

Abstract
The prion hypothesis is strongly supported by the fact that prion infectivity and the pathogenic conformer of prion protein (PrP) are simultaneously propagated in vitro by the serial protein misfolding cyclic amplification (sPMCA). However, due to sPMCA's enormous amplification power, whether an infectious prion can be formed de novo with bacterially expressed recombinant PrP (rPrP) remains to be satisfactorily resolved. To address this question, we performed unseeded sPMCA with rPrP in a laboratory that has never been exposed to any native prions. Two types of proteinase K (PK)-resistant and self-perpetuating recombinant PrP conformers (rPrP-res) with PK-resistant cores of 17 or 14 kDa were generated. A bioassay revealed that rPrP-res17kDa was highly infectious, causing prion disease in wild-type mice with an average survival time of about 172 d. In contrast, rPrP-res14kDa completely failed to induce any disease. Our findings reveal that sPMCA is sufficient to initiate various self-perpetuating PK-resistant rPrP conformers, but not all of them possess in vivo infectivity. Moreover, generating an infectious prion in a prion-free environment establishes that an infectious prion can be formed de novo with bacterially expressed rPrP.—Zhang, Z., Zhang, Y., Wang, F., Wang, X., Xu, Y., Yang, H., Yu, G., Yuan, C., Ma, J. De novo generation of infectious prions with bacterially expressed recombinant prion protein.
Keywords: sPMCA, recombinant PrP, bioassay
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, are a group of fatal neurodegenerative disorders that can be manifested as sporadic, inherited, and infectious disorders. The prion hypothesis postulates that the infectious agent in TSEs is a prion, the scrapie form of prion protein (PrPSc), a pathogenic conformational isoform of the normal host cellular prion protein (PrPC) (1). Because of its self-perpetuating property, PrPSc seeds the conversion of host PrPC, resulting in a replication of the pathogenic PrPSc conformer and the subsequent pathological changes.
The normal PrPC is a cell surface glycoprotein that is mainly α-helical, proteinase K (PK) sensitive, and soluble in mild detergents. In contrast, the disease-associated PrPSc is rich in β-sheet, PK resistant, and highly aggregated (1, 2). The development of the serial protein misfolding cyclic amplification (sPMCA) procedure, a technique involving successive cycles of alternating sonication and incubation (3, 4), has greatly advanced our understanding of the infectious agent. In particular it has been shown that the self-perpetuating PK-resistant prion protein (PrP) conformer and the prion infectivity can be propagated simultaneously by sPMCA with PrPC in the crude brain homogenates (4), PrPC purified from brains or cultured cells (5, 6), baculovirus-derived recombinant PrP (7), or purified bacterially expressed recombinant PrP (rPrP) (8–11), providing unequivocal experimental evidence supporting the prion hypothesis.
We previously showed that a highly infectious prion can be generated by sPMCA with a substrate mixture consisting of bacterially expressed murine rPrP, the synthetic phospholipid 1-palmitoyl-2-oleoylphosphatidylglycerol (POPG), and total RNA isolated from normal mouse liver (8). Similar to disease-associated PrPSc, the sPMCA-generated PK-resistant rPrP (rPrP-res) form contains a C-terminal PK-resistant core, can be propagated indefinitely by sPMCA in vitro, has the capability of infecting susceptible cultured cells, and causes prion disease in wild-type mice. Furthermore, our recent studies showed that infectious prions can be generated by replacing total RNA used in original study (8) with synthetic polyriboadenylic acid (9), or by using synthetic phosphatidylethanolamine as the solitary cofactor (10, 11). These results unambiguously revealed that a highly infectious prion can be generated with bacterially expressed rPrP plus defined synthetic cofactors. However, whether the rPrP-res can be initiated de novo by pure in vitro manipulations remains controversial. This uncertainty is largely due to the facts that the ultrasensitive sPMCA is capable of amplifying minuscule amounts of PrPSc (12–14), and there was limited usage of native prion (diseased mouse brain tissue) in the laboratory that generated the original rPrP-res (8). Because of the significance of de novo prion formation and the availability of a brand new laboratory at East China Normal University in Shanghai, we performed the de novo recombinant prion formation experiment in this new laboratory that has never been exposed to any native prions, eliminating the possibility of potential contamination.
MATERIALS AND METHODS
Reagents
Reagents used in this study included RNA STAT-60 (Tel-Test, Friendswood, TX, USA), Ni-NTA Superflow resin (Qiagen, Hamburg, Germany), polyvinylidene fluoride (PVDF) membrane and ECL reagents (Millipore, Billerica, MA, USA), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-sn-glycerol) (sodium salt; POPG, 16:0–18:1 PG in chloroform; Avanti Polar Lipids, Alabaster, AL, USA), PK (lyophilizate, recombinant, PCR grade; Roche, Indianapolis, IN, USA), and phenylmethanesulfonyl fluoride (PMSF) and isopropyl β-d-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich, St Louis, MO, USA). Other chemicals were purchased from Sango Biotech Co. (Shanghai, China) Antibodies used in this study included 8B4 and M20 anti-PrP antibodies (Santa Cruz Biotechnology, Dallas, TX, USA), SAF32 anti-PrP antibody (Cayman Chemical, Ann Arbor, MI, USA), 8H4 anti-PrP antibody (a generous gift from Dr. Man-Sun Sy, Case Western Reserve University, Cleveland, OH, USA), HRP-conjugated goat anti-mouse IgG antibody (Bio-Rad, Hercules, CA, USA), HRP-conjugated horse anti-goat IgG antibody (Beijing Dingguo Changsheng Biotechnology, Beijing, China), and anti-GFAP antibody (Cell Signaling Technology, Danvers, MA, USA; or Dako, Carpinteria, CA, USA). The rPrP-expressing plasmid pET-22b moPrP23-230 was a generous gift from Dr. Surachai Supattapone (Dartmouth Medical School, Hanover, NH, USA).
Purification of rPrP
The pET22b moPrP23-230-transformed Escherichia coli BL21 (DE3) cells were cultured in 1 L Luria broth medium till OD600 reached 0.5–0.6. Afterward, IPTG was added to reach a final concentration of 1 mM, and the culture was continued for 5 h. Induced E. coli cells were harvested (5000 g, 20 min, 4°C) and stored at −80°C.
For purification, E. coli cells were resuspended in 75 ml of buffer A (10 mM Tris–HCl and 100 mM NaPO4, pH 8.0) and lysed through 4 rounds of 3 min sonication on ice (amplitude=80, 2-s pulse on/1-s pulse off, 10 min incubation on ice between rounds; Misonix sonicator 3000; Misonix Inc., Farmingdale, NY, USA). Inclusion bodies were collected by centrifugation (10,000 g, 30 min, 4°C), resuspended in 75 ml of buffer B (6 M GuHCl, 10 mM Tris–HCl, 100 mM NaPO4, and 10 mM BME, pH 8.0), and sonicated on ice till completely solubilized (4 to 6 rounds of 3 min sonication, amplitude=80, 2-s pulse on/1-s pulse off, 10 min incubation on ice between rounds). After centrifugation (10,000 g, 30 min, 4°C), the supernatant was mixed with 25 ml of Ni–NTA Superflow resin (preequilibrated with buffer B) and stirred for 1 h at room temperature. The resin was then packed into a glass chromatography column and washed with 120 ml of buffer B. Afterward, a 200 ml gradient of buffer B to buffer A was applied to the column (1.5 ml/min), which was followed by a wash with 75 ml buffer C (10 mM Tris–HCl, 100 mM NaPO4, and 50 mM imidazole, pH 8.0). The rPrP was eluted with buffer D (10 mM Tris–HCl, 100 mM NaPO4, and 500 mM imidazole, pH 5.8). The eluted fractions with OD280 ≥ 0.1 were combined, added to dialysis tubing (8000 MWCO) and dialyzed for 1.5 h against buffer E (10 mM NaPO4, pH 5.8, changing buffer every 30 min) and another 1.5 h against ddH2O (changing ddH2O every 30 min). Dialyzed rPrP in ddH2O was kept at −80°C.
Preparation of POPG lipid vesicles and isolation of total RNA
Preparation of POPG vesicle and isolation of total RNA from FVB/NJ mouse liver were performed as described previously (8, 15).
Preparation of sPMCA substrate
After centrifugation at 100,000 g for 1 h at 4°C, the soluble rPrP concentration was determined using the DC protein assay kit (Bio-Rad). Here 864 μl of soluble rPrP (0.46 mg/ml in ddH2O) was mixed with 216 μl of POPG (1 mg/ml in 20 mM Tris–HCl, pH 7.4) in a 1.5-ml RNase-free microcentrifuge tube and incubated at room temperature for 10 min. During the incubation, a mixture of 6420 μl ddH2O, 480 μl of 5% Triton X-100, 960 μl of 10× TN buffer (1.5M NaCl and 100 mM Tris–HCl, pH 7.5), and 19.2 μl of 500 mM EDTA was prepared in a 15-ml centrifuge tube. The rPrP-POPG mixture was transferred to the 15-ml centrifuge tube, thoroughly mixed, and incubated at room temperature for 5 min. After the incubation, 640 μl of mouse liver RNA (2.4 mg/ml in RNase-free H2O) was added. The substrate mixture was thoroughly mixed, portioned into aliquots in RNase-free PCR tubes (90 μl/tube), and stored at −80°C. The concentrations of each component in the sPMCA substrate mixture were determined based on our previous studies of PrP-lipid interaction (16, 17), rPrP-res generation, and propagation (8, 9).
sPMCA instrument setup
A Misonix sonicator S-4000 with a microplate horn was used for sPMCA. All reactions were carried out in 8-strip thin-wall 200-μl PCR tubes that were placed in a homemade rack in the microplate horn. The bottom of tubes were ∼3 mm above the horn surface. The sonicator was connected to a heated Cole-Parmer Polystat circulating water bath (Cole-Parmer, Vernon Hills, IL, USA), and the level of circulating water in the horn was adjusted to cover the reaction mixture in the PCR tubes. Water temperature is set at 39°C in the circulator so that the temperature of water bath in the microplate horn is at 37°C. During the reaction, the microplate horn was covered with a plastic wrap to prevent evaporation. Sonicator was programmed to sonicate for 30 s followed by a 29.5-min incubation, and the amplitude of the sonicator was set at 65. For each round of PMCA, the sonication-incubation cycle repeated 48 times (24 h).
sPMCA
For the first round, 10 μl PBS was added to the substrate. At the end of first round, the reaction product was mixed by pipetting, and 10 μl of reaction product was transferred to a new tube containing 90 μl substrate for the second PMCA round. The same transferring steps were repeated to carry out serial PMCA reactions.
Detection of rPrP-res in sPMCA products
The detection of rPrP-res was performed as described previously (8, 9, 15).
Mouse bioassay
Wild-type CD-1 mice were purchased from the Shanghai Laboratory Animal Center (Shanghai, China), and 6-wk-old male mice were used for all bioassays. Preparation of inocula, intracerebral (i.c.) inoculation, second-round transmission, and biochemical and pathological analyses of inoculated mice were performed as described previously (8, 9, 15). The mouse bioassay was performed at the specific pathogen-free animal facility at East China Normal University, and the i.c. inoculation was performed after mice were completely anesthetized. For all inoculations (including both rPrP-res14kDa and rPrP-res17kDa), the volume of inoculum was always 30 μl/mouse, which equals 300 μl of sPMCA products and contains 12.42 ng of total PrP. All mice were monitored daily, and efforts were made to minimize pain and suffering. All mouse experiments were performed according to the Guidelines on the Humane Treatment of Lab Animals established in 2006 by the Ministry of Science and Technology of China [policy (2006)398] and approved by the Institutional Animal Care and Use Committee of East China Normal University (approval protocol AR2009/10005). Histopathological analyses were performed as described previously (8, 18).
Biosafety
All experiments were performed at designated biosafety level 2 (BSL-2) laboratories at East China Normal University or at Ohio State University, following the U.S. Centers for Disease Control and Prevention guidelines for experiments involving mouse prions (19). All sPMCA products were kept at designated −80°C freezers in BSL-2 labs clearly marked with biohazard signs. All personnel were trained to work in a BSL-2 laboratory.
RESULTS
De novo formation of rPrP-res
The unseeded sPMCA was performed with a substrate mixture consisting of murine rPrP, POPG, and total RNA isolated from normal mouse liver, and the de novo formation of rPrP-res was detected by immunoblot analyses of PK-digested sPMCA products. In a total of 6 tries (4–8 samples and 12–24 rounds of sPMCA per experiment; Table 1), the successful generation of rPrP-res was detected in 3 separated unseeded sPMCA experiments (Fig. 1 and Table 1). The initiation of the rPrP-res conformer appeared to be random, and we detected the first appearance of rPrP-res at different sPMCA rounds, ranging from round 5 to round 18 (Fig. 1, Supplemental Fig. S1, and data not shown). Interestingly, rPrP-res with 2 different sizes of PK-resistant core, 14 kDa (rPrP-res generated in experiment 3) and 17 kDa (rPrP-res generated in experiments 4 and 5), were detected (denoted as rPrP-res14kDa and rPrP-res17kDa). Once formed, both rPrP-res forms are able to propagate indefinitely by sPMCA.
Table 1.
De novo rPrP-res formation by sPMCA
| Experiment | Total samples | Total rounds of PMCA | Samples with de novo rPrP-res formation | Molecular mass of PK-resistant fragment (kDa) |
|---|---|---|---|---|
| 1 | 8 | 24 | 0 | — |
| 2 | 8 | 24 | 0 | — |
| 3 | 8 | 24 | 8 | 14 |
| 4 | 4 | 12 | 4 | 17 |
| 5 | 8 | 20 | 6 | 17 |
| 6 | 4 | 20 | 0 | — |
Figure 1.

De novo formation of rPrP-res. The sPMCA reaction was carried out with a substrate consisting of rPrP, POPG, and total RNA isolated from normal mouse liver. The presence of rPrP-res was determined by subjecting the sPMCA product to 50 μg/ml PK digestion at 37°C for 30 min, followed by immunoblot analysis. A) De novo formation of rPrP-res17kDa detected by the 8H4 antibody. B) De novo formation of rPrP-res14kDa detected by the M20 antibody. Numbers at top indicate sPMCA rounds. C, control (undigested sPMCA substrate). The full-length rPrP detected in lanes 14 and 20 likely resulted from incomplete PK digestion.
Comparing the biochemical properties of rPrP-res17kDa and rPrP-res14kDa
Similar to naturally occurring PrPSc, both rPrP-res forms were aggregated (Fig. 2A, D) and PK resistant (Fig. 2B, E). The PK-resistant rPrP fragments were still detectable after a 200 μg/ml PK digestion at 37°C for 30 min, indicating a relatively high PK resistance. A panel of anti-PrP antibodies was used to probe the PK-resistant fragments of the 2 rPrP-res forms to determine whether they have a C-terminal PK resistance similar to naturally occurring PrPSc (2). For rPrP-res17kDa, the PK-resistant core was not recognized by the 8B4 antibody (recognizing an N-terminal epitope at aa 35–45; refs. 20, 21), but was detected by SAF32, 8H4, and M20 antibodies (Fig. 2C). Since SAF32 antibody recognizes the octarepeat region (22) and the epitopes of 8H4 and M20 antibodies are localized at the C terminus of PrP (21, 22), we concluded that the PK-resistant core of rPrP-res17kDa is similar to that of naturally occurring PrPSc, from the C terminus of the octarepeat region to the C-terminal end of PrP (23). Surprisingly, the PK-resistant core of rPrP-res14kDa was detected only by M20 antibody, but not by any of the other 3 antibodies (Fig. 2F). Considering the size of the PK-resistant fragment (∼14 kDa) and the location of 8H4 epitope (aa 175–185; refs. 20, 21), the inability of this antibody to recognize the PK-resistant core of rPrP-res14kDa likely indicates that the rPrP-res14kDa conformation is different from that of rPrP-res17kDa, resulting in the inaccessibility of 8H4 epitope by immunoblot detection. The conformational-dependent exposure of the 8H4 epitope is consistent with previously reported renaturation of the conformational PrP epitope during the process of electrophoresis and/or immunoblot analysis (24). Nevertheless, the detection by M20 antibody (Fig. 2F) does support the hypothesis that the PK-resistant core of rPrP-res14kDa is also at the C terminus of PrP.
Figure 2.

Biochemical characterization of rPrP-res17kDa and rPrP-res14kDa. A, D) rPrP-res17kDa (A) and rPrP-res14kDa (D) propagated by sPMCA were separated into supernatant (S) and pellet (P) fractions by centrifugation for 1 h at 100,000 g at 4°C. Amounts of rPrP in the supernatant and pellet fractions were determined by immunoblot analysis (lanes 2 and 3). Amounts of rPrP-res in the supernatant and pellet fractions were determined by PK digestion and immunoblot analysis (lanes 4 and 5). T, total rPrP input. B, E) Serial PK digestion of rPrP-res17kDa (B) and rPrP-res14kDa (E). Concentrations of PK were 25, 50, 100, 200, and 400 μg/ml; digestion was carried out at 37°C for 30 min. C, F) rPrP-res17kDa (C) and rPrP-res14kDa (F) were digested with PK and detected by immunoblot analysis with various antibodies as indicated. C, control (undigested sPMCA substrate). rPrP-res17kDa (A, B) and rPrP-res14kDa (D, E) were detected by 8H4 and M20 antibody, respectively.
The rPrP-res17kDa, but not rPrP-res14kDa, causes prion disease in wild-type mice
To determine whether the newly formed rPrP-res forms are capable of causing prion disease in animals, we intracerebrally inoculated wild-type CD-1 mice with rPrP-res14kDa or rPrP-res17kDa. The sPMCA substrate mixture without going through sPMCA or inoculation diluent (1 mg/ml BSA in PBS) was used as the control inoculum. Mice that received i.c. inoculation with rPrP-res17kDa developed neurological signs of prion disease, including clasping, tail plasticity, hypokinesia, kyphosis, and mild ataxia and became listless and cachexic at the terminal stage. The survival times for rPrP-res17kDa-inoculated mice ranged from 161 to 185 days postinoculation (dpi), and the average survival time was 172.3 ± 1.6 dpi (means±se; Fig. 3A and Table 2). None of the control mice that received i.c. inoculation with control inocula developed disease (Fig. 3A). Although the survival time of rPrP-res17kDa-inoculated mice appeared to be longer than the average survival time of 150 ± 2.2 dpi for rPrP-resOSU, the rPrP-res generated in our previous study at Ohio State University (8), it was still within the variation among different prion inoculations (25). More interestingly, i.c. inoculation of rPrP-res14kDa in wild-type CD-1 mice did not cause any signs of disease (Fig. 3B; the last rPrP-res14kDa-inoculated mouse was euthanized at 602 dpi).
Figure 3.
Survival plots. A) Survival plot of CD-1 mice inoculated with rPrP-res17kDa and control inocula, sPMCA substrate, and inoculum diluent (PBS+BSA). B) Survival plot of CD-1 mice inoculated with rPrP-res14kDa and inoculum diluent (PBS+BSA).
Table 2.
Summary of infectivity bioassay in wild-type CD-1 mice
| Inoculum | Diseased/inoculated mice | Survival (dpi) | Average survival (d) |
|---|---|---|---|
| rPrP-res17kDa | 10/10 | 161, 162, 166, 170, 171, 173, 176, 177, 182, 185 | 172.3 ± 1.6 |
| Control 1: sPMCA substrate | 0/5 | >300 | NA |
| Control 2: PBS + BSA | 0/5 | >300 | NA |
| 2nd passage of rPrP-res17kDa: 1% brain homogenates from mice inoculated with i.c. rPrP-res17kDa | 7/7 | 146, 158, 161, 160, 170, 170, 174 | 161.3 ± 1.8 |
| 2nd passage of control mice: 1% brain homogenates from mice inoculated with sPMCA substrate | 0/5 | >250 | NA |
| rPrP-res14kDa | 0/9 | >365 | NA |
| Control: PBS + BSA | 0/5 | >365 | NA |
Average survival values are means ± se. NA, not applicable.
Analyses of mouse brain tissues confirmed the presence of spongiosis, astrogliosis, and PK-resistant PrPSc in the brains of rPrP-res17kDa-inoculated mice (Fig. 4A, B), but not in rPrP-res14kDa-inoculated mouse brains (Fig. 4C, D). Similarly, the aberrant PrP deposit was observed only in rPrP-res17kDa-inoculated mice (data not shown). The glycosylated and glycophosphatidylinositol (GPI)-anchored PrPSc in rPrP-res17kDa-inoculated mouse brains migrated around 19–26 kDa and was readily detected by the 8H4 antibody (Fig. 4B). In contrast, no PrPSc was detected in rPrP-res14kDa-inoculated mouse brains (Fig. 4D). Since the PK-resistant fragment of rPrP-res14kDa was recognized only by M20 antibody (Fig. 2F), we repeated the immunoblot analysis of PK-digested brain homogenates with M20 antibody and confirmed that there was no PK-resistant PrP band in the brain homogenates of rPrP-res14kDa-inoculated mice (Fig. 4E). These results indicate that the rPrP-res14kDa could not seed endogenous PrPC conversion in vivo.
Figure 4.

Characterization of CD-1 mice inoculated with rPrP-res17kDa and rPrP-res14kDa. A) HE and GFAP staining of mouse brains i.c. inoculated with rPrP-res17kDa or control inoculum (PBS + BSA) as indicated. B) PrPSc in rPrP-res17kDa-inoculated mouse brain homogenates was detected by PK digestion and immunoblot analysis with 8H4 antibody. Each number represents an individual inoculated mouse. C = undigested mouse brain homogenate was used as a control. C1 and C2 are brain homogenates prepared from mice intracerebrally inoculated with control inocula, inoculum diluent (PBS+BSA) and sPMCA substrate, respectively. C) HE and GFAP staining of mouse brains intracerebrally inoculated with rPrP-res14kDa or control inoculum (PBS+BSA) as indicated. D) Brain homogenates (BH) prepared from rPrP-res14kDa-inoculated mice (14-kDa BH) and rPrP-res17kDa-inoculated mice (17-kDa BH) were subjected to PK digestion and immunoblot analysis with 8H4 antibody. C, control (undigested mouse brain homogenates). E) PK-resistant PrPSc in the brain homogenates prepared from mice inoculated with rPrP-res17kDa (17-kDa BH) and rPrP-res14kDa (14-kDa BH) was detected by immunoblot analysis with M20 antibody.
Second-round transmission was performed by intracerebrally inoculating wild-type CD-1 mice with 1% brain homogenates prepared from rPrP-res17kDa-inoculated mice. As expected, all mice developed disease, with an average survival time of 161.3 ± 1.8 dpi (Table 2 and Fig. 5A). Spongiosis, astrogliosis, and PK-resistant PrPSc were detected in the brains of mice that received second-round transmission (Fig. 5B, C). None of the mice that received i.c. inoculation of brain homogenate prepared from first-round control mice (which received i.c. inoculation of sPMCA substrate) developed disease (Table 2 and Fig. 5). Collectively, we concluded that the de novo generated rPrP-res17kDa is highly infectious, capable of causing bona fide prion disease in wild-type CD-1 mice. The rPrP-res14kDa, however, is not infectious to wild-type mice.
Figure 5.
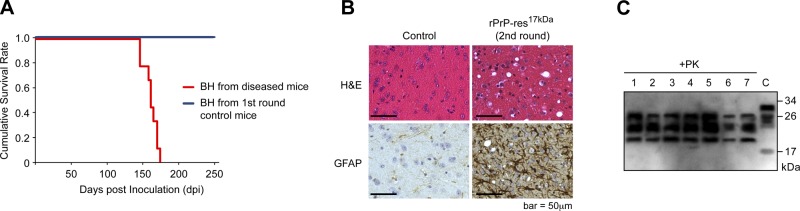
Second-round transmission of rPrP-res17kDa. A) Survival plots of mice inoculated with 1% brain homogenates (BH) prepared from rPrP-res17kDa inoculated mice or 1% brain homogenates (BH) prepared from control mice received inoculation of sPMCA substrate. B) Pathological analysis of mice received second-round transmission. The rPrP-res17kDa -inoculated and control mouse brain sections were stained with hematoxylin and eosin (H&E) or an anti-GFAP antibody as indicated. C) PrPSc in brian homogenates prepared from mice that received second-round transmission of rPrP-res17kDa was detected by PK digestion followed by immunoblot analysis with 8H4 antibody. C, control (undigested mouse brain homogenate).
DISCUSSION
Recent studies have demonstrated the association of prion infectivity with various in vitro generated rPrP conformers, strongly supporting the prion hypothesis (8–11, 26–29). We previously showed that a highly infectious recombinant prion can be generated with rPrP plus POPG and total mouse liver RNA via an unseeded sPMCA reaction (8). To determine whether the infectious rPrP-res can indeed be generated de novo, the current study was carried out in a new prion-free laboratory, eliminating the possibility of contamination. Our results clearly showed that an infectious recombinant prion can be generated de novo, which causes prion disease in wild-type mice with a 100% attack rate and a relatively synchronized survival time.
Compared to rPrP amyloid fiber prepared by various approaches (27–29), the sPMCA-generated recombinant prions generally contain higher infectivity (8–11, 26), suggesting that the sPMCA procedure, cyclic sonication and incubation, contributes to the formation of an infectious prion. The sPMCA was originally developed based on the notion that sonication-fragmented PrPSc aggregates would increase the seeding efficiency (3), which readily explains the PrPSc-seeded prion propagation, but cannot account for the de novo prion formation. We propose that the de novo prion formation in our system is due to the ability of sPMCA to reassemble rPrP aggregates during each cycle of PMCA, i.e., breaking PrP aggregates by sonication and reforming PrP aggregates during the incubation. The sPMCA substrate used in our system contains 2 cofactors, POPG and RNA (8, 30), both of which are known to induce rPrP unfolding and aggregation (16, 17, 31–36). Particularly, we have shown that the rPrP-POPG interaction results in a β-sheeted rPrP conformer with PrPSc-like PK-resistance (16). Thus, the rPrP in our sPMCA substrate mixture is likely in an aggregated and β-sheeted conformational state similar to the infectious PrPSc conformer, but without the seeding capability. When this substrate is subjected to sPMCA, cycles of sonication and incubation will lead to the breakup and reassembly of rPrP aggregates. Because of the lack of seeding during the de novo prion formation process, the PrP reaggregation process is random, resulting in packaging of a variety of PrP aggregates during each PMCA cycle. However, if one of the newly packaged rPrP aggregates possesses the self-perpetuating property, it will act as a seed to guide the rPrP reaggregation and gradually become the dominantly propagated rPrP species, resulting in the formation of a self-perpetuating PrP conformer. This model explains the de novo recombinant prion formation by sPMCA and the randomness of rPrP-res appearance in sPMCA (Fig. 1 and Supplemental Fig. S1). It also accounts for the previous studies showing de novo prion formation with purified PrPC (5) or brain homogenates (37), since both cofactors, lipids and RNA, are present in the sPMCA substrates used in those studies.
Although the lipid- and/or RNA-interacted rPrP in the sPMCA substrate is already in a conformation similar to the final infectious PrP conformation, further rPrP conformational changes during the assembly of self-perpetuating rPrP aggregates cannot be excluded. In fact, the formation of different types of rPrP-res with the same substrate mixture in our current and previous studies (8) supports the hypothesis that there are further rPrP conformation alterations during sPMCA-mediated assembly.
The rPrP-res14kDa generated in this study is clearly self-perpetuating in vitro, but it failed to cause any disease in wild-type mice. The shorter PK-resistant core and the inaccessibility of the 8H4 epitope (Fig. 2F) suggest that the conformation of rPrP-res14kDa is completely different from that of rPrP-res17kDa or rPrP-resOSU. Since the sPMCA substrate mixtures used for de novo formation of rPrP-res14kDa and rPrP-res17kDa were identical, the generation of totally different rPrP-res forms could be due to the randomness of sPMCA-mediated rPrP aggregation process, the particular sonication power output during that experiment (the power output varies despite always setting the amplitude of the sonicator at 65), or the combination of both. If the variation in sonication power output does play a role, it would indicate that different physical treatments can lead to different rPrP conformations (or differently packed rPrP aggregates), which would be consistent with the previous reports showing that different shaking modes generate distinct types of rPrP amyloid fibers (38) and that different sonication powers are required to propagate different prion strains (14).
Because of its ability to amplify minute amounts of the disease-associated PrPSc conformer, sPMCA is considered one of the most promising diagnostic approaches for prion disease and has been demonstrated to sensitively detect PrPSc in body fluids of rodent prion models (39–41). However, the ability of sPMCA to generate infectious prion de novo encourages caution in applying sPMCA for the diagnostic purpose.
Collectively, our results reveal that sPMCA is capable of forming various self-perpetuating rPrP-res conformers de novo, and some of these rPrP conformers, such as rPrP-res17kDa and rPrP-resOSU, are sufficient to cause prion disease in wild-type animals. In addition to understanding the origin of infectious mammalian prions, our findings may also have important implications in understanding the relationship between in vitro self-perpetuating protein conformers and the in vivo prion-like spread of pathology in various human diseases (42–48).
Supplementary Material
Acknowledgments
The authors thank Dr. Man-Sun Sy (Case Western Reserve University, Cleveland, OH, USA) and Dr. Surachai Supattapone (Dartmouth Medical School, Hanover, NH, USA) for providing reagents used in this study.
Financial support for this study was provided by the National Natural Science Foundation of China, projects 31172347 and 30871369; the Ministry of Education of China, project 985; and the U.S. National Institutes of Health, grants R01 NS071035 and R01 NS 060729.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- BSL-2
- biosafety level 2
- dpi
- days postinoculation
- i.c.
- intracerebral
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- PK
- proteinase K
- PMSF
- phenylmethanesulfonyl fluoride
- POPG
- 1-palmitoyl-2-oleoylphosphatidylglycerol
- PrP
- prion protein
- PrPC
- cellular prion protein
- PrPSc
- scrapie form of prion protein
- rPrP
- recombinant prion protein
- rPrP-res
- proteinase K-resistant recombinant prion protein
- PVDF
- polyvinylidene fluoride
- sPMCA
- serial protein misfolding cyclic amplification
- TSE
- transmissible spongiform encephalopathy
REFERENCES
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U. S. A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Caughey B., Chesebro B. (1997) Prion protein and the transmissible spongiform encephalopathies. Trends Cell Biol. 7, 56–62 [DOI] [PubMed] [Google Scholar]
- 3. Saborio G. P., Permanne B., Soto C. (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813 [DOI] [PubMed] [Google Scholar]
- 4. Castilla J., Saa P., Hetz C., Soto C. (2005) In vitro generation of infectious scrapie prions. Cell 121, 195–206 [DOI] [PubMed] [Google Scholar]
- 5. Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U. S. A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Geoghegan J. C., Miller M. B., Kwak A. H., Harris B. T., Supattapone S. (2009) Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Pathog. 5, e1000535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Imamura M., Kato N., Yoshioka M., Okada H., Iwamaru Y., Shimizu Y., Mohri S., Yokoyama T., Murayama Y. (2011) Glycosylphosphatidylinositol anchor-dependent stimulation pathway required for generation of baculovirus-derived recombinant scrapie prion protein. J. Virol. 85, 2582–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang F., Wang X., Yuan C. G., Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang F., Zhang Z., Wang X., Li J., Zha L., Yuan C. G., Weissmann C., Ma J. (2012) Genetic informational RNA is not required for recombinant prion infectivity. J. Virol. 86, 1874–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U. S. A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deleault N. R., Walsh D. J., Piro J. R., Wang F., Wang X., Ma J., Rees J. R., Supattapone S. (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U. S. A. 109, E1938–E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cosseddu G. M., Nonno R., Vaccari G., Bucalossi C., Fernandez-Borges N., Di Bari M. A., Castilla J., Agrimi U. (2011) Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog. 7, e1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saa P., Castilla J., Soto C. (2006) Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281, 35245–35252 [DOI] [PubMed] [Google Scholar]
- 14. Morales R., Duran-Aniotz C., Diaz-Espinoza R., Camacho M. V., Soto C. (2012) Protein misfolding cyclic amplification of infectious prions. Nat. Protoc. 7, 1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang F., Wang X., Ma J. (2011) Conversion of bacterially expressed recombinant prion protein. Methods 53, 208–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang F., Yang F., Hu Y., Wang X., Jin C., Ma J. (2007) Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry 46, 7045–7053 [DOI] [PubMed] [Google Scholar]
- 17. Wang F., Yin S., Wang X., Zha L., Sy M. S., Ma J. (2010) Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry 49, 8169–8176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X., Bowers S. L., Wang F., Pu X. A., Nelson R. J., Ma J. (2009) Cytoplasmic prion protein induces forebrain neurotoxicity. Biochim. Biophys. Acta 1792, 555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. U.S. Centers for Disease Control and Prevention (2009) Prion diseases. In Biosafety in Microbiological and Biomedical Laboratories, 5th Ed HHS Publication No. (CDC) 21–1112 (Closewood L. C., Wilson D. E., eds) Sec. VIII-H, pp 282–289, U.S. Department of Health and Human Services, Washington, DC [Google Scholar]
- 20. Li R., Liu T., Wong B. S., Pan T., Morillas M., Swietnicki W., O'Rourke K., Gambetti P., Surewicz W. K., Sy M. S. (2000) Identification of an epitope in the C terminus of normal prion protein whose expression is modulated by binding events in the N terminus. J. Mol. Biol. 301, 567–573 [DOI] [PubMed] [Google Scholar]
- 21. Yin S., Pham N., Yu S., Li C., Wong P., Chang B., Kang S. C., Biasini E., Tien P., Harris D. A., Sy M. S. (2007) Human prion proteins with pathogenic mutations share common conformational changes resulting in enhanced binding to glycosaminoglycans. Proc. Natl. Acad. Sci. U. S. A. 104, 7546–7551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feraudet C., Morel N., Simon S., Volland H., Frobert Y., Creminon C., Vilette D., Lehmann S., Grassi J. (2005) Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J. Biol. Chem. 280, 11247–11258 [DOI] [PubMed] [Google Scholar]
- 23. Parchi P., Zou W., Wang W., Brown P., Capellari S., Ghetti B., Kopp N., Schulz-Schaeffer W. J., Kretzschmar H. A., Head M. W., Ironside J. W., Gambetti P., Chen S. G. (2000) Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl. Acad. Sci. U. S. A. 97, 10168–10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kang H. E., Weng C. C., Saijo E., Saylor V., Bian J., Kim S., Ramos L., Angers R., Langenfeld K., Khaychuk V., Calvi C., Bartz J., Hunter N., Telling G. C. (2012) Characterization of conformation-dependent prion protein epitopes. J. Biol. Chem. 287, 37219–37232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tamguney G., Giles K., Glidden D. V., Lessard P., Wille H., Tremblay P., Groth D. F., Yehiely F., Korth C., Moore R. C., Tatzelt J., Rubinstein E., Boucheix C., Yang X., Stanley P., Lisanti M. P., Dwek R. A., Rudd P. M., Moskovitz J., Epstein C. J., Cruz T. D., Kuziel W. A., Maeda N., Sap J., Ashe K. H., Carlson G. A., Tesseur I., Wyss-Coray T., Mucke L., Weisgraber K. H., Mahley R. W., Cohen F. E., Prusiner S. B. (2008) Genes contributing to prion pathogenesis. J. Gen. Virol. 89, 1777–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim J. I., Cali I., Surewicz K., Kong Q., Raymond G. J., Atarashi R., Race B., Qing L., Gambetti P., Caughey B., Surewicz W. K. (2010) Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 285, 14083–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Legname G., Baskakov I. V., Nguyen H. O., Riesner D., Cohen F. E., DeArmond S. J., Prusiner S. B. (2004) Synthetic mammalian prions. Science 305, 673–676 [DOI] [PubMed] [Google Scholar]
- 28. Colby D. W., Wain R., Baskakov I. V., Legname G., Palmer C. G., Nguyen H. O., Lemus A., Cohen F. E., DeArmond S. J., Prusiner S. B. (2010) Protease-sensitive synthetic prions. PLoS Pathog. 6, e1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Makarava N., Kovacs G. G., Bocharova O., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., Baskakov I. V. (2010) Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 119, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma J. (2012) The role of cofactors in prion propagation and infectivity. PLoS Pathog. 8, e1002589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kazlauskaite J., Sanghera N., Sylvester I., Venien-Bryan C., Pinheiro T. J. (2003) Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 42, 3295–3304 [DOI] [PubMed] [Google Scholar]
- 32. Kazlauskaite J., Pinheiro T. J. (2005) Aggregation and fibrillization of prions in lipid membranes. Biochem. Soc. Symp. 72, 211–222 [DOI] [PubMed] [Google Scholar]
- 33. Morillas M., Swietnicki W., Gambetti P., Surewicz W. K. (1999) Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem. 274, 36859–36865 [DOI] [PubMed] [Google Scholar]
- 34. Gomes M. P., Millen T. A., Ferreira P. S., e Silva N. L., Vieira T. C., Almeida M. S., Silva J. L., Cordeiro Y. (2008) Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J. Biol. Chem. 283, 19616–19625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adler V., Zeiler B., Kryukov V., Kascsak R., Rubenstein R., Grossman A. (2003) Small, highly structured RNAs participate in the conversion of human recombinant PrP(Sen) to PrP(Res) in vitro. J. Mol. Biol. 332, 47–57 [DOI] [PubMed] [Google Scholar]
- 36. Wang F., Ma J. (2013) Role of lipid in forming an infectious prion? Acta Biochim. Biophys. Sin. (Shanghai) 45, 485–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barria M. A., Mukherjee A., Gonzalez-Romero D., Morales R., Soto C. (2009) De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog. 5, e1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Makarava N., Baskakov I. V. (2008) The same primary structure of the prion protein yields two distinct self-propagating states. J. Biol. Chem. 283, 15988–15996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saa P., Castilla J., Soto C. (2006) Presymptomatic detection of prions in blood. Science 313, 92–94 [DOI] [PubMed] [Google Scholar]
- 40. Castilla J., Saa P., Soto C. (2005) Detection of prions in blood. Nat. Med. 11, 982–985 [DOI] [PubMed] [Google Scholar]
- 41. Gonzalez-Romero D., Barria M. A., Leon P., Morales R., Soto C. (2008) Detection of infectious prions in urine. FEBS Lett. 582, 3161–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aguzzi A. (2009) Cell biology: beyond the prion principle. Nature 459, 924–925 [DOI] [PubMed] [Google Scholar]
- 43. Aguzzi A., Rajendran L. (2009) The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783–790 [DOI] [PubMed] [Google Scholar]
- 44. Brundin P., Melki R., Kopito R. (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Polymenidou M., Cleveland D. W. (2011) The seeds of neurodegeneration: prion-like spreading in ALS. Cell 147, 498–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Polymenidou M., Cleveland D. W. (2012) Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med. 209, 889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prusiner S. B. (2012) Cell biology. A unifying role for prions in neurodegenerative diseases. Science 336, 1511–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soto C. (2012) Transmissible proteins: expanding the prion heresy. Cell 149, 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.