

Abstract
The mechanical overloading of cartilage is involved in the pathophysiology of osteoarthritis (OA) by both biochemical and mechanical pathways. The application of fluid shear stress to chondrocytes recapitulates the earmarks of OA, as evidenced by the release of proinflammatory cytokines (PICs), matrix metalloproteinases (MMPs), and apoptotic factors. Dysregulations or mutations in these genes might directly cause OA in addition to determining the stage at which OA becomes apparent, the joint sites involved, and the severity of the disease and how rapidly it progresses. However, the underlying mechanisms remain unknown. In this review, we propose that the dysregulation of cyclooxygenase-2 (COX-2) is associated with fluid shear stress-induced OA via its metabolic products at different stages of the disease. Indeed, high fluid shear stress rapidly induces the production of PICs and MMPs via COX-2-derived prostaglandin (PG)E2 at the early stage of OA. In contrast, prolonged shear exposure (>12 h) aggravates the condition by concurrently up-regulating the expression of proapoptotic genes and down-regulating the expression of antiapoptotic genes in a 15-deoxy-Δ (12,14)-prostaglandin J2 (15d-PGJ2)-dependent manner at the late stage of disease. These observations may help to resolve long-standing questions in OA progression and provide insight for development of strategies to treat and combat OA.—Wang, P., Guan, P.-P., Guo, C., Zhu, F., Konstantopoulos, K., Wang, Z.-W. Fluid shear stress-induced osteoarthritis: roles of cyclooxygenase-2 and its metabolic products in inducing the expression of proinflammatory cytokines and matrix metalloproteinases.
Keywords: PGE2, 15d-PGJ2, inflammation, apoptosis, chondrocytes
Osteoarthritis (OA) is considered a musculoskeletal disorder that is characterized by the progressive degradation of cartilage, and the mechanisms leading to the progression of cartilage degradation and failure have been extensively investigated. The excessive chronic or repetitive mechanical loading of articular cartilage producing hydrostatic stress, tensile strain, and fluid flow (1) leads to the irreversible calcification of cartilage matrix and OA disease by inducing the expression of proinflammatory cytokines (PICs), matrix metalloproteinases (MMPs), and apoptotic factors in chondrocytes (2). Accumulating in vitro results support the notion that low fluid shear (<10 dyn/cm2) is chondroprotective (3), whereas high shear stress (>10 dyn/cm2) elicits the release of PICs (4), MMPs (3) and apoptotic genes (5). Although the detailed mechanisms remain unclear, cyclooxygenase 2 (COX-2) and its metabolic products [i.e., prostaglandins (PGs)] have been hypothesized to be responsible for mediating the imbalance between anabolic and catabolic metabolism in OA cartilage.
Previous work suggests that COX-2 expression is highly induced in OA-affected cartilage (6). In addition, the prolonged application of high fluid shear to human chondrocytes in vitro recapitulates the gene expression profiles, such as COX-2, associated with OA in vivo (7). Although OA is classified as a noninflammatory joint disease, PGs are believed to play a pivotal role in the pathogenesis and progression of the disease. PGE2 and PGD2 are the major PGs synthesized by chondrocytes, and the secretion of PGE2 and PGD2 [and its dehydration end product 15-deoxy-Δ-(12,14)-prostaglandin J2 (15d-PGJ2)] is markedly higher in OA than in healthy cartilage (8). PGE2 has been implicated in the pain signaling (9), cartilage erosion (10), and inflammation associated with OA and adjuvant-induced arthritis (6, 11). In contrast, the role of PGD2 and 15d-PGJ2 in the metabolism of articular cartilage remains a matter of debate. Although there are investigations suggesting that PGD2 and 15d-PGJ2 have chondroprotective effects on counteracting the interleukin (IL)-1β-mediated induction of MMPs (12), others studies suggest that these molecules have the ability to induce apoptosis in chondrocytes (5, 12). In agreement with these early observations, recent data have demonstrated that PG production in chondrocytes varies with both the zone (depth) of the chondrocyte in the cartilage and with the grade of OA. Of note, PGE2 is detected at the early stage of OA and a short duration of shear stress exposure, whereas 15d-PGJ2 is found at the late stage of OA and accumulates after prolonged application of high fluid shear stress to human chondrocytes (13).
Although PGs have been reported to be responsible for OA progression, the underlying mechanism of this process has yet to be elucidated. Because OA is often a consequence of excessive mechanical forces (1, 2), and given the important roles of PICs (4), MMPs (3), and apoptotic genes (5) in OA progression, we herein summarize the potential mechanisms contributing to the high fluid shear-induced pathogenesis and progression of OA via a COX-2- and PG-dependent process in human chondrocytes. Specifically, we attempt to fill the gaps in our understanding of the pathogenesis of OA by examining the available data with regard the effect of high fluid shear stress on the expression of COX-2 and PGs; the roles of COX-2 and PGs in the expression of PICs, MMPs, and apoptotic genes; and the influence of PICs, MMPs and apoptotic genes on the pathogenesis of the disease. The implications of these findings for the treatment of OA are also discussed.
INVOLVEMENT OF PICS AND MMPS IN THE PATHOGENESIS AND PROGRESSION OF OA
Inflammation is now believed to be involved in the pathogenesis and progression of OA at the early stage of the disease (14). Secreted PICs and MMPs play critical roles in disturbing metabolism, which in turn enhances catabolism in OA joint tissue, and IL-1β (15) and IL-6 (16) appear to be the main mediators involved in the pathophysiology of OA. In addition to IL-1β and IL-6, other proinflammatory cytokines, including IL-8 (17), IL-10 (18, 19), chemokine (C-C motif) ligand 3 (CCL3; ref. 17), interferon (INF)-α4 (19), IFN-γ (19), leukemia inhibitory factor (LIF; ref. 17), and chemokines (19), have also been implicated as being involved in the disease (Table 1). In addition, most MMPs, such as MMP-1 (17), MMP-2 (19), MMP-3 (19), MMP-7 (20), and MMP-13 (17, 19), are markedly induced in OA cartilage, thus mediating matrix degradation in OA cartilage (Table 1).
Table 1.
OA or shear stress induces the expression of PICs and MMPs
| Symbol | Trend |
Experimental model |
||
|---|---|---|---|---|
| OA | SS | OA | SS | |
| Cytokines | ||||
| IL-1β | ↑ | ↑ | Human OA cartilage | Human chondrocytes, THP1, and rabbit vein grafts |
| IL-6 | ↑ | ↑ | Postmenopausal women OA cartilage | Human chondrocytes and THP1 |
| IL-8 | ↑ | ↑ | Human OA cartilage | THP1 and HUVECs |
| IL-10 | ↓ | ↑ | Subchondral bone of OA rat and human OA cartilage | Rabbit vein grafts |
| IL-1 | ↑ | Human early knee OA patients | ||
| IL-18 | ↓ | Subchondral bone of OA rat | ||
| CXCL1 | ↑ | THP1 | ||
| CXCL2 | ↑ | Human OA cartilage | ||
| CCL1 | ↑ | THP1 | ||
| CCL3 | ↑ | Human OA cartilage | ||
| LIF | ↑ | Human OA cartilage | ||
| TNF-α | ↑ | Rat blood leukocytes | ||
| TNFAIP6 | ↑ | Human OA cartilage | ||
| Tnfsf9 | ↑ | Subchondral bone of OA rat | ||
| IFN-α4 | ↓ | Subchondral bone of OA rat | ||
| IFN-γ | ↓ | Subchondral bone of OA rat | ||
| MCP-1 | ↑ | THP1 and HUVECs | ||
| ICAM-1 | ↑ | HUVECs | ||
| VCAM-1 | ↓ | HUVECs | ||
| VEGF A | ↓ | Subchondral bone of OA rat | ||
| E-Selectin | ↓ | HUVECs | ||
| MMPs | OA | SS | OA | SS |
| MMP-1 | ↑ | ↓5 dyn ↑20 dyn | Human OA cartilage | Human chondrocytes and glioma U87 cells |
| Mmp-2 | ↑ | ↑ | Subchondral bone of OA rat | HUVECs, human glioma U87 cells, and saphenous vein organ |
| Mmp-3 | ↑ | OA rat and patients | ||
| MMP-7 | ↑ | Human OA cartilage | ||
| MMP-9 | ↓ | ↑ | Patients with knee OA | HUVECs and human saphenous vein organ |
| Mmp12 | ↑ | Subchondral bone of OA rat | ||
| MMP13 | ↑ | ↓5 dyn ↑20 dyn | Human OA cartilage and subchondral bone of OA rat | Human chondrocytes and MC3T3-E1 |
| Mmp14 | ↑ | Subchondral bone of OA rat | HUVECs | |
| Mmp16 | ↑ | Subchondral bone of OA rat | ||
| Mmp23 | ↑ | Subchondral bone of OA rat | ||
| Mmp24 | ↑ | Subchondral bone of OA rat | ||
| TIMP-1 | ↑ | ↑ | Subchondral bone of OA rat | Human saphenous vein organ |
| TIMP-2 | ↑ | Human saphenous vein organ | ||
HUVEC, human umbilical vein endothelial cell; SS, shear stress.
PICs
PICs are commonly produced and released into the synovial fluid. Many of these PICs are tightly regulated and maintained at low levels for normal homeostasis, and their balance may be disturbed in OA cartilage. Among the PICs in OA, IL-1β, IL-6, LIF, IL-8, and IL-18 are considered to be the major players, whereas IL-10 and IFN-γ are classified as anti-inflammatory cytokines due to their ability to block catabolic cytokines (21). As shown in Table 1, the expression levels of PICs, including IL-1β (15), IL-6 (16), LIF (17), and IL-8 (17), are highly elevated, though such anti-inflammatory cytokines as IL-10 (18, 19) and IFN-γ (19) are clearly suppressed in human or rat OA cartilage. The imbalance between PICs and anti-inflammatory cytokines will eventually contribute to inflammation, resulting in the early onset of OA (Fig. 1). In contrast, IL-18 was not found to be up-regulated in rat OA cartilage (Table 1 and ref. 19), which indicates that IL-18 may not play an important role in the development and progression of OA.
Figure 1.

Imbalance of PICs and MMPs in OA. Levels of PICs, including IL-1β, IL-6, IL-8, TNF-α, CXCL2, and CCL3, are elevated in OA cartilage; in contrast, IFN-γ is down-regulated in OA. Moreover, the expression levels of MMPs, such as MMP-1, MMP-2, MMP-3, MMP-7, MMP-12, MMP-13, MMP-14, MMP-16, MMP-23, and MMP-24, are increased in OA cartilage, whereas MMP-9 is down-regulated in the serum of patients with OA. These actions cause the imbalance of PICs and MMPs, potentially contributing to the physiopathology of OA.
In addition to IL imbalance, accumulating evidence demonstrates that chemokine (C-X-C motif) ligand 2 (CXCL2; ref. 17), CCL3 (17), TNFAIP6 (17), and Tnfsf9 (19) also potentially contribute to the pathogenesis of OA. Among these cytokines, TNF-α isoforms, such as, tumor necrosis factor-α-induced 6 (TNFAIP6; ref. 17) and tumor necrosis factor (ligand) superfamily, member 9 (Tnfsf9; ref. 19), appear to play more important roles in OA disease than CXCL2 (17) and CCL3 (17). In patients with OA, tumor necrosis factor-α (TNF-α) is elevated in synovial fluid, synovial membrane, subchondral bone, and cartilage and can act independently or in concert with other cytokines to initiate and propagate inflammation. For instance, the simultaneous injection of IL-1β and TNF-α into the rabbit knee joint causes more cartilage destruction than cytokine alone (22). As the most popular investigated mediators of OA, IL-1β and TNF-α exert their effects on OA progression by down-regulating the synthesis of major extracellular matrix (ECM) components through inhibition of anabolic activities in chondrocytes (23). For instance, treatment with IL-1β not only reduces type II collagen expression but also down-regulates the expression of aggrecan in chondrocytes, which, in turn, disturb the structural component of cartilage by decreasing the major component of ECM (24). Similar results were also obtained in IL-1β-activated chondrocytes (23), worsening the physiopathology of OA. In addition, the mechanisms of IL-1β and TNF-α in the pathogenesis of OA are likely rather complicated, as a number of studies have demonstrated that IL-1β and TNF-α also stimulate the production of other PICs involved in the pathology of the disease; for example, IL-1β and TNF-α mediate OA progression by inducing the production of IL-6 (25), IL-8 (26), and macrophage chemoattractant protein 1 (MCP-1; ref. 27). Similarly, IL-1β and TNF-α treatment also induce the production of nitric oxide (NO) and reactive oxygen species (ROS), accelerating the effects of IL-1β and TNF-α in cartilage impairment.
MMPs
The proinflammatory and catabolic effects of IL-1β and TNF-α are not only mediated through the activation of other cytokines but also potentiate cartilage degradation by induction of MMPs. Several studies have suggested that IL-1β and TNF-α activate MMPs, including MMP-1, MMP-3, and MMP-13, which are key regulators of cartilage destruction (28–30). In accordance with these investigations, most MMPs, including MMP-1 (17), MMP-2 (19), MMP-3 (19), MMP-7 (20), and MMP-13 (17, 19), are markedly induced in OA cartilage (Table 1). Although the detailed mechanisms are not thoroughly known, these results have clearly indicated that MMPs play important roles in mediating the effects of PICs on ECM degradation in OA cartilage. To reveal the underlying mechanisms, the fluid shear stress technique has been applied to mimic mechanical loading in joint cartilage (Fig. 2).
Figure 2.
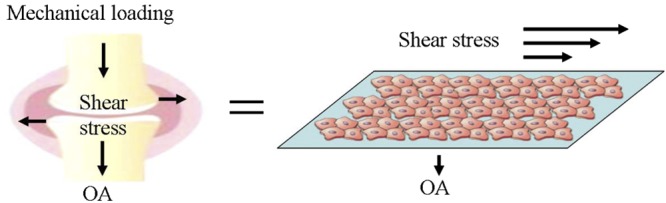
Similarities of fluid shear stress in vivo and in vitro. Fluid shear stress exists in normal joint cartilage and is induced by daily activities. A fluid shear stress experimental model was established in vitro to mimic in vivo fluid shear stress and the in vitro experimental model produces similar fluid shear stress as in vivo.
PICs and MMPs play pivotal roles in mediating shear stress-induced OA progression
In view of the effects of shear stress with regard to inducing the expressions of PICs and MMPs, it is necessary to elucidate their important roles of PICs and MMPs in the development of OA. As a common chronic degenerative disease characterized by the loss of articular cartilage components due to an imbalance between ECM destruction and repair (31), the main clinical sign of OA is joint pain (32). Although the exact mechanism of knee pain is unclear, it is believed to be related to local inflammation of the knee joints, which involves the production of PICs (33). The major PICs, which are generally also catabolic, include IL-1α, IL-1β, TNF-α, LIF, IL-8, IL-17, and IL-18, whereas IL-4, IL-10, IL-11, IL-13, IL-1 receptor antagonist (IL-1ra), and interferon (IFN)-γ are classified as anti-inflammatory cytokines. Moreover, some factors, such as IL-6 and transforming growth factor β1 (TGF-β), may have dual roles.
Among these cytokines, TNF-α and IL-1β are believed to play pivotal roles in the development and progression of OA, as based on considerable evidence from studies in both culture and animal models over the past 2 decades. In cell culture studies, treatment with IL-1β was found to reduce type II collagen expression in chondrocytes isolated from several different species (24). The expression of aggrecan, a major structural component of cartilage, was also reported to be down-regulated in IL-1β-treated chondrocytes (34). Similarly, TNF-α has been shown to suppress the synthesis of proteoglycan, link protein, and type II collagen in chondrocytes (23). In animal models of OA, intra-articular injection of recombinant preparations of IL-1β could be shown to stimulate the destruction of articular cartilage (21). In addition, coinjection of TNF-α with IL-1β elicited more severe damage to articular cartilage than the injection of either cytokine alone (21).
Apart from TNF-α and IL-1β, such PICs as IL-6, LIF, and IL-8 may not by themselves stimulate cartilage destruction but may modulate the effects of the cytokines that are directly catabolic. For instance, IL-1β and TNF-α stimulate the synthesis of IL-6, LIF, and IL-8 in chondrocyte cultures. The involvement of LIF in OA pathogenesis is based on its capacity to stimulate cartilage proteoglycan resorption, MMP synthesis, and NO production. IL-8 potentiates the capacity of TNF-α in PGE2 production by OA synovial fibroblasts (35). Moreover, the presence of IL-8 in the synovial fluid of patients with OA enhances the release of IL-1β, IL-6, and TNF-α (36), potentially aggravating the pathogenesis of OA. However, neither IL-6 nor IL-8 alone is capable of stimulating cartilage degradation directly (37). Similarly, LIF is not able to induce OA directly but has been shown to enhance IL-1β and IL-8 expression in chondrocytes and IL-1β and TNF-α in synovial fibroblasts (38). Conversely, IL-1β and TNF-α have the ability to up-regulate the expression of LIF (39).
The feedback and crosstalk among these cytokines will undoubtedly lead to cartilage destruction and aggravate the course of OA. However, PICs do not directly degrade cartilage collagens and proteoglycans, and the MMP activation cascade is given the greatest attention in PIC-mediated cartilage degradation. As critical factors in OA development, IL-1β alone, or together with TNF-α, stimulates the synthesis of MMP-1, MMP-3, and MMP-13, which have been localized in the regions of cartilage degradation and function as the key regulators of cartilage degradation in OA patients (40). Although the overexpression of MMP-1 is not involved in the cleavage of resident molecules, it is implicated in the degradation of cartilage collagens (41). A recent study showed that a selective collagenase inhibitor against MMP-3 blocked the enhanced cleavage of type II collagen in OA cartilage (41). In addition, MMP-3 also has the capacity to degrade aggrecan; another important collagenase, MMP-13 similarly degrades type II collagen, potentially contributing to OA (41). In addition to these collagenases, other proteinases produced by chondrocytes, such as MMP-2 and MMP-9, may have roles in the further degradation of various matrix components (42). However, the activities of MMPs in stimulating OA are manifested in many ways. According to the above discussion, MMPs have the ability to degrade cartilage, yet the degradation products of cartilage matrix, such as fibronectin fragments, have been shown to stimulate the synthesis of catabolic cytokines by chondrocytes, which may initiate or amplify cartilage destruction in OA (43). Therefore, PICs play key roles in mediating shear stress-induced OA progression by activating MMPs.
FLUID SHEAR STRESS REVEALS THE MECHANISMS BY WHICH OA INDUCES PICS AND MMPS
Fluid shear stress is applied to human chondrocytes to elucidate the regulating mechanisms of PICs and MMPs in the pathogenesis of OA. Indeed, the prolonged application of high fluid shear to human chondrocytes in vitro recapitulates the gene expression profiles associated with OA in vivo (7). As shown in Table 1, the levels of IL-1β (44–46), IL-6 (10, 13, 44, 46, 47), IL-8 (45), IL-10 (44), and TNF-α (48) are significantly elevated in different shear-activated human cell lines (Table 1). More interestingly, these shear-induced PICs are similarly regulated in OA cartilage (Table 1). These data demonstrate that fluid shear stress is an appropriate in vitro model for OA investigation and emphasize the importance of shear stress in the pathogenesis of OA. Moreover, three additional genes, CXCL1 (45), CCL1 (45), and MCP-1 (45, 49), are up-regulated in shear-activated human THP1 monocytes and human umbilical vein endothelial cells (HUVECs; Table 1). Although we could find no similar reports for OA-activated cells, the same family of proteins, including CXCL2 (17) and CCL3 (17), has been found to be up-regulated in human OA cartilage. Therefore, shear stress has been broadly applied to human chondrocytes to reveal the mechanism by which OA induces PICs and MMPs.
Given the abilities of PICs to regulate the expression of MMPs in OA cartilage, it is possible to deduce that MMPs could be elevated by fluid shear stress. As shown in Table 1, the expression of MMP-1 (3), MMP-2 (49), MMP-9, MMP-13 (3), and MMP-14 (50) is induced in response to the exposure to high fluid shear stress. In contrast, relatively low fluid shear stress suppresses the activities of MMP-1 (3) and MMP-13 (3) in human chondrocytes. Although shear-activated MMP-1 and MMP-13 are not regulated in a shear stress-dependent manner, these observations are consistent with the physiopathology of OA, supporting the notion that low fluid shear is chondroprotective (3), whereas high shear stress in vivo impairs or degrades the matrix by eliciting the release of PICs (4) and MMPs in cartilage (3). Unlike MMP-9 suppression in OA-activated cartilage (51), MMP-9 is significantly induced in shear stress-activated HUVECs and human saphenous vein organ culture (3). As described below, our recent investigations in human chondrocytes reconcile these conflicting results (46). Moreover, tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2 are up-regulated by shear stress exposure in human saphenous vein organ culture, and in agreement with these observations, TIMP-1 is also up-regulated in human OA cartilage (19) (Fig. 3). Although TIMP-1 and TIMP-2 are up-regulated in OA- or shear stress-activated chondrocytes, these factors might not play a predominant role in OA as endogenous inhibitors of MMPs.
Figure 3.

Shear stress mediates the regulation of PICs and MMPs in different experimental models. Shear stress induces the expression of IL-1β, IL-6, IL-8, IL-10, TNF-α, CXCL1, CCL1, and MCP-1 in different cells. In addition, shear stress also increases the production of MMP-1, MMP-2, MMP-9, MMP-13, MMP-14, and TIMP-1 in different cell models. Induction of these PICs and MMPs mediates the shear stress-induced pathogenesis of OA.
It is known that PICs and MMPs are up-regulated in OA- or shear stress-activated cartilage; however, most of the signaling pathways that mediated their expression remain unclear. To bridge these gaps, we summarize and address the signaling pathways through which fluid shear stress regulates the synthesis of PICs and MMPs, as based on both our data and the recent data of others (10, 13, 46, 47, 52). The signaling pathways in which these genes are involved and the potential function of the specific proteins they encode could provide some insight into how these genes affect developmental processes and susceptibility to OA.
PICs
As discussed above, data have shown that the levels of IL-1β (44–46), IL-6 (10, 13, 45, 47, 52), IL-8 (45), IL-10 (44), and TNF-α (48) are significantly up-regulated in different shear-activated human cell lines (Table 1). In efforts to reveal the underlying mechanisms, studies have reported that the NF-κB signaling pathway is involved in fluid shear stress-induced IL-8 gene transcriptional activation in HUVECs (53). In addition, inflammatory molecules, such as Toll-like receptor 4 (TLR-4), continue to be reported as potential key molecules in mediating flow shear stress-induced IL-8 gene expression via the activation of NF-κB in HUVECs (54). In agreement with these prior works (53, 54), Cheng et al. (55) further found that several signaling molecules, including tyrosine kinase, G protein, calcium, phospholipase C, and cAMP-dependent protein kinase, play critical roles in low shear stress-induced IL-8 gene expression in human endothelial cells.
As fluid shear stress methods have proven to be useful for revealing the signaling pathways of IL-8 transcriptional regulation in human endothelial cells, high fluid shear stress has also been applied to human chondrocytes. The results have demonstrated that high fluid shear stress induces IL-1β via an EP2 and EP3-dependent pathway in human T/C-28a2 chondrocytes (46). In agreement with these data, Glossop and Cartmell (56) also suggested that fluid shear stress induces the expression of IL-1β in human mesenchymal stem cells, and the EP2 receptor has been reported to be involved in the regulation of IL-1β in human prostate cancer cells (57). Therefore, the above data demonstrate that high fluid shear stress has the ability to regulate IL-1β expression in human chondrocytes.
In addition to IL-1β, it was found that high fluid shear stress is able to regulate IL-6 expression in human chondrocytes. As a multifunctional PIC with a wide array of biological activities in different species, IL-6 has been implicated in pain signaling in OA cartilage (9). IL-6 expression is tightly regulated under physiological conditions; in response to an inflammatory insult, IL-6 levels increase in vivo and then decline to the baseline levels on resolution of the insult. Specifically, IL-6 was significantly induced in human OA cartilage (16) or shear stress-activated human chondrocytes (13, 52) in which superinduction is mediated by complicated signaling pathways. As shown in Fig. 4, IL-6 is up-regulated in a COX-2/PGE2-dependent manner at the early stage of OA (10, 13, 52). Indeed, the concurrent up-regulation of EP2 and down-regulation of EP3 play pivotal roles in mediating PGE2 signals to regulate the expression of IL-6 (13, 52). In agreement with these observations, Chen et al. (58) have recently reported that an EP2 agonist induces IL-6 production in RAW264.7 macrophages and EP4 is also suggested to be a PGE2 receptor to induce IL-6 expression in RAW264.7 macrophages (58). However, human chondrocytes express very low levels of EP4 and lack EP1 receptors, observations that indicate their minor roles in mediating PGE2 signals. More interestingly, PGE2 stimulates IL-6 expression via both EP receptors and TLR4 in human chondrocytes (47). The activation of TLR4 induces the phosphorylation of ERK1/2, PI3-K/Akt, and PKA/CREB, in turn regulating NF-κB-dependent IL-6 synthesis in a time-dependent fashion (13, 52). Conversely, caveolin-1 induction by 15d-PGJ2 accumulation at the late stage of OA exhibits antagonistic effects on IL-6 synthesis (13), which explains why IL-6 is temporally regulated by high fluid shear stress and provides insight into why an IL-6-mediated inflammatory mechanism only appears at the early stage of OA.
Figure 4.

Shear stress reveals the mechanisms by which OA induces PICs and MMPs in human chondrocytes. At the early stage of OA, shear stress mediates the development and progression of OA by inducing the expression of IL-6 and MMP-9 in human chondrocytes. Involvement of COX-2 and its derived PGE2 is implicated in the production of IL-6 and MMP-9, which is mediated by PI3-K-, ERK1/2-, PKA- and JNK-activated NF-κB pathways. In contrast, the apoptotic mechanism shows its effect on the physiopathology of OA at the late stage. Plk1 and Plk3 play pivotal roles in mediating chondrocytic apoptosis via a p53-dependent pathway.
In light of these data, the experimental model of high fluid shear stress mimics the shear stress actions that exist in mechanical loading cartilage and appears to be an excellent in vitro method to reveal the mechanisms by which OA induces PIC expression. In addition to IL-1β and IL-6, shear stress results in its inhibitory effects on TNF-induced JNK activation via MEK5-BMK1 in endothelial cells (59), and oscillatory fluid shear stress has ability to block TNFR1 signaling in osteoblasts (60). Although shear stress has revealed the signaling pathways of PICs in endothelial cells and osteoblasts, the underlying mechanisms for shear stress-induced PICs are still limited to IL-1β and IL-6 in human chondrocytes. In addition to ILs and TNF-α, other cytokines, including MCP-1, are up-regulated, whereas E-selectin is down-regulated in response to fluid shear stress (Table 1). Despite the fact that their secretion has been detected in different experimental models, to the best of our knowledge, the regulating mechanisms have not yet been elucidated. Thus, the discovery of further genetic regulation together with these technical advances will improve the understanding of OA pathogenesis.
MMPs
In the interest of a focused discussion and providing evidence for PICs being the key mediators in the pathogenesis of OA and potentiating cartilage degradation through MMP induction, it is important to elucidate the roles of fluid shear stress on MMP stimulation via PICs. For example, the catabolic effects of IL-1β are usually mediated by inducing the activities of MMP-1, MMP-9, and MMP-13 in OA (61), whereas the production of these MMPs is suppressed by IL-4, IL-10, and IL-13. Although different species of PICs show antagonistic effects on MMP induction, MMPs are always considered to function in the degradation of ECM components. In OA, particularly in disease in the early stage, inflammation of the synovial membrane is observed and may participate in the development of the disease, and destructive MMPs are primarily derived from cartilage. The levels of MMP-1, MMP-3, MMP-13, and MMP-28 in OA cartilage are elevated compared to nonarthritic cartilage and cartilage explants studies suggest that cytokines, such as IL-1β, IL-17, and TNF-α, mechanical compression, and mechanical injury are potential stimulatory factors. The mechanical compression of articular cartilage increases the expression and activation of MMP-2 and MMP-9, and even a single injurious mechanical compression increases the levels of MMP-3 and TIMP-1 (62). However, mechanical injury-induced high fluid shear stress up-regulates IL-1β (44–46), IL-6 (10, 13, 45, 47, 52), IL-8 (45), IL-10 (44), and TNF-α expression in human cell lines. Given the potential roles of PICs in the inductions of MMPs, it is reasonable to consider that shear stress has the ability to up-regulate MMP expression.
As shown in Table 1, at least 10 types of MMPs are elevated in OA joint tissues, with many of them undoubtedly being caused by fluid shear stress, which in turn results in cartilage and bone destruction during OA progression. Sweeney et al. (63) first reported that shear stress increased pro-MMP-2 activity in bovine aortic endothelial cells via the activation of Gi-protein. Furthermore, p38 was reported to play a role in fluid shear stress-induced MMP-2 up-regulation in human periodontal ligament cells (64). In addition, nuclear matrix protein 4/cas interacting zinc finger protein (Nmp4/CIZ) was found to be a transcription factor that mediates fluid shear stress-induced MMP-13 expression in osteoblasts. In addition to reports in other cell lines, more direct evidence was observed in human C28-I2 chondrocytes, suggesting that CBP/p300-interacting transactivator with ED-rich tail 2 (CITED2) mediates MMP-1 and MMP-13 regulation in response to fluid shear stress (3). It was further found that low fluid shear stress suppresses MMP-1 and MMP-13 expression, whereas high fluid shear stress-induced the activities of MMP-1 and MMP-13 in human C28-I2 chondrocytes (3). In addition, ERK1/2 was recently found to be involved in shear activation of MMP-1 in human periodontal ligament cells (64). In agreement with a prior study (3), increased expression of MMP-9 at the early stage of OA was found in high fluid shear-activated human T/C-28a2 chondrocytes (Fig. 4 and ref. 46). More importantly, the antagonistic effects of shear-induced IL-1β and 15d-PGJ2 were revealed to regulate MMP-9 expression in PI3-K-, ERK1/2- and JNK-dependent NF-κB activation in the same cell model (Fig. 4 and ref. 46). These observations clearly indicate that high fluid shear stress will induce cartilage injury in an MMP-dependent manner, thereby mediating ECM degradation. Furthermore, the inflammatory effects of PICs in MMP activation were observed at this stage of disease.
In light of the prior works suggesting the inflammatory roles of PICs in MMP induction, it is important to summarize the current mechanisms of PICs in MMP regulation. Kusano et al. (65) reported that IL-1 and IL-6 induced the expression of MMP-2, MMP-3, MMP-9, and MMP-13, an observation that was associated with bone matrix degradation in mouse calvarias. In accordance with this report, IL-1β was able to induce MMP-9 expression via ERK1/2, p38, JNK, and NF-κB signaling pathways in human tracheal smooth muscle cells (66). Similarly, Okada et al. (67) found that IL-1β increased MMP-9 expression via the ERK1/2-dependent angiotensin II type 1 receptor (AT1R) signaling pathway in adult rat cardiac fibroblasts. In addition to NF-κB, activator protein-1 (AP-1) also plays key roles in IL-1α-stimulated MMP-3 expression in human trabecular meshwork cells (68). More directly and importantly, IL-1β stimulates MMP-2 expression via a PGE2-dependent mechanism in human chondrocytes (69). A Wnt/β-catenin negative feedback loop inhibits IL-1-induced MMP-1, MMP-3, and MMP-13 expression in human chondrocytes (70). Collectively, it is clear that PICs mediate the effects of high fluid shear stress on MMP induction, which, in turn, contributes to the development of OA via ECM degradation.
HIGH FLUID SHEAR STRESS TEMPORALLY INDUCES COX-2 AND ITS METABOLIC PRODUCTS
COX-2 is one of the first genes to be induced in response to mechanical stimulation, and accumulating evidence has demonstrated the relationship between COX-2 and high fluid shear stress in different experimental models. Shear stress was first reported to induce COX-2 expression in endothelial cells. However, several discrete signaling pathways have been implicated as being involved in the genesis of COX-2 synthesis in response to a specific stimulus imposed on cells. For example, COX-2 is reported to be regulated by chemical agonists, such as okadaic acid, via activator protein (AP)-1 and cyclic AMP response element (CRE)-binding proteins in chondrocytic models (71). Accordingly, the COX-2 promoter has been analyzed over the past decade to thoroughly decipher the mechanism involved in shear stress-induced COX-2 expression in human chondrocytes. The promoter of the COX-2 gene contains a TATA box and several regulatory elements, including NF-κB sites, a CCAAT/enhancer-binding protein (C/EBP) motif, Sp1 sites, a CRE motif, AP-1, AP-2, and polyomavirus enhancer activator 3 (PEA-3) sites (72). Different regulatory elements have been demonstrated to regulate COX-2 expression in different cells, including NF-κB binding sites, the C/EBP motif, activating transcription factor/CRE sequences, and the PEA-3 site. Specifically, the C/EBP and AP-1 binding motifs are critically involved in shear stress-associated COX-2 induction in human chondrocytic cells in response to high fluid shear stress, whereas the NF-κB binding site does not mediate shear stress-induced COX-2 expression, even though NF-κB has critical roles in IL-1β-induced COX-2 expression (73). In more detail, c-Jun was found to be the pivotal molecule of the AP-1 complex for mediating COX-2 induction in human chondrocytes (74). However, the expression of COX-2 is not sustained in a highly activated level and will in fact be temporally induced in response to high fluid shear stress and then return to the basal level after a 2 h shear stress exposure (52).
In light of the roles of COX-2 metabolic products in the pathogenesis of OA, the mechanisms of PG regulation will be further addressed in this review. Among the COX-2 metabolic products, PGE2 and PGD2 are the major PGs synthesized by chondrocytes and have important functions in the development of OA. The secretion of PGE2 and PGD2 (and its dehydration end product 15d-PGJ2) is markedly higher in OA than in healthy cartilage. PGD synthase (PGDS) and microsomal PGES-1 (mPGES-1) are responsible for the biosynthesis of PGD2 and PGE2, respectively. Fluid shear stress increases PGE2 release in vascular smooth muscle cells (75), and PGE2 was also found to be induced in renal epithelial cells. In accordance with these prior works, the application of high fluid shear stress to human T/C-28a2 chondrocytes rapidly induced the secretion of PGE2 in an mPGES-1-dependent manner, the level of which then declined below the basal level after a prolonged application of high fluid shear stress (13). In contrast, L-PGDS mRNA and protein levels are below the static control levels after a short exposure of human chondrocytes to high fluid shear stimulation, reaching the maximal levels with a prolonged shear stress application (13). More interestingly, the sequential induction of PGE2 and PGD2 in shear-activated human chondrocytes is consistent with the report of Gilroy et al. (76), which suggest that COX-2-dependent metabolism shifts from PGE2 to PGD2 biosynthesis in the resolution phase of carrageen-induced inflammation in rats. Along these lines, the sequential induction of mPGES-1 and H-PGDS (but not L-PGDS) to mediate the resolution of endotoxin-induced inflammation in mouse heart was reported by Schuligoi et al. (77). In view of these reports, it is possible that PGE2 and PGD2 distinctly exert different roles in OA progression at different stages of OA. Below, we dissect the potential roles of PGE2 and PGD2 in the regulation of PICs, MMPs, and apoptotic genes, which contributes to the pathogenesis of OA.
INVOLVEMENT OF PGE2 IN INDUCING THE EXPRESSION OF PICS AND MMPS AT THE EARLY STAGE OF OA
As the key metabolic product of COX-2 (10, 46) and a molecule with broad biological functions, PGE2 is an important mediator in OA- or shear stress-induced PIC and MMP expression. As shown in Table 2, several studies have reported that PGE2 treatment stimulated IL-1β (78), IL-6 (78, 79), and IL-10 (80) expression in HUVECs, T lymphocytes, macrophages, and dendritic cells. Consistent with these observations, IL-1β, IL-6, and IL-10 are similarly regulated in OA cartilage (Table 1) and shear stress-activated cells (Table 1), implying that OA or fluid shear stress might stimulate IL-1β, IL-6, and IL-10 expression via a COX-2/PGE2 signaling cascade. In addition, IFN-γ was not up-regulated in PGE2-treated human T lymphocytes or PBL cells (ref. 79 and Table 2). As evidenced by the data from OA cartilage or shear stress-treated cells (Table 1), these observations concretely strengthen the fact that the COX-2/PGE2 signaling pathway plays pivotal roles in OA or shear stress induces the synthesis of PICs. However, other PICs, including IL-2 (79), IL-3 (79), IL-4 (79), and TNF-α (79), are suppressed in different PGE2-treated cells, which might indicate that the COX-2/PGE2 signaling pathway is not required in OA- or shear stress-induced PIC synthesis (Table 2).
Table 2.
Antagonistic effects of PGE2 and 15d-PGJ2 on both cytokines and MMPs
| Symbol | Trend |
Experimental model |
||
|---|---|---|---|---|
| PGE2 | 15d-PGJ2 | PGE2 | 15d-PGJ2 | |
| Cytokines | ||||
| IL-1β | ↑ | ↓ | HUVECs | RAW264.7, J774A.1, mouse microglia and astrocytes, and Balb/c mice |
| IL-2 | ↓ | Human T lymphocytes and PBL | ||
| IL-3 | ↓ | Human T lymphocytes | ||
| IL-4 | ↓ | Human T lymphocytes | ||
| IL-6 | ↑ | ↓ | HUVECs and human T lymphocytes | RAW264.7, J774A.1, mouse microglia and astrocytes, and Balb/c mice |
| IL-8 | ↑ | NCI-H 292 and human monocytes/macrophages | ||
| IL-10 | ↑ | Human macrophages and dendritic cells | ||
| IL-12(p70) | ↓ | Balb/c mice | ||
| IL-12(p40) | ↓ | C57BL/6 mice | ||
| TNF-α | ↓ | ↓ | Human T lymphocytes | RAW264.7, J774A.1, THP-1, mouse microglia/astrocytes, Balb/c mice, and C57BL/6 mice |
| IFN-γ | ↓ | Human T lymphocytes and PBL | ||
| TGF-β1 | ↓ | RAW264.7 and J774A.1 | ||
| NO | nM↑μm↓ | Rat MC (RMC) and C57BL/6 mice | ||
| MCP-1 | ↓ | RAW264.7, J774A.1, and mouse microglia/astrocytes | ||
| GM-CSF | ↑ | NCI-H 292 | ||
| MMPs | PGE2 | 15d-PGJ2 | PGE2 | 15d-PGJ2 |
| MMP-1 | ↓ | ↑, ↓ | Human articular chondrocytes and mouse osteoblasts | Human breast cancer cells, synovial fibroblasts, and HMEC-1 |
| MMP-2 | ↓ | Diabetic rats | ||
| MMP-9 | ↑ | ↓ | Human pancreatic cells and mouse dendritic cells | Diabetic rats and human vascular smooth muscle cells |
| MMP-13 | ↓ | Human articular chondrocytes | ||
| MMP-14 | ↑ | HUVECs | ||
| TIMP-1 | ↑ | Pulp fibroblasts | ||
| TIMP-3 | ↓ | Diabetic rats | ||
As shown in Table 2, MMP-9 is stimulated in PGE2-treated human articular chondrocytes and mouse osteoblasts, which is consistent with the data from high fluid shear stress-treated or human OA chondrocytes (46). Moreover, MMP-14 expression increased in PGE2-treated HUVECs (81). As OA and shear stress exposure highly stimulate MMP-14 expression, it is reasonable to hypothesize that MMP-14 is regulated via the COX-2/PGE2 signaling pathway; it is also possible to hypothesize that PGE2 activates MMPs by inducing PICs. More recent results have revealed that high fluid shear stress-induced PGE2 is capable of regulating the expression of IL-1β via EP2 and EP3 receptors in human T/C-28a2 chondrocytes (46). In contrast, MMP-1 (82) and MMP-13 (82) are down-regulated in PGE2-treated human articular chondrocytes and mouse osteoblasts. In addition, low fluid shear stress (5 dyn/cm2) suppresses the expression of MMP-1 and MMP-13 in human C-28I2 chondrocytes (3); however, PGE2 does not always exert catabolic functions and most likely exerts chondroprotective effects by inhibiting the expression of MMP-1 and MMP-13 in human chondrocytes. Nonetheless, these chondroprotective effects of PGE2 are not contradictory with its proinflammatory effects on cartilage degradation via the induction of MMPs, which might be explained by the threshold of PGE2 concentration in joint cartilage. Indeed, PGE2 has been associated with catabolic effects at the nano- to micromolar concentrations produced by arthritic tissues (6) because it suppresses the production of proteoglycans and stimulates ECM degradation. In contrast, low (picomolar) concentrations of PGE2 exert anabolic effects (83), as evidenced by the stimulation of proteoglycan (aggrecan) synthesis (84). Therefore, all the above data demonstrate that PGE2 is conserved to regulate MMP expression, although the regulating pattern is dependent on the concentration of PGE2 in joint cartilage.
15d-PGJ2 INDUCES THE PATHOGENESIS OF OA AT THE LATE STAGE OF THE DISEASE VIA APOPTOSIS BUT NOT ANTI-INFLAMMATORY EFFECTS
The effects of another important COX-2-derived prostaglandin, 15d-PGJ2, on OA remain a matter of debate. 15d-PGJ2 has been reported to induce chondrocytic apoptosis in a dose- and time-dependent manner via a peroxisome proliferator-activated receptor-γ (PPAR-γ)-dependent pathway. Although 15d-PGJ2 has also been shown to have a proapoptotic effect on other cell types, such as endothelial cells, tumor cells, and neurons, separate lines of evidence suggest that the molecule may have chondroprotective effects. For instance, 15d-PGJ2 and PGD2 decrease the production of MMPs (12, 85) suggesting that 15d-PGJ2 and PGD2 have anti-inflammatory effects on OA- or shear stress-induced inflammation. For these reasons, it is necessary to further address the biological functions of 15d-PGJ2 in the pathogenesis of OA via both anti-inflammatory effects and apoptosis.
Anti-inflammatory effects
The antagonistic effects of 15d-PGJ2 on the PGE2-induced synthesis of IL-6 and MMP-9 (46, 47) might indicate anti-inflammatory effects on OA development and progression. As the natural ligand for PPAR-γ, the best studied effects of 15d-PGJ2 are related to the activation of PPAR-γ. Although the consequences of PPAR-γ activation are complicated, the evidence to date suggests that these transcription factors support an anti-inflammatory phenotype. In the presence of 15d-PGJ2, the secretion of many PICs, such as IL-1β (86, 87), IL-6 (87, 88), IL-12(p70) (86), IL-12(p40) (88), TNF-α (86–88), and NO (88, 89), are inhibited (Table 2). More interestingly, 15d-PGJ2 treatment also increases the expression of granulocyte macrophage colony-stimulating factor 2 receptor β (GM-CSF), which synergistically activates PPAR-γ and adds to its anti-inflammatory effects in human airway epithelial cells (NCI-H292) (90). However, other studies suggest that 15d-PGJ2 exerts anti-inflammatory effects on NCI-H292 and human monocytes/macrophages and also increases IL-8 expression (90) and decreases MCP-1 (87). These data demonstrate that 15d-PGJ2 functions as a proinflammatory mediator in NCI-H292 cells, human monocytes, and macrophages. Although the effects of 15d-PGJ2 on inflammatory responses are still in debate, we can at least conclude that most PICs are suppressed by 15d-PGJ2 treatment (Table 2).
15d-PGJ2 also shows its effects by suppressing MMP expression. As shown in Table 2, the synthesis of MMP-1 (91), MMP-2 (92) and MMP-9 (92) is suppressed in different experimental models. These results are clearly in contrast to the data from OA-, shear stress-, or PGE2-treated cells in which the expression of MMP-1, MMP-2, and MMP-9 were highly induced (Tables 1, 2). In this regard, 15d-PGJ2 appears to exert chondroprotective effects and is not involved in OA development. However, 15d-PGJ2 is generally induced at the late stage of OA, accelerating the progression of OA by inducing chondrocyte apoptosis.
Apoptosis
Although 15d-PGJ2 shows antagonistic effects on inflammation, its biological function in OA development and progression cannot be negated. In light of its roles in human articular chondrocyte apoptosis, it is possible that 15d-PGJ2 exerts its effects on OA pathogenesis by inducing apoptosis in chondrocytes. As shown in Table 3, proapoptotic genes, including growth arrest and DNA-damage-inducible (GADD45), caspase 3, cellular FLICE-like inhibitory protein (cFLIP), cytochrome c (CYC), Bax, death receptor 5 (DR5), and p21, are induced in 15d-PGJ2-treated cells. In addition, 15d-PGJ2 treatment is able to down-regulate the expression of antiapoptotic genes, including, cellular inhibitor of apoptosis protein (cIAP)-1 (93), cIAP-2 (93), X-chromosome linked inhibitor of apoptosis protein (XIAP; ref. 93), baculoviral IAP repeat-containing 5 (Survivin; ref. 94), and Bcl-2 (94), in different cells (Table 3), potentially contributing to apoptosis, which plays a key role in the development and progression of OA. In contrast, 15d-PGJ2 treatment also decreases the expression of some proapoptotic genes, including Fas-L (95), caspase 8 (93), caspase 9 (93), and Bcl-xl (96), in different cells (Table 3), and these differences might be species specific, reflecting the fact that 15d-PGJ2 might also have chondroprotective effects.
Table 3.
15d-PGJ2-mediated apoptosis via different target genes
| Symbol | Trend | Experimental model |
|---|---|---|
| Proapoptosis | ||
| Fas-L | ↓ | Human T lymphocytes |
| Gadd45 | ↑ | Human SH-SY5Y cells |
| CCNG1 | ↑ | Human SH-SY5Y cells |
| CTSD | ↑ | Human SH-SY5Y cells |
| CASP3 | ↑ | Human renal proximal epithelial cell |
| CASP8 | ↓ | Human malignant B cells |
| CASP9 | ↓ | Human malignant B cells |
| cFLIP | ↑ | Human malignant B cells |
| CYC | ↑ | Human renal proximal epithelial cell |
| Bcl-xl | ↓ | Human chondrosarcoma, human colon cancer DLD-1 cells, and human chronic myeloid leukemia K562 cells |
| Bax | ↓ or ↑ | Human gastric epithelial cells, human renal proximal epithelial cell, and human myeloid leukemia cells |
| DR5 | ↑ | Human colon adenocarinoma HCT116 |
| p21 | ↑ | Human breast cancer cells |
| Antiapoptosis | ||
| cIAP-1 | ↓ | Human malignant B cells, human vascular endothelial cells |
| cIAP-2 | ↓ | Human malignant B cells |
| XIAP | ↓ | Human malignant B cells, human colon cancer DLD-1 cells, and human chronic myeloid leukemia K562 cells |
| Survivin | ↓ | Human myeloid leukemia cells |
| Bcl-2 | ↓ | Human gastric epithelial cells and human myeloid leukemia cells |
Given that 15d-PGJ2 induces apoptosis in certain experimental models, it is also reported that the prolonged application of high fluid shear will induce human chondrocyte apoptosis in a 15d-PGJ2-dependent manner, thereby contributing to the development of OA (5, 7). The details include the following: 1) exogenously added 15d-PGJ2 diminished the viability of human chondrocytes under static conditions; 2) knockdown of L-PGDS abolished shear-induced chondrocytic apoptosis; 3)15d-PGJ2 accumulation mediated the down-regulation of PKA, which concurrently up-regulated the expression of polo-like kinase (Plk) 1 and down-regulated the expression of Plk3; and 4) the imbalanced alteration of Plk1 and Plk3 resulted in chondrocyte apoptosis via p53 phosphorylation, inducing the expression of tumor protein p53 inducible nuclear proteins (TP53INPs), Fas and Bax (5). In agreement with the above data (5), Shan et al. (97) also found that 15d-PGJ2-induced apoptosis in the chondrocytes in OA patients. These authors further found that such PICs as IL-1β and TNF-α have the ability to induce the release of 15d-PGJ2 from human chondrocytes, leading to their apoptosis via PPARγ, p38, and NF-κB signaling pathways (97). In this respect, IL-1β and TNF-α not only have the ability to induce inflammation at the early stage of OA but also aggravate the progression of OA by inducing chondrocyte apoptosis at the late stage of the disease. Therefore, although we could segregate inflammatory mechanisms from apoptotic mechanisms and separate OA progression into 2 stages, crosstalk between inflammation and apoptosis exists during the course of OA development.
POTENTIAL THERAPEUTIC APPLICATIONS OF COX-2 MODULATED PICS AND MMPS IN SHEAR STRESS-INDUCED OA
Given the pivotal roles of COX-2 and its metabolic products in the pathogenesis of OA, nonsteroidal anti-inflammatory drugs (NSAIDs) are a mainstay of the treatment for patients with OA. Celecoxib and Rofecoxib were developed to treat OA and despite the fact that these drugs were found to improve OA pathogenesis by inhibiting the expression of COX-2 in cartilage (98), research into the mechanism is far behind the clinical applications, severely impeding therapeutic strategies for OA. Thus, summarizing and elucidating the mechanisms of the COX-2 modulation of PICs and MMPs will be helpful in achieving drug combinations to combat OA.
Considering the critical roles of COX-2 and its metabolic products in regulating the expression of PICs and MMPs in the early stage of OA, it is possible that Celecoxib and Rofecoxib suppress OA development by inhibiting the expression of PICs and MMPs. Although there is no direct evidence suggesting that COX-2 inhibition improves the progression of OA by suppressing the expression of PICs and MMPs, in vivo and in vitro models of OA have provided substantial evidence that blocking IL-1β and TNF-α could be beneficial in counteracting the degradative mechanisms associated with OA pathology. For instance, antagonists of IL-1β and TNF-α down-regulate the expression of the major MMPs associated with decreased aggrecan and type II collagen (99). Moreover, licofelone, an inhibitor of COXs, has been developed for potentially combating OA by deactivating MMP-13 (100). These data clearly provide clues that COX-2 inhibitors most likely exert their effects by suppressing the progression of OA in a PIC- and MMP-dependent manner. However, the treatment of OA with COX-2 inhibitors together with antagonists of PICs and MMPs will depend on further mechanistic investigations.
CONCLUSIONS
We herein summarize the mechanisms of PICs, MMPs, and apoptotic factors in mediating the effects of high fluid shear stress on inducing the development and progression of OA. Indeed, increased expression of PICs and MMPs in response to high fluid shear stress is believed to play a pivotal role in the development and progression of OA via a COX-2/PGE2-dependent pathway at the early stage of the disease. At the late stage of OA, 15d-PGJ2 exerts its effects on OA pathogenesis by inducing apoptosis but not an inflammatory mechanism. However, we could not distinctly segregate the inflammatory mechanisms from the apoptotic mechanisms and thoroughly separate OA progression into 2 stages because of the crosstalk between inflammation and apoptosis (Fig. 5). Although we could not completely decipher the mechanisms of OA onset by shear stress, the summary and reconstruction of the signaling network that regulates shear-mediated OA progression, as based on the results from different experimental models, may at least refine our understanding of the individual roles of PICs, MMPs, and apoptotic-related genes in OA pathophysiology. This knowledge will be instrumental in developing strategies to treat and combat OA disorders.
Figure 5.
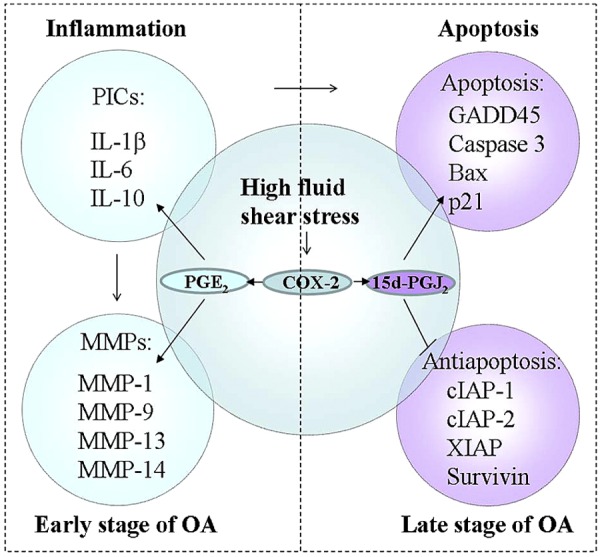
Networks of shear stress-induced OA progression. Mechanisms of PICs and MMPs in mediating high fluid shear stress in OA development are summarized. Indeed, the increased expression of PICs and MMPs in response to high fluid shear stress is believed to play a key role in the development of OA via a COX-2/PGE2-dependent pathway at the early stage of the disease. At the late stage of OA, 15d-PGJ2 exerts its effects on the pathogenesis of OA by inducing apoptosis but not an inflammatory mechanism. However, we could not distinctly segregate the inflammatory mechanism from apoptotic mechanism and thoroughly separate OA progression into 2 stages due to the roles of PICs in apoptosis.
Acknowledgments
This work was supported, in whole or in part, by the U.S. National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) grant RO1 (AR053358), Fundamental Research Funds of China (N120520001 and N120320001), and Liaoning Province High School Talent Support Program of China (LJQ2013029).
Footnotes
- 15d-PGJ2
- 15-deoxy-Δ-(12,14)-prostaglandin J2
- AP
- activator protein
- CCL
- chemokine (C-C motif) ligand
- CCNG1
- cyclin G1
- C/EBP
- CCAAT/enhancer-binding protein
- cFLIP
- cellular FLICE-like inhibitory protein
- cIAP
- cellular inhibitor of apoptosis protein
- COX-2
- cyclooxygenase 2
- CRE
- cyclic AMP response element
- CTSD
- cathepsin D
- CXCL
- chemokine (C-X-C motif) ligand
- CYC
- cytochrome c
- DR5
- death receptor 5
- ECM
- extracellular matrix
- GADD45
- growth arrest and DNA-damage-inducible
- GM-CSF
- granulocyte macrophage colony-stimulating factor 2 receptor β
- HUVEC
- human umbilical vein endothelial cell
- ICAM-1
- intercellular adhesion molecule 1
- IL
- interleukin
- IFN
- interferon
- LIF
- leukemia inhibitory factor
- MCP-1
- macrophage chemoattractant protein 1
- MMP
- matrix metalloproteinase
- mPGES-1
- microsomal prostaglandin E synthase 1
- OA
- osteoarthritis
- PEA-3
- polyomavirus enhancer activator 3
- PG
- prostaglandin
- PGDS
- prostaglandin D synthase
- PIC
- proinflammatory cytokine
- PLK
- polo-like kinase
- PPAR-γ
- peroxisome proliferator-activated receptor-γ
- Survivin
- baculoviral IAP repeat containing 5
- TGF-β1
- transforming growth factor β1
- TIMP
- tissue inhibitor of metalloproteinase
- TLR-4
- Toll-like receptor 4
- TNF-α
- tumor necrosis factor-α
- TNFAIP6
- tumor necrosis factor-α-induced 6
- Tnfsf9
- tumor necrosis factor (ligand) superfamily, member 9
- TP53INP
- tumor protein p53 inducible nuclear protein 1
- VEGF-A
- vascular endothelial growth factor A
- XIAP
- X-chromosome linked inhibitor of apoptosis protein
REFERENCES
- 1. Carter D. R., Beaupre G. S., Wong M., Smith R. L., Andriacchi T. P., Schurman D. J. (2004) The mechanobiology of articular cartilage development and degeneration. Clin. Orthop. Relat. Res., S69–S77 [DOI] [PubMed] [Google Scholar]
- 2. Buckwalter J. A., Martin J. A., Brown T. D. (2006) Perspectives on chondrocyte mechanobiology and osteoarthritis. Biorheology 43, 603–609 [PubMed] [Google Scholar]
- 3. Yokota H., Goldring M. B., Sun H. B. (2003) CITED2-mediated regulation of MMP-1 and MMP-13 in human chondrocytes under flow shear. J. Biol. Chem. 278, 47275–47280 [DOI] [PubMed] [Google Scholar]
- 4. Mohtai M., Gupta M. K., Donlon B., Ellison B., Cooke J., Gibbons G., Schurman D. J., Smith R. L. (1996) Expression of interleukin-6 in osteoarthritic chondrocytes and effects of fluid-induced shear on this expression in normal human chondrocytes in vitro. J. Orthop. Res. 14, 67–73 [DOI] [PubMed] [Google Scholar]
- 5. Zhu F., Wang P., Kontrogianni-Konstantopoulos A., Konstantopoulos K. (2010) Prostaglandin (PG)D(2) and 15-deoxy-delta(12,14)-PGJ(2), but not PGE(2), mediate shear-induced chondrocyte apoptosis via protein kinase A-dependent regulation of polo-like kinases. Cell Death Differ. 17, 1325–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amin A. R., Attur M., Patel R. N., Thakker G. D., Marshall P. J., Rediske J., Stuchin S. A., Patel I. R., Abramson S. B. (1997) Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J. Clin. Invest. 99, 1231–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu F., Wang P., Lee N. H., Goldring M. B., Konstantopoulos K. (2010) Prolonged application of high fluid shear to chondrocytes recapitulates gene expression profiles associated with osteoarthritis. PLoS One 5, e15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jacques C., Sautet A., Moldovan M., Thomas B., Humbert L., Berenbaum F. (1999) Cyclooxygenase activity in chondrocytes from osteoarthritic and healthy cartilage. Rev. Rhum. Engl. Ed. 66, 701–704 [PubMed] [Google Scholar]
- 9. Brenn D., Richter F., Schaible H. G. (2007) Sensitization of unmyelinated sensory fibers of the joint nerve to mechanical stimuli by interleukin-6 in the rat: an inflammatory mechanism of joint pain. Arthritis Rheum. 56, 351–359 [DOI] [PubMed] [Google Scholar]
- 10. Wang P., Zhu F., Konstantopoulos K. (2010) Prostaglandin E2 induces interleukin-6 expression in human chondrocytes via cAMP/protein kinase A- and phosphatidylinositol 3-kinase-dependent NF-kappaB activation. Am. J. Physiol. Cell Physiol. 298, C1445–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Portanova J. P., Zhang Y., Anderson G. D., Hauser S. D., Masferrer J. L., Seibert K., Gregory S. A., Isakson P. C. (1996) Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo. J. Exp. Med. 184, 883–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fahmi H., Di Battista J. A., Pelletier J. P., Mineau F., Ranger P., Martel-Pelletier J. (2001) Peroxisome proliferator–activated receptor gamma activators inhibit interleukin-1beta-induced nitric oxide and matrix metalloproteinase 13 production in human chondrocytes. Arthritis Rheum. 44, 595–607 [DOI] [PubMed] [Google Scholar]
- 13. Wang P., Zhu F., Tong Z., Konstantopoulos K. (2011) Response of chondrocytes to shear stress: antagonistic effects of the binding partners Toll-like receptor 4 and caveolin-1. FASEB J. 25, 3401–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Felson D. T. (2006) Clinical practice. Osteoarthritis of the knee. N. Engl. J. Med. 354, 841–848 [DOI] [PubMed] [Google Scholar]
- 15. Towle C. A., Hung H. H., Bonassar L. J., Treadwell B. V., Mangham D. C. (1997) Detection of interleukin-1 in the cartilage of patients with osteoarthritis: a possible autocrine/paracrine role in pathogenesis. Osteoarthritis Cartilage 5, 293–300 [DOI] [PubMed] [Google Scholar]
- 16. Afzal S., Khanam A. (2011) Serum estrogen and interleukin-6 levels in postmenopausal female osteoarthritis patients. Pak. J. Pharm. Sci. 24, 217–219 [PubMed] [Google Scholar]
- 17. Karlsson C., Dehne T., Lindahl A., Brittberg M., Pruss A., Sittinger M., Ringe J. (2010) Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage 18, 581–592 [DOI] [PubMed] [Google Scholar]
- 18. Lems W. F., den Uyl D. (2012) Exercise-induced changes in interleukin-10 in patients with knee osteoarthritis: new perspectives? Arthritis Res. Ther. 12, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang R., Fang H., Chen Y., Shen J., Lu H., Zeng C., Ren J., Zeng H., Li Z., Chen S., Cai D., Zhao Q. (2012) Gene expression analyses of subchondral bone in early experimental osteoarthritis by microarray. PLoS One 7, e32356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ohta S., Imai K., Yamashita K., Matsumoto T., Azumano I., Okada Y. (1998) Expression of matrix metalloproteinase 7 (matrilysin) in human osteoarthritic cartilage. Lab. Invest. 78, 79–87 [PubMed] [Google Scholar]
- 21. Goldring M. B. (2000) Osteoarthritis and cartilage: the role of cytokines. Curr. Rheumatol. Rep. 2, 459–465 [DOI] [PubMed] [Google Scholar]
- 22. Henderson B., Pettipher E. R. (1989) Arthritogenic actions of recombinant IL-1 and tumour necrosis factor alpha in the rabbit: evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 75, 306–310 [PMC free article] [PubMed] [Google Scholar]
- 23. Saklatvala J. (1986) Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature 322, 547–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chadjichristos C., Ghayor C., Kypriotou M., Martin G., Renard E., Ala-Kokko L., Suske G., de Crombrugghe B., Pujol J. P., Galera P. (2003) Sp1 and Sp3 transcription factors mediate interleukin-1 beta down-regulation of human type II collagen gene expression in articular chondrocytes. J. Biol. Chem. 278, 39762–39772 [DOI] [PubMed] [Google Scholar]
- 25. Guerne P. A., Carson D. A., Lotz M. (1990) IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J. Immunol. 144, 499–505 [PubMed] [Google Scholar]
- 26. Lotz M., Terkeltaub R., Villiger P. M. (1992) Cartilage and joint inflammation. Regulation of IL-8 expression by human articular chondrocytes. J. Immunol. 148, 466–473 [PubMed] [Google Scholar]
- 27. Villiger P. M., Terkeltaub R., Lotz M. (1992) Monocyte chemoattractant protein-1 (MCP-1) expression in human articular cartilage. Induction by peptide regulatory factors and differential effects of dexamethasone and retinoic acid. J. Clin. Invest. 90, 488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lefebvre V., Peeters-Joris C., Vaes G. (1990) Modulation by interleukin 1 and tumor necrosis factor alpha of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochim. Biophys. Acta 1052, 366–378 [DOI] [PubMed] [Google Scholar]
- 29. Mengshol J. A., Vincenti M. P., Coon C. I., Barchowsky A., Brinckerhoff C. E. (2000) Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 43, 801–811 [DOI] [PubMed] [Google Scholar]
- 30. Reboul P., Pelletier J. P., Tardif G., Cloutier J. M., Martel-Pelletier J. (1996) The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J. Clin. Invest. 97, 2011–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Todhunter P. G., Kincaid S. A., Todhunter R. J., Kammermann J. R., Johnstone B., Baird A. N., Hanson R. R., Wright J. M., Lin H. C., Purohit R. C. (1996) Immunohistochemical analysis of an equine model of synovitis-induced arthritis. Am. J. Vet. Res. 57, 1080–1093 [PubMed] [Google Scholar]
- 32. Guccione A. A., Felson D. T., Anderson J. J., Anthony J. M., Zhang Y., Wilson P. W., Kelly-Hayes M., Wolf P. A., Kreger B. E., Kannel W. B. (1994) The effects of specific medical conditions on the functional limitations of elders in the Framingham Study. Am. J. Public Health 84, 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Orita S., Koshi T., Mitsuka T., Miyagi M., Inoue G., Arai G., Ishikawa T., Hanaoka E., Yamashita K., Yamashita M., Eguchi Y., Toyone T., Takahashi K., Ohtori S. (2011) Associations between proinflammatory cytokines in the synovial fluid and radiographic grading and pain-related scores in 47 consecutive patients with osteoarthritis of the knee. BMC Musculoskelet. Disord. 12, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stove J., Huch K., Gunther K. P., Scharf H. P. (2000) Interleukin-1beta induces different gene expression of stromelysin, aggrecan and tumor-necrosis-factor-stimulated gene 6 in human osteoarthritic chondrocytes in vitro. Pathobiology 68, 144–149 [DOI] [PubMed] [Google Scholar]
- 35. Alaaeddine N., Di Battista J. A., Pelletier J. P., Kiansa K., Cloutier J. M., Martel-Pelletier J. (1999) Differential effects of IL-8, LIF (pro-inflammatory) and IL-11 (anti-inflammatory) on TNF-alpha-induced PGE(2)release and on signalling pathways in human OA synovial fibroblasts. Cytokine 11, 1020–1030 [DOI] [PubMed] [Google Scholar]
- 36. Yu C. L., Sun K. H., Shei S. C., Tsai C. Y., Tsai S. T., Wang J. C., Liao T. S., Lin W. M., Chen H. L., Yu H. S., Han S. H. (1994) Interleukin 8 modulates interleukin-1 beta, interleukin-6 and tumor necrosis factor-alpha release from normal human mononuclear cells. Immunopharmacology 27, 207–214 [DOI] [PubMed] [Google Scholar]
- 37. Westacott C. I., Sharif M. (1996) Cytokines in osteoarthritis: mediators or markers of joint destruction? Semin. Arthritis Rheum. 25, 254–272 [DOI] [PubMed] [Google Scholar]
- 38. Villiger P. M., Geng Y., Lotz M. (1993) Induction of cytokine expression by leukemia inhibitory factor. J. Clin. Invest. 91, 1575–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang A., Campbell J. (1993) A simple improvement to the Triton lysis procedure for plasmid isolation. BioTechniques 14, 730. [PubMed] [Google Scholar]
- 40. Ishiguro N., Ito T., Ito H., Iwata H., Jugessur H., Ionescu M., Poole A. R. (1999) Relationship of matrix metalloproteinases and their inhibitors to cartilage proteoglycan and collagen turnover: analyses of synovial fluid from patients with osteoarthritis. Arthritis Rheum. 42, 129–136 [DOI] [PubMed] [Google Scholar]
- 41. Dahlberg L., Billinghurst R. C., Manner P., Nelson F., Webb G., Ionescu M., Reiner A., Tanzer M., Zukor D., Chen J., van Wart H. E., Poole A. R. (2000) Selective enhancement of collagenase-mediated cleavage of resident type II collagen in cultured osteoarthritic cartilage and arrest with a synthetic inhibitor that spares collagenase 1 (matrix metalloproteinase 1). Arthritis Rheum. 43, 673–682 [DOI] [PubMed] [Google Scholar]
- 42. Baici A. (1998) Inhibition of extracellular matrix-degrading endopeptidases: problems, comments, and hypotheses. Biol. Chem. 379, 1007–1018 [PubMed] [Google Scholar]
- 43. Homandberg G. A. (1999) Potential regulation of cartilage metabolism in osteoarthritis by fibronectin fragments. Front. Biosci. 4, D713–730 [DOI] [PubMed] [Google Scholar]
- 44. Jiang Z., Berceli S. A., Pfahnl C. L., Wu L., Goldman D., Tao M., Kagayama M., Matsukawa A., Ozaki C. K. (2004) Wall shear modulation of cytokines in early vein grafts. J. Vasc. Surg. 40, 345–350 [DOI] [PubMed] [Google Scholar]
- 45. Messer R. L., Mickalonis J., Lewis J. B., Omata Y., Davis C. M., Brown Y., Wataha J. C. (2008) Interactions between stainless steel, shear stress, and monocytes. J. Biomed. Mater. Res. A 87, 229–235 [DOI] [PubMed] [Google Scholar]
- 46. Wang P., Zhu F., Konstantopoulos K. (2012) The antagonistic actions of endogenous interleukin-1beta and 15-deoxy-delta12,14-prostaglandin J2 regulate the temporal synthesis of matrix metalloproteinase-9 in sheared chondrocytes. J. Biol. Chem. 287, 31877–31893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang P., Zhu F., Konstantopoulos K. (2011) Interleukin-6 synthesis in human chondrocytes is regulated via the antagonistic actions of prostaglandin (PG)E2 and 15-deoxy-delta(12,14)-PGJ2. PLoS One 6, e27630. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Krizanac-Bengez L., Mayberg M. R., Cunningham E., Hossain M., Ponnampalam S., Parkinson F. E., Janigro D. (2006) Loss of shear stress induces leukocyte-mediated cytokine release and blood-brain barrier failure in dynamic in vitro blood-brain barrier model. J. Cell. Physiol. 206, 68–77 [DOI] [PubMed] [Google Scholar]
- 49. Carroll G., McGloughlin T., O'keeffe L., Callanan A., Walsh M. (2009) Realistic temporal variations of shear stress modulate MMP-2 and MCP-1 expression in arteriovenous vascular access. Cell. Mol. Bioeng. 2, 591–605 [Google Scholar]
- 50. Tajima S., Yokoyama S., Yamamoto A. (1983) Effect of lipid particle size on association of apolipoproteins with lipid. J. Biol. Chem. 258, 10073–10082 [PubMed] [Google Scholar]
- 51. Naito K., Takahashi M., Kushida K., Suzuki M., Ohishi T., Miura M., Inoue T., Nagano A. (1999) Measurement of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases-1 (TIMP-1) in patients with knee osteoarthritis: comparison with generalized osteoarthritis. Rheumatology (Oxford) 38, 510–515 [DOI] [PubMed] [Google Scholar]
- 52. Wang P., Zhu F., Lee N. H., Konstantopoulos K. (2010) Shear-induced interleukin-6 synthesis in chondrocytes: roles of E prostanoid (EP) 2 and EP3 in cAMP/protein kinase A- and PI3-K/Akt-dependent NF-kappaB activation. J. Biol. Chem. 285, 24793–24804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liang F., Hu D. Y., Wang B. Y., Huang N., Wu Q., Chen H. Q. (2005) [Transcriptional activation of interleukin-8 gene induced by shear stress in human umbilical vein endothelial cells]. Zhonghua Nei Ke Za Zhi 44, 421–424 [PubMed] [Google Scholar]
- 54. Liang F., Huang N., Wang B., Chen H. (2002) [Assessment of the role of TLR-4 in shear-stress-induced IL-8 gene transcription activation in vascular endothelial cells by gene mutation and gene transfection technology]. Sheng. Wu Yi Xue Gong Cheng Xue Za Zhi 19, 667–672 [PubMed] [Google Scholar]
- 55. Cheng M., Liu X., Li Y., Tang R., Zhang W., Wu J., Li L., Gang Y., Chen H. (2007) IL-8 gene induction by low shear stress: pharmacological evaluation of the role of signaling molecules. Biorheology 44, 349–360 [PubMed] [Google Scholar]
- 56. Glossop J. R., Cartmell S. H. (2009) Effect of fluid flow-induced shear stress on human mesenchymal stem cells: differential gene expression of IL1B and MAP3K8 in MAPK signaling. Gene Expr. Patt. 9, 381–388 [DOI] [PubMed] [Google Scholar]
- 57. Jiang J., Dingledine R. (2013) Role of prostaglandin receptor EP2 in the regulations of cancer cell proliferation, invasion, and inflammation. J. Pharmacol. Exp. Ther. 344, 360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen B. C., Liao C. C., Hsu M. J., Liao Y. T., Lin C. C., Sheu J. R., Lin C. H. (2006) Peptidoglycan-induced IL-6 production in RAW 264.7 macrophages is mediated by cyclooxygenase-2, PGE2/PGE4 receptors, protein kinase A, I kappa B kinase, and NF-kappa B. J. Immunol. 177, 681–693 [DOI] [PubMed] [Google Scholar]
- 59. Li L., Tatake R. J., Natarajan K., Taba Y., Garin G., Tai C., Leung E., Surapisitchat J., Yoshizumi M., Yan C., Abe J., Berk B. C. (2008) Fluid shear stress inhibits TNF-mediated JNK activation via MEK5-BMK1 in endothelial cells. Biochem. Biophys. Res. Commun. 370, 159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang H., Young S. R., Gerard-O'Riley R., Hum J. M., Yang Z., Bidwell J. P., Pavalko F. M. (2011) Blockade of TNFR1 signaling: A role of oscillatory fluid shear stress in osteoblasts. J. Cell. Physiol. 226, 1044–1051 [DOI] [PubMed] [Google Scholar]
- 61. Roman-Blas J. A., Jimenez S. A. (2006) NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage 14, 839–848 [DOI] [PubMed] [Google Scholar]
- 62. Murphy G., Nagase H. (2008) Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: destruction or repair? Nat. Clin. Pract. Rheumatol. 4, 128–135 [DOI] [PubMed] [Google Scholar]
- 63. Sweeney V., Cummins P. M., Birney Y. A., Cullen J. P., Redmond E. M., Cahill P. A. (2004) Cyclic strain-mediated regulation of endothelial matrix metalloproteinase-2 expression and activity. Cardiovasc. Res. 63, 625–634 [DOI] [PubMed] [Google Scholar]
- 64. Zheng L., Huang Y., Song W., Gong X., Liu M., Jia X., Zhou G., Chen L., Li A., Fan Y. (2012) Fluid shear stress regulates metalloproteinase-1 and 2 in human periodontal ligament cells: involvement of extracellular signal-regulated kinase (ERK) and P38 signaling pathways. J. Biomech. 45, 2368–2375 [DOI] [PubMed] [Google Scholar]
- 65. Kusano K., Miyaura C., Inada M., Tamura T., Ito A., Nagase H., Kamoi K., Suda T. (1998) Regulation of matrix metalloproteinases (MMP-2, -3, -9, and -13) by interleukin-1 and interleukin-6 in mouse calvaria: association of MMP induction with bone resorption. Endocrinology 139, 1338–1345 [DOI] [PubMed] [Google Scholar]
- 66. Liang K. C., Lee C. W., Lin W. N., Lin C. C., Wu C. B., Luo S. F., Yang C. M. (2007) Interleukin-1beta induces MMP-9 expression via p42/p44 MAPK, p38 MAPK, JNK, and nuclear factor-kappaB signaling pathways in human tracheal smooth muscle cells. J. Cell. Physiol. 211, 759–770 [DOI] [PubMed] [Google Scholar]
- 67. Okada M., Yamawaki H., Hara Y. Angiotensin II enhances interleukin-1 beta-induced MMP-9 secretion in adult rat cardiac fibroblasts. J. Vet. Med. Sci. 72, 735–739 [DOI] [PubMed] [Google Scholar]
- 68. Fleenor D. L., Pang I. H., Clark A. F. (2003) Involvement of AP-1 in interleukin-1alpha-stimulated MMP-3 expression in human trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 44, 3494–3501 [DOI] [PubMed] [Google Scholar]
- 69. Choi Y. A., Lee D. J., Lim H. K., Jeong J. H., Sonn J. K., Kang S. S., Baek S. H. (2004) Interleukin-1beta stimulates matrix metalloproteinase-2 expression via a prostaglandin E2-dependent mechanism in human chondrocytes. Exp. Mol. Med. 36, 226–232 [DOI] [PubMed] [Google Scholar]
- 70. Ma B., van Blitterswijk C. A., Karperien M. (2012) A Wnt/beta-catenin negative feedback loop inhibits interleukin-1-induced matrix metalloproteinase expression in human articular chondrocytes. Arthritis Rheum. 64, 2589–2600 [DOI] [PubMed] [Google Scholar]
- 71. Miller C., Zhang M., He Y., Zhao J., Pelletier J. P., Martel-Pelletier J., Di Battista J. A. (1998) Transcriptional induction of cyclooxygenase-2 gene by okadaic acid inhibition of phosphatase activity in human chondrocytes: co-stimulation of AP-1 and CRE nuclear binding proteins. J. Cell. Biochem. 69, 392–413 [PubMed] [Google Scholar]
- 72. Saunders M. A., Sansores-Garcia L., Gilroy D. W., Wu K. K. (2001) Selective suppression of CCAAT/enhancer-binding protein beta binding and cyclooxygenase-2 promoter activity by sodium salicylate in quiescent human fibroblasts. J. Biol. Chem. 276, 18897–18904 [DOI] [PubMed] [Google Scholar]
- 73. Thomas B., Berenbaum F., Humbert L., Bian H., Bereziat G., Crofford L., Olivier J. L. (2000) Critical role of C/EBPdelta and C/EBPbeta factors in the stimulation of the cyclooxygenase-2 gene transcription by interleukin-1beta in articular chondrocytes. Eur. J. Biochem. 267, 6798–6809 [DOI] [PubMed] [Google Scholar]
- 74. Abulencia J. P., Gaspard R., Healy Z. R., Gaarde W. A., Quackenbush J., Konstantopoulos K. (2003) Shear-induced cyclooxygenase-2 via a JNK2/c-Jun-dependent pathway regulates prostaglandin receptor expression in chondrocytic cells. J. Biol. Chem. 278, 28388–28394 [DOI] [PubMed] [Google Scholar]
- 75. Alshihabi S. N., Chang Y. S., Frangos J. A., Tarbell J. M. (1996) Shear stress-induced release of PGE2 and PGI2 by vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 224, 808–814 [DOI] [PubMed] [Google Scholar]
- 76. Gilroy D. W., Colville-Nash P. R., Willis D., Chivers J., Paul-Clark M. J., Willoughby D. A. (1999) Inducible cyclooxygenase may have anti-inflammatory properties. Nat. Med. 5, 698–701 [DOI] [PubMed] [Google Scholar]
- 77. Schuligoi R., Grill M., Heinemann A., Peskar B. A., Amann R. (2005) Sequential induction of prostaglandin E and D synthases in inflammation. Biochem. Biophys. Res. Commun. 335, 684–689 [DOI] [PubMed] [Google Scholar]
- 78. Rola-Pleszczynski M., Stankova J. (1992) Cytokine gene regulation by PGE(2), LTB(4) and PAF. Mediators Inflamm. 1, 5–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sottile A., Venza I., Venza M., Valenti A., Teti D. (1996) PGE2-induced immunoregulation mediated by cytokine production from cultures of human peripheral T lymphocytes. Immunopharmacol. Immunotoxicol. 18, 27–36 [DOI] [PubMed] [Google Scholar]
- 80. Harizi H., Gualde N. (2006) Pivotal role of PGE2 and IL-10 in the cross-regulation of dendritic cell-derived inflammatory mediators. Cell. Mol. Immunol. 3, 271–277 [PubMed] [Google Scholar]
- 81. Alfranca A., Lopez-Oliva J. M., Genis L., Lopez-Maderuelo D., Mirones I., Salvado D., Quesada A. J., Arroyo A. G., Redondo J. M. (2008) PGE2 induces angiogenesis via MT1-MMP-mediated activation of the TGFbeta/Alk5 signaling pathway. Blood 112, 1120–1128 [DOI] [PubMed] [Google Scholar]
- 82. Nishitani K., Ito H., Hiramitsu T., Tsutsumi R., Tanida S., Kitaori T., Yoshitomi H., Kobayashi M., Nakamura T. (2010) PGE2 inhibits MMP expression by suppressing MKK4-JNK MAP kinase-c-JUN pathway via EP4 in human articular chondrocytes. J. Cell. Biochem. 109, 425–433 [DOI] [PubMed] [Google Scholar]
- 83. Martel-Pelletier J., Pelletier J. P., Fahmi H. (2003) Cyclooxygenase-2 and prostaglandins in articular tissues. Semin. Arthritis Rheum. 33, 155–167 [DOI] [PubMed] [Google Scholar]
- 84. Lowe G. N., Fu Y. H., McDougall S., Polendo R., Williams A., Benya P. D., Hahn T. J. (1996) Effects of prostaglandins on deoxyribonucleic acid and aggrecan synthesis in the RCJ 3.1C5.18 chondrocyte cell line: role of second messengers. Endocrinology 137, 2208–2216 [DOI] [PubMed] [Google Scholar]
- 85. Zayed N., Afif H., Chabane N., Mfuna-Endam L., Benderdour M., Martel-Pelletier J., Pelletier J. P., Motiani R. K., Trebak M., Duval N., Fahmi H. (2008) Inhibition of interleukin-1beta-induced matrix metalloproteinases 1 and 13 production in human osteoarthritic chondrocytes by prostaglandin D2. Arthritis Rheum. 58, 3530–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alves C., de Melo N., Fraceto L., de Araujo D., Napimoga M. (2011) Effects of 15d-PGJ(2)-loaded poly(D,L-lactide-co-glycolide) nanocapsules on inflammation. Br. J. Pharmacol. 162, 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Storer P. D., Xu J., Chavis J. A., Drew P. D. (2005) Cyclopentenone prostaglandins PGA2 and 15-deoxy-delta12,14 PGJ2 suppress activation of murine microglia and astrocytes: implications for multiple sclerosis. J. Neurosci. Res. 80, 66–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Alleva D. G., Johnson E. B., Lio F. M., Boehme S. A., Conlon P. J., Crowe P. D. (2002) Regulation of murine macrophage proinflammatory and anti-inflammatory cytokines by ligands for peroxisome proliferator-activated receptor-gamma: counter-regulatory activity by IFN-gamma. J. Leukoc. Biol. 71, 677–685 [PubMed] [Google Scholar]
- 89. Martinez A. E., Sanchez-Gomez F. J., Diez-Dacal B., Oeste C. L., Perez-Sala D. (2012) 15-Deoxy-Delta(12,14)-prostaglandin J2 exerts pro- and anti-inflammatory effects in mesangial cells in a concentration-dependent manner. Inflamm. Allergy Drug Targets 11, 58–65 [DOI] [PubMed] [Google Scholar]
- 90. Chiba T., Ueki S., Ito W., Kato H., Takeda M., Kayaba H., Furue M., Chihara J. (2009) 15-Deoxy-Delta(12,14)-prostaglandin J2 induces IL-8 and GM-CSF in a human airway epithelial cell line (NCI-H292). Int. Arch. Allergy Immunol. 149(Suppl. 1), 77–82 [DOI] [PubMed] [Google Scholar]
- 91. Fahmi H., Pelletier J. P., Di Battista J. A., Cheung H. S., Fernandes J. C., Martel-Pelletier J. (2002) Peroxisome proliferator-activated receptor gamma activators inhibit MMP-1 production in human synovial fibroblasts likely by reducing the binding of the activator protein 1. Osteoarthritis Cartilage 10, 100–108 [DOI] [PubMed] [Google Scholar]
- 92. Pustovrh M. C., Capobianco E., Martinez N., Higa R., White V., Jawerbaum A. (2009) MMP/TIMP balance is modulated in vitro by 15dPGJ(2) in fetuses and placentas from diabetic rats. Eur. J. Clin. Invest. 39, 1082–1090 [DOI] [PubMed] [Google Scholar]
- 93. Piva R., Gianferretti P., Ciucci A., Taulli R., Belardo G., Santoro M. G. (2005) 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis in human malignant B cells: an effect associated with inhibition of NF-kappa B activity and down-regulation of antiapoptotic proteins. Blood 105, 1750–1758 [DOI] [PubMed] [Google Scholar]
- 94. Liu J. J., Huang R. W., Lin D. J., Peng J., Wu X. Y., Lin Q., Pan X. L., Song Y. Q., Zhang M. H., Hou M., Chen F. (2005) Expression of survivin and bax/bcl-2 in peroxisome proliferator activated receptor-gamma ligands induces apoptosis on human myeloid leukemia cells in vitro. Ann. Oncol. 16, 455–459 [DOI] [PubMed] [Google Scholar]
- 95. Cippitelli M., Fionda C., Di Bona D., Lupo A., Piccoli M., Frati L., Santoni A. (2003) The cyclopentenone-type prostaglandin 15-deoxy-delta12,14-prostaglandin J2 inhibits CD95 ligand gene expression in t lymphocytes: interference with promoter activation via peroxisome proliferator-activated receptor-gamma-independent mechanisms. J. Immunol. 170, 4578–4592 [DOI] [PubMed] [Google Scholar]
- 96. Nishida K., Kunisada T., Shen Z. N., Kadota Y., Hashizume K., Ozaki T. (2008) Chondrosarcoma and peroxisome proliferator-activated receptor. PPAR Res. 2008, 250568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shan Z. Z., Masuko-Hongo K., Dai S. M., Nakamura H., Kato T., Nishioka K. (2004) A potential role of 15-deoxy-delta(12,14)-prostaglandin J2 for induction of human articular chondrocyte apoptosis in arthritis. J. Biol. Chem. 279, 37939–37950 [DOI] [PubMed] [Google Scholar]
- 98. Laine L., White W. B., Rostom A., Hochberg M. (2008) COX-2 selective inhibitors in the treatment of osteoarthritis. Semin. Arthritis Rheum. 38, 165–187 [DOI] [PubMed] [Google Scholar]
- 99. Kobayashi M., Squires G. R., Mousa A., Tanzer M., Zukor D. J., Antoniou J., Feige U., Poole A. R. (2005) Role of interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum. 52, 128–135 [DOI] [PubMed] [Google Scholar]
- 100. Boileau C., Pelletier J. P., Tardif G., Fahmi H., Laufer S., Lavigne M., Martel-Pelletier J. (2005) The regulation of human MMP-13 by licofelone, an inhibitor of cyclo-oxygenases and 5-lipoxygenase, in human osteoarthritic chondrocytes is mediated by the inhibition of the p38 MAP kinase signalling pathway. Ann. Rheum. Dis. 64, 891–898’ [DOI] [PMC free article] [PubMed] [Google Scholar]