not display agonist activity in terms of either [Ca]i responses or inward currents at all concentrations tested (1, 10 and 100 μM). Instead, incubation with HTMT decreased the actions induced by histamine in all the neurons studied (Figure 4A,C). The concentration–response relationship of the action of HTMT on the effects of histamine on [Ca]i (Figure 4B) yielded an IC50 of 19 μM and a slope factor of 1.6. Similarly, the concentration–response relationship of the action of HTMT on the inward currents elicited by histamine (100 μM) yielded an IC50 of 16 μM and a slope factor of 1.6 (Figure 4D).
Figure 4.
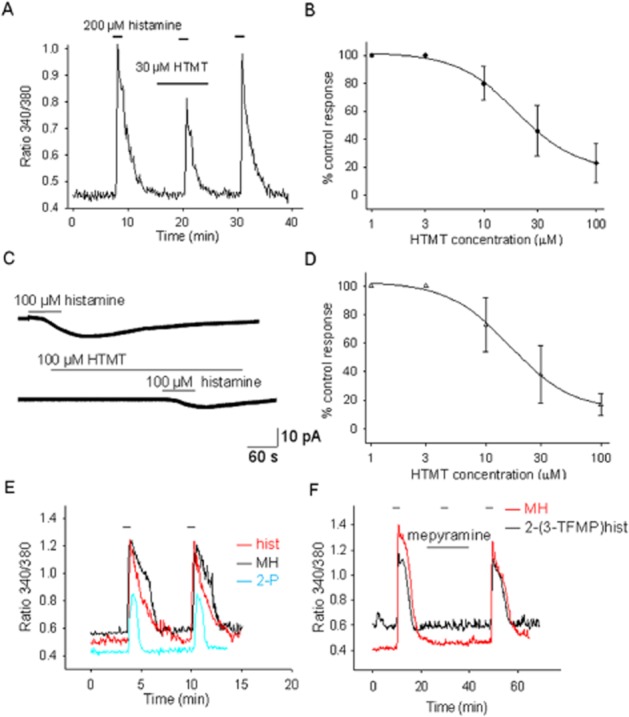
HTMT does not have agonist activity at the H1 receptors expressed in mouse PO/AH neurons but antagonizes histamine effects. (A) [Ca]i responses to histamine (100 μM) before during and after HTMT (30 μM) incubation. The trace represents the average of the responses from five cultured PO/AH neurons. Note that HTMT does not elicit an increase of [Ca]i but decreases the histamine effect. (B) Concentration–response curve for the antagonist activity of HTMT on the [Ca]i responses to histamine (100 μM). Each point represents the average of data collected from 20 different PO/AH neurons. The data were fitted with a logistic function. The fit yielded an IC50 of 19 μM. (C) Inward current activated by histamine (100 μM) before and during HTMT (100 μM) incubation recorded in a cultured PO/AH neurons. HTMT does not activate inward current but decreases the histamine effect. The holding potential was −50 mV. (D) Concentration–response curve for the antagonist activity of HTMT on the inward current activated by histamine (100 μM). Each point represents the average of data collected from six different PO/AH neurons. The data were fitted with a logistic function. The fit yielded an IC50 of 16 μM. (E) Short incubations (40 s) with histamine or H1 receptor agonists do not induce long lasting desensitization. The traces are responses from individual cells and are representative of the average response of PO/AH neurons. (F) Mepyramine (0.1 μM) blocks the responses activated by methylhistaprodifen (MH; 100 μM) and 2-(3-TFMP) histamine (100 μM). The traces are representative responses of individual PO/AH neurons. (E, F) Response was recorded from PO/AH cultures from C57/Bl6 mice. (A–F) TTX (1 μM), CNQX (20 μM), AP-5 (50 μM) and bicuculline (20 μM) were present in the extracellular solution.
Properties of H1 receptors in PO/AH cultures from C57/Bl6 mice
As most transgenic models are on the C57/Bl6 background, we have carried out [Ca]i imaging experiments also in PO/AH cultures from this mouse strain. The results were very similar to those described earlier in the Swiss Webster strain. Histamine increased [Ca]i with an EC50 of 36 μM. Methylhistaprodifen and [2-(3-TFMP]histamine were full agonists (with EC50 of 31 and 40 μM, respectively), while betahistine and 2-pyridylethylamine were only partial agonists (with EC50 of 237 and 61 μM, respectively, and maximal responses of 59 and 68% respectively). HTMT had no agonist activity but antagonized histamine responses with an IC50 of 25 μM. The concentration–response relationships of the five H1 receptor ligands obtained using C57/Bl6 cultures were not statistically different from the corresponding values obtained using cultures of neurons from Swiss Webster mice (P > 0.05, anova). Each concentration–response curve was generated using data from at least 11 neurons. As observed also in cultures from Swiss Webster mice, incubations of short duration (40 s) with histamine or H1 receptor agonists did not induce long lasting desensitization of the H1 receptors (Figure 4E). Also, the responses to methylhistaprodifen and [2-(3-TFMP]histamine were antagonized by either trans-triprolidine (0.3 μM) or mepyramine (0.1 μM) (Figure 4F) in all neurons studied (n = 7 and n = 9 respectively).
Discussion and conclusions
This study provides, for the first time to our knowledge, a pharmacological profile of mouse H1 receptors. Our data, obtained using two functional assays in cultured PO/AH neurons, are compatible with our earlier observations in slices and dissociated cells (Lundius et al., 2010; Tabarean, 2012) and indicate that trans-triprolidine and mepyramine potently antagonize the activation of the mouse H1 receptors expressed in PO/AH neurons by histamine and all the H1 receptor agonists studied. To further ensure that H3 receptors were not involved in the measured effects, we have carried out a set of experiments in the presence of the H3 receptor antagonist thioperamide (3 μM). In the presence of the antagonist, the actions of histamine, or of the other agonists studied, were not changed (data not shown).
All the agonists studied have relatively low potency for these receptors in both mouse strains studied. Surprisingly, histamine has relatively low potency at the mouse H1 receptors, with EC50 of ∼40 μM, and maximal responses are obtained at concentrations of 150 μM or higher. Nevertheless, these concentrations are similar to those utilized in other studies of mouse H1 receptors (Amano et al., 2001; Zhou et al., 2007; Kajihara et al., 2010). The present results are also in line with findings in ventromedial hypothalamic nucleus where histamine responses were blocked by 1 μM mepyramine, while 100 μM betahistine was required to activate a depolarization (Zhou et al., 2007). Although in our previous study in PO slices (Lundius et al., 2010) concentration–response curves were not generated, we observed that in cultured PO/AH neurons, there is a trend for even higher concentrations of agonists being required for activating a response or for reaching maximal responses.
A significant heterogeneity among PO/AH neurons in the same culture in terms of the minimal concentration of histamine required to trigger a response was also observed. Thus, some neurons responded to 3 μM histamine, while others responded only to higher concentrations (10 μM or higher). This discrepancy may be due to variations in the density of H1 receptors expressed by different PO/AH neurons, availability of ‘docked’ G proteins, or of other effector proteins downstream of the H1 receptors. In our functional assays, at the cellular level, it is possible that a response is triggered when a ‘threshold’ level of an intracellular second messenger (e.g. inositol trisphosphate) is reached. This hypothesis has also been raised by studies in which synthetic second messengers were applied intracellularly (Cancela et al., 2002) and by modelling studies (Wacke and Thiel, 2001). Thus, the density of H1 receptors may play a role in determining the minimal concentration of agonist required to elicit a response and therefore influence the apparent potency of an agonist. Such a ‘threshold’ mechanism for triggering a cellular response may also explain why our concentration–response curves were steeper (Hill coefficients of 1.1–1.6) than those obtained from binding studies (∼1).
Methylhistaprodifen is the most potent and selective H1 receptor agonist reported so far in the literature (Elz et al., 2000). At mouse H1 receptors expressed in PO/AH neurons, methylhistaprodifen was a full agonist and had a slightly higher potency than histamine. The slower decay time course of responses to methylhistaprodifen probably reflects the fact that it binds with higher affinity to mouse H1 receptors than histamine and the other agonists studied. 2-(3-TFMP) histamine is the second best H1 receptor agonist synthesized so far in terms of selectivity and potency (Leschke et al., 1995). 2-(3-TFMP) histamine was a full agonist at mouse H1 receptors with potency similar to that of histamine.
2-Pyridylethylamine is a widely used H1 receptor agonist but it appears to have affinity also for H2 receptors (Flynn et al., 1979; Leschke et al., 1995). In our preparation, 2-pyridylethylamine had only partial agonist activity and significantly lower potency than histamine. Because in this preparation H2 receptors are not present (Lundius et al., 2010), the responses can be attributed solely to activation of H1 receptors. Indeed, the responses to 2-pyridylethylamine were blocked by H1 receptor antagonists in all neurons tested. Similarly, betahistine is used in many studies as an H1 receptor agonist; however, it has been reported to be a partial agonist at H1 receptors and an antagonist at H3 receptors (Arrang et al., 1985). In PO/AH neurons, betahistine acted as a partial agonist at H1 receptors with low potency. The faster decay time course of the 2-pyridylethylamine and betahistine responses may reflect the fact that these compounds may bind with lower affinity to H1 receptors than histamine does, or that they induce a fast desensitization of the receptors.
Studies employing mutagenesis of the H1 receptor isoforms have revealed sites involved in the binding of histamine and some H1 receptor ligands (Leurs et al., 1994; 1995; Ohta et al., 1994; Wieland et al., 1999). Although no such data on mouse (m) H1 receptors are available, we can interpret our results by comparing the receptor's sequence with that of the other isoforms. Thus, all the crucial amino acids (Asp107, Thr194, Asn198 and Lys200) are conserved in all species studied, including the mouse. However, numerous studies have shown that the actions of some H1 receptor ligands are isoform specific. Thus, the human isoform is much less sensitive to histamine and histaprodifens than the guinea pig isoform and this difference is, at least in part, attributed to Asn84 in the human H1 receptor which corresponds to a Ser in the guinea pig receptor (Seifert et al., 2003; Bruysters et al., 2005). The mouse H1 receptor shares with the human H1 receptor the Asn84 which may explain the relatively low potency of histamine and of methylhistaprodifen observed in this study.
As the mouse H1 receptor sequence is very similar to that of the rat isoform, it was surprising to find that HTMT had antagonist activity in our mouse preparation, in contrast with its agonist action at rat H1 receptors (Alexander et al., 2011). We would like to note, however, that a similar situation has been found for 1-allyl-8S-lisuride, which is a partial agonist at the human H1 receptor and an antagonist at the guinea pig isoform (Pertz et al., 2006). Discrepancies regarding the pharmacological activity of HTMT among species have also been reported by other studies: it was initially identified as an H2 receptor agonist with 104 times higher affinity than histamine in mouse natural suppressor cells but a poor agonist for both H1 and H2 receptors in guinea pig myocardium (Khan et al., 1986). The specific binding sites for HTMT have not been studied in any H1 receptor isoform. While the mouse H1 receptor sequence displays high homology with the other isoforms, particularly with the rat isoform, it presents some differences in the N-terminal and the E2 loop, regions shown to contribute to the pharmacological properties of the H1 receptor (Strasser et al., 2008). Thus, the mouse H1 receptor presents Ala10 and Ser11, in the N terminal region, amino acids not present in the corresponding positions of the other isoforms. Similarly, in the E2 loop, Thr169 and Leu171 are found only in the mouse H1 receptor. It is therefore possible that the difference observed in the activity of HTMT are due to sequence differences at these sites.
Taken together, our results show that mouse H1 receptors have a distinct pharmacological profile particularly in terms of selective agonists. We identify here two full agonists of the mouse H1 receptor, methylhistaprodifen and 2-(3-TFMP) histamine, with potencies similar to that of histamine that may represent valuable tools for the study of physiological and pathophysiological actions mediated by H1 receptors.
Acknowledgments
This work was supported by the National Institutes of Health Grant NS060799 (I. V. T.).
Glossary
- HTMT
6-[2-(4-imidazolyl)ethylamino]-N-(4-trifluoromethylphenyl)heptanecarboxamide
- PO/AH
preoptic/anterior hypothalamic
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano R, Hiruma H, Nishida S, Kawakami T, Shimizu K. Inhibitory effect of histamine on axonal transport in cultured mouse dorsal root ganglion neurons. Neurosci Res. 2001;41:201–206. doi: 10.1016/s0168-0102(01)00275-9. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Quach TT, Dam Trung Tuong M, Yeramian E, Schwartz JC. Actions of betahistine at histamine receptors in the brain. Eur J Pharmacol. 1985;111:73–84. doi: 10.1016/0014-2999(85)90115-3. [DOI] [PubMed] [Google Scholar]
- Bruysters M, Jongejan A, Gillard M, van de Manakker F, Bakker RA, Chatelain P, et al. Pharmacological differences between human and guinea pig histamine H1 receptors: Asn84 (2.61) as key residue within an additional binding pocket in the H1 receptor. Mol Pharmacol. 2005;67:1045–1052. doi: 10.1124/mol.104.008847. [DOI] [PubMed] [Google Scholar]
- Cancela JM, Van Coppenolle F, Galione A, Tepikin AV, Petersen OH. Transformation of local Ca2+ spikes to global Ca2+ transients: the combinatorial roles of multiple Ca2+ releasing messengers. EMBO J. 2002;21:909–919. doi: 10.1093/emboj/21.5.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazot PL. Advances in histamine pharmacology reveal new drug targets. Br J Pharmacol. 2009;157:1–3. doi: 10.1111/j.1476-5381.2009.00228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazot PL. Therapeutic potential of histamine H3 receptor antagonists in dementias. Drug News Perspect. 2010;23:99–103. doi: 10.1358/dnp.2010.23.2.1475899. [DOI] [PubMed] [Google Scholar]
- Connelly WM, Shenton FC, Lethbridge N, Leurs R, Waldvogel HJ, Faull RL, et al. The histamine H4 receptor is functionally expressed on neurons in the mammalian CNS. Br J Pharmacol. 2009;157:55–63. doi: 10.1111/j.1476-5381.2009.00227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Lian J, Pai N, Huang XF. Reducing olanzapine-induced weight gain side effect by using betahistine: a study in the rat model. J Psychopharmacol. 2012;26:1271–1279. doi: 10.1177/0269881112449396. [DOI] [PubMed] [Google Scholar]
- Elz S, Kramer K, Pertz HH, Detert H, ter Laak AM, Kuhne R, et al. Histaprodifens: synthesis, pharmacological in vitro evaluation, and molecular modeling of a new class of highly active and selective histamine H(1)-receptor agonists. J Med Chem. 2000;43:1071–1084. doi: 10.1021/jm991056a. [DOI] [PubMed] [Google Scholar]
- Flynn SB, Gristwood RW, Owen DA. Differentiation of the roles of histamine H1- and H2-receptors in the mediation of the effects of histamine in the isolated working heart of the guinea-pig. Br J Pharmacol. 1979;65:127–137. doi: 10.1111/j.1476-5381.1979.tb17341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo RX, Anaclet C, Roberts JC, Parmentier R, Zhang M, Guidon G, et al. Differential effects of acute and repeat dosing with the H3 antagonist GSK189254 on the sleep-wake cycle and narcoleptic episodes in Ox-/- mice. Br J Pharmacol. 2009;157:104–117. doi: 10.1111/j.1476-5381.2009.00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas H, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci. 2003;4:121–130. doi: 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–1241. doi: 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- Jin CY, Anichtchik O, Panula P. Altered histamine H3 receptor radioligand binding in post-mortem brain samples from subjects with psychiatric diseases. Br J Pharmacol. 2009;157:118–129. doi: 10.1111/j.1476-5381.2009.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajihara Y, Murakami M, Imagawa T, Otsuguro K, Ito S, Ohta T. Histamine potentiates acid-induced responses mediating transient receptor potential V1 in mouse primary sensory neurons. Neuroscience. 2010;166:292–304. doi: 10.1016/j.neuroscience.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Khan MM, Marr-Leisy D, Verlander MS, Bristow MR, Strober S, Goodman M, et al. The effects of derivatives of histamine on natural suppressor cells. J Immunol. 1986;137:308–314. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leschke C, Elz S, Garbarg M, Schunack W. Synthesis and histamine H1 receptor agonist activity of a series of 2-phenylhistamines, 2-heteroarylhistamines, and analogues. J Med Chem. 1995;38:1287–1294. doi: 10.1021/jm00008a007. [DOI] [PubMed] [Google Scholar]
- Leurs R, Smit MJ, Tensen CP, Ter Laak AM, Timmerman H. Site-directed mutagenesis of the histamine H1-receptor reveals a selective interaction of asparagine207 with subclasses of H1-receptor agonists. Biochem Biophys Res Commun. 1994;201:295–301. doi: 10.1006/bbrc.1994.1701. [DOI] [PubMed] [Google Scholar]
- Leurs R, Smit MJ, Meeder R, Ter Laak AM, Timmerman H. Lysine200 located in the fifth transmembrane domain of the histamine H1 receptor interacts with histamine but not with all H1 agonists. Biochem Biophys Res Commun. 1995;214:110–117. doi: 10.1006/bbrc.1995.2263. [DOI] [PubMed] [Google Scholar]
- Lundius EG, Sanchez-Alavez M, Ghochani Y, Klaus J, Tabarean IV. Histamine influences body temperature by acting at H1 and H3 receptors on distinct populations of preoptic neurons. J Neurosci. 2010;30:4369–4381. doi: 10.1523/JNEUROSCI.0378-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, Hayashi H, Mizuguchi H, Kagamiyama H, Fujimoto K, Fukui H. Site-directed mutagenesis of the histamine H1 receptor: roles of aspartic acid107, asparagine198 and threonine194. Biochem Biophys Res Commun. 1994;203:1096–1101. doi: 10.1006/bbrc.1994.2295. [DOI] [PubMed] [Google Scholar]
- Pertz HH, Gornemann T, Schurad B, Seifert R, Strasser A. Striking differences of action of lisuride stereoisomers at histamine H1 receptors. Naunyn Schmiedebergs Arch Pharmacol. 2006;374:215–222. doi: 10.1007/s00210-006-0111-0. [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M. Histaminergic transmission in the mammalian brain. Physiol Rev. 1991;71:1–51. doi: 10.1152/physrev.1991.71.1.1. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K, Burckstummer T, Pertz HH, Schunack W, Dove S, et al. Multiple differences in agonist and antagonist pharmacology between human and guinea pig histamine H1-receptor. J Pharmacol Exp Ther. 2003;305:1104–1115. doi: 10.1124/jpet.103.049619. [DOI] [PubMed] [Google Scholar]
- Sethi J, Sanchez-Alavez M, Tabarean IV. Loss of histaminergic modulation of thermoregulation and energy homeostasis in obese mice. Neuroscience. 2012;217:84–95. doi: 10.1016/j.neuroscience.2012.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, Seifert R. Ligand-specific contribution of the N terminus and E2-loop to pharmacological properties of the histamine H1-receptor. J Pharmacol Exp Ther. 2008;326:783–791. doi: 10.1124/jpet.108.140913. [DOI] [PubMed] [Google Scholar]
- Tabarean IV. Persistent histamine excitation of glutamatergic preoptic neurons. Plos ONE. 2012;7:e47700. doi: 10.1371/journal.pone.0047700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabarean IV, Conti B, Behrens M, Korn H, Bartfai T. Electrophysiological properties and thermosensitivity of mouse preoptic and anterior hypothalamic neurons in culture. Neuroscience. 2005;135:433–449. doi: 10.1016/j.neuroscience.2005.06.053. [DOI] [PubMed] [Google Scholar]
- Takada M, Li ZK, Hattori T. A direct projection from the tuberomammillary nucleus to the spinal cord in the rat. Neurosci Lett. 1987;79:257–262. doi: 10.1016/0304-3940(87)90439-3. [DOI] [PubMed] [Google Scholar]
- Wacke M, Thiel G. Electrically triggered all-or-none Ca(2)+-liberation during action potential in the giant alga Chara. J Gen Physiol. 2001;118:11–22. doi: 10.1085/jgp.118.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada H. [From biochemistry to pharmacology: the histaminergic neuron system in the brain] Nippon Yakurigaku Zasshi. 1992;99:63–81. doi: 10.1254/fpj.99.63. [DOI] [PubMed] [Google Scholar]
- Wieland K, Laak AM, Smit MJ, Kuhne R, Timmerman H, Leurs R. Mutational analysis of the antagonist-binding site of the histamine H(1) receptor. J Biol Chem. 1999;274:29994–30000. doi: 10.1074/jbc.274.42.29994. [DOI] [PubMed] [Google Scholar]
- Zhou J, Lee AW, Devidze N, Zhang Q, Kow LM, Pfaff DW. Histamine-induced excitatory responses in mouse ventromedial hypothalamic neurons: ionic mechanisms and estrogenic regulation. J Neurophysiol. 2007;98:3143–3152. doi: 10.1152/jn.00337.2007. [DOI] [PubMed] [Google Scholar]