

Abstract
Homologous recombination (HR) is a key process in the repair of double-stranded DNA breaks (DSBs) that can initiate cancer or cell death. Human Bloom's syndrome RecQ-family DNA helicase (BLM) exerts complex activities to promote DSB repair while avoiding illegitimate HR. The oligomeric assembly state of BLM has been a key unresolved aspect of its activities. In this study we assessed the structure and oligomeric state of BLM, in the absence and presence of key HR-intermediate DNA structures, by using single-molecule visualization (electron microscopic and atomic force microscopic single-particle analysis) and solution biophysical (dynamic light scattering, kinetic and equilibrium binding) techniques. Besides full-length BLM, we used a previously characterized truncated construct (BLM642–1290) as a monomeric control. Contrary to previous models proposing a ring-forming oligomer, we found the majority of BLM molecules to be monomeric in all examined conditions. However, BLM showed a tendency to form dimers when bound to branched HR intermediates. Our results suggest that HR activities requiring single-stranded DNA translocation are performed by monomeric BLM, while complex DNA structures encountered and dissolved by BLM in later stages of HR induce partial oligomerization of the helicase.—Gyimesi, M., Pires, R.H., Billington, N., Sarlós, K., Kocsis, Z.S. Módos, K., Kellermayer, M. S. Z., Kovács, M. Visualization of human Bloom's syndrome helicase molecules bound to homologous recombination intermediates.
Keywords: DNA repair, oligomerization, structure, electron microscopy, atomic force microscopy
Human cells are exposed to various physical and chemical DNA-damaging agents causing tens of thousands of DNA lesions in the genome of a single cell per day (1). In the absence of repair mechanisms, cells are prone to cancerous processes or cell death. The most severe form of DNA lesion is the double-stranded break (DSB), which is repaired via complex biochemical pathways (2). Homologous recombination (HR)-based repair mechanisms enable the cell to repair DSBs in an error-free manner. Bloom's syndrome RecQ-family DNA helicase (BLM), one of the human RecQ-family helicases, is a key player in HR-based repair processes. BLM exerts both pro- and antirecombinogenic functions by assisting the DNA-end resection machinery producing 3′-single-stranded DNA (ssDNA) overhangs at DSB sites (3–5), disrupting RAD51 recombinase nucleoprotein filaments, activating strand exchange, and dissolving double Holliday junctions (DHJs) and displacement loops (D loops) (6–11).
Oligomerization of helicases is a central but controversial aspect of the aforementioned complex HR activities. A monomer unit of helicases from superfamilies (SFs) 1–2 contains two RecA domains, while the monomers of SF3-6 helicases contain a single RecA or AAA+ domain (12). As adenosine 5′-triphosphate (ATP) binding and hydrolysis require a pocket formed by two adjacent core domains, SF1-2 helicases can function even in their monomeric form, while SF3-6 helicases must form oligomers (usually hexamers or dodecamers) in order to exert mechanochemical activities. BLM, an SF2 helicase, contains two tandem RecA domains and, thus, it can perform ATPase-dependent enzymatic activities in a monomeric form (13, 14). However, based on size-exclusion chromatography and low-resolution electron microscopy (EM) analyses, it was proposed that BLM forms multimeric ring structures in the absence of DNA (15). A recent study demonstrated that the unwinding activity of BLM reaches optimum at a BLM/substrate molar ratio of 1, which suggests that the monomeric form might be sufficient for this activity, while the branch migration activity requires higher BLM/substrate molar ratios, indicating oligomerization of BLM (16). Another recent study showed that BLM unwinds double-stranded DNA (dsDNA) substrates as a monomer (17). The N-terminal domain of BLM (BLM1–431) was shown to exist as hexa- and dodecamers in solution, indicating the role of this domain in BLM oligomerization (18). It was also shown that a truncated BLM construct lacking the N-terminal domain (BLM642–1290) is monomeric but retains complex activity profile of the full-length enzyme (13, 14).
The detection of various multimeric forms of RecQ-family helicases led to the proposal that these enzymes may perform their activities during different HR subprocesses in different oligomeric states (16, 19–21). In this study we tested this hypothesis by determining the oligomeric state of BLM in various conditions resembling those encountered by the enzyme during different stages of HR. We performed a comprehensive set of high-resolution EM and atomic force microscopy (AFM) single-particle analyses in the presence of different HR-intermediate DNA structures. We complemented these structural analyses with dynamic light scattering (DLS) and solution kinetic and spectroscopic measurements addressing the assembly state of the enzyme and the interaction between active sites.
MATERIALS AND METHODS
Materials, experimental conditions, and data analysis
Unless otherwise stated, all materials were obtained from Sigma-Aldrich (St. Louis, MO, USA). ATP and adenosine 5′-(β,γ-imido)triphosphate (AMPPNP) were purchased from Roche Applied Science (Penzberg, Gemany). 3′-(N-methylanthraniloyl)-2′-deoxyadenosine 5′-diphosphate (mdADP) was from Jena Bioscience (Jena, Germany). mdADP was produced by extensive hydrolysis of mdATP by BLM, in conditions based on the determined enzymatic turnover rates. Oligonucleotides were from VBC-Biotech (Vienna, Austria; see Supplemental Data for sequence information). M13 DNA isolation was carried out as described previously (22). All experiments were carried out at 25°C in an assay buffer containing 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, and 1 mM DTT plus various NaCl concentrations, as specified in Supplemental Table S1, alongside applied protein concentrations (see also Discussion). Protein samples were centrifuged at 4°C at 16,000 g immediately before EM, AFM, and DLS experiments to eliminate aggregates. Unless otherwise stated, DNA concentrations are those of oligo- or polynucleotide molecules (as opposed to constituent nucleotides). Data analysis was performed using OriginPro 8.0 (OriginLab, Northampton, MA, USA) and IgorPro 6.0 and 6.1 (WaveMetrics, Portland, OR, USA) software. Means ± sem are indicated for all values in the text.
Protein expression and purification
Full-length BLM was expressed as described previously (15), with the following modifications. Transformed cells were incubated in selective medium (containing no sugar but 3% (v/v) glycerol and 2% (v/v) dl-lactate) at 30°C. Expression was started with 2% (w/v) galactose, and cultures were further shaken at 20°C for 24 h. Cells were frozen in liquid N2 in large bullets. Bullets were quickly ground in a coffee grinder, and the powder was melted in 15-ml tubes in a room-temperature water bath. Further steps were performed according to the published protocol. The purified sample was frozen and stored in liquid N2 in 40 μl droplets. BLM642−1290 was expressed and purified as described previously (13).
EM
Samples were incubated at room temperature for 10 min in the presence of 2 mM AMPPNP, and 10 μM DNA substrates where indicated. Following incubation, in some cases (as indicated) samples were cross-linked with 2% glutaraldehyde for 10 min at room temperature. Both crosslinked and noncrosslinked samples were diluted before imaging (Supplemental Table S1). Samples were applied as 5-μl drops to UV-treated carbon-coated copper grids and negatively stained with 1% uranyl acetate. Images were recorded using an AMT XR-60 CCD camera (Advanced Microscopy Techniques Corp., Danvers, MA, USA) at a nominal magnification of ×75,000 using a Jeol 1200EX II microscope at 80 kV (Jeol Ltd., Akishima, Japan). Using catalase crystals as a calibration standard, the magnification was determined to correspond to 0.245 nm/pixel. Image processing was performed using SPIDER software (Wadsworth Laboratories, Albany, NY, USA) as described previously (23).
AFM
Samples were treated as described for EM experiments. Following crosslinking, samples were diluted (Supplemental Table S1). Oscillating-mode AFM images were obtained in liquid with an MFP3D AFM instrument (Asylum Research, Santa Barbara, CA, USA) using lever A of Bio-Lever cantilevers (Olympus, Tokyo, Japan) with a typical resonance frequency of 9.1 kHz and a nominal spring constant of 30 pN/nm. Samples were immobilized onto mica modified with glutaraldehyde as described previously (24) and scanned with a free amplitude of ∼0.3 V and an amplitude setpoint of ∼0.2 V. Scans (1×1 and 5×5 μm; (1024×1024 pixels) were performed at scanning frequencies of 0.75 and 0.6 Hz, respectively.
DLS
DLS measurements were carried out using an ALV goniometer (ALV GmbH, Hemsbach, Germany) equipped with a Melles Griot diode-pumped solid-state laser (457.5 nm; CVI Laser Optics, Albuquerque, NM, USA). Samples (200 μl) were irradiated by the laser, and scattered light was detected at 90° relative to the incident light. Autocorrelation functions were calculated by a data acquisition system developed at the Department of Biophysics and Radiation Biology, Semmelweis University. Particle size distributions were determined using the maximum entropy method (25). To avoid overestimation of the contribution of large particles, scattered light intensity values were normalized by the corresponding radii. The shape of the molecules was approximated as spheres to calculate their rh values.
Kinetic and spectroscopic measurements
Fluorescence emission spectra of Cy3-labeled oligonucleotides were recorded in a SPEX FluoroMax spectrofluorometer (Horiba Jobin Yvon, Palaiseau, France) in the presence of 10% glycerol. Cy3 fluorescence was excited at 545 nm and emission was detected between 550–600 nm with slit widths of 2 nm on both sides.
Transient kinetic experiments were carried out in a BioLogic SFM 300/400 stopped-flow apparatus (BioLogic, Grenoble, France). mdADP fluorescence was excited at 280 nm via energy transfer from protein tryptophans and detected through a 400-nm cutoff filter.
Steady-state ATPase activities were measured using a pyruvate kinase/lactate dehydrogenase (PK/LDH) coupled assay (14 U/ml PK, 20 U/ml LDH, 1 mM ATP, 1 mM phosphoenol pyruvate, 200 μM NADH) in the presence of 50 μg/ml bovine serum albumin. NADH absorbance (ε340 nm=6220 M–1cm–1) was followed in a Shimadzu UV-2101PC spectrophotometer (Shimadzu Corp., Kyoto, Japan).
Further details of experimental procedures are provided in Supplemental Data.
RESULTS
Single-particle EM analysis reveals that BLM is predominantly monomeric both in the absence and presence of various DNA structures
We applied EM imaging to assess the oligomerization state of single BLM molecules. Full-length BLM and BLM642–1290 (a monomeric control construct) were incubated in the absence and presence of substrates and then applied to UV-treated carbon-coated copper grids and were negatively stained with uranyl acetate. To prevent oligomer disassembly on dilution, in some cases (as indicated below) we also treated samples with glutaraldehyde crosslinker after appropriate incubation with substrates but preceding dilution.
The global average of noncrosslinked BLM particles imaged in the absence of substrates (apo state) revealed an object ∼13 nm in length, consisting of two lobes with diameters of 9 and 5 nm (Fig. 1A). Classification of particles revealed heterogeneity in appearance, apparently resulting from the flexibility of the connection between the two lobes (Supplemental Fig. S1). The global average of apo-BLM imaged after crosslinking treatment revealed a more detailed shape consisting of several domains (Fig. 1B). The size range of particles (10–15 nm) was similar to those observed in a previous low-resolution EM study (15). However, in contrast to the conclusion of the earlier work, the alignment of the crystal structure of the helicase core of Escherichia coli RecQ (26) onto the BLM image strongly indicated that the observed BLM particles are monomers (Fig. 1C, D). A multimeric ring consisting of 160-kDa BLM molecules would appear several times larger than those observed both in the current and earlier studies.
Figure 1.

Single-particle EM analysis reveals that BLM is predominantly monomeric in the absence and presence of HR-intermediate DNA structures. A, B) Global averages of EM images of BLM particles recorded in the absence of substrates without (A; n=1892) and with (B; n=557) glutaraldehyde crosslinking. Scale bars = 10 nm. Class averages are shown in Supplemental Fig. S1. C) Crystal structure of the 59 kDa helicase core of E. coli RecQ (1OYY.pdb) comprising the N- and C-core RecA (red and blue, respectively), Zn2+-binding (yellow), and winged-helix (green) domains. The black arrow indicates the approximate diameter of the construct (7.4 nm). D) Alignment of the 1OYY crystal structure onto the global average of EM images (B). The alignment strongly indicates that BLM particles of 10–15 nm diameter are monomers. We further propose that the large lobe appearing on the left side of the core is the N-terminal domain, whereas the smaller lobe on the right is the HRDC domain. As the C-terminal domain of BLM is predicted to be unstructured, it may not be visible in EM images. Scale bar = 10 nm. E) Schematic illustration of DNA substrates. Arrows indicate DNA strands pointing toward the 3′-end. F, G) Representative EM images of individual monomeric (F) and dimeric (G) BLM particles recorded after glutaraldehyde crosslinking in the presence of 2 mM AMPPNP and 10 μM DL DNA. Scale bars = 10 nm. H) Distribution of BLM and BLM642–1290 particles between monomeric (black) and oligomeric (white) states in the presence of various substrates as indicated. AMPPNP (2 mM) was present in all experiments involving DNA substrates (10 μM). Results of analyses are shown in Table 1.
It has been proposed that helicases can exert simple activities, such as translocation, along ssDNA in monomeric form, but complex DNA structures, including HR intermediates, may induce formation of oligomers (21). Therefore, we also imaged BLM in the presence of a nonhydrolyzable ATP analog, AMPPNP, plus ssDNA [54-mer oligo-dT (dT54)] or multistranded structures resembling HR intermediates (Fig. 1E). DNA substrates used included a synthetic 4-way junction containing a 12-nt mobile region [X12, mimicking a Holliday junction (HJ)] and immobile D-loop substrates (DL, 3-INV, and 5-INV). DL and 3-INV are D-loop structures containing a 3′-invading strand with a ds or ss lagging strand, respectively (Fig. 1E). In addition, in previous studies it was shown that D loops with a 5′-invading strand are better substrates for BLM than 3′-invading forms (6, 8). As 5′ invasions do not allow subsequent strand extension and halt HR-based repair, the proposed specific disruption of these structures by BLM may contribute to efficient HR. Thus we also used a 5′-invading D-loop structure in our experiments (5-INV, Fig. 1E).
In the absence of substrates, >90% of particles were monomeric (Fig. 1F). Even the monomeric particles showed variability in size and appearance, indicating considerable freedom of domain movements. The size of these particles was slightly smaller than that expected for a 160-kDa protein, suggesting that some parts of the molecules were not visible in certain conditions. We assigned particles that were definitively larger than monomers as oligomers (Fig. 1G, H and Table 1). However, most of the observed oligomers were dimers, and the presence of any larger oligomers was not apparent. As compared to the apo form, the presence of AMPPNP and dT54 decreased the fraction of oligomers (Fig. 1H and Table 1). In the presence of complex DNA structures the predominant form of BLM was still the monomer, however, all D-loop substrates significantly increased the oligomer population to above 10% (Fig. 1H and Table 1). These data demonstrate that, although BLM dominantly adopts the monomeric form in the presence of HR-intermediate DNA structures, the enzyme shows a tendency to form oligomers (mostly dimers) when bound to these DNA structures. Importantly, no ring-like tetra- or hexameric structures were observed in any conditions.
Table 1.
Distribution of monomeric and oligomeric states of BLM and BLM642–1290 in the presence of different substrates, as determined by EM (cf. Fig. 1H)
| Protein | Substrate | Particles counted | Monomer (%) | Oligomer (%) |
|---|---|---|---|---|
| BLM | — | 645 | 95 | 5 |
| AMPPNP | 904 | 99 | 1 | |
| AMPPNP + dT54 | 574 | 98 | 2 | |
| AMPPNP + X12 | 1061 | 96 | 4 | |
| AMPPNP + DL | 697 | 88 | 12 | |
| AMPPNP + 3-INV | 634 | 88 | 12 | |
| AMPPNP + 5-INV | 829 | 88 | 12 | |
| BLM642−1290 | — | 661 | 98 | 2 |
| AMPPNP + 5-INV | 428 | 96 | 4 |
Schematic representation of DNA structures is shown in Fig. 1E.
We performed control experiments using the monomeric BLM642–1290 construct to demonstrate that the observed DNA-induced shift toward larger particles of BLM resulted from oligomerization mediated by protein–protein interactions and not merely from recruitment of multiple protein molecules to one DNA molecule. We found that the fraction of oligomers formed by BLM642–1290 in the 5-INV substrate was <5% (Fig. 1H and Table 1), indicating that BLM oligomerization observed in the presence of D-loop-like DNA structures is mediated by protein-protein interactions involving the N-terminal and/or C-terminal domains.
Single-molecule AFM imaging shows that complex DNA structures induce partial oligomerization of BLM
Scanning mode AFM analysis was used to further study oligomerization of BLM and BLM642–1290 with the same substrate set as for the EM images. We relied on particle height analysis, which can be determined with reasonable accuracy, as opposed to volume calculations affected by cantilever tip contamination (this may vary even during one scan), which may result in apparent widening of particles, and by the convolution of the tip being several orders of magnitude greater than the size of the scanned objects, which again hinders volume determination, especially for small molecules.
Apo-BLM appeared in 3 populations. The smallest-size form (1.1 nm in height) represented 44% of BLM particles, whereas 26% were characterized by a 1.4 nm height and 30% of the particles adopted the largest-size form (2.1 nm in height) (Fig. 2, Table 2, and Supplemental Fig. S2). These heights were smaller than those expected based on EM results. To confirm that the observed size differences between EM and AFM images did not result from protein-specific features, we performed control scans of the apo form of BLM642–1290, as well as γ-globulin, a well-defined monomeric globular protein with molecular mass of 158 kDa (similar to that of BLM, 160 kDa). The majority of 70-kDa BLM642–1290 molecules (86%) were characterized by a 0.9 nm height, and γ-globulin by 0.7 and 1.0 nm heights (Table 2 and Supplemental Fig. S2). These results confirmed that the difference between particle sizes deduced from EM images and the apparent AFM single-particle heights of a given molecular species is systematic.
Figure 2.
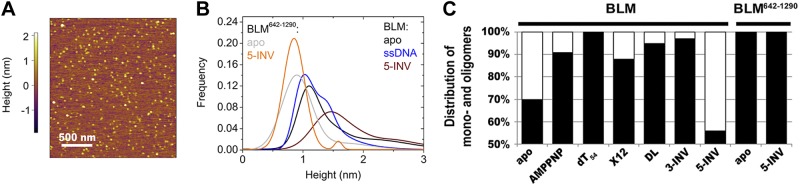
Single-particle AFM analysis corroborates substrate-induced shifts in the distribution of assembly states of predominantly monomeric BLM. A) Representative 2- × 2-μm portion of an AFM height scan of BLM particles in the presence of 2 mM AMPPNP and 10 μM DL DNA. AFM height images recorded in all conditions are shown in Supplemental Fig. S4. B) Gaussian fits to histograms resulting from height analysis of BLM and BLM642–1290 AFM images recorded in the absence (apo) and presence of AMPPNP (2 mM) plus various DNA substrates (10 μM) as indicated (black, apo-BLM; blue, BLM+AMPPNP+dT54; wine, BLM+AMPPNP+5-INV; gray, apo-BLM642–1290; orange, BLM642–1290+AMPPNP+5-INV). All histograms, fits and results of analyses are shown in Table 2 and Supplemental Fig. S2. C) Distribution of AFM-imaged BLM and BLM642–1290 particles between monomeric (black) and oligomeric (white) states in the presence of various substrates as indicated. AMPPNP (2 mM) was present in all experiments involving DNA substrates (10 μM).
Table 2.
Heights, half-widths, and area distributions of major populations of BLM and BLM642–1290 in the presence of different substrates, as determined by AFM (cf. Fig. 2C and Supplemental Fig. S2)
| Protein | Substrate | Particles counted | Height ± half-width (nm) | Area distribution (%) | Mono/oligomer distribution (%) |
|---|---|---|---|---|---|
| BLM | — | 816 | 1.1 ± 0.4 | 44 | 70 |
| 1.4 ± 0.6 | 26 | ||||
| 2.1 ± 1.4 | 30 | 30 | |||
| AMPPNP | 433 | 1.0 ± 0.5 | 59 | 91 | |
| 1.4 ± 0.5 | 32 | ||||
| 2.1 ± 1.1 | 9 | 9 | |||
| AMPPNP + dT54 | 1107 | 1.0 ± 0.5 | 93 | 100 | |
| 1.4 ± 0.3 | 7 | ||||
| — | 0 | ||||
| AMPPNP + X12 | 1081 | 1.0 ± 0.4 | 27 | 88 | |
| 1.5 ± 1.0 | 61 | ||||
| 2.3 ± 0.5 | 8 | 12 | |||
| 3.5 ± 0.8 | 4 | ||||
| AMPPNP + DL | 2117 | 1.2 ± 0.3 | 34 | 95 | |
| 1.4 ± 0.6 | 61 | ||||
| 2.1 ± 0.2 | 5 | 5 | |||
| AMPPNP + 3-INV | 892 | 1.0 ± 0.3 | 10 | 97 | |
| 1.5 ± 0.6 | 87 | ||||
| 2.3 ± 0.2 | 3 | 3 | |||
| AMPPNP + 5-INV | 1633 | 1.4 ± 0.7 | 56 | 56 | |
| 2.2 ± 1.4 | 26 | 44 | |||
| >2.5 | 18 | ||||
| BLM642−1290 | — | 354 | 0.9 ± 0.6 | 86 | 100 |
| 1.7 ± 1.1 | 14 | ||||
| AMPPNP + 5-INV | 309 | 0.9 ± 0.4 | 97 | 100 | |
| 1.6 ± 0.2 | 3 | ||||
| γ-Globulin | — | 517 | 0.7 ± 0.2 | 45 | |
| 1.0 ± 0.5 | 55 |
Schematic representation of DNA structures is shown in Fig. 1E.
EM images showed marked conformational heterogeneity of BLM monomers (Supplemental Fig. S1), suggesting that the 1.1- and 1.4-nm species detected in AFM scans belong to the same assembly state in different conformations or bound to the surface in different orientations. This assignment was also supported by the presence of two peaks in the height distribution of the homogenous γ-globulin sample. Thus, the BLM populations of 1.1 and 1.4 nm height were assigned as monomers, and the populations of characteristic heights > 2 nm were considered as oligomers.
Nucleotide (AMPPNP) binding to BLM increased the abundance of the monomer population to 91% in the absence of DNA, and the oligomer population practically disappeared in the presence of AMPPNP and dT54 (Fig. 2B, C; Table 2; and Supplemental Fig. S2). These observations supported the hypothesis that the monomeric form of BLM dominates in the presence of ssDNA and/or nucleotide substrates.
We also tested the effect of complex DNA structures on the distribution of BLM assembly states. As compared to the results obtained in dT54, the presence of X12, DL, and 3-INV substrates slightly decreased the abundance of monomers, while the 5-INV substrate markedly increased the oligomer population to 44% (Fig. 2B, C; Table 2; amd Supplemental Fig. S2). Table 2 shows that the abundance of the 1.4 nm population increased in the presence of all four complex DNA structures (as compared to that in dT54), suggesting that the 1.1- and 1.4-nm populations represent different conformational states of the BLM monomer rather than different modes of binding to the mica surface. In line with the EM results, no ring-like structures were identified in AFM experiments in any conditions tested.
In control AFM experiments performed with BLM642–1290, the addition of AMPPNP and 5-INV did not cause a shift of the height distribution toward larger particles (Fig. 2B, C; Table 2; and Supplemental Fig. S2). This finding confirmed that the DNA-induced effects in BLM are due to protein–protein interactions and not to the recruitment of multiple enzyme subunits to the same DNA molecule. These observations also supported the hypothesis that BLM oligomerization is mediated by protein-protein interactions involving the N-terminal and/or C-terminal domains.
DLS experiments show that BLM is dominantly monomeric during ATP hydrolysis
We further assessed the oligomerization state of BLM by DLS measurements. Apo-BLM showed a broad size distribution with an average hydrodynamic radius (rh) of 9.4 ± 7.3 nm (median±distribution half-width indicated; Fig. 3 and Table 3). When ATP was added to BLM, a large peak appeared at rh = 3.9 ± 1.3 nm (representing 48% of the total mass of particles), while the population at rh = 12.2 ± 6.7 nm became less abundant (Fig. 3 and Table 3). The positions of the two peaks were similar in the presence of AMPPNP, but the population characterized by the smaller rh value was less abundant than in ATP (Table 3). In control experiments, we obtained an rh of 5.0 ± 2.9 nm for γ-globulin, indicating that the peak below 5 nm can be assigned to the monomeric form of BLM. Also considering that the hydrodynamic radius of a nonspherical particle is smaller than the half of its maximal outer diameter, rh values of 3.9–4.7 nm for monomeric BLM were consistent with the apparent outer diameter of 10–15 nm BLM particles visualized by EM (Fig. 1). We note, however, that the DLS hydrodynamic radii were well above those calculated from AFM height values (Tables 2 and 3), resulting from previously published inherent differences between the two experimental methods (27, 28).
Figure 3.

Distribution of hydrodynamic radii (rh) of BLM determined by DLS. Distributions are shown for apo-BLM (dark gray), BLM + ATP (black) and BLM + AMPPNP + dT54 (light gray). Exponentially modified gaussian functions were fitted to data points. Results of analysis are shown in Table 3.
Table 3.
Hydrodynamic radii (rh) and polydispersity (PD) values of BLM and BLM642–1290 in the absence and presence of different substrates, as determined by DLS
| Substrate |
rh ± PD [nm (% mass)] |
|||
|---|---|---|---|---|
| BLM |
BLM642-1290 |
|||
| No DNA | dT54 | No DNA | dT54 | |
| No nucleotide | 9.4 ± 7.3 | 9.9 ± 3.7 | 3.2 ± 2.1 | 3.5 ± 1.9 |
| ATP | 12.2 ± 6.7 (52%) | 7.8 ± 4.7 | 2.8 ± 2.4 | 3.2 ± 7.4 |
| 3.9 ± 1.3 (48%) | ||||
| AMPPNP | 10.3 ± 7.4 (87%) | 11.7 ± 7.4 (31%) | 3.8 ± 2.2 | 3.8 ± 2.0 |
| 4.7 ± 7.4 (13%) | 5.2 ± 4.8 (69%) | |||
The addition of dT54 alone did not cause a significant change in the size distribution of BLM. The monomeric form of BLM did not appear as a discrete peak even in the presence of ATP and dT54, but the median of the distribution was lower than that in the apo form (Table 3). In the presence of AMPPNP and dT54, a separate peak at 5.2 ± 4.8 nm could be identified, representing as much as 69% of the total mass (Fig. 3 and Table 3). The presence of this peak indicated that a large fraction of BLM molecules adopt the monomeric form in these conditions.
In control experiments, we determined an rh of 3.2 ± 2.1 nm for apo-BLM642–1290 (Table 3). This value is slightly smaller than that determined for bovine serum albumin (4.0±0.5 nm), which is a 66-kDa protein existing in heterogeneous populations of mono-, di- and tetrameric forms. This confirms that BLM642–1290 is monomeric in solution. The results also showed that BLM642–1290 adopted the same monomeric form in the presence of nucleotide and/or DNA substrates (Table 3), supporting the hypothesis that the oligomerization of full-length BLM is mediated by the N-terminal and/or C-terminal domains.
In summary, our DLS measurements confirmed that BLM is in equilibrium between different quaternary structures and/or conformational states in the absence of substrates, but both ATP and AMPPNP induce structural changes that result in the appearance of a significant fraction of smaller, likely monomeric particles. Our DLS results are in line with those of a recent study (17). Nevertheless, in contrast to the conclusion drawn in that study, we propose that the differences in ATP and AMPPNP do not clearly indicate that the hydrolysis of ATP (which process does not take place in AMPPNP) would be required to induce dissociation of BLM oligomers.
BLM occludes a 43-nt stretch on ssDNA
We assessed the binding of BLM to DNA via fluorescence titration of 5′-Cy3-labeled oligonucleotides of different lengths (dT45, dT79, dT100) with BLM (Fig. 4A). The BLM concentration dependence of fluorescence intensities was fitted by a quadratic binding equation (Supplemental Eq. S1), which yielded binding stoichiometries (n; mol BLM/mol oligo-dT) of 1.1 ± 0.1, 1.5 ± 0.2, and 2.5 ± 0.2 for dT45, dT79, and dT100, respectively. From these values, the length of the ssDNA stretch occluded by the helicase (b=L/n, where L=oligo-dT length in nuclotides) was calculated to be 41 ± 4, 54 ± 4 and 41 ± 3 nt in the case of dT45, dT79, and dT100, respectively. We note that the determined occluded site size is not necessarily equivalent to the direct contact site size of the protein on DNA.
Figure 4.

Kinetic and spectroscopic experiments show that BLM occludes 43 nt along ssDNA, translocates as a monomer, and hydrolyzes ATP noncooperatively. A) BLM concentration dependence of relative fluorescence intensities of 100 nM 5′-Cy3-labeled oligonucleotides (solid squares, dT45; open diamonds, dT79; solid circles, dT100). Intensities of protein-free oligonucleotides were taken as unity. Solid lines show best fits based on Supplemental Eq. S1. B) Oligo-dT concentration dependence of the total amplitudes of mdADP fluorescence transients recorded on rapidly mixing a premixture of 0.56 μM BLM and 40 μM mdADP plus increasing concentrations of dT54 (solid squares) and dT90 (open circles) with 2.5 mM ATP in the stopped-flow apparatus (premix concentrations stated). Solid lines show best fits based on Supplemental Eq. S2. Inset: representative trace recorded in the absence of DNA, fitted by a double exponential function. C) ATPase activities of 30 nM BLM and BLM642–1290 (as indicated) in the presence of dT72 (solid columns) and m13 DNA (shaded columns) at the specified DNA/enzyme concentration ratios. D, E) ATP concentration dependence of steady-state ATPase activities of 1 μM BLM in the absence (D) and 15 nM BLM in the presence (E) of 500 nM dT72 at 130 (open circles) and 500 mM (solid squares) NaCl. Parameters obtained from Hill analysis (solid lines) of all data are listed in Table 4.
Earlier we found that the fluorescent ADP analog mdADP exhibits a markedly smaller signal change on binding to BLM642–1290-DNA complexes than to apo-BLM642–1290 (13). This phenomenon can be exploited to determine the DNA-bound fraction of helicase molecules in rapid kinetic mdADP “chasing” experiments. To this end, we preincubated BLM with mdADP plus varying concentrations of ssDNA (dT54 or dT90), and rapidly mixed this premixture with a large excess of ATP in the stopped-flow apparatus to monitor mdADP dissociation transients. Similarly to our previous findings on BLM642–1290 (13), mdADP release was biphasic (Fig. 4B inset), indicating that the conformational change between two ADP bound states proposed for BLM642–1290 is also characteristic of the full-length enzyme. As in BLM642–1290, the oligo-dT concentration dependence of the total amplitude of the fluorescence change could be used to reveal the binding stoichiometries of BLM binding to oligo-dT (mol BLM/mol oligonucleotide; Supplemental Eq. S2), which were 1.3 ± 0.1 and 2.3 ± 0.1 for dT54 and dT90, respectively (Fig. 4B). From these values, occluded site sizes of 42 ± 3 and 39 ± 2 nt were calculated in the case of dT54 and dT90, respectively. These values closely matched those determined in 5′-Cy3-oligo-dT titration assays. Taken together, the results yielded an average occluded site size of 43 ± 2 nt. As expected, the determined occluded site size did not systematically depend on oligo-dT length in the investigated range.
BLM performs ssDNA translocation in a monomeric form
We found that the DNA-activated ATPase activities of full-length BLM and BLM642–1290 were practically identical in the presence of dT72 (Fig. 4C). This result could reflect that BLM is either monomeric or relatively short DNA substrates can bind and equally activate all BLM subunits in a putative oligomeric structure. Thus, if BLM subunits work independently, the monomeric or oligomeric forms cannot be distinguished based on ATPase activities measured in the presence of short DNA substrates. Therefore, we also determined the ATPase activity of BLM and BLM642–1290 in the presence of a long circular ssDNA substrate [m13mp18 phage (m13) DNA]. At low DNA/enzyme concentration ratios leading to full decoration of DNA by the enzyme, a long ssDNA substrate is unlikely to bind and activate all subunits of an oligomer due to steric hindrance. Consequently, if the functional form of BLM was oligomeric, at full decoration the ATPase activity of BLM would be expected to be significantly lower than that of BLM642–1290 under the same conditions. However, we found that the ATPase activities of BLM and BLM642–1290 in m13 DNA were identical even at a low DNA/enzyme concentration ratio of 50 nt/BLM subunit (corresponding to almost fully decorated m13 DNA; Fig. 4C). These results gave further support to the proposition that the functionally active ssDNA-translocating form of BLM is monomeric. We note that an ssDNA strand coiling around a multimeric ring, or ssDNA binding into a central hole of an oligomeric ring could also produce this behavior. However, we excluded these scenarios based on the lack of ring structures in the EM and AFM images (Figs. 1 and 2 and Supplemental Figs. S1 and S2).
The ATPase activities of BLM and BLM642–1290 were generally higher in dT72 than in m13 DNA (Fig. 4C). This difference was similar in the two constructs, and thus we exclude the possibility that it arose from oligomerization effects in BLM. Furthermore, we found that a 54-mer ssDNA oligonucleotide of nonrepetitive sequence (DNA_54mer, described in ref. 13) activated the ATPase activities of BLM and BLM642–1290 to the same level as did m13 DNA, indicating that the difference between dT72 and m13 DNA activation is sequence specific and not influenced by DNA end effects.
BLM subunits show no cooperativity in ATP hydrolysis
To assess cooperativity of ATP hydrolysis by BLM, we performed Hill analysis of the ATP concentration dependence of BLM ATPase activity in the absence and presence of dT72. We performed these assays at a range of NaCl concentrations to mimic all conditions used in other experiments (Fig. 4D, E and Table 4). Determined Hill coefficients (nH) were close to unity and were largely independent of applied conditions (Table 4). The lack of cooperativity in near-physiological conditions reflected independent functioning of BLM active sites.
Table 4.
Kinetic parameters of ATP hydrolysis by BLM in the absence and presence of dT72
| [NaCl] (mM]) | No DNA |
dT72 |
||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KATP (μM) | nH | kcat (s−1) | KATP (μM) | nH | |
| 50 | ND | ND | ND | 15 ± 1 | 17 ± 1 | 0.93 ± 0.04 |
| 130 | 0.18 ± 0.01 | 61 ± 6 | 0.88 ± 0.05 | 18 ± 1 | 22 ± 2 | 0.99 ± 0.05 |
| 500 | 0.25 ± 0.01 | 410 ± 50 | 0.72 ± 0.03 | 17 ± 1 | 94 ± 8 | 0.85 ± 0.04 |
Conditions were as in Fig. 4D, E. Standard errors of the fits are shown. KATP, Michaelis constant for ATP; ND, not determined.
DISCUSSION
Despite the lack of necessity for oligomerization to gain functional active sites, several SF1 and SF2 helicases, including RecQ-family enzymes, have been shown to function in oligomeric assembly states in certain conditions (19–21, 29–33). RecQ1 has been shown to exist as a mixture of lower and higher-order oligomers in the absence of nucleotide. The addition of an ATP analog, adenosine 5′-O-(3-thio)triphosphate (ATPγS), shifted the distribution toward lower-order forms, whereas ssDNA did not influence the equilibrium in either in the absence or in the presence of nucleotide (20). In another study, various DNA substrates induced a switch from the dimeric form of WRN to a tetrameric state in the presence of ATPγS (19).
It was proposed that BLM forms tetra- or hexameric ring structures in the absence of substrates and in the presence of ATPγS via oligomerization mediated by its N-terminal domain (15, 18). In an earlier low-resolution EM study, BLM particles with maximal outer diameters of 11 and 13 nm were assigned as tetra- and hexameric rings, respectively (15). In contrast to this assignment, our more refined EM substructural analysis strongly suggested that BLM particles of 10–15 nm diameter are monomers (Fig. 1 and Supplemental Fig. S1). This conclusion is supported by the comparison of the crystal structure of the 59-kDa helicase core of E. coli RecQ (26), characterized by an outer diameter of ∼7.5 nm, with our EM-imaged particles of 160-kDa BLM (Fig. 1). The maximal diameter of a putative tetra- or hexameric ring of BLM molecules would be expected to have a significantly greater diameter than that observed in EM images. The resolution attained in our study also allowed the exclusion of a ring-like arrangement in any particles observed. The difference of our results from that of the previous low-resolution EM analysis (15) may also originate from the fact that previous EM imaging was carried out at a BLM concentration (1 μM) that resulted in fully saturated surfaces in our AFM experiments, disabling the identification of discrete particles. Our EM and AFM scans were thus performed at markedly lower protein concentrations (1–100 nM; Supplemental Table S1), facilitating the classification of particles and more precise resolution of their substructure.
Notably, the dominantly monomeric nature of BLM was preserved throughout a wide range of NaCl and protein concentrations applied in our imaging experiments (Supplemental Table S1). BLM showed a tendency to aggregate at ionic strengths below 200 mM. Although this process did not affect the DNA-activated ATPase activity (and, thus, the native fold) of the enzyme (Table 4), we performed the crosslinking procedures preceding EM and AFM imaging at 500 mM NaCl to avoid aggregation effects. Notably, however, BLM appeared to be dominantly monomeric in EM images also without crosslinking, in which case the protein was kept at low ionic strength (Supplemental Fig. S1). Our conclusions about the oligomerization properties of BLM are therefore supported by experiments performed in a wide range of conditions.
We assign the central region of the observed monomeric BLM particles as the helicase core comprising the N- and C-core RecA, Zn2+-binding (ZnBD) and winged helix (WH) domains (Fig. 1C, D). This core is flexibly connected to a large lobe that can be assigned as the N-terminal domain (Fig. 1 and Supplemental Fig. S1). Our kinetic and spectroscopic experiments revealed that BLM occludes a 43-nt stretch on ssDNA (Fig. 4A, B), which is ∼3 times larger than that determined in our earlier experiments for the BLM642–1290 construct (14 nt) lacking the large N-terminal and the smaller C-terminal domains (13). In conjunction with the EM substructure of BLM particles (Fig. 1), this difference strongly suggests that the N-terminal and/or C-terminal regions of BLM significantly contribute to DNA binding by the full-length enzyme. The interaction of these regions with DNA is still poorly understood.
Our results showing that BLM interacts dominantly in its monomeric form with various DNA structures encountered during HR-based DNA repair bear important functional consequences. BLM is a key component of DNA end resection machineries (3–5) producing ssDNA overhangs at DSB sites. RAD51 recombinase is recruited to the resected DNA end to form a nucleoprotein filament and perform homology search with a mechanism similar to that used by bacterial RecA (34). RAD51 nucleoprotein filaments can be dissociated by BLM in early phases of HR, which has been suggested as an important step of HR quality control (7). Both the end resection and RAD51 nucleoprotein clearance activities require the ssDNA translocation activity of BLM. In later phases, when the invading DNA strand has found the homologous segment, HR is branched to two major pathways. The DHJ formed in one pathway is dissolved by BLM. In the other pathway (synthesis-dependent strand annealing), the extended D loop is resolved by concomitant helicase and strand annealing activities of BLM (2). Thus, HJs and D loops, resembled by DNA substrates shown in Fig. 1E, are key DNA structures encountered and processed by BLM during DSB repair. In addition, BLM has also been shown to be involved in the regression and reversal of stalled replication forks (35). During these processes, a so-called chicken-foot intermediate is formed, which is also mimicked by the X12 substrate (Fig. 1E). Our results suggest that BLM performs the above complex mechanochemical activities dominantly in a monomeric form, while complex DNA structures resembling HR intermediates induce the formation of a discernible population of dimers. The latter finding suggests that complex DNA structures may be processed more efficiently by the concerted action of two BLM subunits. Our results also show that dimerization is mediated by protein-protein interactions involving BLM's N-terminal and/or C-terminal domains, as the BLM642–1290 construct lacking these regions did not show any tendency for substrate-induced oligomerization (Figs. 1 and 2, Supplemental Fig. S2, and Tables 1–3).
Our results on the interaction of BLM with protein-free DNA substrates do not exclude the possibility that the involvement of BLM-bound partner proteins and/or recombinases bound the DNA strands processed by BLM may affect the oligomerization state of BLM. The site of interaction of BLM with topoisomerase IIIα in the DHJ dissolvasome complex was localized to the N-terminal domain of BLM (36, 37). The binding site for replication protein A (RPA), a key component of the end resection machinery (4) and the DHJ dissolvasome (11), was also mapped to the N-terminal domain of BLM (38). Furthermore, the BLM-RAD51 interaction, which affects D-loop disruption, has been shown to be independently mediated by the N- and C-terminal domains of BLM (39). Thus, the N-terminal domain of BLM, which mediates the oligomerization of the enzyme, is also responsible for various protein–protein interactions, leaving open the possibility of coupling between these phenomena.
It has recently been proposed that the electrostatic repulsion between the C-terminal HRDC domains of the BLM oligomer during DHJ dissolution facilitates DNA strand separation (40). The researchers speculated that 4 BLM monomers bind to the DHJ structure, and thus a dimeric unit could be recruited to each 4-armed branching region (i.e., each HJ). In our experiments, one 4-armed region was present in each complex DNA substrate used, implying that BLM dimers could be recruited to these substrates. In line with this finding, the small oligomeric species observed in our EM and AFM experiments were dominantly dimers (Fig. 1). Taken together, our results shed light on the dynamic nature of the assembly state of BLM during various mechanochemical activities exerted to promote DNA repair.
Supplementary Material
Acknowledgments
The authors thank The Electron Microscopy Core Facility of the U.S. National Heart, Lung, and Blood Institute for the use of electron microscopy facilities.
This work was supported by Norway grants (NNF2-85613 to M.K.); the Hungarian Scientific Research Fund (K71915 and NK81950 to M.K.); TÁMOP (4.2.1/B-09/1/KMR-2010-0003 to M.K.); the Human Frontier Science Program (RGY0072/2010 to M.K.); and the Momentum Program of the Hungarian Academy of Sciences (LP2011-006/2011 to M.K.). M.K. is a Bolyai Fellow of the Hungarian Academy of Sciences. M.G. is a Marie Curie Fellow. This research was realized in the frames of TÁMOP 4.2.4. A/2–11-1–2012-0001, National Excellence Program: elaborating and operating an inland student and researcher personal support system. The project was subsidized by the European Union and cofinanced by the European Social Fund.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- AFM
- atomic force microscopy
- AMPPNP
- adenosine 5′-(β,γ-imido)triphosphate
- ATP
- adenosine 5′-triphosphate
- ATPγS
- adenosine 5′-O-(3-thio)triphosphate
- BLM
- Bloom's syndrome RecQ-family DNA helicase
- D loop
- displacement loop
- DLS
- dynamic light scattering
- DHJ
- double-Holliday junction
- DSB
- double-stranded break
- dsDNA
- double-stranded DNA
- dT54
- 54-mer oligo-dT
- EM
- electron microscopy
- HJ
- Holliday junction
- HR
- homologous recombination
- mdADP
- 3′-(N-methylanthraniloyl)-2′-deoxyadenosine 5′-diphosphate
- SF1-6
- superfamily 1–6
- ssDNA
- single-stranded DNA
REFERENCES
- 1. Lodish H., Berk A., Matsudaira P., Kaiser C. A., Krieger M., Scott M. P., Zipursky S. L., Darnell J. (2004) Molecular Biology of the Cell, 5th Ed., W.H. Freeman, New York [Google Scholar]
- 2. Chu W. K., Hickson I. D. (2009) RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer 9, 644–654 [DOI] [PubMed] [Google Scholar]
- 3. Gravel S., Chapman J. R., Magill C., Jackson S. P. (2008) DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nimonkar A. V., Genschel J., Kinoshita E., Polaczek P., Campbell J. L., Wyman C., Modrich P., Kowalczykowski S. C. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nimonkar A. V., Ozsoy A. Z., Genschel J., Modrich P., Kowalczykowski S. C. (2008) Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. U. S. A. 105, 16906–16911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bachrati C. Z., Borts R. H., Hickson I. D. (2006) Mobile D-loops are a preferred substrate for the Bloom's syndrome helicase. Nucleic Acids Res. 34, 2269–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bugreev D. V., Yu X., Egelman E. H., Mazin A. V. (2007) Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev. 21, 3085–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Brabant A. J., Ye T., Sanz M., German I. J., Ellis N. A., Holloman W. K. (2000) Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry 39, 14617–14625 [DOI] [PubMed] [Google Scholar]
- 9. Wu L., Hickson I. D. (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874 [DOI] [PubMed] [Google Scholar]
- 10. Bugreev D. V., Mazina O. M., Mazin A. V. (2009) Bloom syndrome helicase stimulates RAD51 DNA strand exchange activity through a novel mechanism. J. Biol. Chem. 284, 26349–26359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Plank J. L., Wu J., Hsieh T. S. (2006) Topoisomerase IIIalpha and Bloom's helicase can resolve a mobile double Holliday junction substrate through convergent branch migration. Proc. Natl. Acad. Sci. U. S. A. 103, 11118–11123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singleton M. R., Dillingham M. S., Wigley D. B. (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50 [DOI] [PubMed] [Google Scholar]
- 13. Gyimesi M., Sarlos K., Kovacs M. (2010) Processive translocation mechanism of the human Bloom's syndrome helicase along single-stranded DNA. Nucleic Acids Res. 38, 4404–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Janscak P., Garcia P. L., Hamburger F., Makuta Y., Shiraishi K., Imai Y., Ikeda H., Bickle T. A. (2003) Characterization and mutational analysis of the RecQ core of the bloom syndrome protein. J. Mol. Biol. 330, 29–42 [DOI] [PubMed] [Google Scholar]
- 15. Karow J. K., Newman R. H., Freemont P. S., Hickson I. D. (1999) Oligomeric ring structure of the Bloom's syndrome helicase. Curr. Biol. 9, 597–600 [DOI] [PubMed] [Google Scholar]
- 16. Mazina O. M., Rossi M. J., Deakyne J. S., Huang F., Mazin A. V. (2012) Polarity and bypass of DNA heterology during branch migration of Holliday junctions by human RAD54, BLM, and RECQ1 proteins. J. Biol. Chem. 287, 11820–11832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu Y. N., Bazeille N., Ding X. Y., Lu X. M., Wang P. Y., Bugnard E., Grondin V., Dou S. X., Xi X. G. (2012) Multimeric BLM is dissociated upon ATP hydrolysis and functions as monomers in resolving DNA structures. Nucleic Acids Res. 40, 9802–9814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beresten S. F., Stan R., van Brabant A. J., Ye T., Naureckiene S., Ellis N. A. (1999) Purification of overexpressed hexahistidine-tagged BLM N431 as oligomeric complexes. Protein Expr. Purif. 17, 239–248 [DOI] [PubMed] [Google Scholar]
- 19. Compton S. A., Tolun G., Kamath-Loeb A. S., Loeb L. A., Griffith J. D. (2008) The Werner syndrome protein binds replication fork and holliday junction DNAs as an oligomer. J. Biol. Chem. 283, 24478–24483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muzzolini L., Beuron F., Patwardhan A., Popuri V., Cui S., Niccolini B., Rappas M., Freemont P. S., Vindigni A. (2007) Different quaternary structures of human RECQ1 are associated with its dual enzymatic activity. PLoS Biol. 5, e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vindigni A., Hickson I. D. (2009) RecQ helicases: multiple structures for multiple functions? HFSP J. 3, 153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sambrook J., Russel D. W. (2001) Molecular Cloning, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA [Google Scholar]
- 23. Burgess S., Walker M., Wang F., Sellers J. R., White H. D., Knight P. J., Trinick J. (2002) The prepower stroke conformation of myosin V. J. Cell Biol. 159, 983–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang H., Bash R., Yodh J. G., Hager G. L., Lohr D., Lindsay S. M. (2002) Glutaraldehyde modified mica: a new surface for atomic force microscopy of chromatin. Biophys. J. 83, 3619–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bryan R. K., Fougere P. F. (1990) Maximum Entropy and Bayesian Methods pp. 221–232, Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- 26. Bernstein D. A., Zittel M. C., Keck J. L. (2003) High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 22, 4910–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoo C. M., Starostin N., West P., Mecartney M. L. (2008) A comparison of atomic force microscopy (AFM) and dynamic light scattering (DLS) methods to characterize nanoparticle size distributions. J. Nanopart. Res. 10, 89–96 [Google Scholar]
- 28. Shi C., Paige M. F., Maley J., Loewen M. C. (2009) In vitro characterization of ligand-induced oligomerization of the S. cerevisiae G-protein coupled receptor, Ste2p. Biochim. Biophys. Acta 1790, 1–7 [DOI] [PubMed] [Google Scholar]
- 29. Niedziela-Majka A., Chesnik M. A., Tomko E. J., Lohman T. M. (2007) Bacillus stearothermophilus PcrA monomer is a single-stranded DNA translocase but not a processive helicase in vitro. J. Biol. Chem. 282, 27076–27085 [DOI] [PubMed] [Google Scholar]
- 30. Fischer C. J., Maluf N. K., Lohman T. M. (2004) Mechanism of ATP-dependent translocation of E. coli UvrD monomers along single-stranded DNA. J. Mol. Biol. 344, 1287–1309 [DOI] [PubMed] [Google Scholar]
- 31. Maluf N. K., Fischer C. J., Lohman T. M. (2003) A dimer of Escherichia coli UvrD is the active form of the helicase in vitro. J. Mol. Biol. 325, 913–935 [DOI] [PubMed] [Google Scholar]
- 32. Brendza K. M., Cheng W., Fischer C. J., Chesnik M. A., Niedziela-Majka A., Lohman T. M. (2005) Autoinhibition of Escherichia coli Rep monomer helicase activity by its 2B subdomain. Proc. Natl. Acad. Sci. U. S. A. 102, 10076–10081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matlock D. L., Yeruva L., Byrd A. K., Mackintosh S. G., Langston C., Brown C., Cameron C. E., Fischer C. J., Raney K. D. Investigation of translocation, DNA unwinding, and protein displacement by NS3h, the helicase domain from the hepatitis C virus helicase. Biochemistry 49, 2097–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Forget A. L., Kowalczykowski S. C. (2012) Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 482, 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ralf C., Hickson I. D., Wu L. (2006) The Bloom's syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 281, 22839–22846 [DOI] [PubMed] [Google Scholar]
- 36. Wu L., Davies S. L., North P. S., Goulaouic H., Riou J. F., Turley H., Gatter K. C., Hickson I. D. (2000) The Bloom's syndrome gene product interacts with topoisomerase III. J. Biol. Chem. 275, 9636–9644 [DOI] [PubMed] [Google Scholar]
- 37. Hu P., Beresten S. F., van Brabant A. J., Ye T. Z., Pandolfi P. P., Johnson F. B., Guarente L., Ellis N. A. (2001) Evidence for BLM and topoisomerase IIIalpha interaction in genomic stability. Hum. Mol. Genet. 10, 1287–1298 [DOI] [PubMed] [Google Scholar]
- 38. Doherty K. M., Sommers J. A., Gray M. D., Lee J. W., von Kobbe C., Thoma N. H., Kureekattil R. P., Kenny M. K., Brosh R. M., Jr. (2005) Physical and functional mapping of the replication protein A interaction domain of the Werner and Bloom syndrome helicases. J. Biol. Chem. 280, 29494–29505 [DOI] [PubMed] [Google Scholar]
- 39. Wu L., Davies S. L., Levitt N. C., Hickson I. D. (2001) Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 276, 19375–19381 [DOI] [PubMed] [Google Scholar]
- 40. Kim Y. M., Choi B. S. (2010) Structure and function of the regulatory HRDC domain from human Bloom syndrome protein. Nucleic Acids Res. 38, 7764–7777 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.