

Abstract
BACKGROUND AND PURPOSE
Podocyte injury plays a key role in the development of diabetic nephropathy (DN). We have recently shown that 11R-VIVIT, an inhibitor of cell-permeable nuclear factor of activated T-cells (NFAT), attenuates podocyte apoptosis induced by high glucose in vitro. However, it is not known whether 11R-VIVIT has a protective effect on DN, especially podocyte injury, under in vivo diabetic conditions. Hence, we examined the renoprotective effects of 11R-VIVIT in diabetic db/db mice and the possible mechanisms underlying its protective effects on podocyte injury in vivo and in vitro.
EXPERIMENTAL APPROACH
Type 2 diabetic db/db mice received i.p. injections of 11R-VIVIT (1 mg·kg−1) three times a week and were killed after 8 weeks. Immortalized mouse podocytes were cultured under different experimental conditions.
KEY RESULTS
11R-VIVIT treatment markedly attenuated the albuminuria in diabetic db/db mice and also alleviated mesangial matrix expansion and podocyte injury. However, body weight, food and water intake, and glucose levels were unaffected. It also attenuated the increased NFAT2 activation and enhanced urokinase-type plasminogen activator receptor (uPA receptor) expression in glomerulor podocytes. In cultured podocytes, the increased nuclear accumulation of NFAT2 and uPA receptor expression induced by high glucose treatment was prevented by 11R-VIVIT or NFAT2-knockdown; this was accompanied by improvements in the filtration barrier function of the podocyte monolayer.
CONCLUSIONS AND IMPLICATIONS
The NFAT inhibitor 11R-VIVIT might be a useful therapeutic strategy for protecting podocytes and treating DN. The calcinerin/NFAT2/uPA receptor signalling pathway should be exploited as a therapeutic target for protecting podocytes from injury in DN.
Keywords: 11R-VIVIT, diabetic nephropathy, podocyte, activate nuclear factor of activated T-cells, uPAR
Introduction
Diabetic nephropathy (DN) is an important and common complication of type 2 diabetes leading to end-stage renal disease. Much of the morbidity and mortality of diabetes can be attributed to nephropathy (Ibrahim and Hostetter, 1997). The clinical manifestations of DN are characterized by persistent albuminuria, thickness of glomerular basement membrane (GBM), accumulation of the extracellular matrix, which results in glomerulosclerosis and progressive renal dysfunction. The mechanisms underlying DN pathogenesis are still far from being fully understood. Current therapies for DN in clinical settings are largely symptomatic treatments, which include lowering blood glucose levels, controlling cholesterol and blood pressure and preventing complications (Parving et al., 2001; Remuzzi et al., 2002; Wolf and Ritz, 2003; Vejakama et al., 2012). Although these strategies are widely used for the treatment of DN, their renoprotective effects are not sufficient to prevent the progression of DN to end-stage renal disease (Choudhury et al., 2010; Seaquist and Ibrahim, 2010; Coca et al., 2012). Therefore, there is a need for the development of new drugs for DN based on a new mechanism of action.
Podocytes are terminally differentiated cells that reside on the outer surface of the GBM and play a key role in maintaining the structure and function of the glomerular filtration barrier. Accumulating evidence suggests that glomerular podocytes play a pivotal role in the pathogenesis of diabetic kidney disease (Wolf et al., 2005; Li et al., 2007). Exploration of the mechanisms of podocyte injury in diabetic conditions will be critical for the development of novel preventive and therapeutic approaches to DN.
The nuclear factor of activated T-cells (NFAT), which is the substrate for calcineurin (CaN), represents a family of Ca2+ dependent transcription factors. Although originally thought to be largely restricted to cells of the immune system, abundant evidence now indicates that NFAT family members are expressed in non-immune cells with some expressed ubiquitously (Rao et al., 1997; Benedito et al., 2005). NFAT has been shown to regulate heart valve development, skeletal muscle and smooth muscle cell differentiation and vascular development (Chin et al., 1998; Ranger et al., 1998; Graef et al., 2001; Larrieu et al., 2005). In addition, recent studies have demonstrated that the activation of NFAT is involved in renal tubular cell apoptosis, podocyte injury and glomerulosclerosis (Lin et al., 2010; Wang et al., 2010; 2011). We also found that high glucose (HG) stimulation activates NFAT2 in podocytes in vitro. Pretreatment with a cell permeable NFAT inhibitor 11R-VIVIT (Aramburu et al., 1998) completely blocked NFAT2 nuclear accumulation induced by HG. In addition, the apoptosis effects induced by HG were also attenuated by concomitant treatment with 11R-VIVIT in cultured podocytes (Li et al., 2013). These results suggest that NFAT2 is involved in the podocyte injury associated with DN, and 11R-VIVIT might have renoprotective effects in DN and podocyte injury.
Based on the above findings, in this study we investigated the potential therapeutic role of 11R-VIVIT on the progression of DN in diabetic db/db mice, which lack the hypothalamic leptin receptor; this is a genetic model of type 2 diabetes that exhibits clinical and histological features of DN similar to the human disease (Cohen et al., 1996; Sharma et al., 2003). We also explored the underlying molecular mechanisms in these experimental animal models and in vitro.
Methods
Animals
Male C57BL/KsJ db/db mice and their age-matched wild-type (BKS) mice were purchased from Model Animal Research Center of Nanjing University, and housed in the animal centre of Zhongshan School of Medicine, Sun Yat-sen University. The former strain of mice is obese and known to develop type 2 diabetes, followed by diabetic kidney disease. All mice were provided with a standard diet and water ad libitum and were maintained in a temperature (23 ± 2°C) – and humidity (55 ± 5%) – controlled room with a 12-h light, 12-h dark cycle. The total number of mice used was 15. At 12 weeks of age, half of the db/db mice were randomly chosen to receive 11R-VIVIT, a special inhibitor of NFAT (Merck KGaA, Darmstadt, Germany). The 11R-VIVIT was injected i.p. to the db/db mice (n = 5) (1 mg·kg−1), three times a week for 8 weeks. The other half of the db/db and five BKS mice were given the same volume of PBS without 11R-VIVIT. Fasting blood glucose was measured in tail-vein blood, weekly, using a one touch ultra glucometer and Test Strip (Lifescan, Milpitas, CA, USA) after 6 h of fasting. Body weight was obtained at 1-week intervals. For urine collection, individual mice were caged once every 2 weeks in a metabolic cage for 24 h. After 8 weeks of treatment, mice were anaesthetized (ketamine; 70 mg·kg−1 i.p.) and blood samples were obtained from the retro-orbital venous plexus for determination of the plasma concentration of creatinine. Heart, liver and kidney tissues were weighed. The samples were frozen and kept in liquid nitrogen until use. Animal care and experiments were performed in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010), and the review board of the Guangdong General Hospital granted ethical permission for this study. Renal tissues from patients with DN and renal cell carcinoma (adjacent normal tissues) were respectively obtained after patients gave written consent. The patient's baseline characteristics were given in Supporting Information Table S1. The study of patients was conducted in accordance with the Second Helsinki Declaration and approved by the Ethics Committee for Guangdong General Hospital.
Blood and urine analysis
Urinary albumin or urinary and serum creatinine were measured using mouse albumin-specific ELISA and creatinine kits (Cusabio Biotech, Wuhan, China) respectively, according to the manufacturer's instructions. Proteinuria was expressed as μg albumin mg−1 creatinine.
Renal histology analysis
Paraffin embedded tissues were cut at 4 μm, perpendicular to the long axis of the kidney, and stained with periodic acid Schiff (PAS) for light microscopic examination. Sections were coded and read by an observer unaware of the experimental group. In each animal of the three experimental groups, more than 20 glomeruli were used and averaged for morphometric analysis. To quantify mesangial expansion, using Image-Pro Plus 6.0 (Media Cybernetics, Georgia Avenue, MD, USA) programme, the area of the PAS-positive and nuclei-free material in the mesangium (mesangial area) was factored by the glomerular tuft area to arrive at the fraction of mesangial matrix.
Podocyte number counting
The number of podocytes per glomerulus was determined based on methods described previously (Wang et al., 2005; Toyonaga et al., 2011). Sections, each 4 μm, of formalin-fixed, paraffin embedded tissue were deparaffinized and hydrated with water. Tissue sections were placed in preheated 10 mM Tris-buffered saline with 1.0 mM EDTA (pH 9.0) and heated for 20 min at 100°C in a water bath. After cooling to room temperature (RT), slides were rinsed in de-ionized water and then immersed in 3% H2O2 to quench endogenous peroxidase activity. After being washed, slides were incubated with rabbit polyclonal antibody Wilm's tumour antigen 1 (WT1; sc-192, Santa Cruz, CA, USA) for 60 min at RT, then washed and immunoperoxidase staining was performed using the Envision Plus system for rabbit primary antibodies (Dako Cytomation, Carpinteria, CA, USA) according to the manufacturer's instructions. The slides were counterstained with haematoxylin and permanently mounted before examination by light microscopy. The number of podocytes per glomerulus is the average number of nuclei stained in 20 randomly selected glomeruli.
Immunocytochemistry
For immunofluorescent labelling, 4 μm sections were washed once with PBS, permeabilized with 0.5% Triton X-100 in PBS, and incubated with 5% BSA for 20 min before further incubation with one of the primary antibodies [synaptopodin (N-14), sc-21536; urokinase-type plasminogen activator receptor (uPAR; FL-290), sc-10815; NFATc1, sc-13033; WT1, sc-15421; Santa Cruz Biotech] for 2 h at RT. For double labelling, sections were washed three times with PBS for 5 min and one of the secondary antibodies [FITC-donkey anti-goat IgG (H + L); Texas Red-donkey anti-goat IgG (H + L); Protein Tech Group, Inc, Chicago, IL, USA 1:50; goat anti rabbit Alexa Fluor 555; goat anti rabbit Alexa Fluor488, Cell Signaling Technology, Shanghai, China, 1:1000] was applied for 1 h. Images were captured with confocal microscopy (LeicaSP5-FCS, Wetzlar, Germany). All images were analysed by two investigators blinded to the identity of the samples.
Electron microscopy
Tissue samples of the kidney were cut into 1 mm cubes and fixed for 4 h at 4°C in 2.5% glutaraldehyde in 0.1 mol·L−1 phosphate buffer. They were postfixed in 1% buffered osmium tetroxide, dehydrated through graded ethanols and embedded in Epon. Thin sections (80 nm) were cut with a diamond knife, collected on 300-mesh copper or nickel grids, and double stained with uranyl acetate and lead citrate before examination using an electron microscope (H-700, Hitachi, Tokyo, Japan). Six photographs were taken in each glomerulus with a magnification of 10 000. Using the Image-Pro Plus 6.0 programme, five points of glomerular basement membrane picked up in each photograph and the thickness was measured; 20–30 measurements were averaged on 2–3 glomeruli per specimen.
Cell culture and treatment
The conditionally immortalized mouse podocyte cell line was kindly provided by Dr Peter Mundel (Massachusetts General Hospital, Boston, MA, USA). Cells were grown in 12 μg·mL−1 type-I collagen (Shengyou Biotechnology, Hangzhou, China) -coated flasks at the permissive temperature (33°C) in RPMI-1640 (Gibco BRL, Gaithersburg, MD, USA) supplemented with 10% FBS (Gibco BRL) and 50 U·mL−1 γ-interferon (IFN-γ, ProSpec, Ness Ziona, Israel). To induce differentiation, podocytes were reseeded and cultured at 37°C in the absence of IFN-γ (non-permissive condition) for 10 to 14 days. After differentiation of podocytes was confirmed by the identification of synaptopodin, a podocyte differentiation marker, cells were synchronized into quiescence by growing cells in serum-free DMEM (Gibco BRL) for 24 h, and then treated with normal glucose (NG, 5.3 mM), HG (30 mM) or NG (5.3 mM) plus mannitol (24.7 mM; osmolality control) for 48 h before experimentation. For inhibition experiments, 11R-VIVIT at concentrations of 100 nM was added to cells treated with HG (30 mM) for 48 h. Each reaction was repeated in triplicate.
Immunofluorescence of cultured podocytes
After being subjected to various treatments, podocytes were fixed with 4% paraformaldehyde at −20°C for 20 min, and then were blocked with 5% BSA for 20 min at RT to block non-specific binding. Then, the cells were incubated with rabbit anti-NFAT2 antibody (Santa Cruz, 1:50) overnight at 4°C. After three washes with PBS, cells were incubated with the goat anti rabbit Alexa Fluor 555(Cell Signaling Technology) for 1 h at RT, and then double stained with 4′-6- diamidino-2-phenylindole (DAPI) for 5 min to visualize the nuclei. After being washed, the slides were mounted with antifade mounting medium (Beyotime Institute of Biotechnology, Shanghai, China). Photomicrographs were taken with confocal microscopy (LeicaSP5-FCS). All images were analysed by two investigators blinded to the identity of the samples.
Transfection of small interfering RNA
The small interfering RNA (siRNA) sequences that target NFAT2 and control siRNA were synthesized by Invitrogen: NFAT2-siRNA sequence (sense: 5′-GCCAUAACUUUCUGCAAG ATT-3′; antisense: 5′-UCUUGCAGAAAGUUAUGGCTT-3′); control siRNA sequence (sense: 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense: 5′-ACGUGACACGUUCGGAGAATT-3′). After a 24-h incubation with NFAT2-siRNA or Control-siRNA, the cells were then treated with 30 mM glucose for an additional 48 h. Cells that were not transfected and were exposed to 5.3 mM glucose or 30 mM glucose or 5.3 mM glucose plus 24.7 mM mannitol for 48 h were considered to be the controls. Transfection was processed in 6-well plates or 50 cm2 culture plates with siRNAs using Lipofectamine 2000 (Invitrogen) transfection reagent protocols.
Real-time quantitative RT-PCR
Total RNA from cultured podocytes was extracted with Trizol Reagent according to the manufacturer's instructions (Invitrogen). Then, complementary DNA was synthesized from 2 μg of total RNA using the reverse transcriptase SuperScript (Takara Biotechnology, Dalian, China). Subsequently, the cDNA was subjected to quantitative RT-PCR (qRT-PCR) using Power SYBR Green PCR Master Mix (Takara Biotechnology). Each real-time PCR reaction consisted of 2 μL diluted RT product, 10 μL SYBR Green PCR Master Mix (2×) and 250 nM forward and reverse primers in a total volume of 20 μL. Reactions were carried out on 7500 qRT-PCR System (Applied Biosystems, Foster, CA, USA). The amplification conditions comprised an initial denaturation step at 95°C for 10 min, followed by amplification of the target DNA for 40 cycles of 95°C (5 s) and 60°C (30 s), and extension at 72°C (40 s). The primers used for real-time PCR were as follows: Plaur, forward 5′-AAGCCTGCAATG CCGCTATC-3′; reverse 5′-GGGTGTAGTTGCAACACTTCAG GA-3′; GAPDH forward 5′-TGTGTCCGTCGTGGATCTGA-3′, reverse 5′-TTGCTGTTGAAGT CGCAGGAG-3′. Each reaction was amplified in triplicate and the fold change in expression of each gene was calculated using the 2−△△Ct method, using GAPDH mRNA as an internal control.
Protein extraction and Western blot analysis
Podocytes subjected to different experimental conditions were lysed with RIPA lysis buffer. The samples were centrifuged and the supernatants were collected as total cell extracts. Nuclear and cytoplasmic proteins from cells were extracted using the Nuclear and Cytoplasmic Protein Extraction kit (Beyotime Institute of Biotechnology) according to the manufacturer's instructions. Protein concentration was quantified by the Bradford Assay kit (Beyotime Institute of Biotechnology). An aliquot of cell lysates containing 30 μg of protein was separated on 10% SDS-polyacrylamide gels, and then transferred to PVDF membranes (Amersham Biosciences, CA, USA) by electroblotting. After being blocked, the membranes were incubated overnight at 4°C with the following primary antibodies: (i) mouse monoclonal antibodies to NFAT2 (Santa Cruz, 1:400); (ii) rabbit polyclonal antibody to histone 3 (Abcam, Cambridge, USA; 1:250). (iii) rabbit polyclonal antibodies to uPA receptor (Santa Cruz, 1:400). (iv) mouse monoclonal antibodies to GAPDH (Santa Cruz, 1:500). After being washed, the horseradish peroxidase conjugated Goat Anti-Mouse IgG or Goat Anti-Rabbit IgG (Jackson Immuno Research, West Grove, PA, USA, 1: 10 000) was added and the samples incubated for 1 h at RT. The immunoblots were washed and immersed in ECL Plus Western Blotting Detection Reagents (Pluslight, Forevergen, China), and then exposed to X-ray film (Kodak, Rochester, NY, USA). The bands of the resulting autoradiographs were quantified densitometrically using Bandscan software. Protein expression was quantified as the ratio of specific band to histone 3 (nuclear fractions) or GAPDH.
Albumin influx assay
A simple albumin influx assay was adapted to evaluate the filtration barrier function of podocyte monolayer, as described previously (Rico et al., 2005; Li et al., 2008; Qiu et al., 2012). Briefly, podocytes (5 × 103) were seeded onto the collagen-coated transwell filters (3-μm pore; Corning, New York, USA) in the top chamber and cultured under differentiating conditions. After 10 days, podocytes were deprived of serum (starved) overnight, incubated with or without 11R-VIVIT for 30 min, then incubated with or without 30 mM glucose for 48 h. Cells were washed twice with PBS supplemented with 1 mM MgCl2 and 1 mM CaCl2 to preserve the cadherin-based junctions. The top chamber was then refilled with 0.15 mL of RPMI 1640 and the bottom chamber with 1 mL of RPMI 1640 supplemented with 40 mg·mL−1 BSA and the samples incubated at 37°C for 6 h, and a small aliquot of medium from the top chamber was collected for albumin concentration measurement by using a bicinchoninic acid protein assay kit (Biocolor Bioscience & Technology Company, Shanghai, China).
Statistical analysis
All values are expressed as mean ± SEM. Statistical analysis was performed using the statistical package SPSS for Windows Ver. 13.0 (SPSS, Inc., Chicago, IL, USA). Multiple comparisons among the groups were conducted by one-way ANOVA with least significant difference- test or Tamhane's-test. P-values <0.05 were considered significant.
Results
To establish the potential role of NFAT2 in the pathophysiology of DN, we investigated the effect of the NFAT inhibitor 11R-VIVIT in db/db mice, a typical 2 type diabetes animal model, and followed the developing DN. After 8 weeks treatment, animals were killed and various biochemical and histological parameters were evaluated. In addition, the possible mechanisms underlying the protective effects of 11R-VIVIT on podocyte injury were also investigated in these experimental animal models and in vitro in cultured podocytes.
Metabolic characteristics of db/db and BKS mice
Metabolic characteristics of the experimental mice are shown in Table 1. Body, kidney and liver weights were significantly heavier in diabetic db/db mice than in non-diabetic BKS control mice. Kidney-to-body weight ratio differed significantly between BKS and db/db mice, since the diabetic db/db mice were found to weigh more. Heart weights did not differ between the db/db mice and the BKS mice. The db/db mice exhibited hyperglycaemia associated with obesity throughout the experimental periods (12–20 weeks of age). We measured blood glucose in all the experimental animals once a week over this period, blood glucose in diabetic db/db mice was markedly higher than in BKS mice. Eight weeks of treatment with 11R-VIVIT, a cell-permeable NFAT inhibitor, at a dose of 1 mg·kg−1 body weight, i.p., three times a week significantly suppressed the increase in kidney weight so that it remained at the same weight as that of the non-diabetic kidney, but it did not affect blood glucose levels, body weight, food and water intake in either diabetic db/db mice or non-diabetic BKS (Table 1).
Table 1.
Influences of 11R-VIVIT on physiological parameters in db/db and BKS mice
| BKS + PBS | db/db + PBS | db/db + 11R-VIVIT | |
|---|---|---|---|
| Fasting glucose(mmol·L−1) | 7.0 ± 0.2 | 30.8 ± 0.9* | 29.8 ± 1.1* |
| Food intake (mL) | 4.2 ± 0.1 | 9.8 ± 0.8* | 9.2 ± 0.1* |
| Water intake (mL) | 4.3 ± 0.1 | 14.4 ± 0.6* | 13.3 ± 0.4* |
| Body weight (g) | 25.1 ± 0.4 | 50.5 ± 1.6* | 49.3 ± 1.1* |
| Kidney weight (g) | 0.30 ± 0.01 | 0.44 ± 0.01* | 0.33 ± 0.02△ |
| Liver weight (g) | 1.13 ± 0.05 | 2.83 ± 0.26* | 2.82 ± 0.18* |
| Heart weight (g) | 0.13 ± 0.01 | 0.13 ± 0.01 | 0.13 ± 0.01 |
| Kidney/body weight (mg·g−1) | 12.12 ± 0.28 | 8.69 ± 0.44* | 7.00 ± 0.37*△ |
Data shown are mean ± SEM, n = 5. *P < 0.0.01 versus BKS + PBS; △P < 0.05 versus db/db + PBS.
Urinary albumin excretion and renal function
Urinary albumin excretion, which is one of the parameters of glomerular dysfunction in diabetes, was measured. Urinary albumin excretion (μg·mg−1 creatinine) was significantly increased in db/db mice as compared with that in BKS mice at the age of 20 weeks (Figure 1A). The NFAT inhibitor 11R-VIVIT (i.p.) significantly attenuated the increase in urinary albumin excretion rates in db/db mice (Figure 2). Creatinine clearance, which is generally considered as a marker of renal function, was determined to evaluate the effect of 11R-VIVIT on renal function in db/db mice. The creatinine clearance of db/db mice was lower than that of BKS mice at 20 weeks of age. The creatinine clearance of the 11R-VIVIT-treated db/db group was markedly increased compared with the vehicle-treated db/db control after 8 weeks of treatment (Figure 1B).
Figure 1.
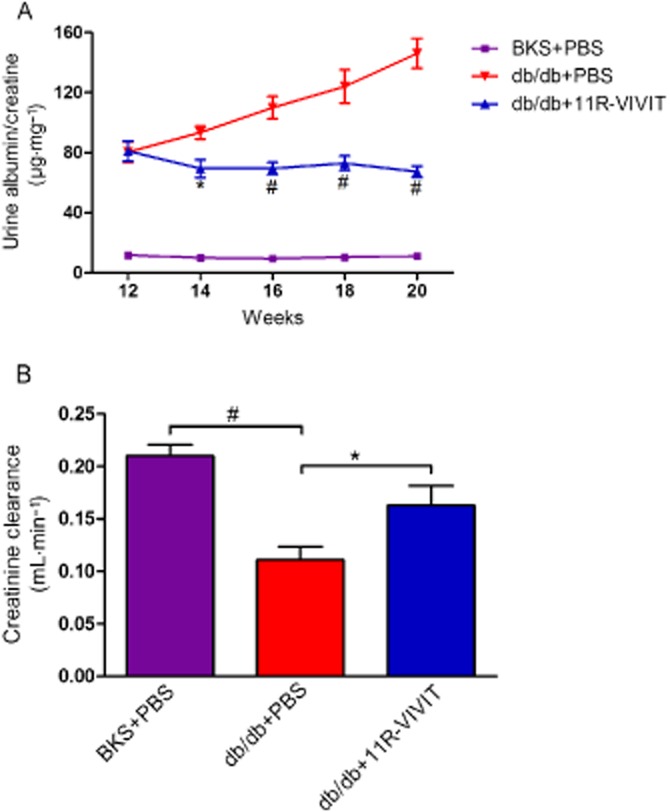
Effect of urinary albumin excretion and kidney function in db/db mice. The graphs show urinary albumin excretion (μg·mg−1 creatinine) (A), creatinine clearance (B) in vehicle-treated BKS mice, vehicle treated db/db mice and 11R-VIVIT treated db/db mice. A 24-h pooled urine sample was collected in a metabolic cage at 12, 14, 16, 18 and 20 weeks of age for each mouse. Urinary albumin or urinary and serum creatinine were measured using a competitive ELISA. Data are shown as means ± SEM. n = 5. *P < 0.05, #P < 0.01 versus db/db control.
Figure 2.
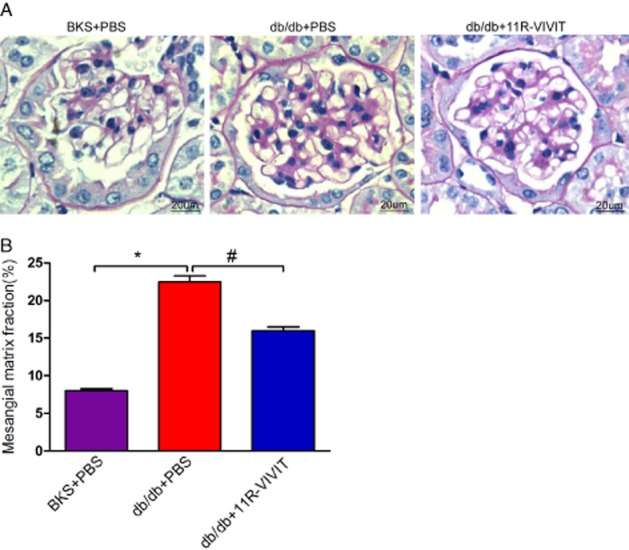
Effects of 11R-VIVIT on mesangial expansion. (A) Representative photomicrographs of periodic acid–Schiff staining of glomeruli of BKS + PBS, db/db + PBS, db/db+11R-VIVIT mice at 20 weeks. (B) Semiquantitative analyses of mesangial expansion at 20 weeks (n = 5). Original magnification ×400. Data are mean ± SEM. *P < 0.01 versus BKS + PBS; #P < 0.01 versus db/db + PBS.
Renal histological examination
Diabetic nephropathy in humans is histologically characterized by hypertrophy of glomerular, expansion of glomerular mesangium. Representative light micrographs of glomeruli are shown in Figure 2A. The mesangial matrix expansion was evident in glomeruli of diabetic db/db mice. PAS-positive mesangial matrix areas in db/db mice were substantially enlarged compared with BKS mice. The fraction of mesangial matrix further confirmed these results (Figure 2B). In contrast, these renal pathological parameters were markedly ameliorated in 11R-VIVIT treated db/db mice, after 8 weeks, compared with vehicle-treated db/db mice.
Glomerular basement membrane and podocyte foot process
The kidney ultrastructure was further examined by electron microscopy. As shown in Figure 3A-a, normal morphology of the glomerular filtration barrier including GBM and podocyte foot process was seen in non-diabetic BKS. As expected, chronic diabetes caused GBM thickening and foot process effacement (Figure 3A-b, 3B). Interestingly, the extent of foot process effacement and GBM thickening was more marked in the diabetic db/db mice (Figure 3A-b) than in the db/db mice treated with 11R-VIVIT (Figure 3A-c). The impairment of the glomerular filtration barrier is consistent with the more severe albuminuria observed in db/db mice (Figure 1).
Figure 3.
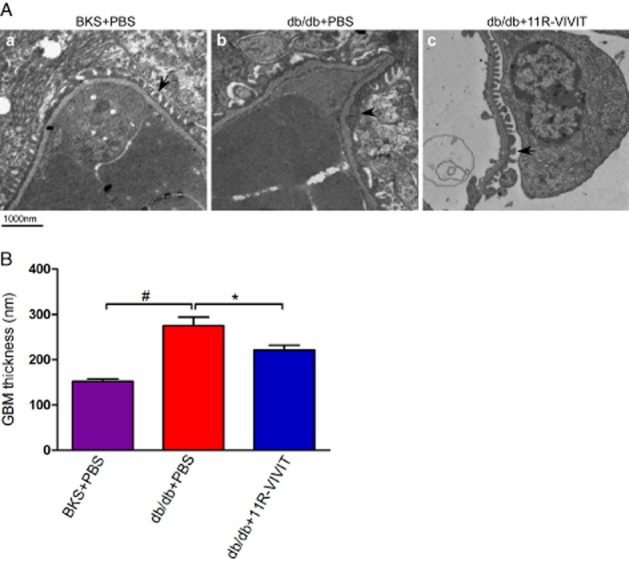
11R-VIVIT attenuated glomerular basement membrane (GBM) thickening and podocyte foot process effacement. Kidney samples from non-diabetic BKS control mice treated with PBS and db/db mice treated with PBS or 11R-VIVIT were subjected to electron microscopic analyses. (A) Representative electron micrographs show GBM thickening and foot process effacement in the PBS treated diabetic kidney. The morphological injury was attenuated in diabetic mice that received 11R-VIVIT. Scale bar, 1000 nm. (B) Effect of the different treatments on the thickness of the GBM. Data are mean ± SEM. #P < 0.01 versus BKS + PBS; *P < 0.01 versus db/db + PBS.
Podocyte numbers
Podocytes have a key role in maintaining the integrity of the glomerular filtration barrier, and diabetic renal injury is known to lead to loss of podocytes. We examined the podocytes in glomeruli by immunostaining with anti-WT1 antibody, a podocyte-specific marker. As shown in Figure 4A, B, there was a marked reduction in glomerular WT1-positive cells in glomeruli of db/db mice relative to BKS mice (P < 0.01, 10.85 ± 0.81 vs. 16.75 ± 0.95). This reduction in WT1-positive cells was significantly ameliorated in 11R-VIVIT-treated db/db mice (P < 0.05, 13.8 ± 0.86 vs. 10.85 ± 0.81, Figure 4B). These results indicate that 11R-VIVIT prevents podocyte depletion in diabetic animal models.
Figure 4.
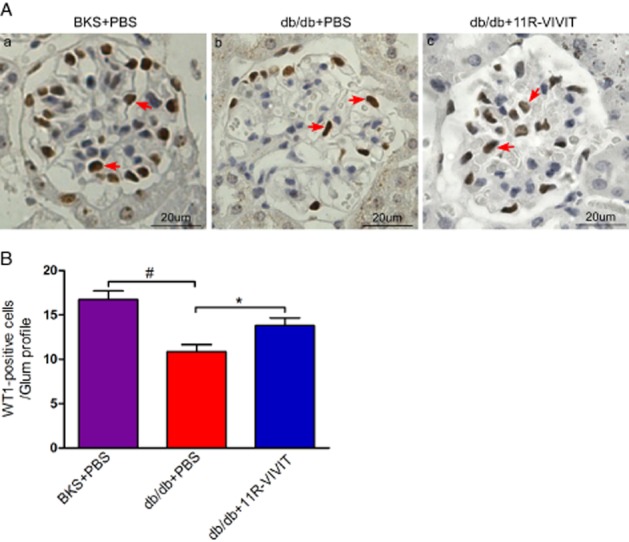
11R-VIVIT partially restores podocyte number. (A) Typical images of WT1 stained glomeruli from BKS, db/db and db/db+11R-VIVIT mice. Arrows point to WT-1-positive cells. Magnification ×400. (B) Quantification of podocyte markers WT-1-positive cells per glomerular section. The number of WT1-stained nuclei was obtained from 20 glomeruli per kidney section from five mice per group. Data are means ± SEM. *P < 0.05, #P < 0.01 versus db/db + PBS.
NFAT2 and uPAR expression in glomerular podocyte
To explore the underlying mechanism of 11R-VIVIT's effects, we studied the protein expression of NFAT2 and uPA receptors in the glomeruli podocyte of diabetic db/db and BKS mice. Two podocyte-specific markers, WT1 and synaptopodin, were also used in this present study. Figure 6A, B demonstrates the effect of 11R-VIVIT on NFAT2 and uPA receptor protein levels using immunofluorescent staining. As shown in Figure 5A, NFAT2 activation was markedly increased in the glomerular podocytes of the diabetic db/db mice compared with BKS mice. Treatment with 11R-VIVIT (8 weeks) inhibited the nuclear accumulation of NFAT2. Similar results were observed for uPA receptor expression (Figure 5B). Immunofluorescent staining also exhibited a restoration of WT1 and synaptopodin expression in the 11R-VIVIT treated group, which is consistent with the notion that 11R-VIVIT restores the podocyte integrity in DN mice.
Figure 6.
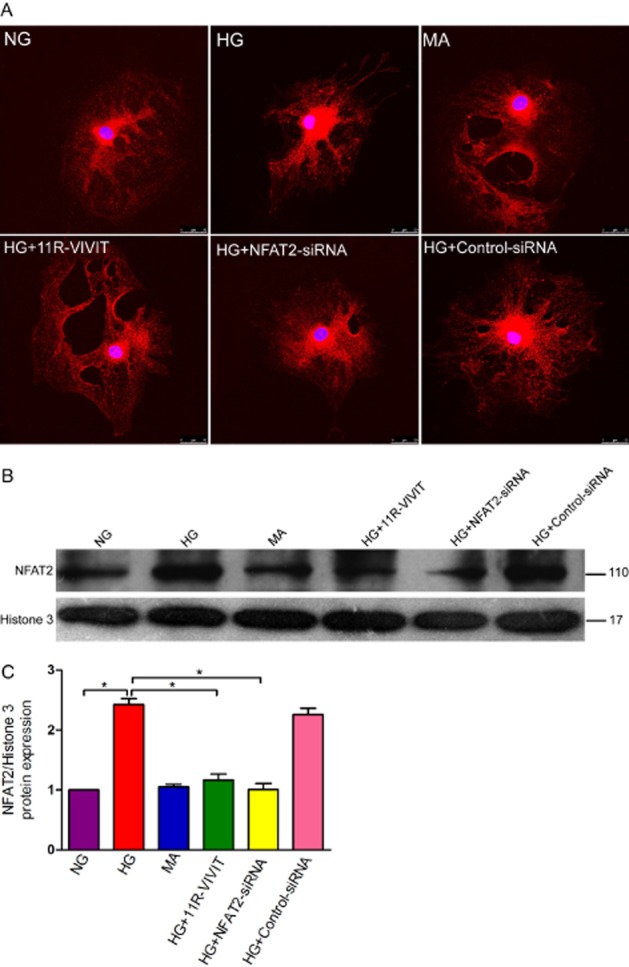
Effects of 11R-VIVIT and NFAT2 gene knockdown on the expression of NFAT2 in podocytes treated with high glucose. (A) Confocal images of podocytes showing the expression of NFAT2 (red) and DAPI-stained nuclei (blue). (B) High glucose treatment increases the expression of NFAT2 detected by immunoblot in a nuclear extract of podocytes. Histone 3 was used as a nuclear protein control. (C) Densitometric analysis of three repetitions of the experiment shown in (C). Values are expressed as the mean ± SEM. *P < 0.01 versus HG. Note NG, normal glucose (5.3 mM) group; HG, high glucose group (30 mM); MA, normal glucose (5.3 mM) + mannitol (24.7 mM) group, as an osmolality control; HG+11R-VIVIT, high glucose (30 mM) + 11R-VIVIT(100 nM, a cell permeable NFAT inhibitor) group; HG + NFAT2-siRNA, high glucose (30 mM) + NFAT2-siRNA (50 nM) group; HG + control-siRNA, high glucose (30 mM) + control- siRNA (50 nM), as a negative control group. High glucose increased the nuclear localization of NFAT2 in podocytes, and this effect was blocked by either 11R-VIVIT, a cell permeable inhibitor of NFAT, or NFAT2-siRNA.
Figure 5.
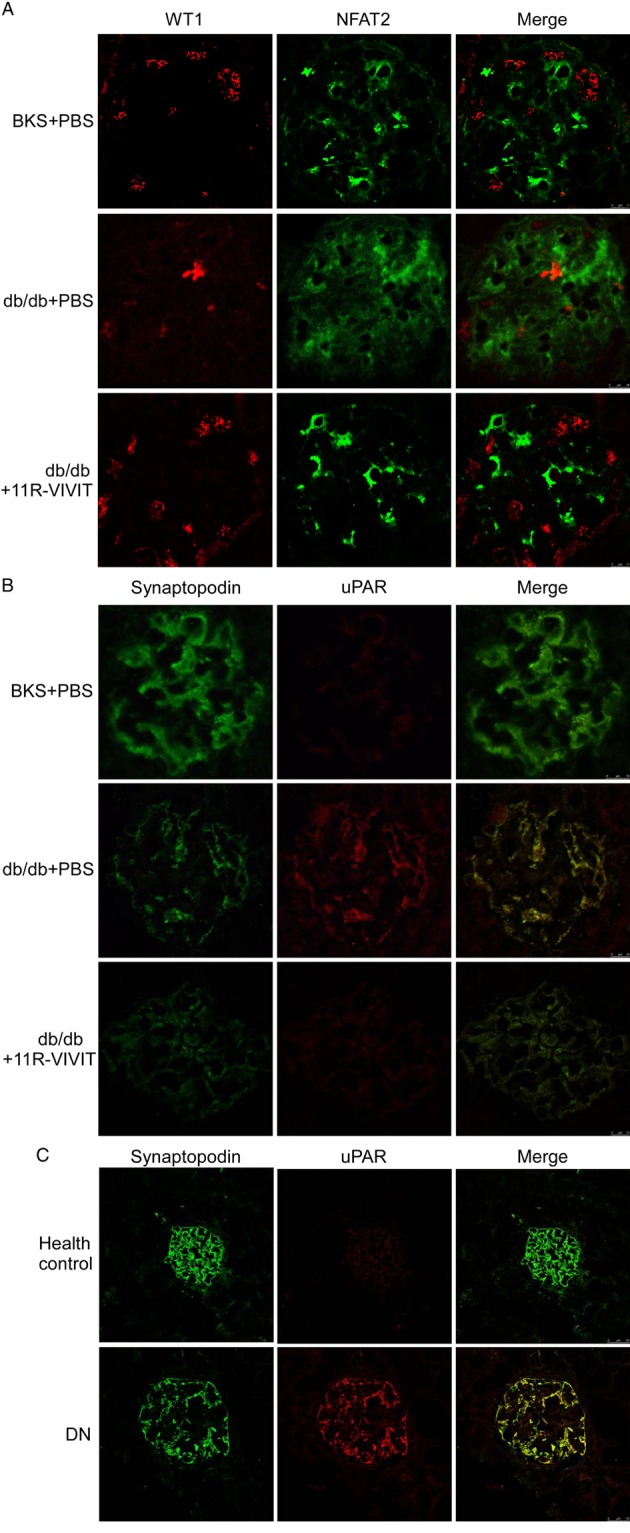
NFAT inhibitor (11R-VIVIT) inhibited NFAT2 activation and uPA receptor expression in glomerular podocytes. (A) Double immunofluorescent staining of NFAT2 (green), WT1 identified podocytes (red), and merged images in the kidney from non-diabetic mice or diabetic animals. Although NFAT2 was faintly observed in non-diabetic glomeruli, increased immunoreactivity for NFAT2 was observed in the diabetic mice. Co-staining for NFAT2 (green) demonstrates that NFAT2 activation is markedly inhibited in glomerular podocytes of 11R-VIVIT-treated diabetic mice. Original magnification ×400. (B) Double immunofluorescent staining of uPA receptors (red), synaptopodin identified podocytes (green), and merged images (yellow) in the kidney from non-diabetic mice or diabetic animals. The protein expression of uPA receptors (red) was significantly increased in diabetic glomeruli. Co-staining for uPA receptors (red) demonstrates that uPA receptor expression was restored in glomerular podocytes of 11R-VIVIT treated diabetic mice, original magnification ×400. (C) Double immunofluorescent staining of uPA receptors (red), synaptopodin identified podocytes (green), and merged images in the kidney from patients with DN or healthy control (normal renal tissues from patients with renal cell carcinoma). Co-staining for uPA receptors (red) demonstrates that their expression was markedly increased in glomerular podocytes of DN patients. Original magnification ×200.
Using immunofluorescent staining, we also investigated the expression of uPA receptors in glomerular podocytes of patients with DN. Consistent with the results from db/db mice, in patients with DN, the protein expression of uPA receptors was also markedly increased in glomerular podocytes (Figure 5C).
Effects of 11R-VIVIT and NFAT2 knockdown on the expression of NFAT2 in podocytes treated with high glucose
We have previously reported that high glucose treatment increases the nuclear localization of NFAT2, and that this effect is blocked by concurrent treatment with 11R-VIVIT (Li et al., 2013). To further confirm these effects, 11R-VIVIT or NFAT2-siRNA was used. As shown in Figure 6, 11R-VIVIT and NFAT2-siRNA significantly reduced the expression of NFAT2 in high glucose-treated podocytes (P < 0.01).
Effects of 11R-VIVIT and NFAT2 knockdown on the expression of uPA receptors in podocytes treated with high glucose
To show that endogenous NFAT2 is able to mediate the mRNA of the gene for uPA receptors (Plaur) and protein expression of uPA receptors in cultured podocytes under high glucose conditions in vitro, the NFAT inhibitor 11R-VIVIT or NFAT2-siRNA was used. As shown in Figure 7, NFATc1-siRNA-treated podocytes showed a significant reduction in both the mRNA and protein expression of uPA receptors compared to control cells (P < 0.01). Similar results were also observed in podocytes treated with 11R-VIVIT. Based on these results, it is likely that NFAT2 is involved in the regulation of uPA receptor expression in high glucose- treated podocytes.
Figure 7.
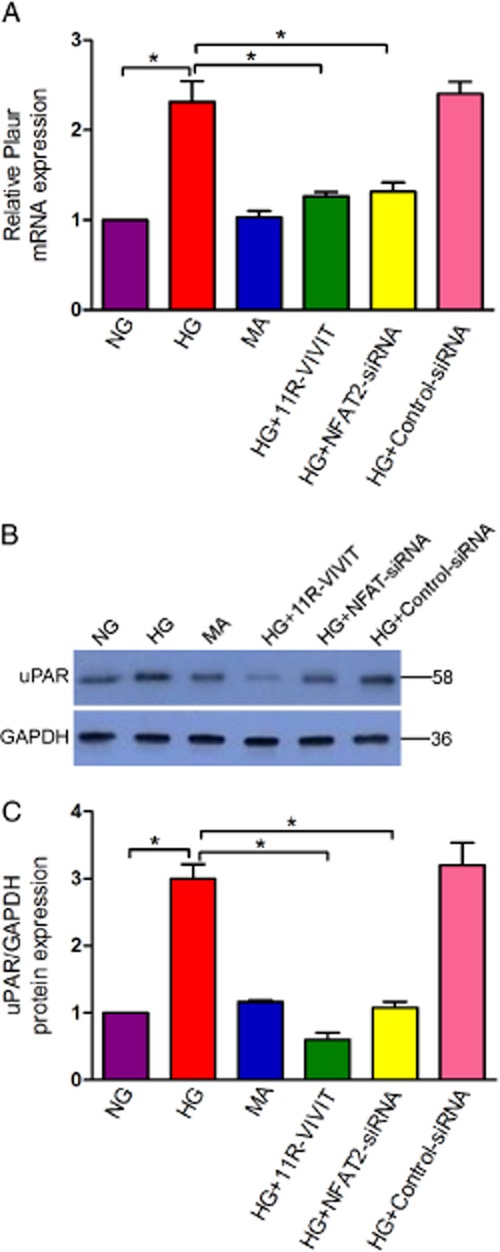
Effects of 11R-VIVIT and NFAT2 gene knockdown on the expression of the uPA receptor (Plaur) gene in podocytes treated with high glucose. (A)The mRNA level of uPA receptors was examined using real-time quantitative RT-PCR analysis, and GAPDH mRNA was used as an internal control. Quantitative data were calculated by 2-△△ CT. (B) The protein level of uPA receptors was analysed using Western blot analysis(n = 3). (C) Densitometry analysis of three repetitions of the experiment shown in (B). Note NG, normal glucose (5.3 mM) group; HG, high glucose group(30 mM); MA, normal glucose (5.3 mM) + mannitol(24.7 mM)group, as an osmolality control; HG+11R-VIVIT, High glucose (30 mM)+ 11R-VIVIT(100 nM, a cell permeable NFAT inhibitor) group; HG + NFAT2-siRNA, High glucose (30 mM) + NFAT2-siRNA (50 nM) group; HG + control-siRNA, High glucose (30 mM) + control-siRNA (50 nM), as a negative control group. All Values are expressed as the mean ± SEM. *P < 0.01 versus HG.
Albumin influx assay
To examine the functional consequence of glucose-induced podocyte injury and the effects of NFAT2 inhibition on glucose-induced podocyte injury in vitro, we examined the filtration barrier function of podocytes using a cellular permeability influx assay system. The albumin flux rate across the differentiated podocyte monolayer was measured. Differentiated podocytes in the presence or absence of 11R-VIVIT or NFAT2-siRNA were incubated with or without glucose (30 mM) for 48 h to induce podocyte injury; the media were collected and subjected to albumin assay. As shown in Figure 8, compared with the controls, high glucose incubation resulted in a marked albumin influx across the podocyte monolayer. However, 11R-VIVIT and NFAT2-siRNA significantly attenuated this high glucose-induced albumin influx. This result suggests that inhibition of NFAT2 attenuated the high glucose-induced filtration barrier dysfunction of podocyte monolayer.
Figure 8.
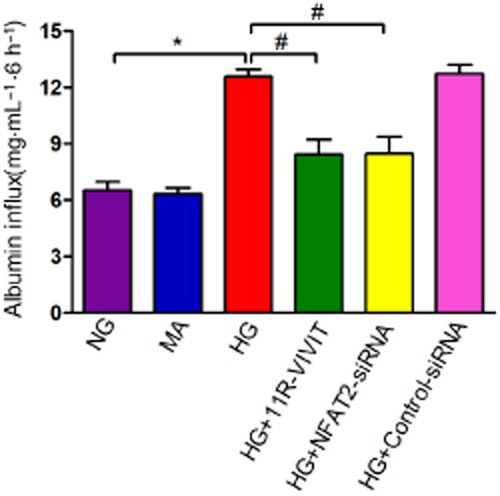
11R-VIVIT attenuates the high glucose-induced dysfunction of the filtration barrier in podocytes. Graphic presentation shows the albumin flux rate across the differentiated podocyte monolayer in each group after 6 h of treatment. Values (mg·mL−1) are means ± SEM. *P < 0.05 versus NG, #P < 0.05 versus HG.
Discussion
DN is one of the most important health problems worldwide, and this is likely to worsen to critical levels in the next decades (Atkins and Zimmet, 2010). Recently, increasing evidence has been obtained demonstrating that podocytes play an important role in the pathogenesis of proteinuria and progression of DN (Wharram et al., 2005; Wolf et al., 2005; Li et al., 2007; Fogo, 2011). Therefore, the development of novel and innovative therapeutic strategies targeted at the causative mechanisms of DN, especially for podocyte injury, has become increasingly urgent.
Recently, a NFAT inhibitor peptide (11R-VIVIT) was developed based on the conserved CaN docking site within the NFAT family member. This peptide interferes selectively with the CaN-NFAT interaction without affecting CaN phosphatase activity (Aramburu et al., 1999). In the present study, we demonstrated, for the first time, that this cell permeable NFAT inhibitor has a protective effect on renal injury, specifically on podocytes injury of DN in db/db mice. The in vitro results demonstrated that the dysfunction of the filtration barrier of podocytes induced by high glucose was significantly alleviated after treatment with 11R-VIVIT, which further supports the in vivo results. In addition, our results also indicate that these protective effects of 11R-VIVIT are probably as a result of its ability to prevent the increased expression of uPA receptors and NFAT2 activation under diabetic conditions.
Urinary albumin excretion has been regarded as one of the first signs of glomerular damage in overt diabetic renal disease. As previously reported (Sugaru et al., 2006), in this study, the urinary albumin excretion increased significantly throughout the study period in db/db mice. However, the treatment with 11R-VIVIT lowered the elevated levels of urinary albumin excretion in db/db mice, suggesting 11R-VIVIT has a renoprotective effect in this DN model. Furthermore, 11R-VIVIT did not affect the blood glucose levels or body weight of these mice, indicating that its renoprotective effect is independent of its hypoglycaemic action.
In addition to the increase in urinary albumin excretion, the most notable renal pathological findings in DN are an expansion of glomerular mesangium and thickening of GBM. In the present study we observed both accelerated mesangial expansion and GBM thickening in glomeruli of db/db mice compared with BKS mice. The treatment with 11R-VIVIT inhibited this mesangial expansion and GBM thickening observed in the diabetic mice. Consistent with the effects on urinary albumin and histological changes seen in db/db mice, marked changes in creatinine clearance were also observed; creatinine clearance is generally considered to be a marker of renal function. After treatment with 11R-VIVIT, the creatinine clearance was markedly increased in db/db mice compared to those in control mice. These data indicated that 11R-VIVIT can inhibit mesangial expansion and improve renal function, at least for creatinine clearance.
The glomerular podocyte is known to play a critical role in kidney structure and urinary filtration. The podocyte foot process surrounds the outside of the GBM and the gaps between adjacent podocyte foot processes form slit diaphragms (filtration slits). A series of recent studies have suggested that podocyte injury plays a key role in the development of DN (Wolf et al., 2005; Li et al., 2007). Drugs that have beneficial effects on podocytes can improve our ability to treat DN. In the present study, its was shown that, after treatment with 11R-VIVIT, the number of podocytes was increased, foot process effacement ameliorated, suggesting the protective effects of 11R-VIVIT against podocyte injury. In addition, in in vitro cultured podocytes, we also demonstrated that the filtration barrier dysfunction of podocytes induced by high glucose was significantly alleviated after 11R-VIVIT treatment. This further confirms that 11R-VIVIT protects against podocyte injury under high glucose conditions.
To explore the possible mechanisms underlying the protective effects of 11R-VIVIT on podocyte injury, we investigated the protein expression of transcription factors NFAT2 and uPA receptors both in the experimental animal models and in cultured podocytes under high glucose conditions.
NFAT2 is one member of the NFAT family of transcription factors (Rao et al., 1997). NFAT are regulated primarily at the level of their subcellular localization through the actions of serine/threonine phosphatase CaN. In resting cells, NFAT family members are normally located in the cytoplasm in a hyperphosphorylated latent form. However, following activation CaN directly dephosphorylates NFAT proteins, they are imported into the nucleus and increased intrinsic DNA binding activity occurs; this can be specifically inhibited by cyclosporin A. Once located in the nucleus, NFAT are then free to bind to their target promoter elements and activate the transcription of specific NFAT target genes, either alone or in combination with other nuclear partners (Rao et al., 1997). The NFAT is expressed in many cell types and contributes to diverse cellular functions (Chin et al., 1998; Ranger et al., 1998; Graef et al., 2001; Benedito et al., 2005; Larrieu et al., 2005). Recently, a role for NFAT in podocyte signalling was implicated in cultured cells using a luminescent NFAT promoter (Schlondorff et al., 2009; Nijenhuis et al., 2011). Results from a recent study also suggest that podocyte-specific overexpression of NFAT2 leads to albuminuria and glomerulosclerosis (Wang et al., 2010). In addition, we recently demonstrated that HG stimulation activates NFAT2 in a time- and dose-dependent manner in cultured podocytes. Pretreatment with 11R-VIVIT (100 nM) completely blocked NFAT2 activation. In the present study, we demonstrated that NFAT2 activation was markedly increased in glomerular podocytes of the diabetic db/db mice compared with BKS mice. However, after treatment with 11R-VIVIT, this effect was significantly inhibited in glomerular podocyte of the diabetic db/db mice. In the in vitro cultured podocytes, we further found that 11R-VIVIT (100 nM) or NFAT2 gene knockdown blocked NFAT2 activation induced by HG.
The uPA receptor is a multidomain glycoprotein tethered to the cell membrane with a glycosyl-phosphatidylinositol anchor. The uPA receptor was originally identified as the binding site for the extracellular protease urokinase-type plasminogen activator on the cell surface. However, the uPA receptor interacts with many other proteins such as vitronectin, uPA receptor-associated proteins and integrins (Smith and Marshall, 2010). The uPA receptor is associated with cell motility of podocytes (Wei et al., 2008; Zhang et al., 2012b) and is highly expressed in motile cells. Recently, it has been reported that uPA receptor signalling in glomerular podocytes leads to foot process effacement and proteinuria (Wei et al., 2008; Zhang et al., 2012a,b). In the present study, we found that uPA receptor protein expression was significantly increased in glomerular podocytes of the diabetic db/db mice. Similar results were also obtained in patients with DN. However, 11R-VIVIT treatment decreased the expression of uPA receptors in glomerular podocytes of diabetic db/db mice. This observation was further supported by the results obtained in the cell culture experiments under high glucose conditions. In previous studies, we identified uPA receptors as NFAT target genes (Zhang et al., 2012a), and showed that HG increases the intracellular Ca2+ concentration [Ca2+]i, leading to the activation of CaN, and subsequent nuclear accumulation of NFAT2 (Li et al., 2013). On the basis of these findings, we propose that the CaN/NFAT2/uPA receptor signalling pathway is involved in podocyte injury of DN and the protective effects of 11R-VIVIT on podocyte injury are probably produced through inhibition of NFAT2 activation and decreased uPAR protein expression (Figure 9).
Figure 9.
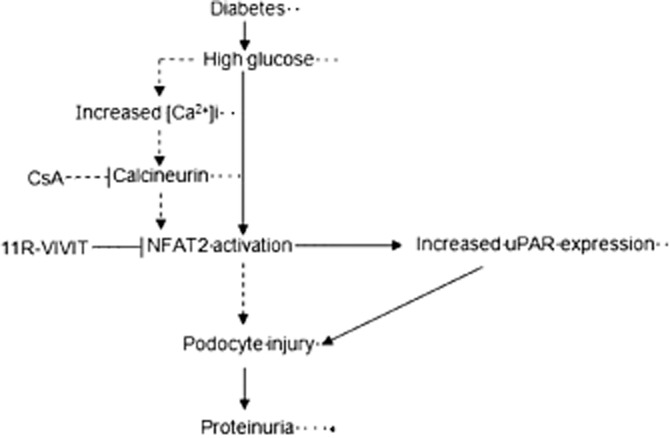
The proposed mechanism for 11R-VIVIT-induced protection against podocyte injury of diabetic nephropathy in db/db mice. Solid arrows represent steps supported by the data from the present study, whereas dashed arrows represent steps supported by our previously published data.
In summary, 11R-VIVIT, a new cell-permeable NFAT inhibitor, has beneficial effects on the diabetic kidney as it decreased urinary albumin excretion, improved kidney function, attenuated glomerular matrix expansion and podocyte injury, and this renoprotective effect in db/db mouse was found to be independent of its hypoglycaemic action. In addition, 11R-VIVIT was found to inhibit the HG-induced NFAT2 activation and increased expression of uPA receptors both in vivo and in vitro. Our data suggest that 11R-VIVIT may be a promising new therapeutic strategy for protecting podocytes and the treatment of DN. The CaN/NFAT2/uPA receptor signalling pathway should be exploited as a therapeutic target for protecting against podocyte injury in DN.
Acknowledgments
We are extremely grateful to all the patients who donated renal samples. This study was supported by the National Natural Science Foundation (81270784; 81170683).
Glossary
- CaN
calcineurin
- DN
diabetic nephropathy
- GBM
glomerular basement membrane
- HG
high glucose
- NFAT
nuclear factor of activated T-cells
- NG
normal glucose
- PAS
periodic acid Schiff
- uPA
urokinase-type plasminogen activator
- WT1
Wilm's tumour antigen 1
Conflict of interest
None of the authors has any potential financial conflict of interest related to this manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1 The baseline characteristics of patients involved in this study.
References
- Aramburu J, Garcia-Cozar F, Raghavan A, Okamura H, Rao A, Hogan PG. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol Cell. 1998;1:627–637. doi: 10.1016/s1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- Atkins RC, Zimmet P. World Kidney Day Steering Committee. Diabetic kidney disease: act now or pay later. J Am Soc Hypertens. 2010;4:3–6. doi: 10.1016/j.jash.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A, et al. The transcription factor NFAT3 mediates neuronal survival. J Biol Chem. 2005;280:2818–2825. doi: 10.1074/jbc.M408741200. [DOI] [PubMed] [Google Scholar]
- Chin ER, Olson EN, Richardson JA, Yang Q, Humphries C, Shelton JM, et al. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998;12:2499–2509. doi: 10.1101/gad.12.16.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury D, Tuncel M, Levi M. Diabetic nephropathy, a multifaceted target of new therapies. Discov Med. 2010;10:406–415. [PubMed] [Google Scholar]
- Coca SG, Ismail-Beigi F, Haq N, Krumholz HM, Parikh CR. Role of intensive glucose control in development of renal end points in type 2 diabetes mellitus: systematic review and meta-analysis intensive glucose control in type 2 diabetes. Arch Intern Med. 2012;172:761–769. doi: 10.1001/archinternmed.2011.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MP, Clements RS, Hud E, Cohen JA, Ziyadeh FN. Evolution of renal function abnormalities in the db/db mouse that parallels the development of human diabetic nephropathy. Exp Nephrol. 1996;4:166–171. [PubMed] [Google Scholar]
- Fogo AB. The targeted podocyte. J Clin Invest. 2011;121:2142–2145. doi: 10.1172/JCI57935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca (2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- Ibrahim HN, Hostetter TH. Diabetic nephropathy. J Am Soc Nephrol. 1997;8:487–493. doi: 10.1681/ASN.V83487. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrieu D, Thiébaud P, Duplàa C, Sibon I, Thézé N, Lamazière JM. Activation of the Ca (2+) / calcineurin/NFAT2 pathway controls smooth muscle cell differentiation. Exp Cell Res. 2005;310:166–175. doi: 10.1016/j.yexcr.2005.07.021. [DOI] [PubMed] [Google Scholar]
- Li JJ, Kwak SJ, Jung DS, Kim JJ, Yoo TH, Ryu DR, et al. Podocyte biology in diabetic nephropathy. Kidney Int Suppl. 2007;106:S36–S42. doi: 10.1038/sj.ki.5002384. [DOI] [PubMed] [Google Scholar]
- Li R, Zhang L, Shi W, Zhang B, Liang X, Liu S, et al. NFAT2 mediates high glucose-induced glomerular podocyte apoptosis through increased Bax expression. Exp Cell Res. 2013;319:992–1000. doi: 10.1016/j.yexcr.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 2008;172:299–308. doi: 10.2353/ajpath.2008.070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Sue YM, Chou Y, Cheng CF, Chang CC, Li HF, et al. Activation of a nuclear factor of activated T-lymphocyte-3(NFAT3) by oxidative stress in carboplatin-mediated renal apoptosis. Br J Pharmacol. 2010;161:1661–1676. doi: 10.1111/j.1476-5381.2010.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179:1719–1732. doi: 10.1016/j.ajpath.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parving HH, Hovind P, Rossing K, Andersen S. Evolving strategies for renoprotection: diabetic nephropathy. Curr Opin Nephrol Hypertens. 2001;10:515–522. doi: 10.1097/00041552-200107000-00006. [DOI] [PubMed] [Google Scholar]
- Qiu WJ, Zhou Y, Jiang L, Fang L, Chen L, Su WF, et al. Genipin inhibits mitochondrial uncoupling protein 2 expression and ameliorates podocyte injury in diabetic mice. PLoS ONE. 2012;7:e41391. doi: 10.1371/journal.pone.0041391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, et al. The transcription factor NFATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Remuzzi G, Schieppati A, Ruggenenti P. Clinical practice. Nephropathy in patients with type 2 diabetes. N Engl J Med. 2002;346:1145–1151. doi: 10.1056/NEJMcp011773. [DOI] [PubMed] [Google Scholar]
- Rico M, Mukherjee A, Konieczkowski M, Bruggeman LA, Miller RT, Khan S, et al. WT1-interacting protein and ZO-1 translocate into podocyte nuclei after puromycin aminonucleoside treatment. Am J Physiol Renal Physiol. 2005;289:F431–F441. doi: 10.1152/ajprenal.00389.2004. [DOI] [PubMed] [Google Scholar]
- Schlondorff J, Del CD, Carrasquillo R, Lacey V, Pollak MR. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol. 2009;296:C558–C569. doi: 10.1152/ajpcell.00077.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaquist ER, Ibrahim HN. Approach to the patient with type 2 diabetes and progressive kidney disease. J Clin Endocrinol Metab. 2010;95:3103–3110. doi: 10.1210/jc.2010-0504. [DOI] [PubMed] [Google Scholar]
- Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284:F1138–F1144. doi: 10.1152/ajprenal.00315.2002. [DOI] [PubMed] [Google Scholar]
- Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- Sugaru E, Nakagawa T, Ono-Kishino M, Nagamine J, Tokunaga T, Kitoh M, et al. SMP-534 ameliorates progression of glomerular fibrosis and urinary albumin in diabetic db/db mice. Am J Physiol Renal Physiol. 2006;290:F813–F820. doi: 10.1152/ajprenal.00357.2005. [DOI] [PubMed] [Google Scholar]
- Toyonaga J, Tsuruya K, Ikeda H, Noguchi H, Yotsueda H, Fujisaki K, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant. 2011;26:2475–2484. doi: 10.1093/ndt/gfq750. [DOI] [PubMed] [Google Scholar]
- Vejakama P, Thakkinstian A, Lertrattananon D, Ingsathit A, Ngarmukos C, Attia J. Reno-protective effects of renin-angiotensin system blockade in type 2 diabetic patients: a systematic review and network meta-analysis. Diabetologia. 2012;55:566–578. doi: 10.1007/s00125-011-2398-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Fields TA, Pazmino K, Dai Q, Burchette JL, Howell DN, et al. Activation of Galpha q-coupled signaling pathways in glomerular podocytes promotes renal injury. J Am Soc Nephrol. 2005;16:3611–3622. doi: 10.1681/ASN.2005020167. [DOI] [PubMed] [Google Scholar]
- Wang L, Chang JH, Paik SY, Tang Y, Eisner W, Spurney RF. Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo. Mol Endocrinol. 2011;25:1376–1386. doi: 10.1210/me.2011-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, et al. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2010;21:1657–1666. doi: 10.1681/ASN.2009121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- Wolf G, Ritz E. Diabetic nephropathy in type 2 diabetes prevention and patient management. J Am Soc Nephrol. 2003;14:1396–1405. doi: 10.1097/01.asn.0000065639.19190.cf. [DOI] [PubMed] [Google Scholar]
- Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–1634. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- Zhang B, Shi W, Ma J, Sloan A, Faul C, Wei C, et al. The calcineurin-NFAT pathway allows for urokinase receptor-mediated beta3 integrin signaling to cause podocyte injury. J Mol Med (Berl) 2012a;90:1407–1420. doi: 10.1007/s00109-012-0960-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Xie S, Shi W, Yang Y. Amiloride off-target effect inhibits podocyte urokinase receptor expression and reduces proteinuria. Nephrol Dial Transplant. 2012b;27:1746–1755. doi: 10.1093/ndt/gfr612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.