

Abstract
Humanin (HN) is a 24-aa polypeptide that offers protection from Alzheimer's disease and myocardial infarction, increases insulin sensitivity, improves survival of β cells, and delays onset of diabetes. Here we examined the acute effects of HN on insulin secretion and potential mechanisms through which they are mediated. Effects of a potent HN analog, HNGF6A, on glucose-stimulated insulin secretion (GSIS) were assessed in vivo and in isolated pancreatic islets and cultured murine β cell line (βTC3) in vitro. Sprague-Dawley rats (3 mo old) that received HNGF6A required a significantly higher glucose infusion rate and demonstrated higher insulin levels during hyperglycemic clamps compared to saline controls. In vitro, compared to scrambled peptide controls, HNGF6A increased GSIS in isolated islets from both normal and diabetic mice as well as in βTC3 cells. Effects of HNGF6A on GSIS were dose dependent, K-ATP channel independent, and associated with enhanced glucose metabolism. These findings demonstrate that HNGF6A increases GSIS in whole animals, from isolated islets and from cells in culture, which suggests a direct effect on the β cell. The glucose-dependent effects on insulin secretion along with the established effects on insulin action suggest potential for HN and its analogs in the treatment of diabetes.—Kuliawat, R., Klein, L., Gong, Z., Nicoletta-Gentile, M., Nemkal, A. Cui, L., Bastie, C., Su, K., Huffman, D., Surana, M., Barzilai, N., Fleischer, N., Muzumdar, R. Potent humanin analog increases glucose-stimulated insulin secretion through enhanced metabolism in the β cell.
Keywords: hyperglycemic clamps, pancreatic islets, diabetes
humanin (hn) is a 24-aa polypeptide (1) that belongs to a novel group of biologically active peptides encoded in short open reading frames (sORFs) in the mitochondrial genome (2). Although thought to be transcribed from the 16S region of the mitochondrial RNA, multiple nuclear loci that code HN-like peptides have also been identified (3). While the exact site of origin of HN remains to be established, the biological effects of HN have been clearly demonstrated in many tissues, both in vivo and in vitro. Despite the significant biological effects, HN fits only some of the conventional criteria of a biologically active peptide. Though HN requires its extracellular secretion and acts on the cells from outside (4), the identity of a cell surface receptor for HN has been elusive. Reports that formyl peptide receptor-like 1 (FPRL1; ref. 5) or FPRL1 and FPRL2 (6) may act as a functional receptor for HN have been challenged by observations that HN activity is not exclusively mediated by these receptors and that HN may exert biological activity by binding to a complex or complexes involving cytokine receptors CNTFR/WSX-1/gp130 (7). Ikonen et al. (8) have reported that HN directly interacts with insulin-like growth factor-binding protein-3 (IGFBP-3), a carrier protein of insulin-like growth factor-I (IGF-I) in the circulation. This interaction alters HN activity and suggests that IGFBP-3 could be one important regulator of HN function in vivo (8). Several intracellular pathways have been proposed to mediate the effects of HN and are likely to depend of cell type (9–11). In addition, HN immunoreactivity has been shown to change with age (12) and pathology in serum, and in tissues such as testis, colon, and diseased and healthy brain tissue (13). In view of the still-unelucidated aspects of HN regulation and function, we and others have focused on the biochemical, cellular, and physiological functions of extracellularly added synthetic HN in vivo and in vitro (12, 14, 15).
Experiments have indicated that primary structure (16, 17) and the ability to self-dimerize (18) are essential for HN bioactivity. Molecular manipulations, such as single amino acid substitutions, have been shown to result in HN analogs with altered biological activities, which allows the construction of potent HN derivatives. Recently, it was shown that the potency in biological activities of S14G-HN (HNG), a synthetic derivative of HN (19), are attributable to the differences in stability (20).
HN and analogs have been shown to have significant biological effects both in vivo and in vitro. HN and its analogs offer protection in Alzheimer disease (AD)-related insults in neurons and smooth muscle vascular cells as well as in vivo models of AD (15, 19, 21). In addition, HNG offers protection in neurons in response to a variety of insults. including ischemia, prion-induced apoptosis, and chemical insults (14, 22). HNG has been shown to decrease infarct size following stroke (19) and to improve memory following AD and scopolamine in rodent models (21, 23). We have recently shown that HNG decreases infarct size and improves cardiac function in a model of myocardial ischemia-reperfusion (14).
HN and its analogs play an important role in glucose homeostasis. We previously demonstrated that HN and its potent non-IGFBP-3 binding analog (HNGF6A; ref. 12) improve insulin sensitivity under hyperinsulinemic-euglycemic clamps (12). Another group has shown that daily injections with HN for 6 wk improved survival of β cells and delayed the onset of diabetes in a nonbese diabetic (NOD) mouse model of type 1 diabetes (24). Interestingly, though HNGF6A significantly reduced blood glucose levels in Zucker diabetic fatty (ZDF) rats (12), insulin levels in these animals did not decrease along with hypoglycemia as expected, which leads us to hypothesize that HNGF6A may have acute, independent effects on insulin secretion. To systematically characterize the role of HNGF6A on insulin secretion, we tested the direct effects of exogenous HNGF6A on insulin secretion in vivo and in vitro.
MATERIALS AND METHODS
All studies were conducted in accordance with the U.S. National Institutes of Health Principles of Laboratory Animal Care (NIH publication no. 85–23, rev. 1985; http://grants1.nih.gov/grants/olaw/references/phspol.htm), and the study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Albert Einstein College of Medicine.
Hyperglycemic clamp
Male Sprague-Dawley rats (3 mo old, n=10, 5/group) were subjected to 2 h of moderate hyperglycemia (11 mM). Briefly, 25% glucose was infused intravenously to raise the plasma glucose concentration acutely to 11 mM. The glucose infusion rate (GIR) was then varied to maintain the plasma glucose concentration at this level for 2 h. A total dose of 60 μg of HNGF6A (sequence: MAPRGASCLLLLTGEIDLPVKRRA; Genemed Custom Peptide Synthesis, San Antonio, TX, USA) was administered per animal; one-third was given as a bolus, and the remainder was administered as a continuous infusion at the rate of the 0.07 mg/kg/h over 2 h. Plasma samples for insulin were obtained at 2-min intervals for the first 10 min and at 10 min intervals throughout the study. Samples for C peptide were collected every 30 min. The area under the curve (AUC) of the first-phase insulin response was calculated by the trapezoid rule, using 0-, 2-, 4-, 6-, 8-, and 10-min samples with the formula (i0 + i2)/2 × 0.5 + (i2 + i4)/2 × 0.5 + (i4 + i6)/2 × 0.5 + (i6 + i8)/2 × 0.5 + (i8 + i10)/2 × 0.5. Insulin levels and GIRs were averaged over the last 1 h of the clamp.
Intravenous glucose tolerance test (IVGTT) to assess first-phase insulin secretion
Indwelling catheters were placed in C57BL6/N mice (n=6/group) in the jugular vein for collection of blood samples without stress during the IVGTT study. After recovery, mice were subjected to i.v. injection of vehicle or HNGF6A (2 mg/kg body weight) 10 min before the injection of 2 g/kg glucose load, and blood samples were collected from mice at 0, 2, 5, 10, 20, and 30 min for estimation of blood glucose and insulin.
Analytical procedures for in vivo studies
Plasma glucose was measured by the glucose oxidase method (Glucose Analyzer II; Beckman Instruments, Inc., Palo Alto, CA, USA), and plasma insulin was measured by ELISA using rat or mouse insulin standards. Plasma C-peptide levels were assayed using ELISA kit from Millipore (Billerica, MA, USA).
Islet isolation
Islets from 25- to 30-g male C57BL6/J wild-type (WT) and diabetic db/db mice were isolated using standard collagenase digestion, as described previously (25), and were cultured overnight in RPMI medium supplemented with 10% FBS plus antibiotics. Prior to experiments, islets were transferred to MilliCell-PCF culture plate filter inserts (Millipore) at a density of ∼10–20 islets/insert. The inserts were placed within individual wells of a 24-well cell culture plate, and each well was filled with 1 ml of DME and 5 mM glucose. After a 6 h preincubation at 5 mM glucose, the inserts were transferred to new wells containing 0.5 ml volume of medium, and islets were challenged with glucose (5 or 16 mM) and HNGF6A (50 ng/ml) as indicated. Medium was collected from beneath inserts; islets were floated by a rapid application of 0.5 ml of PBS added to the inserts. Islets were pelleted and then lysed by sonication as described previously (26). An antiprotease cocktail containing aprotinin (1 mU/ml), leupeptin (0.1 mM), pepstatin (10 μM), EDTA (5 mM), and diisopropylfluorophospbate (1 mM) was added to the collected medium and cell lysates. Insulin levels in the medium were measured using the Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem Inc., Downers Grove, IL, USA). The repeatability of all findings was confirmed by performing each experiment ≥3 times.
Cell culture
Mouse pancreatic β cells (βTC3 line) were maintained in DMEM containing 10% heat-inactivated FBS, d-glucose (4500 μg/ml), and glutamine (4 mM) at 37°C in a humidified environment with 5% CO2. The initial studies probing the effects of HNGF6A were done at glucose concentrations of 0, 5 and 16 mM glucose levels. Subsequent studies were all done at 16 mM glucose levels. For the dose-response study, βTC3 cells were treated with either 24-aa scrambled peptide (SP; sequence: FRGGETRARAMPLIDLSPLCLLKV) or varying doses of HNGF6A between 5 and 500 ng/ml, and insulin in the medium was assessed at 2 h. For the time-course study, βTC3 cells were treated for the indicated times with 50 ng/ml of either SP or HNGF6A. Insulin levels were assayed in the media using the Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem Inc., Downers Grove, IL, USA).
Western blot
βTC3 cells and mouse pancreas were homogenized in 1× RIPA buffer (50 mM Tris, pH7.4; 150 mM NaCl; 1% Triton X-100; and 0.1% SDS) containing 1× proteinase inhibitors (Roche, Indianapolis, IN, USA). Proteins (20 μg) were resolved on precast gradient SDS-PAGE and then transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline with Tween 20 (TBS-T) for 1 h at room temperature. Primary antibodies against CNTFRα, GP130, WSX-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Rattin (GeneTex, Irvine, CA, USA) and β-actin (Sigma, St. Louis, MO, USA) diluted in TBS-T containing 5% of BSA were added to the membranes and incubated overnight at 4°C. The membranes were subsequently washed with TBS-T 3 times for 10 min and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The signals were detected by the SuperSignal West Dura extended duration substrate (Thermo Scientific, Rockford, IL, USA).
Glucose transporter 2 (GLUT2) translocation
βTC3 cells were seeded on coverslips in a 6-well plate 1 d before transfection. Cells transfected with GLUT2-GFP construct (kindly provided by Dr. Jeffery Pessin, Albert Einstein College of Medicine) were incubated in medium, without glucose, overnight, and then treated with 50 ng/ml HNGF6A or SP in 16 mM glucose for 15 min. After 2 washes with cold PBS, cells were fixed using 4% formaldehyde for 20 min. Images were taken using Leica SP2 confocal microscopy (Leica Microsystems, Buffalo Grove, IL, USA).
Glucokinase (GCK) activity assay
Glucose phosphorylation rate was measured by a well-established fluorimetric assay, as described previously (27). In brief, SP- or HNGF6A (50 ng/ml)-treated βTC3 cells were homogenized in 500 μl of cold lysis buffer (10 mM HEPES, pH 7.4; 250 mM sucrose; 2 mM EDTA; 1 mM DTT; 1.5 mM MgCl2; and 10 mMKCl) by 10 passages through a 22-gauge needle. The soluble cytosolic fractions collected from centrifugation at 105 g and 250 μg of this soluble cytosolic protein were added to 100 μl assay buffer (50 mM HEPES, pH 7.7; 100 mM KCl; 10 mM MgCl2; 15 mM β-mercaptoethanol; 0.5 mM NAD+; 5 mM ATP; 10 μg/ml G6PDH; and 0.05% BSA) containing 0.5 or 50 mM glucose and incubated at 37°C for 1 h. A reaction mix with cytosol protein, glucose, and assay buffer without ATP was also created for background subtraction. Relative GCK activity was calculated by subtracting values of hexokinase (collected from 0.5 mM glucose reactions) from values of total kinases (collected from 50 mM glucose reactions).
Glucose oxidation assay
βTC3 cells were grown in 6-well plates for 24 h. Cells were rinsed with warm PBS twice after 60 min of treatment with SP or HNGF6A and then incubated with 1 ml of incubation buffer (M199 medium without phenol red, complemented with 5 mM glucose and 2 mM pyruvate) containing 1 uCi/ml of 14C-U-glucose for 1 h. After incubation, 900 μl of medium was collected, and CO2 in the medium was trapped using 200 μl of 3 N NaOH for 3 h. The 14CO2 was measured by scintillation counting. Oxidation of glucose is expressed in nanomoles of oxidized glucose per incubation time per milligram of protein.
Measurement of mitochondrial transmembrane potential (ΔΨm)
Mitochondrial transmembrane potential was investigated using the fluorochrome dye JC-10 (Enzo Life Sciences, Ann Arbor, MI, USA). βTC3 cells were seeded in 96-well plates and grown to ∼70% confluence. The cells were treated for 30 min at 37°C, 10% CO2 with growth medium with or without HNGF6A (50 ng/ml) and H2O2 (40 μM) to challenge the β cell. This medium was aspirated and replaced with 100 μl/well of 1× Hanks buffered salt solution containing 20 mM HEPES and 20 μM JC-10, and the plate was incubated for an additional 30 min at 37°C. The wells were rinsed 3× with a wash buffer (1× HBSS, 10 mM HEPES, and 0.02 mg/100 ml d-glucose). The final aliquot of wash buffer was left in the wells, and the fluorescent signal was read on a Molecular Devices SpectraMax M5e scanning spectrophotometer, using 490 and 525 nm as green excitation and emission wavelengths, respectively (cutoff 515 nm), and 490 and 590 nm as red excitation and emission wavelengths, respectively (cutoff 570 nm). The ratio of the reading at 590 nm to that at 525 nm (590:525 ratio) was considered as the relative ΔΨm value.
Bioluminescent assay for cellular ATP
βTC3 cells were treated for the indicated time with or without HNGF6A (50 ng/ml), and ATP levels were measured with a bioluminescent assay kit (FL-AA; Sigma). Briefly, cells were grown in 12-well plates, washed with PBS, and dispersed in 85 mM sodium citrate. An aliquot of each sample was removed for cell counting for normalization, and the rest was brought to 2.3% with TCA to arrest ATP metabolism and for storage of samples at 4°C. Just prior to the assay, each sample was neutralized with Tris acetate EDTA buffer (0.1 M Tris, adjusted to pH 7.75 with acetic acid, and 2 mM EDTA), boiled for 3 min, and placed on ice. Samples were placed in black, clear-bottomed 96-well plates in duplicate, and the diluted luciferase reagent was injected as luminescence readings were taken by a FluoStar Optima multimode microplate reader (BMG Labtech, Ortenberg, Germany). Cells were also studied in the presence of aminooxyacetate (AOA), a malate-aspartate shuttle inhibitor. Insulin levels were assessed at 1 h in the presence or absence of HNGF6A after preincubation for 2 h with AOA.
Intracellular calcium measurements
Intracellular calcium levels were determined using the calcium indicator Fura-2/AM (Invitrogen, Carlsbad, CA, USA). Cells were seeded 3 d before the assay in a black, 96-well plate and grown to 70% confluence. The cells were washed with PBS and loaded with 25 μM Fura-2-AM in phenol-red free medium with no FBS for 1 h at 37°C in CO2 incubator. After washing the cells with PBS and aspirating the wash, the plate was placed on ice and growth medium (100 μl) containing the control or HNGF6A (50 ng/ml) was added to the test wells. Low and high standards in the form of buffers A and B, the latter containing 10 mM ionomycin (C-3008; Calcium Calibration Buffer Kit; Life Technologies, Grand Island, NY, USA) were used in 4 wells each. The plate was read for the designated time period at 37 C in a fluorescence plate reader with Softmax software (Molecular Devices Corp., Sunnyvale, CA, USA) to obtain the 340/380 nm fluorescence ratio (ratio of ion-bound to ion-free indicator). Intracellular calcium concentrations were determined as described previously (28).
RESULTS
HNGF6A increases glucose-stimulated insulin secretion (GSIS) in vivo
To determine the effects of HNGF6A on GSIS in vivo, we performed the gold standard hyperglycemic clamps in 3-mo-old male Sprague-Dawley rats. Control and HNGF6A-treated groups were matched in terms of age, gender, body weight, and basal glucose and insulin levels. During the in vivo hyperglycemic clamps, glucose levels were acutely elevated to and maintained at 11 mM. The coefficient of variation of glucose levels during the clamps was 2.98 and 2.55% in HNGF6A and control groups, respectively. As seen in Fig. 1A, B, animals that received HNGF6A (0.07 mg/kg/h) required a significantly higher GIR during the clamp to maintain euglycemia (55.1±0.7 vs. 42.5±1.0 mg/kg/min in HNGF6A vs. controls, respectively, P<0.0001) compared to saline-treated controls. The GIR was significantly different from ∼30 min in HNGF6A-treated animals compared to controls and remained significantly different throughout the duration of the clamp. The insulin levels were higher in the HNGF6A-treated animals (Fig. 1C) throughout the duration of the study. When insulin levels were averaged during the last hour of the clamp under steady-state conditions, insulin levels in the HNGF6A-treated animals were 2-fold higher than in controls (9.9±2.0 vs. 4.61±0.8 ng/ml in HNGF6A vs. controls, respectively, P<0.001; Fig. 1D). HNGF6A did not significantly increase first-phase insulin secretion in vivo in Sprague-Dawley rats (Fig. 1E, F; P=0.34). First-phase insulin secretion in response to HNGF6A was further tested in the C57BL6/N mouse model using IVGTT. After the administration of glucose (2 g/kg), blood glucose and insulin concentration increased in 2 min as expected. Though a trend was apparent, no significant increase was found in first-phase insulin levels in the HNGF6A group over the 30-min period (Fig. 1H, I, P=0.51). The AUC for C peptide tended to be higher in HNGF6A-treated animals (2575.7±360.36 vs. 1871.26±300.63 in HNGF6A vs. controls, respectively, P=0.14) compared to controls (Fig. 1G).
Figure 1.
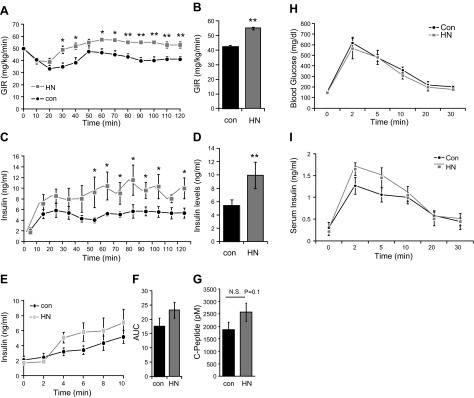
Effects of HNGF6A on GSIS in vivo. A–D) GIR (A, time course; B, average) and insulin levels (C, time course; D, average) in hyperglycemic clamp in young Sprague-Dawley rats (n=18). E–I) Time course (E) and AUC (F) of the effects of HNGF6A on first-phase insulin secretion; C-peptide levels (G); glucose levels (H); and first-phase insulin response (I) in C57BL6/N mice during the IVGTT. Data are presented as means ± sem. *P < 0.05, **P < 0.01.
HNGF6A augments insulin secretion in isolated islets of WT and db/db mice
Impaired GSIS is observed in islets obtained from diabetic mice and humans (29, 30). To confirm the outcomes of the hyperglycemic clamp studies and to extend these observations to an experimental model of diabetes, for the next experiments we isolated islets from 3-mo-old WT and db/db mice. Direct addition of HNGF6A to islets was chosen to avoid any confounding variables introduced by whole-animal studies. The amount of insulin released from db/db islets was lower relative to WT controls under glucose-stimulated (16 mM) conditions (Fig. 2A). As shown, the exposure of islets to 16 mM glucose plus HNGF6A increased insulin release by 3-fold in islets from WT and 2.5-fold in islets from diabetic mice (Fig. 2A).
Figure 2.

Effects of HNGF6A on insulin secretion in islets isolated from WT and db/db diabetic mice and βTC3 cells. A) Insulin levels in the islets isolated from 10- to 12-wk-old C57BLK6/J mice and db/db mice. B) Expression of Rattin (homologue of HN in rodents) and receptors, including GP130, CNTFRa, and WSX-1 in βTC3 cells and mouse pancreas. C) Insulin levels in βTC3 cells treated with or without HNGF6A in the presence or absence of 5 and 16 mM glucose. D) Dose-dependent effects of HNGF6A on insulin secretion. Experiments were repeated ≥3 times; data are presented as means ± sem. *P < 0.05, **P < 0.01 (n=12).
HNGF6A increases GSIS in βTC3 cells
Whole-mouse pancreas and mouse βTC3 cells express endogenous rattin (homologue of HN) in addition to the various receptor components (Fig. 2B). To test whether exogenously added HNGF6A directly affects insulin secretion from the β cells, we characterized basal- and glucose-induced insulin release in the glucose-sensitive βTC3 cells (31, 32). βTC3 cells were incubated in the presence of various concentrations of glucose, and insulin released into medium during a 2-h challenge with or without HNGF6A was measured. An HNGF6A-dependent increase in insulin secretion was seen only in the presence of 16 mM glucose and not in the absence of glucose or at normal glucose levels of 5 mM (Fig. 2C). To identify the optimal dose of HNGF6A, we tested various concentrations of HNGF6A at 16 mM glucose (Fig. 2D). We observed maximal effects at a HNGF6A concentration of 50 ng/ml (Fig. 2D). This HNGF6A concentration (50 ng/ml) was used in all subsequent in vitro experiments unless noted otherwise.
HNGF6A enhances glucose sensing in βTC3 cells
GLUTs and GCK comprise the glucose sensing system, allowing β cells must sense a change in extracellular glucose and initiate GSIS. GLUT2 is the major isoform responsible for glucose transport in β cells, and glucose phosphorylation is the rate-limiting step. To test whether HNGF6A affects GLUT2 translocation, we performed GLUT2 translocation assays in cultured βTC3 cells. Within 15 min, HNGF6A markedly increased membrane-localized GLUT2 (Fig. 3A). This effect was dependent on the presence of glucose (Fig. 3A). GCK activity in HNGF6A-treated β cells was 1.5-fold higher within 15 min of treatment compared to the control β cells (Fig. 3B).
Figure 3.

Effects of HNGF6A on glucose metabolism. A) GLUT2 translocation in βTC3 cells treated with or without HNGF6A in the presence of no glucose (0 mM) or high (16 mM) concentrations of glucose. B) Effects of HNGF6A on glucose phosphorylation rate (GCK activity) in βTC3 cells. C) Effects of HNGF6A on glucose oxidation in βTC3 cells. D) Effects of HNGF6A on intracellular ATP levels in βTC3 cells. E) Time-course effects of HNGF6A on insulin secretion in βTC3 cells. F, G) effects of HNGF6A on intracellular ATP levels (F) and insulin secretion (G) with the presence of AOA an inhibitor of TCA cycle. H) Effects of HNGF6A on mitochondrial membrane potential in βTC3 cells. Experiments were repeated ≥3 times (n=12); data are presented as means ± sem. *P < 0.05.
HNGF6A-induced GSIS is linked to an increase in cellular ATP production
Increased plasma glucose levels initiate increased glucose uptake, glycolysis, and mitochondrial metabolism by the β cell. Cytosolic and mitochondrial reactions generate ATP, and each of these discrete sources of ATP plays an important role in regulating insulin exocytosis (33). Mitochondrial ATP generation, in particular, plays a key role in coupling glucose metabolism with insulin secretion (34). To further explore the potential mechanisms by which HNGF6A may regulate insulin secretion, we analyzed glucose oxidation and ATP production in βTC3 cells. As shown in Fig. 3C, overall glucose oxidation rate, as estimated using 14C-U-glucose, was enhanced by 60% (P<0.05) with HNGF6A treatment in BTC3 cells compared to controls, and a significant increase in ATP production (Fig. 3D) was noted at 60 min incubation (Fig. 3D). Consistent with the insulin secretion data, no significant increase was found in ATP levels when β cells were incubated in 5 mM glucose (data not shown).
To determine the relationship between ATP production and insulin secretion, we studied the time-dependent effects of HNGF6A on insulin exocytosis. Glucose and HNGF6A concentrations were adjusted to 16 mM and 50 mg/ml, respectively, and the insulin released during time periods ranging from 15 to 60 min was measured (Fig. 3E). Although a trend toward HNGF6A-augmented insulin secretion was seen at 15 and 30 min, significant increase was observed at the 60-min time point (Fig. 3E), coinciding with the time point when marked increases in ATP levels were noted (Fig. 3D). Mitochondrial metabolism, as evidenced by increased ATP, is associated with production of reactive oxygen species that could affect mitochondrial integrity. We determined mitochondrial membrane potential (MMP) as a measure of mitochondrial membrane integrity and demonstrate that HNGF6A preserved MMP in the β cells (Fig. 3F).
To investigate the role of mitochondria in HNGF6A-mediated effects, βTC3 cells were pretreated with AOA, an inhibitor of aspartate aminotransferases of the malate-aspartate NADH shuttle (35). A 2-h pretreatment of βTC3 cells with AOA attenuated ATP production in both control and HNGF6A treated cells (Fig. 3G). Along with a reduction in ATP production, AOA also blocked HNGF6A-dependent effects on insulin secretion (Fig. 3H).
Effects of HNGF6A are associated with increase in intracellular calcium and are KATP channel-independent
Several reports in the literature have convincingly demonstrated that glucose regulates insulin secretion by a dual, KATP-dependent and KATP-independent mechanism, and the triggering Ca2+ signal is necessary for both phases of glucose-induced insulin secretion (30). In the presence of 16 mM glucose, HNGF6A gradually increased intracellular Ca2+ levels (Fig. 4A), and these increases in Ca2+ levels become apparent at 60 min (Fig. 4A).
Figure 4.

Effects of HNGF6A are associated with increase in intracellular calcium and are KATP-channel independent. A) Intracellular calcium levels in βTC3 cells grown in 16 mM glucose and treated with or without HNGF6A at 30, 60, and 120 min. B) Effects of HNGF6A on insulin section in the presence of 400 μM diazoxide, a potassium channel activator. Experiments were repeated ≥3 times for calcium (n=16 each) and twice for diazoxide (n=16 each); data are presented as means ± sem. *P < 0.05.
To specifically explore the role of KATP channels in mediating the effects of HNGF6A, we treated cells with diazoxide, a KATP channel opener. As expected, treatment of βTC3 cells with diazoxide significantly decreased insulin secretion. Treatment with diazoxide reduced but did not abolish the HNGF6A-augmented insulin secretion (Fig. 4B).
DISCUSSION
In studies described here, we report novel effects of an HN analog, HNGF6A, on glucose-stimulated-insulin secretion in vivo and in vitro. We show that the ability of HNGF6A to increase insulin secretion is glucose dependent, coupled to ATP production in the β cell, and is KATP-channel independent. Furthermore, these effects on insulin secretion are demonstrable in islets isolated from both WT and diabetic mice.
Based on the model systems used and the in vivo data, it is apparent that the effects of HNGF6A on GSIS demonstrated here are independent of the effects of HNGF6A on insulin action (12) and are distinct from the effects demonstrated with chronic HN treatment in NOD mice, where a decrease in apoptosis and improvement in β-cell survival (24) contributed to improved glucose homeostasis. The effects of HNGF6A on insulin secretion were seen at ∼40 min under hyperglycemic-clamp conditions and persisted throughout the duration of the study. Consistent with the insulin levels, the AUC of the C-peptide tended to be higher in the HNGF6A-treated group. This finding suggests that the higher levels of insulin are the result of increased insulin release from the β cells. Our observation that HNGF6A increased insulin release in response to hyperglycemia in healthy animals in vivo prompted us to test the effects of HNGF6A on diabetic islets, where chronic hyperglycemia is associated with impaired insulin secretion. Indeed, impaired glucose utilization and β-cell metabolism resulting in reduced ATP production are critical pathophysiological features of islets in type 2 diabetes (24). We demonstrate that HNGF6A increases insulin secretion in islets isolated from db/db mice, suggesting that HNGF6A mitigates some of the metabolic abnormalities present in islets in type 2 diabetes.
The in vivo experiments and studies on the islets were done under a stimulating glucose concentration of 16 mM. To understand the effects further, including glucose dependency, direct effects on β cells, and mechanisms, we utilized the well-characterized, glucose-sensitive βTC3 cells. Analogous to what has been reported for the incretin glucagon-like peptide 1 (GLP-1; ref. 36), HNGF6A-augmented insulin secretion was seen only in the presence of hyperglycemia. This observation strongly suggests that glucose metabolism plays a key role in enhanced insulin release by HNGF6A. In addition, this finding also has important clinical implications, as insulin secretagogues, such as the sulfonylurea glibenclamide, that induce insulin release at 2.8 mM (∼40 mg%, hypoglycemia) and 16.8 mM glucose increase the risk of hypoglycemia (37). HNGF6A, through its glucose-sensitive insulin release, could provide a treatment option in diabetes.
The slow kinetics we observed in response to HNGF6A, both in vivo and in vitro, point to the possibility that HNGF6A-enhanced insulin release may be mediated through a time-delayed, KATP-channel-independent mechanism through glucose metabolism. It is well established that increased glucose uptake and metabolism leading to ATP generation are critical for coupling glucose metabolism with insulin secretion (38). Our work demonstrates that exogenously added HNGF6A increased the translocation of GLUT2 and increases GCK activity in β cells. GLUTs and GCK are two major components of a glucose-sensing system in pancreatic β cells and are prerequisites for glucose-stimulated insulin release (38, 39). Activators of GCK have been shown to improve mitochondrial bioenergetics and, therefore, GSIS in islets from both diabetic mice and human islets (40). GCK is linked closely to onset of type 2 diabetes in humans (41), and a mutation in the GCK gene causes early onset of non-insulin-dependent diabetes mellitus (42). Despite the presence of hexokinase, deficiency of GCK in transgenic mice impairs β-cell response specifically to glucose and not the sulfonylurea glibenclamide (43).
Treatment with HNGF6A enhanced glucose sensing and oxidation in β cells. Whereas HNGF6A-enhanced glucose sensing was rather rapid (within 15 min), the earliest time point with a significant HNGF6A-dependent increase in cellular ATP levels was delayed (between 30 min to 1 h, in vitro) and correlated well with the kinetics of insulin secretion in vivo (40 min) and in vitro (1 h). The delayed kinetics provides support for additional regulation beyond the observed GCK activation in the ATP turnover and insulin secretion in response to HNGF6A. β cells use substrate cycling and cytosolic-mitochondria shuttle systems to increase insulin secretion in proportion to glucose (44). The physiological importance of the two NADH shuttle systems in the β cells have been explored in detail (45), and the malate/aspartate shuttle has been shown to play a predominant role in insulin release (46, 47). In our study, AOA inhibition of the malate-aspartate shuttle resulted in loss of both effects of HNGF6A: increased ATP production and insulin secretion. Despite increased ATP production, which will be associated with elevated ROS, HNGF6A preserved MMP. These observations tightly link mitochondrial metabolism and integrity to the ability of HNGF6A to increase insulin secretion.
The generally accepted cascade in GSIS from the β cell is that increase in cytosolic ATP or in the ratio of ATP to ADP closes KATP channels, which leads to plasma membrane depolarization and Ca2+ entry into the cytosol through activation of voltage-dependent Ca2+ channels (VDCCs). The rise in cytosolic Ca2+ concentrations is thought to finally trigger insulin exocytosis. Insulin secretion is biphasic, with a rapid KATP-channel-dependent initial phase followed by a prolonged, KATP-channel-independent augmentation phase of release (29). The ability of HNGF6A to increase insulin secretion even in the presence of diazoxide is consistent with an effect on the augmentation phase and through KATP-channel-independent mechanisms. This finding is also supported by our observations on the lack of an effect on first phase insulin secretion during hyperglycemic clamp as well as IVGTT in vivo.
In summary, the potent effects of HNGF6A on insulin secretion, taken together with the established effects on insulin action, offers a role for HN and its analogs in the treatment of type 2 diabetes. Moreover, our studies, which demonstrate the link between the novel mitochondria-associated peptide HN and specific aspects of glucose uptake and cellular metabolism in the β cell and insulin secretion, provide evidence for the existence of apparent cellular networks among genes, proteins, and metabolites, which, when perturbed, can contribute to disease (48–50). Further work aimed at the exploration of the full spectrum of metabolic couplers should greatly aid our understanding of the mechanism through which HNGF6A increases insulin secretion in health and disease. HNGF6A offers a novel therapeutic option for treatment of diabetes.
Acknowledgments
The authors thank Dr. Natalia V. Cheshenko (Department of Pediatrics, Albert Einstein College of Medicine) for assistance with determination of intracellular calcium concentrations.
This work was supported by grants from the U.S. National Institutes of Health, K08-AG-027462, K08-AG-027462-03S1, and R01-AG-035114 to R.H.M.; P30-AG-038072 to R.K.; R01-AG-618381 and P30-AG-038072 to N.B.; K99/R00 award AG-037574 to D.M.H.; and by the Core laboratories of the Albert Einstein Diabetes Research and Training Center (DK 20541) and Einstein Nathan Shock Center, Cellular and Tissue Aging Core P30-AG-038072.
Disclosure: N.B. is a consultant and a shareholder of CoHBar.
Footnotes
- AOA
- aminooxyactetate
- AUC
- area under the curve
- GCK
- glucokinase
- GIR
- glucose infusion rate
- GLUT
- glucose transporter
- GSIS
- glucose-stimulated insulin secretion
- HN
- humanin
- HNG
- S14G-humanin
- IGF-1
- insulin-like growth factor 1
- IGFBP-3
- insulin-like growth factor binding protein 3
- IVGTT
- intravenous glucose tolerance test
- MMP
- mitochondrial membrane potential
- NOD
- nonobese diabetic
- SP
- scrambled peptide
- WT
- wild type
REFERENCES
- 1. Hashimoto Y., Niikura T., Tajima H., Yasukawa T., Sudo H., Ito Y., Kita Y., Kawasumi M., Kouyama K., Doyu M., Sobue G., Koide T., Tsuji S., Lang J., Kurokawa K., Nishimoto I. (2001) A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proc. Natl. Acad. Sci. U. S. A. 98, 6336–6341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tinoco A. D., Saghatelian A. (2011) Investigating endogenous peptides and peptidases using peptidomics. Biochemistry 50, 7447–7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maximov V., Martynenko A., Hunsmann G., Tarantul V. (2002) Mitochondrial 16S rRNA gene encodes a functional peptide, a potential drug for Alzheimer's disease and target for cancer therapy. Med. Hypotheses 59, 670–673 [DOI] [PubMed] [Google Scholar]
- 4. Yamagishi Y., Hashimoto Y., Niikura T., Nishimoto I. (2003) Identification of essential amino acids in Humanin, a neuroprotective factor against Alzheimer's disease-relevant insults. Peptides 24, 585–595 [DOI] [PubMed] [Google Scholar]
- 5. Le Y., Gong W., Tiffany H. L., Tumanov A., Nedospasov S., Shen W., Dunlop N. M., Gao J. L., Murphy P. M., Oppenheim J. J., Wang J. M. (2001) Amyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J. Neurosci. 21, RC123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harada M., Habata Y., Hosoya M., Nishi K., Fujii R., Kobayashi M., Hinuma S. (2004) N-Formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochem. Biophys. Res. Commun. 324, 255–261 [DOI] [PubMed] [Google Scholar]
- 7. Hashimoto Y., Kurita M., Aiso S., Nishimoto I., Matsuoka M. (2009) Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Mol. Biol. Cell 20, 2864–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ikonen M., Liu B., Hashimoto Y., Ma L., Lee K. W., Niikura T., Nishimoto I., Cohen P. (2003) Interaction between the Alzheimer's survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100, 13042–13047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hashimoto Y., Suzuki H., Aiso S., Niikura T., Nishimoto I., Matsuoka M. (2005) Involvement of tyrosine kinases and STAT3 in Humanin-mediated neuroprotection. Life Sci. 77, 3092–3104 [DOI] [PubMed] [Google Scholar]
- 10. Hashimoto Y., Tsuji O., Niikura T., Yamagishi Y., Ishizaka M., Kawasumi M., Chiba T., Kanekura K., Yamada M., Tsukamoto E., Kouyama K., Terashita K., Aiso S., Lin A., Nishimoto I. (2003) Involvement of c-Jun N-terminal kinase in amyloid precursor protein-mediated neuronal cell death. J. Neurochem. 84, 864–877 [DOI] [PubMed] [Google Scholar]
- 11. Niikura T., Tajima H., Kita Y. (2006) Neuronal cell death in Alzheimer's disease and a neuroprotective factor, humanin. Curr. Neuropharmacol. 4, 139–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muzumdar R. H., Huffman D. M., Atzmon G., Buettner C., Cobb L. J., Fishman S., Budagov T., Cui L., Einstein F. H., Poduval A., Hwang D., Barzilai N., Cohen P. (2009) Humanin: a novel central regulator of peripheral insulin action. PLoS One 4, e6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tajima H., Niikura T., Hashimoto Y., Ito Y., Kita Y., Terashita K., Yamazaki K., Koto A., Aiso S., Nishimoto I. (2002) Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer's disease-related insults. Neurosci. Lett. 324, 227–231 [DOI] [PubMed] [Google Scholar]
- 14. Muzumdar R. H., Huffman D. M., Calvert J. W., Jha S., Weinberg Y., Cui L., Nemkal A., Atzmon G., Klein L., Gundewar S., Ji S. Y., Lavu M., Predmore B. L., Lefer D. J. (2010) Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler. Thromb. Vasc. Biol. 30, 1940–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jung S. S., Van Nostrand W. E. (2003) Humanin rescues human cerebrovascular smooth muscle cells from Abeta-induced toxicity. J. Neurochem. 84, 266–272 [DOI] [PubMed] [Google Scholar]
- 16. Terashita K., Hashimoto Y., Niikura T., Tajima H., Yamagishi Y., Ishizaka M., Kawasumi M., Chiba T., Kanekura K., Yamada M., Nawa M., Kita Y., Aiso S., Nishimoto I. (2003) Two serine residues distinctly regulate the rescue function of Humanin, an inhibiting factor of Alzheimer's disease-related neurotoxicity: functional potentiation by isomerization and dimerization. J. Neurochem. 85, 1521–1538 [DOI] [PubMed] [Google Scholar]
- 17. Arakawa T., Hirano A., Shiraki K., Niikura T., Kita Y. (2011) Advances in characterization of neuroprotective peptide, humanin. Curr. Med. Chem. 18, 5554–5563 [DOI] [PubMed] [Google Scholar]
- 18. Hashimoto Y., Terashita K., Niikura T., Yamagishi Y., Ishizaka M., Kanekura K., Chiba T., Yamada M., Kita Y., Aiso S., Matsuoka M., Nishimoto I. (2004) Humanin antagonists: mutants that interfere with dimerization inhibit neuroprotection by Humanin. Eur. J. Neurosci. 19, 2356–2364 [DOI] [PubMed] [Google Scholar]
- 19. Xu X., Chua C. C., Gao J., Hamdy R. C., Chua B. H. (2006) Humanin is a novel neuroprotective agent against stroke. Stroke 37, 2613–2619 [DOI] [PubMed] [Google Scholar]
- 20. Arakawa T., Niikura T., Tajima H., Kita Y. (2006) The secondary structure analysis of a potent Ser14Gly analog of antiAlzheimer peptide, Humanin, by circular dichroism. J. Pept. Sci. 12, 639–642 [DOI] [PubMed] [Google Scholar]
- 21. Tajima H., Kawasumi M., Chiba T., Yamada M., Yamashita K., Nawa M., Kita Y., Kouyama K., Aiso S., Matsuoka M., Niikura T., Nishimoto I. (2005) A humanin derivative, S14G-HN, prevents amyloid-beta-induced memory impairment in mice. J. Neurosci. Res. 79, 714–723 [DOI] [PubMed] [Google Scholar]
- 22. Sponne I., Fifre A., Koziel V., Kriem B., Oster T., Pillot T. (2004) Humanin rescues cortical neurons from prion-peptide-induced apoptosis. Mol. Cell. Neurosci. 25, 95–102 [DOI] [PubMed] [Google Scholar]
- 23. Mamiya T., Ukai M. (2001) [Gly(14)]-Humanin improved the learning and memory impairment induced by scopolamine in vivo. Br. J. Pharmacol. 134, 1597–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoang P. T., Park P., Cobb L. J., Paharkova-Vatchkova V., Hakimi M., Cohen P., Lee K. W. (2010) The neurosurvival factor Humanin inhibits beta-cell apoptosis via signal transducer and activator of transcription 3 activation and delays and ameliorates diabetes in nonobese diabetic mice. Metabolism 59, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kinasiewicz A., Juszczak M., Pachecka J., Fiedor P. (2004) Pancreatic islets isolation using different protocols with in situ flushing and intraductal collagenase injection. Physiol. Res. 53, 327–333 [PubMed] [Google Scholar]
- 26. Rhodes C. J., Halban P. A. (1987) Newly synthesized proinsulin/insulin and stored insulin are released from pancreatic B cells predominantly via a regulated, rather than a constitutive, pathway. J. Cell Biol. 105, 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davidson A. L., Arion W. J. (1987) Factors underlying significant underestimations of glucokinase activity in crude liver extracts: physiological implications of higher cellular activity. Arch. Biochem. Biophys. 253, 156–167 [DOI] [PubMed] [Google Scholar]
- 28. Cheshenko N., Del Rosario B., Woda C., Marcellino D., Satlin L. M., Herold B. C. (2003) Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 163, 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Straub S. G., Sharp G. W. (2002) Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes/Metabol Res. Rev. 18, 451–463 [DOI] [PubMed] [Google Scholar]
- 30. Henquin J. C., Ishiyama N., Nenquin M., Ravier M. A., Jonas J. C. (2002) Signals and pools underlying biphasic insulin secretion. Diabetes 51(Suppl. 1), S60–S67 [DOI] [PubMed] [Google Scholar]
- 31. Efrat S., Linde S., Kofod H., Spector D., Delannoy M., Grant S., Hanahan D., Baekkeskov S. (1988) Beta-cell lines derived from transgenic mice expressing a hybrid insulin gene-oncogene. Proc. Nat. Acad. Sci. 85, 9037–9041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fleischer N., Chen C., Surana M., Leiser M., Rossetti L., Pralong W., Efrat S. (1998) Functional analysis of a conditionally transformed pancreatic beta-cell line. Diabetes 47, 1419–1425 [DOI] [PubMed] [Google Scholar]
- 33. Jensen M. V., Joseph J. W., Ronnebaum S. M., Burgess S. C., Sherry A. D., Newgard C. B. (2008) Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 295, E1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prentki M. (1996) New insights into pancreatic beta-cell metabolic signaling in insulin secretion. Eur. J. Endocrinol. 134, 272–286 [DOI] [PubMed] [Google Scholar]
- 35. Eto K., Tsubamoto Y., Terauchi Y., Sugiyama T., Kishimoto T., Takahashi N., Yamauchi N., Kubota N., Murayama S., Aizawa T., Akanuma Y., Aizawa S., Kasai H., Yazaki Y., Kadowaki T. (1999) Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science 283, 981–985 [DOI] [PubMed] [Google Scholar]
- 36. Cunningham B. A., Richard A. M., Dillon J. S., Daley J. T., Civelek V. N., Deeney J. T., Yaney G. C., Corkey B. E., Tornheim K. (2003) Glucagon-like peptide 1 and fatty acids amplify pulsatile insulin secretion from perifused rat islets. Biochem. J. 369, 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshida S., Ohishi T., Matsui T., Tanaka H., Oshima H., Yonetoku Y., Shibasaki M. (2010) Novel GPR119 agonist AS1535907 contributes to first-phase insulin secretion in rat perfused pancreas and diabetic db/db mice. Biochem. Biophys. Res. Commun. 402, 280–285 [DOI] [PubMed] [Google Scholar]
- 38. Matschinsky F. M. (1996) Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 45, 223–241 [DOI] [PubMed] [Google Scholar]
- 39. Gong Z., Muzumdar R. H. (2012) Pancreatic function, type 2 diabetes, and metabolism in aging. Int. J. Endocrinol. 2012, 320482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doliba N. M., Qin W., Najafi H., Liu C., Buettger C. W., Sotiris J., Collins H. W., Li C., Stanley C. A., Wilson D. F., Grimsby J., Sarabu R., Naji A., Matschinsky F. M. (2012) Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics. Am. J. Physiol. Endocrinol. Metab. 302, E87–E102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Froguel P., Vaxillaire M., Sun F., Velho G., Zouali H., Butel M. O., Lesage S., Vionnet N., Clement K., Fougerousse F., Tanizawa Y., Weissenbach J., Beckmann J. S., Lathrop G. M., Passa P., Permutt M. A., Cohen D. (1992) Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 356, 162–164 [DOI] [PubMed] [Google Scholar]
- 42. Vionnet N., Stoffel M., Takeda J., Yasuda K., Bell G. I., Zouali H., Lesage S., Velho G., Iris F., Passa P., Froguel P., Cohen D. (1992) Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature 356, 721–722 [DOI] [PubMed] [Google Scholar]
- 43. Terauchi Y., Sakura H., Yasuda K., Iwamoto K., Takahashi N., Ito K., Kasai H., Suzuki H., Ueda O., Kamada N., Jishage K., Komeda K., Noda M., Kanazawa Y., Taniguchi S., Miwa I., Akanuma Y., Kodama T., Yazaki Y., Kadowaki T. (1995) Pancreatic beta-cell-specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J. Biol. Chem. 270, 30253–30256 [DOI] [PubMed] [Google Scholar]
- 44. Cline G. W. (2011) Fuel-stimulated insulin secretion depends upon mitochondria activation and the integration of mitochondrial and cytosolic substrate cycles. Diabetes Metab. J. 35, 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eto K., Suga S., Wakui M., Tsubamoto Y., Terauchi Y., Taka J., Aizawa S., Noda M., Kimura S., Kasai H., Kadowaki T. (1999) NADH shuttle system regulates K(ATP) channel-dependent pathway and steps distal to cytosolic Ca(2+) concentration elevation in glucose-induced insulin secretion. J. Biol. Chem. 274, 25386–25392 [DOI] [PubMed] [Google Scholar]
- 46. Ishihara H., Nakazaki M., Kanegae Y., Inukai K., Asano T., Katagiri H., Yazaki Y., Kikuchi M., Miyazaki J., Saito I., Oka Y. (1996) Effect of mitochondrial and/or cytosolic glycerol 3-phosphate dehydrogenase overexpression on glucose-stimulated insulin secretion from MIN6 and HIT cells. Diabetes 45, 1238–1244 [DOI] [PubMed] [Google Scholar]
- 47. MacDonald M. J., Marshall L. K. (2000) Mouse lacking NAD+-linked glycerol phosphate dehydrogenase has normal pancreatic beta cell function but abnormal metabolite pattern in skeletal muscle. Arch. Biochem. Biophys. 384, 143–153 [DOI] [PubMed] [Google Scholar]
- 48. Nurse P. (2003) Systems biology: understanding cells. Nature 424, 883. [DOI] [PubMed] [Google Scholar]
- 49. Vidal M., Cusick M. E., Barabasi A. L. (2011) Interactome networks and human disease. Cell 144, 986–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matschinsky F. M., Zelent B., Doliba N. M., Kaestner K. H., Vanderkooi J. M., Grimsby J., Berthel S. J., Sarabu R. (2011) Research and development of glucokinase activators for diabetes therapy: theoretical and practical aspects. Handb. Exp. Pharmacol. 203, 357–401 [DOI] [PubMed] [Google Scholar]