

Abstract
Organ injury in sepsis is initially characterized by dysfunction without cell death and structural damage, and thus with the ability to recover organ function. Adaptive metabolic responses to sepsis can prevent bioenergetic failure and death. These studies were aimed at investigating the influence of sepsis on mitochondrial homeostasis, focusing on removal of dysfunctional mitochondria and restitution of a healthy mitochondrial population. These data demonstrate decreased hepatic oxidative phosphorylation by 31 ± 11% following murine cecal ligation and puncture (CLP) at 8 h and 34 ± 9% following LPS treatment in vitro at 12 h (P<0.05). In addition, there was a loss of mitochondrial membrane potential. Mitochondrial density and number initially decreased (relative area per micrograph of 64±10% at baseline vs. 39±13% at 8 h following LPS; P<0.05) and was associated with an increase in autophagy and mitophagy. CLP-induced markers of mitochondrial biogenesis and mitochondrial number and density recovered over time. Furthermore, these data suggest that mitochondrial biogenesis was dependent on an autophagy and mitochondrial DNA/Toll-like receptor 9 (TLR9) signaling pathway. These results suggest that hepatocyte survival and maintenance of function in sepsis is dependent on a mitochondrial homeostasis pathway marked by mitophagy and biogenesis.—Carchman, E. H., Whelan, S., Loughran, P., Mollen, K., Stratamirovic, S., Shiva, S., Rosengart, M. R., Zuckerbraun, B. S. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver.
Keywords: lipopolysaccharide, Toll-like receptor 4, VPS34, peroxisome proliferator-activated receptor-γ coactivator, PGC-1α
Sepsis syndrome from infection is a leading cause of morbidity and mortality in the United States and worldwide. Mortality in sepsis is often secondary to the development of multiple organ dysfunction. Interestingly, initial organ dysfunction in sepsis is characterized by decreased oxidative phosphorylation and a shutdown of cellular function without cell death and permanent structural damage (1, 2). Studies suggest that this initial loss of function and decrease in aerobic respiration are protective adaptive responses that prevent cell death, thus allowing for return of function over time. An inhibition of respiration has been demonstrated in muscle biopsies from patients with sepsis, suggesting that this is relevant to human biology.
Inhibition of oxidative phosphorylation can result in the increased production of mitochondria-generated reactive oxygen species, which can act as second messengers to induce adaptive cell signaling responses (3). However, inhibition of oxidative phosphorylation has potential deleterious consequences, including decreased production of adenosine triphosphate (ATP), loss of mitochondrial membrane potential, and bioenergetic failure. In a long-term experimental sepsis model, decreased mitochondrial respiration was observed, and progression to bioenergetic failure was associated with death (1).
To prevent cell death, mechanisms to remove damaged organelles are necessary. Autophagy, and specifically mitophagy, is one such preserved cellular process to remove dysfunctional mitochondria (4). The process of autophagy involves the formation of autophagosomes, which then fuse with lysosomes to form what is known as autophagolysosomes. This involves a complex interplay of critical autophagic proteins, including LC3, which, when lapidated, binds to autophagosomes and can be used as a marker of autophagy. Autophagy has been shown to be up-regulated in the liver and other organs in the setting of sepsis (5, 6). In addition to removal of dysfunctional mitochondria, cellular processes to restore a viable mitochondrial population that allow the cell and organ to recover are necessary. Several studies have illustrated that mitochondrial biogenesis, or the process of generating new mitochondria, improves survival in sepsis. In animal modes of sepsis, enhanced biogenesis was shown to improve survival, and inhibition of mitochondrial biogenesis worsened outcome (7–9). This has borne out in human studies, in which eventual survivors of severe sepsis were observed to respond early to critical illness with mitochondrial biogenesis (10).
Toll-like receptors (TLRs) are pattern recognition receptors that recognize various products from a given pathogen or from danger-associated molecular pattern molecules (11). TLR9 is localized to the endocytic compartments and homodimerizes and recognizes unmethylated cytosine-phosphodiester-guanine (CpG)-containing DNA motifs present in viral and bacterial DNA (12). In B cells, there has been evidence of recruitment of TLR9 receptor to autophagosome-like compartments. With inhibition of autophagy, mobilization of TLR9 to the autophagosome does not occur (13). Mitochondria, which are phylogenetically similar to bacteria, contain unmethylated CpG motifs within the mitochondrial genome and can activate TLR9 signaling (14).
The purpose of these investigations was to test the hypothesis that sepsis or lipopolysaccharide (LPS) induces mitochondrial injury that then requires removal of dysfunctional mitochondria followed by a replacement of healthy mitochondrial population via biogenesis. Furthermore, that degradation of mitochondria via autophagy, promotes biogenesis via TLR9 signaling. Mechanistic links between autophagy and biogenesis are not well characterized.
MATERIALS AND METHODS
Cell culture
Primary mouse hepatocytes were harvested from C57BL/6, tlr4−/−tlr9CpG1/CpG1 mice, as described previously (15, 16). Cells were utilized on d 1–3 after harvest. Autophagy was inhibited with vacuolar protein sorting 34 (VPS34) siRNA (50 μM), and TLR9 was inhibited with siRNA (50 μM) (Invitrogen, Grand Island, NY, USA). LPS treatment in all hepatocyte experiments was utilized at a concentration of 100 ng/ml, and cells were treated from 0–24 h (Sigma, St Louis, MO, USA). Cells were maintained at 37°C and 5% CO2. In some experiments, tetramethylrhodamine methyl ester (TMRM) or mitotracker green (Invitrogen) was added 30 min prior to fixing cells with paraformaldehyde.
Human primary hepatocytes were obtained from the laboratory of Dr. Stephen C. Strom (University of Pittsburgh). Briefly, all tissues were collected with informed consent from patients undergoing scheduled liver resection procedures performed for a number of different forms of neoplastic disease. Residual liver tissue not needed for diagnostic purposes was transported to the laboratory from the operating rooms in ice-cold Eagle's minimum essential medium (Lonza, Walkersville, MD, USA) and perfused within 90 min of removal. The tissue dissociation and subsequent hepatocyte isolation procedures were performed as described previously (17). Cell viability, assessed by the trypan blue exclusion method, and plating efficiency procedures were performed as described previously (17).
Mitochondrial DNA extraction and treatment
Liver from C57BL/6 mice was homogenized (20 mM Tris, 0.33 M sucrose, and 0.2 mM EDTA, pH 7.5). The homogenate was then spun at 4 C at 800 rpm for 10 min to remove cellular debris. The supernatant was then spun at 8000 rpm for 10 min, and the pellet was saved to represent the mitochondrial fraction. This pellet was then processed using the Wizard Genomic DNA purification kit (Promega, Madison, WI, USA) per the manufacturer's protocol. The DNA concentration was then quantified. Mitochondrial DNA was premixed with a liposome preparation (Lipofectamine 2000; Promega), and cells were exposed to a final concentration of 5 μg/ml. Control preparations in these experiments consisted of liposome without the addition of DNA.
Cecal ligation and perforation
Animal protocols were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. The experiments were performed in adherence to the U.S. National Institutes of Health guidelines on the use of laboratory animals. Cecal ligation and perforation (CLP) was performed on C57BL/6 (Jackson Laboratories, Bar Harbor, ME, USA), tlr4−/−, or tlr9CpG1/CpG1 male mice, ages 6–8 wk, weighing 20–25 g. These animals were anesthetized with pentobarbital sodium (70 mg/kg i.p.). A 2-cm midline laparotomy was performed, and the cecum was identified. Stool was then milked to the tip of the cecum, and it was subsequently ligated 1 cm from the tip with a 2-0 silk tie. The cecum was then perforated with a 22-gauge needle and returned into the abdomen. The musculature and skin were closed with a running 2-0 silk suture. Sham-treated animals underwent laparotomy and bowel manipulation without ligation or perforation. Tissue and blood collection occurred at 1–24 h after CLP. No antibiotics were utilized, and animals had free access to food and water preoperatively and postoperatively.
In vivo siRNA
Autophagy was inhibited in vivo through the use of siRNA specific to VPS34 (Invitrogen; 50 μg/kg). This was administered via hydrodynamic tail vein injection, where the siRNA was delivered in a volume of 2 ml of lactated Ringer's solution and given 3 d prior to CLP. The rapid injection of this large volume creates significant pressure to help promote siRNA uptake. Scramble siRNA (50 μg/kg) was used as a control, again via hydrodynamic tail vein injection.
Immunohistochemistry
Cells were fixed on coverslips with paraformaldehyde for 15 min and then rinsed with cold PBS. Liver tissue from mice was removed after perfusion with cold PBS and 2% paraformaldehyde. Tissue was then placed in 2% paraformaldehyde for 1 h and then switched to 30% sucrose in distilled water solution for 12 h. The tissue was then slowly frozen in 2-methylbutane. Tissue sections were obtained at 7 μm and were then stained for LC3 (Novus, St. Charles, MO, USA) to monitor autophagy, TLR9 (Cell Signaling Technologies, Danvers, MA, USA), or peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α; Abcam, Cambridge, MA, USA). Images were taken with the Zeiss 510 inverted confocal microscope (Carl Zeiss, Oberkochen, Germany).
Western blot analysis
Primary mouse hepatocytes were washed with cold PBS, collected in lysis buffer, sonicated, and spun (10,000 g for 15 min), and then the supernatant was transferred to a new tube. Protein concentrations were determined using the BCA protein assay kit (Pierce Biotechnology, Rockford, IL). Samples were then mixed with loading buffer and run on a SDS-polyacrylamide gel. This gel was then transferred to a cellulose membrane. The membrane was blocked in 5% milk for 1 h and then incubated in primary antibodies. Antibodies utilized were LC3 (Novus Biologicals, Littleton, CO, USA), p62, TLR9 (Cell Signaling Technology, Danvers, MA, USA), PGC-1α, nuclear respiratory factor 1 (NRF-1), β-actin (Abcam, Cambridge, MA, USA), or oxidative phosphorylation cocktail (Mitosciences, Eugene, OR, USA). Membranes were then washed in TBS-Tween 20 (TBST) for 30 min and then placed in secondary antibody for 1 h and then washed for 1 h in TBST prior to being developed using chemiluminescence substance (Thermo Scientific, Rockford, IL, USA).
Electron microscopy
For electron microscopy experiments, mice were euthanized and then perfused with cold PBS, followed by 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), and processed for transmission electron microscopy (TEM), as described previously (18). After dehydration, thin sections were stained with uranyl acetate and lead citrate for observation under a JEM 1011CX electron microscope (Jeol, Peabody, MA, USA). Images were acquired digitally from a randomly selected pool of 10–15 fields under each condition. For in vitro experiments, hepatocytes were fixed with the above paraformaldehyde and glutaraldehyde mixture for 1 h and then switched to PBS and imaged as above. Mitochondrial density and autophagosome density measurements were measured using ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA) by measuring the percentage surface area of mitochondria or autophagosomes, respectively, of the total nonnuclear area per micrograph. For each condition, 4 micrographs were read per sample, and each condition was repeated in triplicate.
PCR
Cells were cultured as described. RNA was prepared by utilizing a silica gel-based membrane method using the RNeasy Midi Kit (Qiagen, Valencia, CA, USA), according to the manufacturer's instructions. An on-column DNase digestion using RNase-free DNase (Qiagen) was performed to rid the samples of genomic DNA. RNA (1 μg) was used to generate cDNA using oligo dT primers and Omniscript (Qiagen) reverse transcriptase. PCR reaction mixtures were prepared using SYBR Green PCR master mix (PE Applied Biosystems, Foster City, CA, USA). SYBR Green 2-step real-time RT-PCR for VPS34, PGC-1α, transcription factor A mitochondrial (Tfam), NRF1, and β-actin was performed as described. All samples were run in duplicate. The level of gene expression for each sample was normalized to β-actin mRNA expression using the comparative Ct method.
Mitochondrial complex IV activity
Complex IV was measured in whole-cell lysates by monitoring the oxidation of reduced cytochrome c. Activity was determined colorimetrically by following the oxidation of reduced cytochrome c as an absorbance decrease at 550 nm. Potassium cyanide was used to determine specificity of oxidation by complex IV.
Oxygen consumption
Hepatocytes were plated at 20,000 cells on XF24 cell culture plates (Seahorse Biosciences, North Billerica, MA, USA) in a final volume of 250 μl. Hepatocytes were then treated with LPS (100 ng/ml) at various time points. These cells were then rinsed with unbuffered DMEM, placed in 37°C incubator without CO2 for 60 min, and then loaded onto the XF24 instrument. Oxygen consumption rates and extracellular acidification rates were measured. Each condition was run in quadruplicate, and each well was read 8 times. Experiments were repeated 3 times.
Mitochondrial membrane potential measurement
TMRM is a potentiometric, cell-permeable fluorescent indicator that accumulates in the highly negatively charged interior of mitochondria. A loss of ΔΨm causes TMRM to leak from mitochondria, resulting in a loss of fluorescence intensity. Hepatocytes were loaded with TMRM at a concentration of 25 nM for 45 min prior to determination of fluorescence. Live imaging of hepatocytes was then performed by confocal laser scanning microscopy with the application of live time series TMRM fluorescence by illumination at 514 nm and detection at 570 nm.
Statistics
The data were then analyzed in SigmaPlot (Systat Software, San Jose, CA, USA) utilizing 1-way ANOVA with Tukey post hoc analysis for significance. Data are presented as means ± sem and graphically shown as means + se[scap]m unless otherwise specified. Oxygen consumption rate and complex IV activity data are represented as box-and-whisker plots demonstrating the distribution of the data from the least and greatest values, the overall median value, and the medians for the upper and lower halves of the data.
RESULTS
Experimental sepsis or LPS results in decreased hepatic oxidative phosphorylation
The influence of CLP in vivo and LPS in vitro on aerobic respiration was determined in hepatic tissue and hepatocytes, respectively. Several studies have illustrated that sepsis results in decreased oxidative phosphorylation, and it has been hypothesized that this is the mechanism behind the cytopathic dysoxia that occurs in sepsis (19, 20). Oxygen consumption rates at 6 h were significantly decreased in whole-liver homogenates from mice that underwent CLP vs. sham operation (Fig. 1A). A time course of LPS treatment of hepatocytes from 0 to 24 h resulted in decreasing oxygen consumption rates that normalized over time (Fig. 1B). Mitochondrial complex IV activity was also decreased in vitro following LPS treatment (Fig. 1C). The influence of sepsis or LPS on mitochondrial respiration can result in changes in mitochondrial integrity and other functions. Previous data demonstrate that LPS leads to initial mitochondrial depolarization followed by normalization over time (5), and this was confirmed by TMRM staining (Supplemental Fig. S1). In addition, mitochondrial morphology is significantly influenced in the setting of sepsis, demonstrating swollen, dysmorphic mitochondria compared to sham treatment, as determined by TEM (Fig. 1D).
Figure 1.

Experimental sepsis or LPS decreases hepatic aerobic respiration. A) Whole-liver homogenates from C57BL6 mice (n=6/group) that underwent CLP demonstrated decreased oxygen consumption rates compared to mice that underwent sham treatment. *P < 0.05 vs. sham treatment. B) Similarly, LPS treatment of primary hepatocytes in vitro resulted in decreased oxygen consumption rates (h 4–12), which normalized by 24 h. *P < 0.05. C) Hepatocytes treated with or without LPS for 8 h were harvested as whole-cell lysates, and complex IV activity was measured, demonstrating a decrease in activity following LPS treatment. *P < 0.01. D) Hepatocyte mitochondrial morphology demonstrated swollen, rounded mitochondria following LPS treatment (6 h) compared to untreated controls.
Experimental sepsis or LPS leads to changes in mitochondrial density and biogenesis
The increase in autophagy and mitophagy in sepsis may be a mechanism to remove dysfunctional mitochondria. Mitotracker staining and TEM demonstrated decreases in hepatocyte mitochondrial density following LPS treatment consistent with such a hypothesis (Fig. 2A). Quantification of the relative mitochondrial density on electron micrographs revealed a decrease from baseline that was maximal at the 8-h time point (64±10% at baseline vs. 39±13% at 8 h following LPS; P<0.05). In addition, there was a return to normal density within 24 to 48 h. Mitochondrial biogenesis may account for this restoration of mitochondrial density. It has previously been demonstrated that biogenesis is increased in the setting of sepsis (7, 9, 10). The influence of LPS on mitochondrial biogenesis was determined. Treatment of hepatocytes with LPS resulted in increased expression of the mitochondrial biogenesis related transcription factors PGC-1α, NRF-1, and TFAM-1, as determined by rtPCR (Fig. 2B), as well as increased protein levels of components of the electron transport chain (Fig. 2C). LPS induction of these transcription factors was also confirmed in human hepatocytes (Supplemental Fig. S2). Increases in PGC-1α protein levels were also seen in hepatocytes following LPS treatment (Fig. 2D) or in livers following cecal ligation and puncture (Fig. 2E).
Figure 2.

LPS treatment or experimental sepsis results in increased mitochondrial biogenesis. A) Mitochondrial density was determined by mitotracker staining (green) or by TEM. Mitotracker staining was diminished at 8 h and recovered by 48 h (scale bar=25 μm). TEM was performed to confirm this decrease in mitochondrial density (scale bar=2 μm); given mitotracker staining may change with mitochondrial functional status or mitochondrial depolarization. Mitochondrial density was determined by measuring mean relative mitochondrial area per 5 micrographic fields at each time point with or without LPS, and was repeated 3 times. At 8 h, mitochondrial density was decreased by 43 ± 12% (P<0.05) compared to non-LPS treated control hepatocytes. B) rtPCR of the transcription factors PGC-1α, NRF-1, and TFAM-1 were all increased at an 8-h time point following LPS treatment compared to non-LPS-treated cells. *P < 0.05. C) Western blotting for components of oxidative phosphorylation (complexes I-V; oxphos cocktail; Mitosciences) also demonstrated increased levels following LPS treatment (24 h). D, E) Immunocytochemistry and immunohistochemistry for PGC-1α demonstrated increased levels of this protein, which is critical to mitochondrial biogenesis, following LPS treatment in vitro (8 h; D) or experimental sepsis (E). Scale bars = 25 μm.
Mitochondrial biogenesis is increased by a TLR4, autophagy-dependent pathway
Others and we have previously demonstrated that sepsis induces autophagic signaling (5, 21). Because of the dependence of LPS signaling on the TLR4 pathway, the influence of this pattern recognition receptor on sepsis-induced hepatic autophagy and biogenesis were investigated. Hepatocytes from wild-type and tlr4−/− mice were treated with and without LPS. LPS-induced autophagy as demonstrated by increased LC3 staining and decreased p62 staining in wild-type but not in tlr4−/− cells, as determined by Western blot analysis (Fig. 3A). Similar results were demonstrated by determining the number of LC3 puncta per cell by immunocytochemistry (Fig. 3B). Wild-type or tlr4−/− mice were randomized to sham or cecal ligation and puncture groups. Autophagy was increased in wild-type but not in tlr4−/− mice, as determined by staining for LC3 by immunohistochemistry (Fig. 3C) and electron microscopy for autophagosomes (Fig. 3D). Similarly, experimental sepsis-induced PGC-1α staining in wild-type but not in tlr4−/− mice (Fig. 3C).
Figure 3.

LPS or experimental sepsis-induced autophagy or biogenesis depends on TLR4 expression. A, B) Autophagic signaling, which was increased in sepsis or following LPS, was decreased in mice deficient in the pattern recognition receptor TLR4. This was demonstrated in vitro by maintained levels of p62, as well as a lack of LC3 induction following LPS treatment in tlr4−/− hepatocytes (8 h) as determined by Western blotting (A). In addition, LC3 puncta quantification by immunohistochemistry demonstrated an increase from a mean of 2.3 ± 1.6 vs. 13.4 ± 14.5 in wild-type hepatocytes (B). No significant induction was seen in tlr4−/− hepatocytes (3.2±1.4 vs. 4.1±2.0). *P < 0.05. C, D) Furthermore, TLR4 deficiency prevented CLP-induced LC3 protein increase in vivo (C) or autophagosome induction (D) as determined by electron microscopy (2±1.6 vs. 8.1±2.6, control vs. CLP in wild-type, P<0.05; 2.4±2.1 vs. 3.4±2.4, control vs. CLP in tlr4−/− mice). In addition, TLR deficiency prevented CLP-induced increases in markers of biogenesis, as demonstrated by PGC-1α levels determined by immunohistochemistry (C). Scale bar = 20 μm. *P < 0.05.
The potential influence of autophagy on biogenesis was next investigated. Autophagy was inhibited in vitro and in vivo with siRNA specific to the autophagy-inducing protein VPS34, which is a class III PI3-kinase (22). Studies have previously demonstrated that knockdown of this protein effectively inhibits sepsis-induced autophagy and increases cell death (5). VPS34 siRNA, but not control siRNA, inhibited LPS or CLP-induced PGC-1α, NRF1, and Tfam levels (Fig. 4A, B). The dependence of LPS-induced biogenesis on autophagic signaling was also suggested by decreased levels of proteins of oxidative phosphorylation (Fig. 4C), as well as decreased mitochondrial DNA content (Fig. 4D).
Figure 4.

Sepsis-induced biogenesis is dependent on autophagic signaling. A) LPS-induced mitochondrial biogenesis-associated transcription factors were decreased following VPS34 siRNA treatment. *P < 0.05 vs. control + scramble siRNA; #P < 0.05 vs. LPS + scramble siRNA; all at 8-h time point following LPS. B) CLP- or LPS (8 h)-induced PGC-1α levels (red staining) were diminished following VPS34 siRNA treatment. Scale bar=20 μm. C) VPS34 siRNA treatment prevented LPS-induced increases in protein levels of components of mitochondrial oxidative phosphorylation. D) Dependence of LPS-induced mitochondrial biogenesis on autophagic signaling was also suggested by a lack of induction of mitochondrial DNA following VPS34 siRNA treatment (24 h). *P < 0.05 vs. control siRNA; #P < 0.05 vs. control VPS34 siRNA.
Autophagy regulates mitochondrial biogenesis via TLR9 signaling
The mechanisms connecting autophagic signaling to the regulation of biogenesis were next investigated. Previous studies have demonstrated that autophagosomes colocalize with the pattern recognition receptor TLR9 (13, 23). A potential ligand for this receptor includes mitochondrial DNA. Thus, it is possible that mitochondrial DNA processed via autophagy or mitophagy activates TLR9 signaling to influence cellular processes, such as mitochondrial biogenesis. To investigate this, TLR9 protein levels were investigated following LPS signaling with and without knockdown of VPS34. LPS treatment induced expression of TLR9 protein, and this was diminished by VPS34 siRNA (Fig. 5A). Conversely, TLR9 siRNA treatment did not prevent LPS-induced increases in VPS34 levels and autophagic signaling (data not shown). However, TLR9 siRNA treatment prevented LPS-induced increases in mitochondrial biogenesis-associated transcription factors, as demonstrated by rtPCR, Western blotting, and immunocytochemistry (Fig. 5B–D). In addition, the addition of isolated hepatic mitochondrial DNA and liposomes led to increased biogenesis, and this effect was inhibited by TLR9 knockdown or receptor mutation (Supplemental Fig. S3). In vivo studies in wild-type mice or mice with mutant TLR9 receptors (Tlr9CpG1/CpG1 mice) demonstrated that cecal ligation and puncture induces autophagic signaling in both strains of mice but fails to induce biogenesis in the TLR9 mutant mice (Fig. 5E). Moreover, hepatic injury was exacerbated in these mice at 24 h, as demonstrated by serum ALT levels (147±31 U/L in wild-type vs. 301±61 U/L in tlr9CpG1/CpG1; P<0.05). Together, these data suggest that LPS and TLR9 signaling are necessary for these molecules to induce mitochondrial biogenesis.
Figure 5.

LPS-induced hepatic biogenesis is dependent on TLR9 signaling. A) LPS treatment induced TLR9 expression in hepatocytes treated with control siRNA but not with VPS34 siRNA (4 h). B–D) TLR9-specific siRNA prevented LPS induced increases in the biogenesis related transcription factors PGC-1α, NRF-1, and Tfam, as determined by immunocytochemistry (B), rtPCR (C), and Western blotting (D), all at 8 h following LPS. *P < 0.05 vs. control + scramble siRNA; #P < 0.05 vs. LPS + scramble siRNA. E) CLP increased PGC-1α protein levels in wild-type mice but failed to induce this protein in the TLR9 mutant (tlr9CpG1/CpG1) mice. Scale bars = 20 μm.
DISCUSSION
In summary, these data demonstrate that sepsis results in decreased hepatic oxygen consumption and oxidative phosphorylation. This is associated with decreased mitochondrial membrane potential and changes in mitochondrial morphology. In vitro, these data illustrate a dynamic quantitative change in mitochondrial density over time. In addition, there is both increased autophagy and mitophagy, as well as mitochondrial biogenesis. Sepsis- or LPS-induced mitochondrial biogenesis is dependent on a TLR4-autophagy-TLR9 signaling pathway (Fig. 6).
Figure 6.
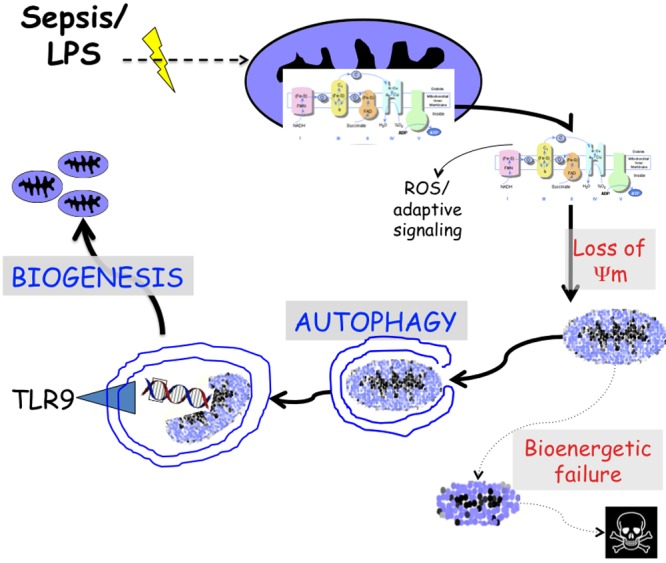
Schema and hypothesis for cycle of mitochondrial homeostasis in sepsis. Mitochondrial oxidative phosphorylation is inhibited in sepsis. As a consequence, mitochondrial signaling, including generation of reactive oxygen species (ROS), initiates adaptive responses. However, this also leads to loss of membrane potential and if left unmitigated results in bioenergetic failure and cell death. Autophagy and mitophagy act as an adaptive signaling pathway to remove and process damaged mitochondria. This includes the processing of mitochondrial DNA to the pattern recognition receptor TLR9, which is necessary to induce mitochondrial biogenesis and restore a healthy mitochondrial population.
Several previous studies have demonstrated that sepsis results in a decrease in oxidative phosphorylation (1, 2). These data are supported by findings in this article. It has been suggested that such a decrease, which can result in a loss of normal cell and organ function, acts to prevent cell death and irreversible structural damage and permanent organ dysfunction. However, inhibition of the mitochondrial electron transport chain has many potential consequences. One such consequence includes the generation of reactive oxygen species, primarily from complexes I and III (24, 25). These reactive oxygen species may serve as second messengers to induce critical cell signaling to regulate adaptive cellular responses. To the contrary, these mitochondria-generated reactive oxygen species may also lead to deleterious oxidative injury in the mitochondria and beyond. Furthermore, inhibition of the electron transport chain can lead to loss of membrane potential. These damaged mitochondria can themselves potentiate cellular injury, leading to decreased ATP production and bioenergetic failure and release of proapoptotic proteins, such as cytochrome c.
We and others have demonstrated that autophagy and mitophagy are increased in the liver in sepsis (5, 6). These findings are confirmed in these current investigations. Autophagy may be one of the ways that the cell removes dysfunctional mitochondria to prevent cell injury (4). However, with the removal of damaged mitochondria, ongoing processes to replace the mitochondrial population would be necessary to prevent death and allow for recovery of cell function (10). This may include mitochondrial fission and fusion, as well as the generation of new mitochondrial populations via mitochondrial biogenesis. Interestingly, increased mitochondrial biogenesis has been associated with survival in sepsis (10).
This article shows that the induction of autophagy and biogenesis is dependent on TLR4 signaling. This is not surprising, as this pattern recognition receptor is well known to be activated by many ligands, including LPS (26). A novel finding of this article is the dependence of LPS-induced biogenesis on autophagic signaling. With inhibition of the proximal autophagy inducing class III PI3-kinase, VPS34, both autophagy and biogenesis are inhibited. This proposes that the process of mitochondrial regeneration is, in part, regulated by one of the processes regulating removal of damaged mitochondria, setting up for a dynamic feedback system to maintain a viable mitochondrial population.
Autophagosomes are double-membraned vesicles that form and fuse with lysosomes to process the contents of the autophagosome. In the process of mitophagy, dysfunctional or damaged mitochondria are tagged by a PINK1/parkin-dependent process to initiate the formation of an autophagosome (27, 28). Interestingly, similar machinery and processes can be utilized to target intracellular bacteria as an innate immune response to result in destruction of these bacteria (29–31). The similarity of bacteria and mitochondria, both of which contain DNA with unmethylated CpG motifs, suggests a possible common cell-signaling pathway. TLR9 is a pattern recognition receptor that has these CpG motifs as its ligand. TLR9 has been shown in B cells to mobilize to autophagolysosomes (13). Thus, this pathway was investigated as a possible link between the autophagic processing of mitochondria and the induction of biogenesis. This hypothesis is supported by the finding that TLR9 knockdown or receptor mutation results in inhibition of sepsis or LPS-induced biogenesis.
Release of mtDNA has been previously suggested to potentiate injury via TLR9 signaling. Hauser et al. (32) suggested that mtDNA acts as a circulating danger associated molecular pattern molecule to activate TLR9 and exacerbate injury. Imaeda et al. (33) demonstrated in a model of acetaminophen liver toxicity that released DNA from damaged cells activated TLR9 and inflammasome signaling. These studies suggest that TLR9 signaling would exacerbate injury, as opposed to our data that suggest that this pathway is protective. A difference may be the source of the TLR9 ligand and how and where the receptor/ligand interaction takes place. An additional study has demonstrated in experimental sepsis, the mice deficient in TLR9 demonstrated decreased mortality in sepsis (34). This current study utilizes a different strain of mice with mutation of the TLR9 receptor, which may account for differences. Furthermore, a lack of TLR9 may exacerbate outcome in experimental models secondary to the immunological consequences and altered pathogen defense mechanisms associated with deletion of this receptor.
The autophagic processing of mitochondrial DNA has also been investigated. Oka et al. (35) illustrated that mtDNA that escapes autophagy accounted for inflammation in the setting of cardiomyopathy. Nakahira et al. (36) showed in macrophages that LPS and ATP treatment stimulated autophagy and, in the absence of autophagy, suggested that dysfunctional mitochondria release their DNA to activate the NALP3 inflammasome. These results are similar in that autophagy of damaged or dysfunctional mitochondria served as an adaptive response to prevent cellular inflammation. The data presented in the current article builds on this and suggest that controlled autophagic processing of mtDNA to TLR9 is a protective stimulus to promote mitochondrial biogenesis and allow for recovery.
In summary, sepsis or LPS-induced autophagy acts to protect against cell damage and injury. This process may involve the dynamic process of removal of dysfunctional mitochondria and restitution of a healthy mitochondrial population via biogenesis. TLR9, at least in part, is necessary for mitochondrial biogenesis and may be activated when mtDNA is presented as a ligand for this receptor via autophagic signaling.
Supplementary Material
Acknowledgments
This work is supported by U.S. National Institutes of Health grants R01 GM082830 (B.S.Z.) and GM082852 (M.R.R.), U.S. Veterans Affairs Merit Award 1I01BX000566 (B.S.Z.), and U.S. Department of Defense grant DM102439 (B.S.Z.).
Author contributions: E.H.C. and B.S.Z., study concept and design; E.H.C., S.P.W., and P.L., acquisition of data; E.H.C., M.R.R., S.Sh., K.P.M., and B.S.Z., analysis and interpretation of data; E.H.C., S.P.M., and B.S.Z., drafting of the manuscript; E.H.C. and B.S.Z., critical revision of the manuscript for important intellectual content and statistical analysis; B.S.Z., acquisition of funding; S.St., technical support. The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ATP
- adenosine triphosphate
- CLP
- cecal ligation and perforation
- CpG
- cytosine-phosphodiester-guanine
- LPS
- lipopolysaccharide
- NRF1
- nuclear respiratory factor 1
- PGC-1α
- peroxisome proliferator-activated receptor-γ coactivator-1α
- TEM
- transmission electron microscopy
- TFAM
- transcription factor A mitochondrial
- TLR
- Toll-like receptor
- TMRM
- tetramethylrhodamine methyl ester
- VPS34
- vacuolar protein sorting 34
REFERENCES
- 1. Brealey D., Karyampudi S., Jacques T. S., Novelli M., Stidwill R., Taylor V., Smolenski R. T., Singer M. (2004) Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R491–R497 [DOI] [PubMed] [Google Scholar]
- 2. Singer M., De Santis V., Vitale D., Jeffcoate W. (2004) Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet 364, 545–548 [DOI] [PubMed] [Google Scholar]
- 3. Zuckerbraun B. S., Chin B. Y., Bilban M., d'Avila J. C., Rao J., Billiar T. R., Otterbein L. E. (2007) Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 21, 1099–1106 [DOI] [PubMed] [Google Scholar]
- 4. Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carchman E. H., Rao J., Loughran P. A., Rosengart M. R., Zuckerbraun B. S. (2011) Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology 53, 2053–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Watanabe E., Muenzer J. T., Hawkins W. G., Davis C. G., Dixon D. J., McDunn J. E., Brackett D. J., Lerner M. R., Swanson P. E., Hotchkiss R. S. (2009) Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab. Invest. 89, 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piantadosi C. A., Withers C. M., Bartz R. R., Macgarvey N. C., Fu P., Sweeney T. E., Welty-Wolf K. E., Suliman H. B. (2011) Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J. Biol. Chem. 286, 16374–16385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reynolds C. M., Suliman H. B., Hollingsworth J. W., Welty-Wolf K. E., Carraway M. S., Piantadosi C. A. (2009) Nitric oxide synthase-2 induction optimizes cardiac mitochondrial biogenesis after endotoxemia. Free Radic. Biol. Med. 46, 564–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lancel S., Hassoun S. M., Favory R., Decoster B., Motterlini R., Neviere R. (2009) Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 329, 641–648 [DOI] [PubMed] [Google Scholar]
- 10. Carre J. E., Orban J. C., Re L., Felsmann K., Iffert W., Bauer M., Suliman H. B., Piantadosi C. A., Mayhew T. M., Breen P., Stotz M., Singer M. (2010) Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am. J. Respir. Crit. Care Med. 182, 745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rubartelli A., Lotze M. T. (2007) Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 28, 429–436 [DOI] [PubMed] [Google Scholar]
- 12. Barber G. N. (2011) Cytoplasmic DNA innate immune pathways. Immunol. Rev. 243, 99–108 [DOI] [PubMed] [Google Scholar]
- 13. Chaturvedi A., Dorward D., Pierce S. K. (2008) The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 28, 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collins L. V., Hajizadeh S., Holme E., Jonsson I. M., Tarkowski A. (2004) Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 75, 995–1000 [DOI] [PubMed] [Google Scholar]
- 15. Kim Y. M., Talanian R. V., Billiar T. R. (1997) Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. 272, 31138–31148 [DOI] [PubMed] [Google Scholar]
- 16. West M. A., Keller G. A., Cerra F. B., Simmons R. L. (1985) Killed Escherichia coli stimulates macrophage-mediated alterations in hepatocellular function during in vitro coculture: a mechanism of altered liver function in sepsis. Infect. Immun. 49, 563–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gramignoli R., Green M. L., Tahan V., Dorko K., Skvorak K. J., Marongiu F., Zao W., Venkataramanan R., Ellis E. C., Geller D., Breite A. G., Dwulet F. E., McCarthy R. C., Strom S. C. (2012) Development and application of purified tissue dissociation enzyme mixtures for human hepatocyte isolation. Cell Transplant. 21, 1245–1260 [DOI] [PubMed] [Google Scholar]
- 18. Stolz D. B., Zamora R., Vodovotz Y., Loughran P. A., Billiar T. R., Kim Y. M., Simmons R. L., Watkins S. C. (2002) Peroxisomal localization of inducible nitric oxide synthase in hepatocytes. Hepatology 36, 81–93 [DOI] [PubMed] [Google Scholar]
- 19. Creery D., Fraser D. D. (2002) Tissue dysoxia in sepsis: getting to know the mitochondrion. Crit. Care Med. 30, 483–484 [DOI] [PubMed] [Google Scholar]
- 20. Crouser E. D., Julian M. W., Blaho D. V., Pfeiffer D. R. (2002) Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit. Care Med. 30, 276–284 [DOI] [PubMed] [Google Scholar]
- 21. Waltz P., Carchman E. H., Young A. C., Rao J., Rosengart M. R., Kaczorowski D., Zuckerbraun B. S. (2011) Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 7, 315–320 [DOI] [PubMed] [Google Scholar]
- 22. Funderburk S. F., Wang Q. J., Yue Z. (2010) The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bertin S., Samson M., Pons C., Guigonis J. M., Gavelli A., Baque P., Brossette N., Pagnotta S., Ricci J. E., Pierrefite-Carle V. (2008) Comparative proteomics study reveals that bacterial CpG motifs induce tumor cell autophagy in vitro and in vivo. Mol. Cell. Proteomics 7, 2311–2322 [DOI] [PubMed] [Google Scholar]
- 24. Piantadosi C. A. (2012) Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 1820, 712–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Erusalimsky J. D., Moncada S. (2007) Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler. Thromb. Vasc. Biol. 27, 2524–2531 [DOI] [PubMed] [Google Scholar]
- 26. Beutler B., Hoebe K., Du X., Ulevitch R. J. (2003) How we detect microbes and respond to them: the Toll-like receptors and their transducers. J. Leukoc. Biol. 74, 479–485 [DOI] [PubMed] [Google Scholar]
- 27. Wang Y., Nartiss Y., Steipe B., McQuibban G. A., Kim P. K. (2012) ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 8, 1462–1476 [DOI] [PubMed] [Google Scholar]
- 28. McBride H. M. (2008) Parkin mitochondria in the autophagosome. J. Cell Biol. 183, 757–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delgado M., Singh S., De Haro S., Master S., Ponpuak M., Dinkins C., Ornatowski W., Vergne I., Deretic V. (2009) Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 227, 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Levine B., Deretic V. (2007) Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7, 767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deretic V. (2005) Autophagy in innate and adaptive immunity. Trends Immunol. 26, 523–528 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C. J. (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Imaeda A. B., Watanabe A., Sohail M. A., Mahmood S., Mohamadnejad M., Sutterwala F. S., Flavell R. A., Mehal W. Z. (2009) Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Invest. 119, 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Plitas G., Burt B. M., Nguyen H. M., Bamboat Z. M., DeMatteo R. P. (2008) Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J. Exp. Med. 205, 1277–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oka T., Hikoso S., Yamaguchi O., Taneike M., Takeda T., Tamai T., Oyabu J., Murakawa T., Nakayama H., Nishida K., Akira S., Yamamoto A., Komuro I., Otsu K. (2012) Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.