

Abstract
Ubiquitination is an important mechanism for the regulation of diverse cellular functions, including proteolysis and DNA repair. The human MutS family protein hMSH4 functions in meiotic recombinational DNA double-strand break (DSB) repair. It was previously observed that hMSH4 interacts with the von Hippel-Lindau binding protein 1 (VBP1), a partner of the VHL ubiquitin E3 ligase as well as a subunit of the prefoldin complex. In this study we address how ubiquitination regulates the homeostasis of hMSH4 in the human embryonic kidney cell line HEK293T. We demonstrate that VBP1 targets hMSH4 for degradation and identify a new VBP1 binding partner, p97, an AAA+ ATPase involved in protein degradation and DNA damage response. VBP1, VHL, and p97 coexist in the hMSH4 immunocomplex and regulate the polyubiquitination of hMSH4. Furthermore, the results of this study demonstrate that VBP1 acts together with p97 to regulate hMSH4 degradation. Overall, this study has revealed a molecular mechanism by which VBP1 controls the levels of hMSH4 by ubiquitination in mitotic cells. Such a mechanism may be important for controlling the role of hMSH4 in regulating homologous recombination and nonhomologous DNA end joining-mediated DSB repair in human cells.—Xu, Y., Her, C. VBP1 facilitates proteasome and autophagy-mediated degradation of MutS homologue hMSH4.
Keywords: ubiquitination, VHL, p97, DSB repair
MutS homologue 4 (MSH4) belongs to the mismatch repair protein family and is essential for meiotic recombination, gamete formation, and fertility (1). Loss of MSH4 in Saccharomyces cerevisiae and Caenorhabditis elegans results in chromosome nondisjunction during meiosis due to a severe reduction in meiotic crossover events (2, 3). Msh4 deficiency in mice causes aberrant chromosome pairing during zygotene and subsequent germ cell loss (4). However, mouse Msh4 colocalizes with meiotic chromosomes from leptonema to pachynema, indicating its role in the process of homologous recombination (4, 5). Like its eukaryotic homologs, hMSH4 interacts with hMSH5 (6–10), and the resulting heterocomplex is suggested to bind to and stabilize recombinational DSB repair intermediates, such as Holliday junction structures (11). Consistent with a potential role for hMSH4 in the regulation of double-strand break (DSB) repair, we recently revealed that hMSH4 exerts an inhibitory effect on nonhomologous end joining (NHEJ)-mediated DSB repair (12). Besides its interaction with hMSH5, hMSH4 also associates with other recombination proteins, including hRAD51/hDMC1 (13), hMLH1 (14), and hMLH3 (15). In addition, the expression pattern of hMSH4 in human tissues is different from that of hMSH5 (6, 16–18), indicating that hMSH4 and hMSH5 might be regulated independently of each other.
Interestingly, it has been shown that hMSH4 interacts with VBP1 (16), a binding partner of the Von Hippel-Lindau (VHL) ubiquitin E3 ligase (19, 20), also known as GIM2/PAC10 in yeast, and a subunit of the prefoldin complex (21). VBP1 is involved in the formation of the VHL-elongin B-elongin C complex (VBC), in which the assembly of VBC requires the chaperonin TRiC/CCT (22–24), and the latter is often associated with prefoldin (25). VBP1 is also known to promote the formation and assembly of α- and γ-tubulins (26) while VHL is reported to associate with and stabilize microtubules (27), indicating that VBP1 and VHL share similar functions. Furthermore, VHL is well known to target the α subunit of hypoxia-induced factor for proteasome-mediated degradation (28), thereby raising the possibility that VBP1 may function in protein degradation pathways. In fact, it has been shown that VBP1 bridges the interaction between HIV-1 integrase and VHL, targeting integrase for destruction (29). Recently Drosophila VBP1 homologue Mgr is found to regulate tubulin degradation together with VHL (30).
The aforementioned degradation of HIF1α also requires the participation of p97 (31). Specifically, studies show that UBXD7 interacts with both CUL2/VHL ligase and HIF1α, and recruits p97 to HIF1α for proteasome-mediated degradation (31, 32). The homohexameric AAA+ ATPase p97, also called VCP and Cdc48 in yeast, performs a wide range of cellular functions, including protein degradation, cell cycle regulation, membrane fusion, and autophagy (33, 34). One of the p97-mediated protein degradation pathways requires the UFD1-NPL4 heterodimer, in which the p97-UFD1-NPL4 complex recognizes the ubiquitin chains of the substrates and shuttles the substrates to the proteasome (35). Accumulating evidence has indicated that p97 mediates the regulation of both DNA replication and repair (36–39).
In this study we report a role for VBP1 in facilitating hMSH4 destruction through multiple proteolytic pathways. We identify p97 as a novel binding partner of VBP1 and show that VBP1, VHL, and p97 function in concert to regulate hMSH4 polyubiquitination and degradation.
MATERIALS AND METHODS
Expression vectors
The generation of the mammalian expression constructs Myc-VBP1, VBP1-DsRed, Flag-hMSH4, Myc-hMSH4, Myc-hMSH4sv, and Myc-hMSH4aa267–936 were previously described (16, 40, 41). VBP1 was also cloned into the pcDNA6/V5-HisB vector to generate 3xFlag-VBP1. To generate shRNA constructs, VBP1 shRNA sh-1 (GGAAGACCTTGACTTTCTTCGAG) and VBP1 shRNA sh-2 (CAAGGATGACTCTACCAAGAACA) oligonucleotides were annealed and cloned into the pmH1P-neo plasmid as described previously (42). HA-ubiquitin constructs (WT, K48, and K63) were gifts from Dr. K.-L. Lim. Myc-p97 and Myc-p97EQ were gifts from Dr. K. Ramadan (University of Zürich-Vetsuisse, Zurich, Switzerland). Flag-VHL was a gift from Dr. M. Matsumoto (Kyushu University, Fukuoka, Japan).
Antibodies and reagents
Anti-Myc antibody (631206) and anti-GFP antibody (632375) were purchased from Clontech (Mountain View, CA, USA). Anti-DsRed antibody (632397) and anti-VHL antibody (556347) were purchased from BD Biosciences (San Jose, CA, USA). Anti-HA antibody (MMS-101P) was purchased from Covance (Princeton, NJ, USA). Anti-α-tubulin antibody (T6199) and anti-Flag M2 antibody (F3165) were from Sigma-Aldrich (St. Louis, MO, USA). Anti-VBP1 antibody (sc-19837) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-VHL (2738), anti-p97 (2648), and anti-LC3A/B (4108) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Purified anti-hMSH4 antibody was described previously (16). Cycloheximide, MG-132, leupeptin, NH4Cl, and chloroquine (CQ) were purchased from Sigma-Aldrich. siRNAs were purchased from Qiagen (Valencia, CA, USA).
Cell culture and transfection
HEK293T and U2OS cells were maintained in DMEM/high-glucose (HyClone, Logan, UT, USA) supplemented with 5% FBS, 5% NBS, 100 U/ml penicillin and 100 μg/ml streptomycin. Most plasmid DNA transfections were carried out using standard calcium phosphate method. The siRNA transfections were carried out by the use of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. VBP1 sh-2 was transfected into HEK293T cells using Amaxa Nucleofector (Lonza Group, Basel, Switzerland).
Western blotting and immunoprecipitation
Cells were lysed in CelLytic M cell lysis reagent (Sigma-Aldrich) supplemented with 1X protease inhibitor cocktail (Thermo Scientific, Waltham, MA, USA). The preparation of HEK293T whole-cell extracts was described previously (12). The protein concentrations of each lysate were measured using the Bradford protein assay reagent (Bio-Rad, Richmond, CA, USA) on a 96-well plate reader (PerkinElmer, Boston, MA, USA). Proteins were separated by SDS-PAGE, transferred onto nitrocellulose membranes, and immunoblotted with indicated antibodies. For immunoprecipitation, cells were lysed in 500 μl lysis reagent. Ninety percent of the total lysates were incubated with 5 μg antibodies for 1.5 h and with protein A/G agarose beads (Invitrogen) for 1 h. The beads were then washed 3 times with Nonidet P-40 lysis buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 10 mM EDTA; and 0.5% Nonidet P-40) and once with RIPA buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, 1% Nonidet P-40, 0.1% SDS, and 0.5% sodium deoxycholate). Immunocomplexes were boiled in Laemmli sample buffer for 10 min before they were subjected to SDS-PAGE. Images were captured either by X-ray films or by Fujifilm LAS 4000 (linear mode; Fuji Photo Film Ltd, Elmsford, NY, USA). Quantifications of all the band intensities were performed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
Protein degradation assay
Cells were transfected with a construct encoding Myc-hMSH4 in the presence or absence of Myc-VBP1. For half-life studies, cycloheximide (100 μg/ml) was added to the medium 24 h post-transfection. Cells were harvested and lysed for immunoblotting analysis at various time points.
In vivo ubiquitination assay
Cells were transfected with HA-Ub and Myc-hMSH4. At 24 h post-transfection, cells were lysed, and immunoprecipitation was performed with an anti-HA antibody and an anti-hMSH4 antibody for reverse immunoprecipitation. Immunocomplexes were resolved by SDS-PAGE, and Western blotting was performed with indicated antibodies. Knockdown of VBP1 was performed 48 h before transfection.
Immunofluorescence microscopy
Cells were grown on glass coverslips, fixed in 3% paraformaldehyde for 12 min, permeabilized in 100% methanol for 10 min at −20°C, blocked in PBS with 5% FBS, and incubated with primary antibody overnight (1:200). After washing with PBS, cells were incubated with Alexa 488 or 555-conjugated secondary antibodies (1:500). Images were captured using a Leica Leitz DMRB microscope (Leica Microsystems, Wetzlar, Germany).
RESULTS
VBP1 mediates hMSH4 protein turnover
It is known that VBP1 plays a role in VHL-mediated protein degradation and interacts with hMSH4 (16, 29, 30), raising the possibility that the hMSH4 and VBP1 interaction may facilitate hMSH4 degradation. To test that, an shRNA construct encoding VBP1 sh-2 was used to silence VBP1 in HEK293T cells (Fig. 1A and Supplemental Fig. S1A). RNAi-mediated depletion of VBP1 resulted in a significant increase in the amount of overexpressed as well as endogenous hMSH4 protein (Fig. 1A, B). Similar results have also been obtained by the use of U2OS cells, indicating that this observation is not cell line specific (Supplemental Fig. S1B). Interestingly, similar levels of hMSH4 elevation could be achieved by the proteasome inhibitor MG-132, in which the effects of VBP1 silencing were largely masked (Fig. 1C), suggesting that VBP1 might act together with the proteasome pathway to facilitate hMSH4 degradation. Consistent with this, silencing of VHL by RNAi also led to the accumulation of hMSH4 protein (Fig. 1D), indicating that the VBP1-VHL pathway mediates hMSH4 degradation. However, as illustrated later in this study, hMSH4 degradation facilitated by high levels of VBP1 cannot be suppressed by MG-132, indicating the involvement of different downstream mechanisms.
Figure 1.

The VBP1-VHL pathway regulates the stability of hMSH4. A) HEK293T cells were transfected with VBP1 shRNA sh-2 or control shRNA constructs using the Nucleofector. After 48 h, cells were transfected with equal amounts of Myc-hMSH4. Immunoblotting was carried out as indicated. Expression construct encoding GFP was used as a control for transfection efficiency, and α-tubulin was used as a loading control. Quantifications were performed using the ImageJ software. Data are presented as means ± sd from 3 independent experiments. *P < 0.05. B) Cell lysates were prepared from HEK293T and HEK293T cells stably expressing VBP1 shRNA sh-2 and subjected to immunoblotting. C) HEK293T cells were transfected with indicated plasmids as described in A. At 24 h post-transfection of Myc-hMSH4, cells were treated with the proteasome inhibitor MG-132 for 12 h before collection. D) HEK293T cells were transfected with VHL siRNA using Lipofectamine 2000. After 48 h, cells were transfected with Myc-hMSH4 before they were subjected to immunoblotting.
VBP1 interacts with p97
It has been reported that p97 facilitates protein turnover and functions together with VHL in mediating HIF1α degradation (31, 32). To test whether VBP1-mediated hMSH4 degradation involves p97, we examined the interaction between VBP1 and p97. The results of our coimmunoprecipitation analysis indicated that VBP1 interacted with p97 in vivo (Fig. 2A). Furthermore, this interaction is independent of p97 ATPase activity, as the p97 dominant-negative mutant E578Q did not affect the interaction between VBP1 and p97 (Supplemental Fig. S2A and refs. 43, 44). We further validated their interaction in HEK293T whole cell extracts. Both forward and reverse coimmunoprecipitation experiments confirmed that endogenous VBP1 interacts with p97 (Fig. 2B), while both VHL and p97 proteins were detectable in VBP1 immunoprecipitates (Fig. 2C). Furthermore, VHL interacted with VBP1 and p97 (Supplemental Figs. S2B and S3A), indicating that VBP1, VHL, and p97 act coordinately in the same complex.
Figure 2.

VBP1 interacts with p97. A) HEK293T cells were transfected to express 3xFlag-VBP1 and Myc-p97. Cells were lysed 24 h post-transfection, and 10% of cell lysates were saved as input. VBP1 was immunoprecipitated with an anti-Flag antibody, and p97 was examined using an anti-Myc antibody. Co-IP, coimmunoprecipitation. B) Whole-cell extracts (WCE) of HEK293T cells were prepared, and immunoprecipitation was carried out using either anti-VBP1 or anti-p97 antibody. IgG was used as a negative control. Asterisk indicates Ig light chain. C) Both parental HEK293T cells and those transfected with Myc-VBP1 were treated with 5 μM MG-132 for 12 h to block proteasome-mediated protein degradation. Cell lysates were then subjected to immunoprecipitation using an anti-Myc antibody. Endogenous p97 and VHL were analyzed in the VBP1 immunocomplex.
hMSH4 associates with the VBP1-VHL-p97 complex
To test whether hMSH4 is a substrate of the VBP1-VHL-p97 complex, we first validated the interaction between hMSH4 and VBP1 in human cells (Fig. 3A). Then we revealed that both VHL and p97 were present in the hMSH4 immunoprecipitates (Fig. 3B), and hMSH4 interacted with both overexpressed and endogenous VHL (Supplemental Fig. S3).
Figure 3.

hMSH4 interacts with the VBP1-VHL-p97 complex. A) Interaction between hMSH4 and VBP1 was analyzed in HEK293T cells by reciprocal coimmunoprecipitation (co-IP) as indicated. B) VHL and p97 coexist in the hMSH4 immunocomplex. HEK293T cells transfected with empty vector or Myc-hMSH4 were subjected to immunoprecipitation using an anti-Myc antibody. C) VBP1 modulates the interaction between hMSH4 and p97. HEK293T cells were first transfected with control shRNA or VBP1 shRNA sh-2 to knock down VBP1 and were then transfected with Myc-hMSH4. At 24 h post-transfection, cells were treated with MG-132 and subjected to immunoprecipitation using an anti-Myc antibody, and immunoblotting was carried out as indicated. D) HEK293T cells were transfected with p97 siRNA using Lipofectamine 2000 before transfection with Myc-hMSH4. The protein level of hMSH4 was examined by immunoblotting.
Furthermore, we analyzed the effect of VBP1 on the hMSH4 and p97 interaction. Specifically, MG-132 was used to equalize the amounts of hMSH4 protein in the control and VBP1 knockdown cells. Interestingly, the results of image quantification indicate that VBP1 knockdown lead to an ∼20% reduction on the interaction between hMSH4 and p97 (Fig. 3C), raising the possibility that VBP1 may modulate the bridging between hMSH4 and p97. Moreover, depletion of p97 using RNAi resulted in an accumulation of hMSH4 protein (Fig. 3D). Together, these results demonstrate that the VBP1-VHL-p97 complex recognizes and binds to hMSH4 protein, thereby harnessing hMSH4 protein turnover.
VBP1 promotes hMSH4 polyubiquitination and degradation
To explore the mechanisms by which VBP1 regulates the stability of hMSH4 protein, we utilized 2 hMSH4 variants, hMSH4sv and hMSH4aa267–936, to determine whether the hMSH4-VBP1 interaction played a role in the process. The hMSH4sv, a splicing variant lacking the carboxyl-terminal helix-turn-helix motif, is able to interact VBP1, whereas hMSH4aa267–936 is deficient (7, 16). We first confirmed that the hMSH4sv variant interacts with VBP1, while the N-terminal deletion mutant hMSH4aa267–936 does not interact with VBP1 (Fig. 4A). Clearly the levels of hMSH4sv protein accumulation increased on VBP1 depletion; however, under the same experimental conditions, the levels of hMSH4aa267–936 protein did not respond to VBP1 silencing (Fig. 4B). These results indicate that VBP1-mediated degradation of hMSH4 requires a physical interaction with hMSH4. We then tested the stability of hMSH4 protein in VBP1-silenced cells, in which we found that loss of VBP1 could extend the half-life of hMSH4 (Fig. 4C). Because both VHL and p97 were known to participate in the process of protein ubiquitination, we speculated that VBP1 might play a role in the polyubiquitination of hMSH4. As shown in Fig. 4D and Supplemental Fig. S4A, RNAi-mediated knockdown of VBP1 reduced the formation of polyubiquitinated hMSH4 by ∼40%. Consistent with the function of VHL as an E3 ligase, VHL knockdown resulted in a 50% reduction of polyubiquitinated hMSH4 (Fig. 4D). On the other hand, RNAi-mediated knockdown of p97 led to a 1.5-fold increase of hMSH4 polyubiquitination (Fig. 4D), suggesting the levels of hMSH4 polyubiquitination are ultimately determined by the coordinated actions of the VBP1-VHL-p97 complex. This result is compatible with the view that p97 facilitates the transfer of polyubiquitinated proteins to degradation machineries such as the proteasome (44). In addition, we determined whether the formation of polyubiquitin chains on hMSH4 was mediated through Lys48 of ubiquitin, a common signal for protein degradation. Specifically we expressed hMSH4 protein together with HA-tagged K48 and K63 ubiquitin, ubiquitin polypeptides possessing only K48 or K63 lysine residue (45), and we examined the levels of hMSH4 polyubiquitination. To our surprise, hMSH4 polyubiquitination could be in the forms of either K48- or K63-linked ubiquitin chains, and knockdown of VBP1 reduced both K48 and K63 mediated polyubiquitination (Fig. 4E). Of note, increasing evidence indicates that, in addition to signaling, polyubiquitination through K63-linked ubiquitin chains can also mediate protein degradation (46, 47). Taken together, these data support the scenario that VBP1 functions at an early step in the process of hMSH4 polyubiquitination and degradation.
Figure 4.

VBP1 promotes hMSH4 polyubiquitination and degradation. A) Interaction of VBP1 with the hMSH4 mutants hMSH4sv or hMSH4aa267–936. HEK293T cells were transfected with 3xFlag-VBP1 together with one of the 2 hMSH4 mutants Myc-hMSH4sv or Myc-hMSH4aa267–936. Immunoprecipitation was carried out using an anti-Flag antibody. B) Effects of VBP1 knockdown on the protein levels of hMSH4sv or hMSH4aa267–936. HEK293T cells were transfected with a control shRNA or the VBP1 shRNA sh-2 before they were transfected to express the hMSH4 mutants. The protein levels of Myc-hMSH4sv or Myc-hMSH4aa267–936 were analyzed by immunoblotting and quantified by the ImageJ software. Data are presented as means ± sd. NS, no significant difference. *P < 0.05. C) Effect of VBP1 knockdown on the levels of hMSH4 protein. HEK293T cells transfected with a control shRNA or VBP1 shRNA sh-2 were transfected with Myc-hMSH4. After 24 h, approximately equal numbers of cells were seeded into 6-cm dishes in the presence of cycloheximide (100 μg/ml). Cells were collected and subjected to Western blotting at various time points as indicated. D) VBP1, VHL, or p97 was silenced in HEK293T cells as indicated. Cells were then transfected with Myc-hMSH4 and HA-Ub, treated with MG-132 for 12 h, and subjected to immunoprecipitation using an anti-HA antibody. Immunoblotting was carried out as indicated. E) Same procedure as in D, except that HA-Ub was replaced by HA-Ub K48 or K63. Asterisks mark nonspecific bands.
VBP1 is codegraded with hMSH4
While investigating the role of VBP1 in the process of hMSH4 degradation, we observed that the levels of endogenous VBP1 were significantly reduced in cells expressing both hMSH4 and ubiquitin in comparison to cells expressing ubiquitin alone (Fig. 5A). A similar observation was also made in cells expressing hMSH4 alone (Fig. 5A, bottom panel). These results suggest that hMSH4 degradation is concomitantly associated with the down-regulation of VBP1. Interestingly, the reduction of VBP1 protein could be rescued by MG-132 treatment, indicating that the reduction of VBP1 is dependent on the proteasome pathway. Consistent with this, the levels of polyubiquitinated VBP1 were elevated under such conditions (Fig. 5B). These results support the idea that the polyubiquitination and degradation of hMSH4 is intrinsically associated with VBP1 polyubiquitination and destruction. It is conceivable that this codegradation mechanism may play a role in fine-tuning hMSH4 levels in cells; e.g., reduction of VBP1 is expected to up-regulate hMSH4. We further observed that the interaction between hMSH4 and VBP1 was abolished following VBP1 ubiquitination, but this interaction was not affected when only hMSH4 was ubiquitinated (Supplemental Fig. S5), suggesting that VBP1 ubiquitination can abrogate the action of VBP1 in promoting hMSH4 degradation.
Figure 5.
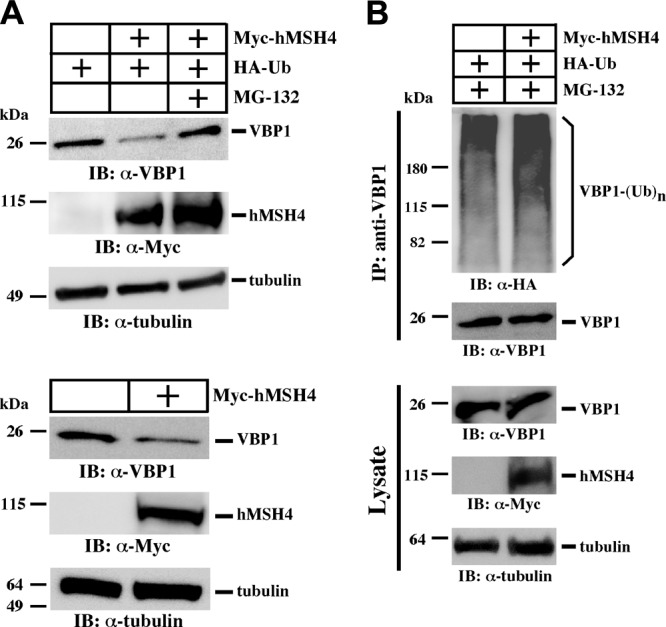
VBP1 is codegraded with hMSH4. A) HEK293T cells were transfected with Myc-hMSH4 and HA-Ub and treated with MG-132. Cells transfected with Myc-hMSH4 alone were used as additional controls. Levels of VBP1 and hMSH4 were analyzed by Western blotting. B) HEK293T cells were transfected with HA-Ub and Myc-hMSH4 or empty vector; they both were treated with MG-132. Immunoprecipitation was carried out using an anti-VBP1 antibody. Levels of ubiquitinated VBP1 were analyzed by immunoblotting with an anti-HA antibody.
VBP1 exerts a biphasic effect on the levels of hMSH4
To evaluate how up-regulation of VBP1 affects the abundance of hMSH4, we transfected cells with hMSH4 and VBP1 expression constructs and analyzed protein levels under overexpression conditions. Intriguingly, the results demonstrated that VBP1 exerted a dose-dependent effect on the levels of hMSH4 (Fig. 6A and Supplemental Fig. S6). Low expression levels of VBP1 did not lead to the reduction of hMSH4, whereas high levels of VBP1 decreased hMSH4. This effect was not observed in a similar experiment where GFP was expressed with VBP1, suggesting that this effect is hMSH4 specific (Fig. 6A). RT-PCR analysis confirmed that VBP1-mediated hMSH4 regulation was not due to changes in hMSH4 transcription (Fig. 6A). In addition, we found that high levels of VBP1 predominantly enhance K63-linked polyubiquitination of hMSH4, whereas K48-linked polyubiquitination is not affected (Supplemental Fig. S7). These observations indicate that the regulation of hMSH4 by VBP1 is controlled in a stoichiometric fashion. In light of this, all subsequent experiments were performed under the same VBP1 transfection conditions that promote hMSH4 degradation.
Figure 6.

Overexpression of VBP1 triggers autophagy-mediated degradation of hMSH4. A) HEK293T cells were transfected with Flag-hMSH4 along with different amounts of Myc-VBP1. At 24 h post-transfection, cell lysates were prepared for immunoblotting analysis. RT-PCR was performed separately to examine the levels of hMSH4 mRNA. Quantifications of hMSH4 protein levels were performed using the ImageJ software. HEK293T cells transfected with GFP and different amounts of Myc-VBP1 were subjected to immunoblotting as a specificity control. B) HEK293T cells stably expressing Flag-hMSH5 were transfected with Flag-hMSH4 and high amounts of Myc-VBP1 and subjected to immunoblotting. C) HEK293T cells were transfected with Myc-hMSH4, Myc-hMSH4sv, or Myc-hMSH4aa267–936 together with high amounts of Myc-VBP1. At 24 h post-transfection, approximately equal numbers of cells were seeded into 6-cm dishes in the presence of cycloheximide (100 μg/ml). At various indicated time points, cells were collected for Western blotting analysis. D–F) HEK293T cells were transfected with Flag-hMSH4 and one of the VBP1 (D), VHL (E), or p97 (F) constructs. At 24 h post-transfection, cells were treated with MG-132 for 12 h or NH4Cl for 24 h as indicated and subjected to immunoblotting. G) U2OS cells transfected with Flag-hMSH4 and with either empty vector or Myc-VBP1 were treated with 50 μM CQ for 3 h. Cells were then fixed and stained with anti-Flag (green) and anti-LC3 (red) antibodies. Arrows indicate the colocalization of hMSH4 with LC3; arrowheads highlight hMSH4 puncta that are not associated with LC3. Scale bars = 50 μm. Quantification of the association between hMSH4 puncta and LC3 is also provided. More than 100 cells were counted. Error bars represent sd from 2 independent experiments. H) HEK293T and HEK293T cells transfected with Myc-VBP1 were subjected to immunoblotting for LC3. Where indicated, cells were treated with 50 μM CQ for 3 h.
Similar to the observations described above, hMSH4 degradation under VBP1 overexpression is dependent on their interaction. It is known that hMSH5 and VBP1 can compete with one another for binding to hMSH4 (16); therefore the increased levels of hMSH5 should be expected to block the effect of VBP1 on hMSH4. Indeed, the levels of hMSH4 remained unchanged with VBP1 overexpression in the stable cell line expressing hMSH5 (HEK293T/flag-hMSH5; Fig. 6B), suggesting that hMSH5 stabilizes hMSH4 by blocking the interaction between hMSH4 and VBP1 (Supplemental Fig. S8). Consistent with these observations, VBP1 overexpression led to significant reductions of hMSH4 and hMSH4sv protein levels at 3 h post-CHX treatment, whereas the levels of hMSH4aa267–936 were unchanged (Fig. 6C), likely attributable to the lack of interaction between hMSH4aa267–936 and VBP1 (Fig. 4A). Together, the physical interaction between hMSH4 and VBP1 is required for hMSH4 degradation.
VBP1 mediates hMSH4 degradation through proteasome and autophagy pathways
We next set out to identify the pathway by which overexpressed VBP1 mediates hMSH4 degradation. Surprisingly, VBP1-mediated hMSH4 degradation could not be completely reversed by the addition of MG-132, whereas treating of these cells with NH4Cl, an effective lysosomal proteolysis inhibitor (48), led to a significant accumulation of hMSH4 protein (Fig. 6D). To confirm that this effect of VBP1 was related to VHL and p97, we further analyzed VHL and p97 in this process. Although hMSH4 degradation-mediated by VHL overexpression could be partially blocked by either MG-132 or NH4Cl (Fig. 6E), only NH4Cl could halt hMSH4 degradation triggered by p97 overexpression (Fig. 6F). These results raised the possibility that VBP1 acts in the same pathway as p97 to mediate hMSH4 degradation, in which autophagy is predominantly utilized for hMSH4 degradation when VBP1 levels are high. The demonstration that hMSH4 underwent VBP1-mediated autophagy-dependent degradation was recapitulated with another autophagy inhibitor CQ (Supplemental Fig. S9).
To further validate the involvement of autophagy in VBP1-mediated hMSH4 degradation, we examined the colocalization of hMSH4 and LC3; the latter is widely used as a marker for autophagosomes (48–50). In cells treated with CQ, ∼50% of the hMSH4 puncta were associated with LC3 (Fig. 6G). Furthermore, elevation of VBP1 levels significantly enhanced the colocalization between hMSH4 and LC3 (Fig. 6G), suggesting the involvement of VBP1 in the autophagic turnover of hMSH4. In addition, the amount of the lipidated form of LC3 (LC3-II), a common marker for autophagosome (48), was increased in VBP1-overexpressed cells (Fig. 6H). As controls, similar levels of LC3-II induction were observed in CQ-treated cells independent of VBP1 overexpression (Fig. 6H). Taken together, these results demonstrate that elevated levels of VBP1 predominantly mediate hMSH4 protein degradation through the autophagy-lysosome system.
Finally, we analyzed time-dependent hMSH4 protein decay and the effects of 3 protein degradation inhibitors MG-132, CQ, and leupeptin. When protein synthesis was inhibited by CHX in cells with endogenous levels of VBP1, the turnover of hMSH4 protein had a half-life of ∼8 h (Fig. 7A). Addition of any of the 3 protein degradation inhibitors (MG-132, CQ, or leupeptin) could prolong the half-life of hMSH4 protein, confirming that the proteasome and autophagy pathways control the degradation of hMSH4 proteins. However, in cells with high levels of VBP1, only CQ could reduce the degradation rate of hMSH4 (Fig. 7B). This result is consistent with our finding that CQ rescues VBP1-mediated autophagy-dependent degradation of hMSH4.
Figure 7.

Multiple pathways mediate hMSH4 degradation. Levels of hMSH4 proteins were determined in triplicates under various conditions as indicated. Immunoblots were quantified, and the data are presented as graphs with sd. A) HEK293T cells were transfected with Myc-hMSH4. At 24 h post-transfection, cells were treated with cycloheximide (100 μg/ml) together with MG-132 (5 μM) CQ (50 μM) or leupeptin (100 μM) for 8, 16, and 24 h. Cell lysates were prepared at indicated time points and analyzed by immunoblotting. Images were quantified by the use of the ImageJ software, and levels of hMSH4 were normalized to the levels of α-tubulin. All levels of hMSH4 were compared to those of the t = 0 controls. B) HEK293T cells were transfected with both Myc-hMSH4 and Myc-VBP1, followed by the same drug treatments as described in A. Cells were harvested at 0, 4, and 8 h. C) A working model for VBP1-dependent proteasome- and autophagy-mediated hMSH4 degradation is proposed.
DISCUSSION
The degradation of polyubiquitinated proteins is largely governed by mechanisms involved with the proteasome and/or the autophagy pathways. Ubiquitin-mediated protein degradation is also known to play important roles in many cellular processes, including DNA damage repair, immune response, cell survival, and programmed cell death (51). It is noteworthy that inhibition of autophagy-based protein degradation is currently being explored as a potential strategy to sensitize cancer cells to conventional anticancer therapies (52).
The results of this study support a scenario in which the levels of hMSH4 protein are monitored and controlled by ubiquitin-mediated protein degradation (Fig. 7C). Specifically, we demonstrated that the binding of VBP1 to hMSH4 recruits the VHL-associated E3 ligase complex, triggering the polyubiquitination of hMSH4. Then VBP1 remains to interact with hMSH4-Ub and facilitates the recruitment of p97 to the complex. p97 recognizes hMSH4-Ub conjugates and targets them for degradation, during which VBP1 is also ubiquitinated, presumably by VHL, and targeted for turnover. In these processes VBP1 functions as a bridge between hMSH4, VHL, and p97. Alternatively, VBP1 polyubiquitination may serve as a mechanism to block VBP1 and hMSH4 interaction, thereby facilitating hMSH4 up-regulation. Although the molecular basis underlying the concentration-dependent biphasic effect of VBP1 on the stability of hMSH4 is presently unknown, it is conceivable that a moderate increase of VBP1 might trigger the recruitment of other prefoldin subunits to form hMSH4-prefoldin complexes that may interfere with proteasome-mediated degradation (53). However, a further increase of VBP1 will certainly shift the balance toward autophagy-mediated hMSH4 degradation.
Polyubiquitinated proteins, commonly produced by the ubiquitin E1-E2-E3 enzyme system, are often targeted to the 26S proteasome for proteolysis (54). The E3 ligase generally provides the substrate specificity in the process. One large class of the E3s is the Cullin-RING ligase (CRL) family, which contains 7 CRL E3 ligases (55). Each CRL utilizes its own set of adaptors and substrate recognition proteins for different clients. In the case of CUL2, within the VBC complex Elongin B/C serves as the adaptor, and VHL serves as the recognition protein (56). To date, only a few proteins are known to be recognized directly by the VBC complex as substrates (28). Recently 2 studies revealed that VBP1 could act as an adaptor molecule in VHL-promoted degradation of target proteins (29, 30). Our data suggest that VBP1 plays a similar role toward the degradation of hMSH4, thereby extending the spectrum of CUL2/VHL substrates.
The role of p97 was extensively studied in endoplasmic reticulum-associated protein degradation, in which misfolded protein substrates are polyubiquitinated and recognized by p97 for retrotranslocation to the proteasome (44). This requires the p97 substrate-recruiting cofactor UFD1-NPL4. Most p97-associated substrate-recruiting cofactors possess ubiquitin-binding domains, like UFD1 and p47, but p97 also interacts with substrate-processing cofactors to control the degree of ubiquitination of substrates (33, 57). A variety of UBX (structurally similar to ubiquitin) domain-containing cofactors are being identified as new adaptors of p97 (31). They all contain the UBX domain but may lack the ubiquitin-associated domain for ubiquitin binding. Our data indicate that VBP1 is a novel cofactor of p97. This is consistent with a previous observation that yeast Pac10 and Cdc48, homologs of human VBP1 and p97, coexist in the same complex (58). Although VBP1 has only the prefoldin domain, it functions like a UBX protein to directly bind to the protein substrate hMSH4. Nevertheless, more studies are needed to gain a better understanding of the molecular interplay between p97 and VBP1.
Another interesting observation of the present study is that hMSH4 polyubiquitin chains can form by either Lys48 or Lys63 linkages. It is commonly accepted that different polyubiquitin chain linkages determine the fates of modified proteins (59). Generally, Lys48- and Lys11-polyubiquitin chains are signals for degradation by the proteasome (60), whereas Lys63-linked chains mediate cellular signaling and autophagic-lysosomal clearance of inclusions (45, 46, 50). Our data are compatible with the model that VBP1-VHL promotes Lys48-linked polyubiquitination of hMSH4, which is then recognized for proteasomal degradation. Conversely VBP1-mediated Lys63-linked polyubiquitination of hMSH4 may be targeted by p97 for autophagic-lysosomal degradation. This is supported by the observation that, under high VBP1 overexpression conditions, the levels of hMSH4 can be rescued by NH4Cl and CQ but not by MG-132 treatment. Therefore, it is plausible that VBP1 may control the switching from proteasome to autophagy by partnering with p97, a protein that has been previously implicated in autophagy-mediated protein degradation (35, 61). A similar scenario has been proposed in the degradation of active Src in cancer cells, in which FAK deficiency led to the activation of autophagy and autophagic turnover of active Src (62). However, it is also possible that high levels of VBP1 preferentially promote Lys63-linked polyubiquitination of hMSH4, which is then recognized by p62 for autophagy-lysosomal degradation (63, 64).
Intriguingly, our data show that VBP1 commits “suicide” in the process of hMSH4 degradation, possibly representing a mechanism by which the up-regulation of hMSH4 can be easily achieved. It is particularly pertinent that the increased expression of the hMSH4 counterpart in mice is well correlated with the down-regulation of VBP1 at the onset of male meiosis (40), during which Msh4 plays an important role in the process of recombinational DSB repair (4). In mitotic cells, however, recombination has to be strictly regulated as recombination-based DSB repair between repetitive elements can lead to chromosome translocation and loss of heterozygosity, thereby generating genomic instability and consequentially promoting cancer development (65). In addition, hMSH4 exhibits a strong suppressive effect on NHEJ-mediated DSB repair (12). Our observations support a scenario that the levels of hMSH4 are differentially regulated in meiotic and mitotic processes, keeping NHEJ activity at low levels during meiosis is necessitated for the maintenance of genomic integrity, while efficient NHEJ in mitotic cells is essential for survival. Hence, an in-depth understanding of the molecular events involved with the regulation of hMSH4 may create a new dimension toward the development of effective strategies for sensitizing cancer cells to genotoxic therapies.
Supplementary Material
Acknowledgments
The authors thank K.-L. Lim (National Neuroscience Institute, Singapore), K. Ramadan (University of Zürich-Vetsuisse, Zurich, Switzerland), and M. Matsumoto (Kyushu University, Fukuoka, Japan) for providing reagents, L. Chu for preparation of the HEK293T whole-cell extracts, and other members of the C.H. laboratory for helpful discussions.
This work was supported in part by U.S. Public Health Service grant GM084353 (C.H.) from the U.S. National Institute of General Medical Sciences.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- CQ
- chloroquine
- CRL
- Cullin-RING ligase
- DSB
- double-strand break
- MSH4
- MutS homologue 4
- NHEJ
- nonhomologous end joining
- VBC
- VHL-elongin B-elongin C complex
- VHL
- Von Hippel-Lindau
REFERENCES
- 1. Handel M. A., Schimenti J. C. (2010) Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat. Rev. Genet. 11, 124–136 [DOI] [PubMed] [Google Scholar]
- 2. Ross-Macdonald P., Roeder G. S. (1994) Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Cell 79, 1069–1080 [DOI] [PubMed] [Google Scholar]
- 3. Zalevsky J., MacQueen A. J., Duffy J. B., Kemphues K. J., Villeneuve A. M. (1999) Crossing over during Caenorhabditis elegans meiosis requires a conserved MutS-based pathway that is partially dispensable in budding yeast. Genetics 153, 1271–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kneitz B., Cohen P. E., Avdievich E., Zhu L., Kane M. F., Hou H., Jr., Kolodner R. D., Kucherlapati R., Pollard J. W., Edelmann W. (2000) MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 14, 1085–1097 [PMC free article] [PubMed] [Google Scholar]
- 5. Lenzi M. L., Smith J., Snowden T., Kim M., Fishel R., Poulos B. K., Cohen P. E. (2005) Extreme heterogeneity in the molecular events leading to the establishment of chiasmata during meiosis Iin human oocytes. Am. J. Hum. Genet. 76, 112–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bocker T., Barusevicius A., Snowden T., Rasio D., Guerrette S., Robbins D., Schmidt C., Burczak J., Croce C. M., Copeland T., Kovatich A. J., Fishel R. (1999) hMSH5: a human MutS homologue that forms a novel heterodimer with hMSH4 and is expressed during spermatogenesis. Cancer Res. 59, 816–822 [PubMed] [Google Scholar]
- 7. Her C., Zhao N., Wu X., Tompkins J. D. (2007) MutS homologues hMSH4 and hMSH5: diverse functional implications in humans. Front. Biosci. 12, 905–911 [DOI] [PubMed] [Google Scholar]
- 8. Pochart P., Woltering D., Hollingsworth N. M. (1997) Conserved properties between functionally distinct MutS homologs in yeast. J. Biol. Chem. 272, 30345–30349 [DOI] [PubMed] [Google Scholar]
- 9. Snowden T., Shim K. S., Schmutte C., Acharya S., Fishel R. (2008) hMSH4-hMSH5 adenosine nucleotide processing and interactions with homologous recombination machinery. J. Biol. Chem. 283, 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winand N. J., Panzer J. A., Kolodner R. D. (1998) Cloning and characterization of the human and Caenorhabditis elegans homologs of the Saccharomyces cerevisiae MSH5 gene. Genomics 53, 69–80 [DOI] [PubMed] [Google Scholar]
- 11. Snowden T., Acharya S., Butz C., Berardini M., Fishel R. (2004) hMSH4-hMSH5 recognizes Holliday junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol. Cell 15, 437–451 [DOI] [PubMed] [Google Scholar]
- 12. Chu Y. L., Wu X., Xu Y., Her C. (2013) MutS homologue hMSH4: interaction with eIF3f and a role in NHEJ-mediated DSB repair. Mol. Cancer 12, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neyton S., Lespinasse F., Moens P. B., Paul R., Gaudray P., Paquis-Flucklinger V., Santucci-Darmanin S. (2004) Association between MSH4 (MutS homologue 4) and the DNA strand-exchange RAD51 and DMC1 proteins during mammalian meiosis. Mol. Hum. Reprod. 10, 917–924 [DOI] [PubMed] [Google Scholar]
- 14. Santucci-Darmanin S., Walpita D., Lespinasse F., Desnuelle C., Ashley T., Paquis-Flucklinger V. (2000) MSH4 acts in conjunction with MLH1 during mammalian meiosis. FASEB J. 14, 1539–1547 [DOI] [PubMed] [Google Scholar]
- 15. Santucci-Darmanin S., Neyton S., Lespinasse F., Saunieres A., Gaudray P., Paquis-Flucklinger V. (2002) The DNA mismatch-repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum. Mol. Genet. 11, 1697–1706 [DOI] [PubMed] [Google Scholar]
- 16. Her C., Wu X., Griswold M. D., Zhou F. (2003) Human MutS homologue MSH4 physically interacts with von Hippel-Lindau tumor suppressor-binding protein 1. Cancer Res. 63, 865–872 [PubMed] [Google Scholar]
- 17. Her C., Doggett N. A. (1998) Cloning, structural characterization, and chromosomal localization of the human orthologue of Saccharomyces cerevisiae MSH5 gene. Genomics 52, 50–61 [DOI] [PubMed] [Google Scholar]
- 18. Paquis-Flucklinger V., Santucci-Darmanin S., Paul R., Saunieres A., Turc-Carel C., Desnuelle C. (1997) Cloning and expression analysis of a meiosis-specific MutS homolog: the human MSH4 gene. Genomics 44, 188–194 [DOI] [PubMed] [Google Scholar]
- 19. Tsuchiya H., Iseda T., Hino O. (1996) Identification of a novel protein (VBP-1) binding to the von Hippel-Lindau (VHL) tumor suppressor gene product. Cancer Res. 56, 2881–2885 [PubMed] [Google Scholar]
- 20. Brinke A., Green P. M., Giannelli F. (1997) Characterization of the gene (VBP1) and transcript for the von Hippel-Lindau binding protein and isolation of the highly conserved murine homologue. Genomics 45, 105–112 [DOI] [PubMed] [Google Scholar]
- 21. Vainberg I. E., Lewis S. A., Rommelaere H., Ampe C., Vandekerckhove J., Klein H. L., Cowan N. J. (1998) Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 93, 863–873 [DOI] [PubMed] [Google Scholar]
- 22. Feldman D. E., Spiess C., Howard D. E., Frydman J. (2003) Tumorigenic mutations in VHL disrupt folding in vivo by interfering with chaperonin binding. Mol. Cell 12, 1213–1224 [DOI] [PubMed] [Google Scholar]
- 23. Feldman D. E., Thulasiraman V., Ferreyra R. G., Frydman J. (1999) Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol. Cell 4, 1051–1061 [DOI] [PubMed] [Google Scholar]
- 24. McClellan A. J., Scott M. D., Frydman J. (2005) Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 121, 739–748 [DOI] [PubMed] [Google Scholar]
- 25. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 26. Geissler S., Siegers K., Schiebel E. (1998) A novel protein complex promoting formation of functional alpha- and gamma-tubulin. EMBO J. 17, 952–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hergovich A., Lisztwan J., Barry R., Ballschmieter P., Krek W. (2003) Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat. Cell Biol. 5, 64–70 [DOI] [PubMed] [Google Scholar]
- 28. Kaelin W. G., Jr. (2008) The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 8, 865–873 [DOI] [PubMed] [Google Scholar]
- 29. Mousnier A., Kubat N., Massias-Simon A., Segeral E., Rain J. C., Benarous R., Emiliani S., Dargemont C. (2007) von Hippel Lindau binding protein 1-mediated degradation of integrase affects HIV-1 gene expression at a postintegration step. Proc. Natl. Acad. Sci. U. S. A. 104, 13615–13620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Delgehyr N., Wieland U., Rangone H., Pinson X., Mao G., Dzhindzhev N. S., McLean D., Riparbelli M. G., Llamazares S., Callaini G., Gonzalez C., Glover D. M. (2012) Drosophila Mgr, a Prefoldin subunit cooperating with von Hippel Lindau to regulate tubulin stability. Proc. Natl. Acad. Sci. U. S. A. 109, 5729–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexandru G., Graumann J., Smith G. T., Kolawa N. J., Fang R., Deshaies R. J. (2008) UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 134, 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Den Besten W., Verma R., Kleiger G., Oania R. S., Deshaies R. J. (2012) NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat. Struct. Mol. Biol. 19, 511–516, S511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jentsch S., Rumpf S. (2007) Cdc48 (p97): a “molecular gearbox” in the ubiquitin pathway? Trends Biochem. Sci. 32, 6–11 [DOI] [PubMed] [Google Scholar]
- 34. Ye Y. (2006) Diverse functions with a common regulator: ubiquitin takes command of an AAA ATPase. J. Struct. Biol. 156, 29–40 [DOI] [PubMed] [Google Scholar]
- 35. Meyer H., Bug M., Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 36. Acs K., Luijsterburg M. S., Ackermann L., Salomons F. A., Hoppe T., Dantuma N. P. (2011) The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 18, 1345–1350 [DOI] [PubMed] [Google Scholar]
- 37. Franz A., Orth M., Pirson P. A., Sonneville R., Blow J. J., Gartner A., Stemmann O., Hoppe T. (2011) CDC-48/p97 coordinates CDT-1 degradation with GINS chromatin dissociation to ensure faithful DNA replication. Mol. Cell 44, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meerang M., Ritz D., Paliwal S., Garajova Z., Bosshard M., Mailand N., Janscak P., Hubscher U., Meyer H., Ramadan K. (2011) The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 13, 1376–1382 [DOI] [PubMed] [Google Scholar]
- 39. Raman M., Havens C. G., Walter J. C., Harper J. W. (2011) A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol. Cell 44, 72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee T. H., Yi W., Griswold M. D., Zhu F., Her C. (2006) Formation of hMSH4-hMSH5 heterocomplex is a prerequisite for subsequent GPS2 recruitment. DNA Repair (Amst.) 5, 32–42 [DOI] [PubMed] [Google Scholar]
- 41. Yi W., Wu X., Lee T. H., Doggett N. A., Her C. (2005) Two variants of MutS homolog hMSH5: prevalence in humans and effects on protein interaction. Biochem. Biophys. Res. Commun. 332, 524–532 [DOI] [PubMed] [Google Scholar]
- 42. Vo A. T., Zhu F., Wu X., Yuan F., Gao Y., Gu L., Li G. M., Lee T. H., Her C. (2005) hMRE11 deficiency leads to microsatellite instability and defective DNA mismatch repair. EMBO Rep. 6, 438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dalal S., Rosser M. F., Cyr D. M., Hanson P. I. (2004) Distinct roles for the AAA ATPases NSF and p97 in the secretory pathway. Mol. Biol. Cell 15, 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ye Y., Meyer H. H., Rapoport T. A. (2003) Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tan J. M., Wong E. S., Kirkpatrick D. S., Pletnikova O., Ko H. S., Tay S. P., Ho M. W., Troncoso J., Gygi S. P., Lee M. K., Dawson V. L., Dawson T. M., Lim K. L. (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17, 431–439 [DOI] [PubMed] [Google Scholar]
- 46. Lim K. L., Lim G. G. (2011) K63-linked ubiquitination and neurodegeneration. Neurobiol. Dis. 43, 9–16 [DOI] [PubMed] [Google Scholar]
- 47. Saeki Y., Kudo T., Sone T., Kikuchi Y., Yokosawa H., Toh-e A., Tanaka K. (2009) Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J. 28, 359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kirkin V., Lamark T., Sou Y. S., Bjorkoy G., Nunn J. L., Bruun J. A., Shvets E., McEwan D. G., Clausen T. H., Wild P., Bilusic I., Theurillat J. P., Overvatn A., Ishii T., Elazar Z., Komatsu M., Dikic I., Johansen T. (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516 [DOI] [PubMed] [Google Scholar]
- 50. Kirkin V., McEwan D. G., Novak I., Dikic I. (2009) A role for ubiquitin in selective autophagy. Mol. Cell 34, 259–269 [DOI] [PubMed] [Google Scholar]
- 51. Komander D., Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 [DOI] [PubMed] [Google Scholar]
- 52. Kreuzaler P., Watson C. J. (2012) Killing a cancer: what are the alternatives? Nat. Rev. Cancer 12, 411–424 [DOI] [PubMed] [Google Scholar]
- 53. Miyazawa M., Tashiro E., Kitaura H., Maita H., Suto H., Iguchi-Ariga S. M., Ariga H. (2011) Prefoldin subunits are protected from ubiquitin-proteasome system-mediated degradation by forming complex with other constituent subunits. J. Biol. Chem. 286, 19191–19203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hua Z., Vierstra R. D. (2011) The cullin-RING ubiquitin-protein ligases. Annu. Rev. Plant Biol. 62, 299–334 [DOI] [PubMed] [Google Scholar]
- 55. Hotton S. K., Callis J. (2008) Regulation of cullin RING ligases. Annu. Rev. Plant Biol. 59, 467–489 [DOI] [PubMed] [Google Scholar]
- 56. Kamura T., Maenaka K., Kotoshiba S., Matsumoto M., Kohda D., Conaway R. C., Conaway J. W., Nakayama K. I. (2004) VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 18, 3055–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Richly H., Rape M., Braun S., Rumpf S., Hoege C., Jentsch S. (2005) A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120, 73–84 [DOI] [PubMed] [Google Scholar]
- 58. Gavin A. C., Bosche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J. M., Michon A. M., Cruciat C. M., Remor M., Hofert C., Schelder M., Brajenovic M., Ruffner H., Merino A., Klein K., Hudak M., Dickson D., Rudi T., Gnau V., Bauch A., Bastuck S., Huhse B., Leutwein C., Heurtier M. A., Copley R. R., Edelmann A., Querfurth E., Rybin V., Drewes G., Raida M., Bouwmeester T., Bork P., Seraphin B., Kuster B., Neubauer G., Superti-Furga G. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147 [DOI] [PubMed] [Google Scholar]
- 59. Ye Y., Rape M. (2009) Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jin L., Williamson A., Banerjee S., Philipp I., Rape M. (2008) Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133, 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tresse E., Salomons F. A., Vesa J., Bott L. C., Kimonis V., Yao T. P., Dantuma N. P., Taylor J. P. (2010) VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sandilands E., Serrels B., McEwan D. G., Morton J. P., Macagno J. P., McLeod K., Stevens C., Brunton V. G., Langdon W. Y., Vidal M., Sansom O. J., Dikic I., Wilkinson S., Frame M. C. (2012) Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat. Cell Biol. 14, 51–60 [DOI] [PubMed] [Google Scholar]
- 63. Long J., Gallagher T. R., Cavey J. R., Sheppard P. W., Ralston S. H., Layfield R., Searle M. S. (2008) Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J. Biol. Chem. 283, 5427–5440 [DOI] [PubMed] [Google Scholar]
- 64. Wooten M. W., Geetha T., Babu J. R., Seibenhener M. L., Peng J., Cox N., Diaz-Meco M. T., Moscat J. (2008) Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J. Biol. Chem. 283, 6783–6789 [DOI] [PubMed] [Google Scholar]
- 65. Moynahan M. E., Jasin M. (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.