

Abstract
Large conductance voltage- and calcium-activated potassium (BK) channels are highly expressed in airway smooth muscle (ASM). Utilizing the ovalbumin (OVA) and house dust mite (HDM) models of asthma in C57BL/6 mice, we demonstrate that systemic administration of the BK channel agonist rottlerin (5 μg/g) during the challenge period reduced methacholine-induced airway hyperreactivity (AHR) in OVA- and HDM-sensitized mice (47% decrease in peak airway resistance in OVA-asthma animals, P<0.01; 54% decrease in HDM-asthma animals, P<0.01) with a 35–40% reduction in inflammatory cells and 20–35% reduction in Th2 cytokines in bronchoalveolar lavage fluid. Intravenous rottlerin (5 μg/g) reduced AHR within 5 min in the OVA-asthma mice by 45% (P<0.01). With the use of an ex vivo lung slice technique, rottlerin relaxed acetylcholine-stimulated murine airway lumen area to 87 ± 4% of the precontracted area (P<0.01 vs. DMSO control). Rottlerin increased BK channel activity in human ASM cells (V50 shifted by 73.5±13.5 and 71.8±14.6 mV in control and asthmatic cells, respectively, both P<0.05 as compared with pretreatment) and reduced the frequency of acetylcholine-induced Ca2+ oscillations in murine ex vivo lung slices. These findings suggest that rottlerin, with both anti-inflammatory and ASM relaxation properties, may have benefit in treating asthma.—Goldklang, M. P., Perez-Zoghbi, J. F., Trischler, J., Nkyimbeng, T., Zakharov, S. I., Shiomi, T., Zelonina, T., Marks, A. R., D'Armiento, J. M., Marx, S. O. Treatment of experimental asthma using a single small molecule with anti-inflammatory and BK channel-activating properties.
Keywords: airway smooth muscle, airway contractility, MAP kinases, ERK
Asthma is characterized by airway inflammation, hyperresponsiveness, and reversible airflow obstruction (1). Despite current therapies, including β-adrenergic agonists, steroids, antihistamines, antileukotrienes, anticholinergics, and phosphodiesterase inhibitors, many patients suffer from repeated asthmatic attacks (2). Moreover, side effects limit use of the current therapeutics in patients with asthma (1). Therefore, novel therapeutics targeting both inflammation and airway smooth muscle (ASM) contractility pathways are needed.
ASM contraction is activated by elevation of the intracellular Ca2+ concentration ([Ca2+]i). Plasma membrane depolarization increases Ca2+ influx via voltage-gated Ca2+ channels. The [Ca2+]i is also increased by influx via voltage-independent Ca2+ channels and release from the endoplasmic reticulum (ER)/sarcoplasmic reticulum (SR) via intracellular Ca2+ release channels [ryanodine receptors (RyRs) and inositol 1,4,5-trisphosphate receptors (IP3Rs); refs. 3, 4]. SR and mitochondrial Ca2+ release and reuptake result in Ca2+ oscillations, the frequency of which correlates with the degree of airway contraction (4). The smooth muscle membrane potential also modulates airway contractility by regulating the activity of M3 and M2 muscarinic receptors, independent of their endogenous ligands (3, 5), and by altering the Ca2+ sensitivity of the contractile proteins via activation of Rho kinase (6, 7). Depolarization-evoked excitation of the M3 muscarinic receptor activates phospholipase C, which results in elevation of inositol 1,4,5 trisphosphate (IP3), thereby activating IP3Rs on the ER/SR, causing intracellular Ca2+ release and muscle contraction. Thus, depolarizing the ASM cell membrane potential affects airway contraction via multiple Ca2+-dependent processes, including Ca2+ influx, Ca2+ release from SR stores, and Ca2+ sensitization of the contractile proteins.
K+ channels expressed in ASM hyperpolarize the plasma membrane and inhibit voltage-dependent Ca2+ influx through the plasma membrane, limiting the contraction of smooth muscle (8). The activity of large conductance voltage- and Ca2+-activated K+ (BK) channels profoundly influences the plasma membrane potential. BK β1 regulatory subunits, which associate with the pore-forming α-subunit, shift the V50 for channel activation negatively, priming the channel for activation by intracellular Ca2+ (9, 10). Without the BK β1-regulatory subunit, BK channels do not appropriately sense RyR-mediated Ca2+ sparks leading to diminished BK channel activity and membrane depolarization (11). In an African-American asthmatic population, a BK β1-subunit polymorphism (C818T; R140W) that reduces BK channel activity is associated with a significant decline in forced expiratory volume in 1 s (FEV1; ref. 12). Mice lacking the BK β1-subunit also exhibit increased tracheal constriction, and tracheal smooth muscle isolated from these mice have increased Ca2+ influx after cholinergic stimulation and increased resting [Ca2+]i compared with wild-type mice (11).
Since asthma is marked by both airway hypercontractility and inflammation (13) and rottlerin is both anti-inflammatory and a potent BK channel agonist (13, 14), we tested the effects of acute and long-term systemic administration of rottlerin on methacholine (MCh)-induced airway reactivity and pulmonary inflammation associated with the ovalbumin (OVA) and house dust mite (HDM) murine models of experimental aeroallergen asthma.
MATERIALS AND METHODS
Isolation of tracheal myocytes
The Animal Care and Use Committees of Columbia University College of Physicians and Surgeons and the Texas Tech University Health Sciences Center each approved all animal experiment protocols performed in their respective facilities. The tracheal tube was isolated by cutting below the pharynx and above the primary bronchus bifurcation. The dorsal muscle layer was cut away from the hyaline cartilage rings and minced into ∼1-mm pieces in a low-Ca2+ HEPES-buffered solution (in mM: 140 NaCl, 4.7 KCl, 1.13 MgCl2, 10 HEPES, 10 glucose, 0.1 CaCl2, and 1 mg/ml BSA, pH 7.4). The tissues were digested with 0.5 mg/ml papain (Roche, Indianapolis, IN, USA) and 1 mg/ml dithiothreitol (Sigma-Aldrich, St. Louis, MO, USA) for 13 min at 37°C. The tissue was then washed and further digested with 1 mg/ml collagenase H and 1 mg/ml collagenase II (Sigma-Aldrich) for 25 min at 37°C. The tissue was washed 3 times in low-Ca2+ solution and gently triturated to disperse single tracheal myocytes.
Cultured human ASM cells
Primary human ASM cell lines were obtained from Drs. Elizabeth Townsend and Charles Emala (Columbia University; ref. 15). A primary cell line of human ASM cells isolated from patients with asthma was obtained from Lonza (Walkersville, MD, USA). Cells were grown to ∼80% confluence in Gibco phenol red-free DMEM/F-12 medium (Invitrogen, Grand Island, NY, USA) supplemented with 10% fetal bovine serum and antibiotics. Cells were then passaged into 6-well culture plates with poly-l-lysine precoated coverslips (BD BioCoat precoated glass coverslips, 12 mm round; BD Biosciences, San Jose, CA, USA) for whole-cell current and membrane potential recordings.
Whole-cell current and membrane potential recording
Standard whole-cell, ruptured patch-clamp technique was used to measure BK channel currents, as described previously (13). Free Ca2+ was 1 μM, calculated using the online program WebmaxC Standard (http://www.stanford.edu/∼cpatton/webmaxcs.htm). For membrane potential measurements, a whole-cell perforated patch-clamp configuration was used. The following solutions were used: extracellular solution (in mM): 134 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4); and intracellular solution (in mM): 75 KCl, 64 K-aspartate, 2 Mg-ATP, 8 NaCl, 1 EGTA, 0.85 CaCl2, and 10 HEPES (pH 7.2). Data were acquired at room temperature using an Axopatch 200B amplifier (Axon Instruments, Sunnyvale, CA, USA) at 5 kHz using a Digidata 1322A and pClamp 9 software (Axon Instruments). The liquid junction potential (−9 mV) was corrected. Pipette resistance was 2–5 MΩ when filled with the pipette solution. The series resistance was not compensated, since the series resistance (<15 MΩ) was much smaller than the cell input resistance (∼1.0 GΩ). Cell capacitance was 25.2 ± 4.0 pF (n=7). Only cells with a seal resistance >2 GΩ were recorded and analyzed.
OVA-asthma model
Groups of 8-wk-old C57BL/6 female mice were sensitized with intraperitoneal injections of 100 μg grade V chicken OVA (Sigma-Aldrich), mixed with 2 mg aluminum hydroxide in saline on d 0 and again on d 7. Control mice were injected with sterile endotoxin-free PBS. On alternate days from d 14 through 22, mice received a 20 min aerosol challenge of either 2% (w/v) OVA in PBS or endotoxin-free PBS using a lumiscope 6610 ultrasonic nebulizer (Lumiscope, East Rutherford, NJ, USA). Rottlerin (Tocris Biosciences, Bristol, UK), suspended in DMSO at a stock concentration of 10 mM, was diluted with PBS to a final concentration of 0.5 μg/μl and injected at a dose of 5 μg/g (100 μg/mouse). PBS mixed with DMSO was administered to control mice. Invasive airway measurements including airway resistance were obtained during a graded MCh (acetyl-β-methylcholine chloride; Sigma-Aldrich) challenge performed on d 23. In the group receiving a single dose of intravenous rottlerin, rottlerin or vehicle was injected via the tail vein 5 min before airway measurements.
HDM asthma model
Groups of female C57BL/6 mice were exposed to 40 μg of purified HDM extract whole protein (Greer Laboratories, Lenoir, NC, USA) without exogenous adjuvant in 25 μl of saline intranasally 5 d/wk for 3 consecutive weeks (16). Isoflurane 2–5% inhalation was utilized for anesthesia before intranasal HDM administration. Rottlerin 5 μg/g (100 μg/mouse) and placebo were administered 3×/wk via intraperitoneal injection.
Measurement of airway responsiveness in vivo
After sedation with ketamine (100 mg/kg i.p.) and xylazine (5 mg/kg i.p.), the neck was dissected, and the trachea was cannulated with an 18-gauge beveled tracheal tube. The mouse was then placed on a mouse ventilator with a tidal volume of 0.2 ml and frequency of 150 breaths/min. After 3–5 min of equilibration on the ventilator, a graded MCh challenge was initiated, and airway resistance was measured invasively using the Buxco Resistance/Compliance system with restrained whole-body plethysmography (rWBP; Buxco PLY4111, Wilmington, NC, USA; ref. 17). Changes in airway resistance were calculated from peak values after each dose of MCh and compared with baseline values. The measurements of airway resistance for the chronic and acute rottlerin experiments were acquired using different Buxco system manifolds.
Quantification of rottlerin concentration in plasma and lung tissue
Mice were injected with rottlerin (5 μg/g; 100 μg/mouse) every other day for a total of 4 doses. After euthanasia by isoflurane and CO2 inhalation, the thorax was opened. Whole blood was obtained by ventricular puncture, and serum was separated after centrifugation at 2000 g for 10 min. The lungs were dissected and flash-frozen in liquid nitrogen. Mouse plasma and lung tissue were kept at −80°C before analysis. Rottlerin was quantified using 100 μl of plasma or ∼150 mg of lung tissue. The tissue was homogenized in 500 μl of water using a Tissue-Tearor (Biospec Products, Inc., Bartlesville, OK, USA). Proteins from plasma and lung homogenates were precipitated with acetonitrile/methanol (4:1). After being vortexed for 60 s, the samples were centrifuged (14,000 g for 10 min). The supernatant was evaporated with nitrogen and resolubilized with 100 μl of 70% methanol and 0.1% formic acid. Each sample (20 μl) was injected onto a Atlantis dC18 3 μm 2.1 × 50 mm column (40°C; Waters, Milford, MA, USA) using an Acquity 2795 HPLC (Waters) with the initial conditions 70% of 0.1% formic acid in methanol (0.5 ml/min) and ramped linearly to 98% methanol with 0.1% formic acid over 5 min. The column was cleaned with 100% methanol with 0.1% formic acid for 5 min and then reequilibrated to the initial conditions for 2 min (total run time: 12 min).
Rottlerin was detected with a MicroMass Quattro Micro tandem mass spectrometer (Waters) with positive electrospray ionization. The drugs were quantified using multiple reaction monitoring of the H+ ion with the transition 517.2 to 335.1 (collision energy: 15 V; cone: 20 V). Spiked plasma was used to create a standard curve, which was linear from 1 to 1000 ng/ml with a limit of quantification (LOQ) and limit of detection (LOD) of 1.0 and 0.5 ng/ml, respectively. Quantification of rottlerin in both plasma and lung tissue was calculated relative to the spiked plasma standard curve. The MS conditions were as follows: desolvation temperature, 300°C; source temperature, 120°C; desolvation gas flow, 500 L/h; cone gas flow, 50 L/h; capillary, 4.0 kV. Rottlerin was detectable in the plasma at all time points (rottlerin plasma concentration of 2.9 μg/ml 2 h after 4th dose, 4.45 ng/ml 24 h after 4th dose, and 0.77 ng/ml trough before 4th dose) and not detected in the lung at the 48 h trough measurement (153.3 ng/g tissue 2 h after 4th dose, 3.85 ng/g tissue 24 h after 4th dose, and undetectable trough level before the 4th dose), leading to the choice of a 48-h dosing interval.
OVA-specific IgE ELISA
After measurement of airway resistance, mice were euthanized, and their serum was collected and stored at −80°C until analysis. Serum levels of OVA-specific IgE were measured by sandwich ELISA (R&D Systems, Minneapolis, MN, USA).
HDM-specific IgG1 ELISA
HDM-specific IgG1 ELISA was performed on plasma samples as per the methods outlined by Johnson et al. (16) with two small modifications. Maxi-Sorp plates (Sigma-Aldrich) were coated overnight with 1 μg HDM in carbonate-bicarbonate buffer. Plates were washed and incubated with the ABC Vectastain kit (Vector Laboratories, Burlingame, CA, USA).
Differential cell counts in the bronchoalveolar lavage (BAL) fluid
BAL was performed by injection of 1 ml saline through a tracheal cannula into the lung. Cells in the BAL fluid were centrifuged and resuspended in cold PBS. For differential cell counts, cytospin preparations were made and stained with the DIFF stain kit (IMEB, Inc., San Marcos, CA, USA). Following coverslip placement, 200 cells were counted and differentiated by standard morphology and staining characteristics.
Cytokine ELISA
IL-4, IL-5, and IL-13 ELISAs (R&D Systems) were performed according to the manufacturer's instructions. The detection limits of the ELISAs were 60 pg/ml for IL-4, 32 pg/ml for IL-5, and 15 pg/ml IL-13.
Protein extraction and immunoblotting
Dissected lungs from mice were flash-frozen in liquid nitrogen for later analysis. Sections of lung were homogenized in 600 μl of RIPA lysis buffer system with PMSF, protease inhibitor cocktail, and sodium orthovanadate (Santa Cruz Biotechnology, Dallas, TX, USA). Following homogenization, the lung lysates were centrifuged (14,000 g for 10 min) and protein concentration was determined by bicinchoninic acid protein assay (Pierce BCA Protein Assay Kit; Thermo Scientific, Rockford, IL, USA). Lung lysates (40 μg) from each group were subjected to Western blot analysis. Cell Signaling Technology (Danvers, MA, USA) rabbit polyclonal antibodies against phospho-extracellular signal-regulated kinase (ERK; T202/Y204, 1:500), total ERK (9102, 1:2500), phospho-c-Jun N-terminal kinase (JNK; T183/Y185, 1:1000), and total JNK (9252, 1:1000) were used following the manufacturer's instructions. Chemiluminescence from Western blots was visualized using the ImageQuant LAS 4000 Series imager (GE Healthcare Life Sciences, Pittsburgh, PA, USA), and densitometry was performed using Carestream Molecular Imaging Software (Carestream Health, Rochester, NY, USA).
Histology
The lung samples were fixed with formalin, embedded in paraffin wax, and sliced into 4-μm-thick sections. After deparaffinization, sections were stained with hematoxylin and eosin (H&E; Sigma-Aldrich) and photographed using a light microscope.
Preparation of lung slices
Murine lung slices were prepared and imaged as previously reported (4, 18, 19) with modifications. Female C57BL/6 mice, 8–12 wk old, were euthanized with an intraperitoneal injection of sodium pentobarbital (40 mg/kg). The chest cavity was opened, and the trachea was exposed and cannulated with an intravenous catheter (20-gauge Intima; BD Biosciences). The lungs were inflated with 1.4 ± 0.1 ml of 2% agarose (low-melting temperature agarose; USB Corp., Santa Clara, CA, USA) in sterile HBSS followed by 0.2 ml of air to flush the agarose out of the airways and into the distal alveolar space. The agarose was gelled by cooling the animal at 4°C for 20 min. Lung lobes were separated and trimmed near to the main bronchus to create a base. Each lobe was transferred to the specimen syringe tube of a tissue slicer (Compresstome VF-300; Precisionary Instruments, Greenville, NC, USA), embedded in agarose, and prepared for sectioning as per manufacturer's instructions. Sections (140 μm) were prepared starting at the peripheral edge of each lung lobe, and 15–20 sections containing small terminal airways with a diameter of 100–300 μm were collected in sterile HBSS. The sections were incubated overnight in low-glucose DMEM (Invitrogen) supplemented with antibiotics at 37°C and 10% CO2. All lung sections were used within 48 h.
Measurement of the airway contractile responses
Hanks' balanced salt solution was supplemented with 20 mM HEPES buffer and adjusted to pH 7.4 (sHBSS). Stock solutions were diluted in sHBSS at the final concentration on the same day of use, and the concentration of vehicle (DMSO) never exceeded 0.1%. Lung slices were mounted on a cover glass of a custom-made perfusion chamber, held in place with a small sheet of nylon mesh, and perfused with the experimental solutions as described previously (4, 18). All experiments were performed at room temperature. The chamber was placed on the stage of an inverted phase-contrast microscope (Diaphot TMD; Nikon, Tokyo, Japan), and lung slices were imaged with a ×10 objective. Digital images were recorded to a hard drive in time lapse (0.5 Hz) using a CCD camera (KP-M1A; Hitachi, Tokyo, Japan), frame grabber (Picolo; Euresys, San Juan Capistrano, CA, USA), and image acquisition software (Video Savant; IO Industries, London, ON, Canada). The area of the airway lumen was calculated from each image using custom-written script in Video Savant that distinguishes the lumen from the surrounding tissue. The lumen area was normalized to the area before stimulation, and the changes in lumen area were plotted against time.
Measurements of intracellular Ca2+
Approximately 10–12 lung slices were incubated for 50 min at 30°C in 2 ml of sHBSS supplemented with 20 μM Oregon Green 488 BAPTA-1, acetoxymethyl ester (Invitrogen) that has been dissolved in 20 μl of dry DMSO plus 5 μl of 20% Pluronic F-127 in DMSO. Subsequently, the slices were transferred to 2 ml sHBSS and incubated for 50 min at 30°C to allow deesterification of the acetoxymethyl ester group. Lung slices were mounted on a cover glass of a custom-made perfusion chamber and perfused with the experimental solutions as described previously (4, 18). Fluorescence (488 nm excitation and 510–530 nm emission) imaging was performed using a custom-made video-rate confocal microscope at 15 Hz (19). Experiments were performed at room temperature. Changes in fluorescence intensity were obtained for each image by selecting regions of interest (ROIs) ranging from 5 to 7 pixel2. Fluorescence values were expressed as a fluorescence ratio (F/F0) normalized to the initial fluorescence (F0).
Statistical analysis
All statistical analyses were performed using Prism 5.0d software (GraphPad, San Diego, CA, USA). Comparisons between 2 groups were made using a Student's t test, with values of P < 0.05 considered statistically significant. Comparisons between ≥3 groups were made using an ANOVA with Sidak's post hoc analysis for relevant comparisons, with values of P < 0.05 considered statistically significant.
RESULTS
Rottlerin activates BK channels and hyperpolarizes the plasma membrane in murine ASM
We examined whether the endogenous BK channels in freshly isolated tracheal smooth muscle cells from C57BL/6 mice could be activated by rottlerin. Step depolarizations were applied from a holding potential of −80 mV in whole-cell patch-clamp configuration with an intracellular recording solution containing 1 μM free Ca2+. Rottlerin (1 μM) significantly shifted the V50 for BK channel activation by 73 ± 24 mV in the hyperpolarizing direction (n=3, P<0.05; Fig. 1A), consistent with our prior observations in vascular smooth muscle cells (13).
Figure 1.

Rottlerin activates BK channels and hyperpolarizes the plasma membrane potential. A) Representative conductance-voltage (G-V) curves from whole-cell voltage-clamp recordings of acutely isolated mouse tracheal smooth muscle cells before (circles) and after (triangles) 1 μM rottlerin (n=3). Pipette solution contained 1 μM free Ca2+. Rottlerin shifted the V50 for BK channel activation by 73 ± 24 mV in the hyperpolarizing direction. B) Representative trace of membrane potential of acutely isolated mouse ASM cells before and after 1 μM rottlerin and 10 μM paxilline. Inset: graph of membrane potential (means±se) before and after rottlerin and paxilline. ***P < 0.001; ANOVA with Sidak's post hoc analysis; n = 7.
The balance between depolarizing and repolarizing currents determines the plasma membrane potential of ASM cells. We hypothesized that the activation of BK channels by rottlerin was sufficient to induce hyperpolarization of the plasma membrane of the freshly isolated tracheal smooth muscle. The membrane potential was measured using the amphotericin-perforated current-clamp configuration. Rottlerin hyperpolarized the plasma membrane potential by 40 ± 8 mV, which was reversed by inhibiting BK channels with paxilline (n=7, P<0.001; Fig. 1B). Taken together, these results suggest that rottlerin-induced activation of BK channels is sufficient to potently hyperpolarize the membrane potential of ASM cells.
Acute administration of rottlerin reduces MCh-induced airway hyperreactivity (AHR) in a murine OVA-asthma model
Based on the effects of rottlerin on ASM plasma membrane potential and the prior demonstration that the membrane potential can affect ASM contractility (3), we hypothesized that rottlerin could reverse the AHR of asthma. Mice received an intraperitoneal injection of OVA mixed with aluminum hydroxide or PBS (control) on d 0 and 7 and on alternate days from d 14 through 22, and 20 min aerosol challenge of either PBS (control) or OVA was administered using an ultrasonic nebulizer (Fig. 2A and refs. 20–22). We measured the airway resistance (RL) in control and OVA-sensitized mice in response to MCh on d 23 and recorded changes in airway resistance from peak values after each MCh dose using a Buxco forced-maneuvers pulmonary function testing system and restrained whole-body plethysmography via tracheal cannulation. Rottlerin (5 μg/g) was injected via the tail vein 5 min before airway resistance measurements (Fig. 2A). Mice sensitized and challenged with OVA receiving a single injection of rottlerin demonstrated a 45% reduction in MCh-induced airway resistance (at 50 mg/ml) compared with a control group of OVA-sensitized mice that received a single injection of PBS (n=10/group, P<0.001; Fig. 2B). The rapid attenuation of MCh-induced AHR in the OVA-asthma model by rottlerin suggests that this effect is independent of anti-inflammatory properties of rottlerin (14, 23).
Figure 2.

Acute intravenous administration of rottlerin reverses MCh-induced AHR in OVA-asthma mice. A) Protocol for OVA-asthma model and rottlerin dosing. B) Graph of airway resistance vs. MCh concentration in nonsensitized (control) or OVA-sensitized and challenged mice. Mice underwent graded MCh challenge 5 min after tail-vein injection of 5 μg/g rottlerin or PBS on d 23 of the protocol. Data are presented as means ± se; n = 10/group. *P < 0.05, **P < 0.01, ***P < 0.001 for OVA + PBS vs. OVA + rottlerin.
Long-term administration of rottlerin reduces airway resistance and inflammation in an OVA-asthma model
Mice were administered rottlerin (5 μg/g) via intraperitoneal injection every other day from d 14 through 22 during the OVA-challenge (Fig. 3A). At 24 h after the last rottlerin injection, airway measurements were obtained. The rottlerin-treated OVA asthma mice exhibited a 49% decrease in MCh-induced airway resistance (at 50 mg/ml) compared with PBS-treated OVA asthma animals (n=20/group, P<0.001; Fig. 3B). Similar effects were observed at lower concentrations of MCh (Fig. 3B). Despite the near normalization of airway resistance by rottlerin the OVA-sensitized mice treated with rottlerin had a significant increase in OVA-specific IgE levels (serum OVA specific IgE levels, means±se; nonsensitized, PBS treated: 396.1±18.2, nonsensitized, rottlerin treated: 367.2±16.3, OVA sensitized, PBS treated: 892.6±24.9, OVA sensitized, rottlerin treated: 769.2±46.6 ng/ml).
Figure 3.

Long-term rotterlin administration attenuates MCh-induced AHR in OVA-asthma mice. A) Protocol for OVA-asthma model and dosing of rottlerin. B) Graph of airway resistance vs. MCh concentration, measured on d 24 in nonsensitized (control) and OVA-sensitized and challenged mice. PBS and rottlerin intraperitoneal injections were administered at the times as shown in A. Data are presented as means ± se; control + PBS (n=17 mice), control + rottlerin (n=17 mice), OVA + PBS (n=20 mice), and OVA + rottlerin (n=20 mice). **P < 0.01, ***P < 0.001 for OVA + PBS vs. OVA + rottlerin.
OVA sensitization and challenge induced a marked peribronchial and perivascular infiltration of inflammatory cells in C57BL/6 mice (Fig. 4A) as shown previously (20, 21, 24). Rottlerin-treated OVA-asthma mice exhibited less pulmonary inflammatory cell infiltration compared with PBS-treated OVA-asthma mice. Lungs from the nonsensitized/challenged mice and from the asthmatic mice were lavaged with PBS and total, and differential cell counts were determined in the BAL fluid. The total number of inflammatory cells increased in the lungs following OVA sensitization and challenge (Fig. 4B). Rottlerin significantly reduced the total number of inflammatory cells in OVA-sensitized mice (P<0.001 for OVA+PBS vs. OVA + rottlerin) and did not affect the number of inflammatory cells in the lungs of nonsensitized control mice (Fig. 4B).
Figure 4.
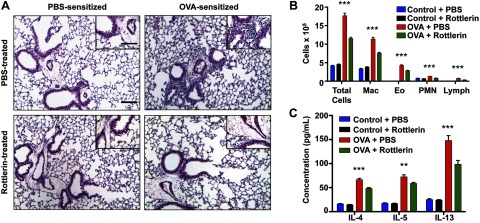
Long-term rottlerin administration attenuates peribronchial and perivascular inflammation in OVA-asthma mice. A) H&E staining of lungs from control and OVA-asthma animals. Images are representative of similar results from 5–6 mice for each experimental condition. Scale bars = 100 μm; inset: 50 μm. B) Graph of total cell count and differential of BAL fluid in the 4 treatment groups. Macrophage (Mac), eosinophil (Eo), PMN, and lymphocyte (lymph) counts are shown. Data are expressed as means + se. ***P < 0.001, OVA + PBS vs. OVA + rottlerin. C) Quantification of the Th2 cytokines IL-4, IL-5, and IL-13 in BAL fluid from control and OVA-sensitized mice treated with PBS or rottlerin. Data are expressed as means ± se. **P < 0.01, ***P < 0.001 for OVA + PBS vs. OVA + rottlerin.
Asthma is characterized by abnormal Th2 immune responses to antigens resulting in the production of cytokines, including IL-4, IL-5, and IL-13, followed by recruitment and activation of eosinophils and mast cells in the airway (25, 26). Rottlerin treatment significantly reduced the Th2 cytokines IL-4, IL-5, and IL-13 in BAL fluid of OVA-asthma mice compared with PBS-treated OVA-asthma mice (P<0.001 for IL-4 and IL-13, P<0.01 for IL-5; Fig. 4C).
Long-term administration of rottlerin attenuates AHR in a HDM model of asthma
HDM is a common allergen that is responsible for the development of asthma in ∼10% of the population (16). Exposure to HDM extract in mice causes persistent airway inflammation, airway hyperresponsiveness, and airway remodeling making this an important model for the study of asthma (16, 27).
C57BL/6 mice were exposed to either purified HDM extract (40 μg of whole protein in 25 μl of saline) or PBS intranasally for 5 d/wk for 3 wk (Fig. 5A; n=9–12 mice/group). Rottlerin was administered via intraperitoneal injection 3×/wk for the 3-wk period. Rottlerin-treated, HDM-sensitized mice exhibited a 53% decrease in MCh induced airway resistance (at 50 mg/ml) compared with PBS-treated HDM asthma animals (P<0.001; Fig. 5B). Airway measurements were obtained 24 h after the last dose of rottlerin. Similar to the OVA-sensitized rottlerin-treated animals, we observed a marked reduction in BAL cell count, as well as peribronchial and perivascular inflammation in the rottlerin-treated HDM-sensitized animals as compared with PBS-treated HDM-sensitized animals (Fig. 5C, D). Despite the significant reduction in airway resistance and inflammation by rottlerin, the HDM-sensitized mice treated with rottlerin had a significant increase in serum HDM specific IgG1 levels (optical density at 450 nm, means±se; nonsensitized, PBS-treated: 9.5±9.7, nonsensitized, rottlerin-treated: 6.4±7.5, HDM-sensitized, PBS-treated: 1024.6±118.7, HDM-sensitized, rottlerin- treated: 1117.9±132.4, P<0.001 as compared with nonsensitized mice, P>0.05 HDM-sensitized, PBS-treated as compared with HDM-sensitized, rottlerin-treated mice).
Figure 5.
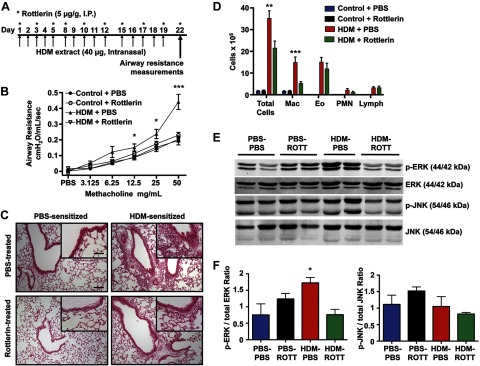
Rottlerin attenuates peribronchial and perivascular inflammation and MCh-induced AHR in an HDM model of asthma. A) Protocol for asthma induction and rottlerin administration using the HDM model. B) Graph of airway resistance vs. MCh concentration performed on d 22 following initiation of HDM protocol (n=9–12 mice/group). Data are expressed as means ± se. *P < 0.05, ***P < 0.001 for HDM + PBS vs. HDM + rottlerin and control + PBS. C) Lungs from control and HDM-sensitized and challenged mice were H&E stained and photographed using a light microscope. Images are representative of similar results from 4 mice/experimental condition. Scale bars = 100 μm; inset: 50 μm. D) Graph of total cell count and differential of BAL fluid in the 4 treatment groups. Data are expressed as means ± se. **P < 0.01, ***P < 0.001 for HDM + PBS vs. HDM + rottlerin. E) Representative Western blot from whole-lung homogenates of nonsensitized and HDM-sensitized mice treated with either PBS or rottlerin. F) Densitometry analysis of Western blots in E. Data are expressed as means + se. *P < 0.05, HDM + PBS vs. HDM + rottlerin; n = 3–4 mice/group.
Activation of the mitogen-activated protein (MAP) kinase ERK is a critical event in asthma pathogenesis and is associated with airway hyperresponsiveness, inflammation, and ASM cell proliferation (28, 29). The MAP kinase JNK is implicated in T cell and eosinophilic inflammatory responses but not airway hyperresponsiveness (30). ERK and JNK phosphorylation lead to the activation of kinase activity. ERK phosphorylation was increased by 129% (P<0.05) in whole-lung homogenates from HDM-sensitized asthmatic mice compared with controls, whereas JNK phosphorylation was not changed. Chronic rottlerin administration caused a 56% reduction in ERK phosphorylation, but not JNK phosphorylation, in whole-lung homogenates from HDM-sensitized mice (Fig. 5E, F).
Rottlerin inhibits acetylcholine (ACh)-induced smooth muscle cell contraction and Ca2+ oscillations in the small airways of lung slices
Lung slices were stimulated with the agonist ACh, which rapidly contracted the pulmonary small airways. The addition of rottlerin induced airway relaxation in a concentration-dependent fashion to the prestimulated level (Fig. 6 and Supplemental Video S1). Thus, rottlerin causes direct smooth muscle relaxation in small airways. Next, the effect of rottlerin on ACh-induced Ca2+ oscillations in ASM cells using confocal microscopy (Fig. 7A) was examined. In response to ACh, ASM cells displayed an increase in Ca2+ oscillations, as described previously (4). In the continued presence of ACh, the subsequent addition of rottlerin caused a progressive decrease in the frequency of the Ca2+ oscillations compared with vehicle-treated lung slices (Fig. 7B, C and Supplemental Video S2); the reduced frequency of Ca2+ oscillations correlated with the relaxation of the airway. These results confirm that rottlerin causes ASM relaxation in part by inhibiting agonist-induced Ca2+ oscillations.
Figure 6.

Rottlerin induces dose-dependent small airway relaxation in lung slices. A) Representative phase-contrast images: 1) airway in a mouse lung slice at rest, 2) airway following stimulation of airway contraction with 100 nM ACh, and 3) airway after relaxation induced by 5 μM rottlerin in the presence of ACh. Images were taken at the times indicated by corresponding numbers in B. B) Representative changes in airway lumen cross-sectional area vs. time showing the contraction in response to ACh and subsequent relaxation induced by rottlerin in the continued presence of ACh. C) Quantification of airway relaxation calculated after 50 min of exposure to DMSO or rottlerin obtained from experiments similar to those shown in B. Airway relaxation was calculated as the increase in lumen area divided by the maximal contraction induced by ACh. Data are means + se of 5 airways from different slices obtained from 3 mice for each rottlerin concentration and vehicle; ***P < 0.001 vs. DMSO control.
Figure 7.

Rottlerin reduces agonist-induced Ca2+ oscillations in ASM cells. A) Representative fluorescence confocal image of part of an airway in a lung slice showing epithelial cells (EPCs) lining the airway lumen, underlying smooth muscle cells (SMCs) and a selected 7- × 7-pixel ROI within a SMC for the measurements of the changes in Oregon green fluorescence plotted in B. B) Ca2+ oscillations induced by the continuous presence of 100 nM ACh were not altered with the addition of DMSO vehicle control. C) Ca2+ oscillations induced by 100 nM ACh were inhibited by 5 μM rottlerin in the continuous presence of ACh. Representative data from 5 experiments using 3 mice.
Rottlerin activates BK channels and hyperpolarizes the membrane potential in human control and asthmatic ASM cells
To determine whether rottlerin can be effective in humans, BK currents were recorded from ASM cells isolated from patients without (normal) and with asthma (Fig. 8). Similar to our findings in murine cells rottlerin (0.5 μM) significantly shifted the V50 for BK channel activation by 73.6 ± 13.5 mV in control cells (n=5, P<0.01, Fig. 8A) and 71.8 ± 14.6 mV in asthmatic cells (n=5, P<0.01, Fig. 8B). Therefore, rottlerin was effective in activating BK channels in asthmatic human ASM cells.
Figure 8.

Rottlerin activates BK channels in human ASM cells. A) Normalized G-V curves from whole-cell voltage-clamp recordings of primary human ASM cells from patients without asthma, before (open circles) and after (solid circles) 0.5 μM rottlerin (n=5 cells). Pipette solution contained 1 μM free Ca2+. Rottlerin shifted the V50 for BK channel activation by −73.6 ± 13.5 mV (P<0.01; paired 2-tailed Student's t test). B) Same experiments as in A except smooth muscle cells from a patient with asthma (n=5 cells). Rottlerin shifted the V50 for BK channel activation by −71.8 ± 14.6 mV (P<0.01). Insets: representative current traces elicited by pulses from –80 to +100 mV.
DISCUSSION
The present study identifies rottlerin as a potential therapeutic that targets both airway hypercontractility and pulmonary inflammation. Utilizing the OVA and HDM models of aeroallergen asthma, we show that rottlerin attenuates MCh-induced AHR and inflammatory cell infiltration of the lung. Rottlerin did not inhibit the sensitization to OVA or HDM. Th2 cytokines and ERK activation phosphorylation were decreased in the rottlerin-treated asthmatic mice, likely explaining the reduced inflammatory cell infiltration in the lungs. The marked reduction in airway resistance immediately after a single intravenous injection of rottlerin and the inhibition of ACh-induced airway contraction by rottlerin in ex vivo lung slices suggest that the effect of rottlerin on airway contractility is in part direct and not solely dependent on immunomodulation. The profound reduction in airway reactivity by rottlerin in the chronically treated asthmatic animals is therefore likely due to a combined effect on inflammation and airway contractility.
Rottlerin hyperpolarizes ASM membrane potential and directly inhibits airway contractility
ASM contraction is mediated primarily by activation of M2 and M3 muscarinic cholinergic receptors. MCh inhibits BK channel activity by both a Gβγ-mediated mechanism and activation of the phospholipase C/PKC pathway (31). Conversely, inhibition of BK channels enhances cholinergic-induced airway contraction (11, 32). Since BK channels oppose cholinergic M2 receptor-mediated depolarization and activation of voltage-dependent Ca2+ channels in tracheal smooth muscle cells (33), rottlerin may augment the BK channel-mediated inhibition of M2 signaling reducing airway contraction.
How might hyperpolarizing the membrane potential of ASM reduce airway contractility in asthmatic animals and in ex vivo lung slices? Hyperpolarization of the membrane potential inhibits voltage-dependent Ca2+ channels thereby reducing contractility. The role of voltage-dependent Ca2+ channels in ASM relaxation is controversial, however, since the level of depolarization of ASM cells may be insufficient to activate the high-voltage Ca2+ channels. Moreover, Ca2+ channel blockers are not effective therapeutics for asthma (34). Alternatively, T-type voltage-dependent Ca2+ channels, which are expressed in ASM, could contribute to depolarization-mediated contraction (35). Voltage-dependent Ca2+ influx could refill the internal Ca2+ stores essential for Ca2+ oscillations (4), rather than directly affecting myofilament Ca2+ (33).
The effects of hyperpolarization on airway contractility may also be independent of Ca2+ influx. Rottlerin-mediated hyperpolarization may inhibit the voltage-dependent activation of M3 muscarinic receptors, which act via Gq and phospholipase C, and generate IP3 to induce Ca2+ release from IP3Rs and further Ca2+ release via RyRs due to local Ca2+-induced Ca2+ release (3). Rottlerin-mediated hyperpolarization also may inhibit the voltage-dependent activation of Rho kinase (6, 7).
ASM cell contraction and small airway bronchoconstriction are regulated by Ca2+ oscillation frequency, which consists of cyclic SR Ca2+ release and reuptake (4). The onset of the inhibition of rottlerin of agonist-induced airway contraction is slower in the ex vivo lung slice assay than after intravenous injection through a tail vein in asthmatic mice for unclear reasons. However, the ex vivo experiments were performed at room temperature as compared with body temperature and the hydrophobicity of rottlerin likely impedes its transport into the lung slice preparation.
The effects of rottlerin on airway contractility could also be due to inhibition of kinases including PKC (36–38) or uncoupling of mitochondrial respiration from oxidative phosphorylation (39). The rottlerin-mediated activation of BK channels and the resultant hyperpolarization of the ASM membrane potential, however, are not dependent on modulation of cellular signaling pathways.
Rottlerin reduces inflammatory cell infiltration in asthma
Long-term administration of rottlerin during the period of OVA and HDM challenge reduced peribronchial and perivascular inflammatory cell infiltration as well as the Th2-specific cytokine production characteristic of these models. Rottlerin inhibits histamine release (23), reduces asthmatic eosinophil migration (40), and decreases monocyte chemotactic factor-1 (MCP-1) release (41). In vitro, phorbol myristate acetate (PMA)-induced phosphorylation of ERK-1 and ERK-2 in T cells from peripheral blood is blocked by rottlerin (42). Rottlerin also decreases the HDM-mediated activation of ERK in human monocytic THP-1 cells (43).
ERK phosphorylation, which is observed in the airway epithelium and smooth muscle cells of patients with asthma (44), correlates with disease severity. ERK phosphorylation is associated with ASM hyperresponsiveness and proliferation (28, 29, 45), leukotriene C4 synthesis (46, 47), IL-13- and IL-4-mediated eotaxin release from ASM cells (48), eotaxin-stimulated eosinophil degranulation (49), and granulocyte-macrophage colony-stimulating factor (GM-CSF) release (50). Pharmacological inhibition of MEK1/2, the upstream activator of ERK1/2, blocked airway inflammation in a mouse model of acute asthma (51). In addition, the ERK inhibitor U0126 has been examined in the ovalbumin model of asthma and similar to rottlerin, intraperitoneal administration decreased Th2 cytokines, total BAL cell count, airway hyperresponsiveness, and ERK phosphorylation in lung homogenates (51). ERK1-deficient mice also fail to develop experimental asthma (52). We showed that rottlerin inhibited HDM induced ERK phosphorylation in vivo, which correlated with a reduction in airway responsiveness and lung inflammation. In the HDM model, JNK phosphorylation was not increased consistent with variable activation in asthma (53, 54). Taken together, rottlerin reduces Th2 cytokines in the asthmatic lungs likely via the inhibition of ERK phosphorylation and activity.
Rottlerin is a natural compound isolated from the powder (kamala) covering the capsules of Mallotus phillippinensis (the monkey-faced tree). Kamala has been used for centuries in India as an anthelmintic agent against tapeworm (55), suggesting excellent safety when administered in this form. In an animal model of Parkinson's disease, a dose 4 times higher than the one used in this study was administered orally or via intraperitoneal injection to C57/BL mice without side effects (56).
Current acute therapies for asthma include β-adrenergic agonists, which enhance cardiac chronotropy, thereby limiting their use in patients with cardiovascular comorbidities. Since BK channels do not play a role in modulating the electrophysiological properties of the heart, direct activation of BK channels by rottlerin, which hyperpolarizes the ASM plasma membrane potential and inhibits Ca2+ oscillations, may be an alternative to β-adrenergic agonists in mediating acute airway relaxation. Rottlerin was well tolerated in our animal studies and effectively limited airway constriction and pulmonary inflammation in both the OVA and HDM models of asthma. Rottlerin targets airway contraction through BK channel activation and suppresses experimental asthmatic airway inflammation by inhibiting ERK phosphorylation and production of Th2 cytokines. Our results suggest that rottlerin, a compound with both bronchodilatory and anti-inflammatory properties, warrants further investigation as a potential therapeutic agent in asthma.
Supplementary Material
Acknowledgments
The authors thank Yong-Xiao Wang and members of the Wang laboratory at Albany Medical College (Albany, NY, USA) for help with the isolation of murine ASM cells. The authors thank Drs. Charles Emala and Elizabeth Townsend for providing human ASM cells and for thoughtful critiques of this work. The authors thank Jacquelyn Trice for assistance with the lung slice experiments, Sarah Marks for assistance with cell culture and histology, and Dr. Amy Simon for advice about the HDM model. The authors thank Drs. Tiffany Thomas and Serge Cremers (Columbia University Biomarkers Core Lab, Irving Institute for Clinical and Translational Research; U.S. National Institutes of Health grant UL1 TR000040) for assistance with the liquid chromatography-mass spectrometry work.
This study was supported by funds from an American Asthma Foundation grant (to A.R.M.); U.S. National Institute of Heart, Lung, and Blood grants P01-HL-081172 (to S.O.M., A.R.M., and J.M.D.), R01-HL-086936 (to J.M.D.), T32-HL-007854 (to M.P.G.), and T32-HL-007343 (to M.P.G.); American Heart Association grant 11SDG5670050 (to J.F.P.-Z.); American Lung Association grant RG-196192-N (to J.F.P.-.Z); and Flight Attendant Medical Research Institute Young Clinical Scientist Award 103236 (to J.M.D. and M.P.G.). Columbia University, on behalf of S.O.M., S.I.Z., A.R.M., and J.M.D., has filed a patent for the use of rottlerin in activating BK channels and treating asthma.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ACh
- acetylcholine
- AHR
- airway hyperreactivity
- ASM
- airway smooth muscle
- BAL
- bronchoalveolar lavage
- BK channel
- large conductance voltage- and calcium-activated potassium channel
- [Ca2+]i
- intracellular calcium concentration
- ER
- endoplasmic reticulum
- ERK
- extracellular signal-regulated kinase
- FEV1
- forced expiratory volume in 1 s
- H&E
- hematoxylin and eosin
- HDM
- house dust mite
- IP3
- inositol 1,4,5-trisphosphate
- IP3R
- inositol 1,4,5-trisphosphate receptor
- JNK
- c-Jun N-terminal kinase
- MAP
- mitogen activated protein
- MCh
- methacholine
- OVA
- ovalbumin
- ROI
- region of interest
- RyR
- ryanodine receptor
- sHBSS
- HEPES buffer-supplemented Hanks' balanced salt solution
- SR
- sarcoplasmic reticulum
REFERENCES
- 1. Fanta C. H. (2009) Asthma. N. Engl. J. Med. 360, 1002–1014 [DOI] [PubMed] [Google Scholar]
- 2. Janssen L. J., Killian K. (2006) Airway smooth muscle as a target of asthma therapy: history and new directions. Respir. Res. 7, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu Q. H., Zheng Y. M., Korde A. S., Yadav V. R., Rathore R., Wess J., Wang Y. X. (2009) Membrane depolarization causes a direct activation of G protein-coupled receptors leading to local Ca2+ release in smooth muscle. Proc. Natl. Acad. Sci. U. S. A. 106, 11418–11423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perez J. F., Sanderson M. J. (2005) The frequency of calcium oscillations induced by 5-HT, ACH, and KCl determine the contraction of smooth muscle cells of intrapulmonary bronchioles. J. Gen. Physiol. 125, 535–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ben-Chaim Y., Tour O., Dascal N., Parnas I., Parnas H. (2003) The M2 muscarinic G-protein-coupled receptor is voltage-sensitive. J. Biol. Chem. 278, 22482–22491 [DOI] [PubMed] [Google Scholar]
- 6. Janssen L. J., Tazzeo T., Zuo J., Pertens E., Keshavjee S. (2004) KCl evokes contraction of airway smooth muscle via activation of RhoA and Rho-kinase. Am. J. Physiol. Lung Cell. Mol. Physiol. 287, L852–L858 [DOI] [PubMed] [Google Scholar]
- 7. Liu C., Zuo J., Pertens E., Helli P. B., Janssen L. J. (2005) Regulation of Rho/ROCK signaling in airway smooth muscle by membrane potential and [Ca2+]i. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L574–L582 [DOI] [PubMed] [Google Scholar]
- 8. Kotlikoff M. I., Kamm K. E. (1996) Molecular mechanisms of beta-adrenergic relaxation of airway smooth muscle. Annu. Rev. Physiol. 58, 115–141 [DOI] [PubMed] [Google Scholar]
- 9. Butler A., Tsunoda S., McCobb D. P., Wei A., Salkoff L. (1993) mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science 261, 221–224 [DOI] [PubMed] [Google Scholar]
- 10. Knaus H. G., Garcia-Calvo M., Kaczorowski G. J., Garcia M. L. (1994) Subunit composition of the high conductance calcium-activated potassium channel from smooth muscle, a representative of the mSlo and slowpoke family of potassium channels. J. Biol. Chem. 269, 3921–3924 [PubMed] [Google Scholar]
- 11. Semenov I., Wang B., Herlihy J. T., Brenner R. (2006) BK channel beta1-subunit regulation of calcium handling and constriction in tracheal smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L802–L810 [DOI] [PubMed] [Google Scholar]
- 12. Seibold M. A., Wang B., Eng C., Kumar G., Beckman K. B., Sen S., Choudhry S., Meade K., Lenoir M., Watson H. G., Thyne S., Williams L. K., Kumar R., Weiss K. B., Grammer L. C., Avila P. C., Schleimer R. P., Burchard E. G., Brenner R. (2008) An african-specific functional polymorphism in KCNMB1 shows sex-specific association with asthma severity. Hum. Mol. Genet. 17, 2681–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zakharov S. I., Morrow J. P., Liu G., Yang L., Marx S. O. (2005) Activation of the BK (SLO1) potassium channel by mallotoxin. J. Biol. Chem. 280, 30882–30887 [DOI] [PubMed] [Google Scholar]
- 14. Springael C., Thomas S., Rahmouni S., Vandamme A., Goldman M., Willems F., Vosters O. (2007) Rottlerin inhibits human T cell responses. Biochem. Pharmacol. 73, 515–525 [DOI] [PubMed] [Google Scholar]
- 15. Townsend E. A., Thompson M. A., Pabelick C. M., Prakash Y. S. (2010) Rapid effects of estrogen on intracellular Ca2+ regulation in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L521–L530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson J. R., Wiley R. E., Fattouh R., Swirski F. K., Gajewska B. U., Coyle A. J., Gutierrez-Ramos J. C., Ellis R., Inman M. D., Jordana M. (2004) Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am. J. Respir. Crit. Care Med. 169, 378–385 [DOI] [PubMed] [Google Scholar]
- 17. Henderson W. R., Jr., Tang L. O., Chu S. J., Tsao S. M., Chiang G. K., Jones F., Jonas M., Pae C., Wang H., Chi E. Y. (2002) A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am. J. Respir. Crit. Care Med. 165, 108–116 [DOI] [PubMed] [Google Scholar]
- 18. Perez-Zoghbi J. F., Bai Y., Sanderson M. J. (2010) Nitric oxide induces airway smooth muscle cell relaxation by decreasing the frequency of agonist-induced Ca2+ oscillations. J. Gen. Physiol. 135, 247–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanderson M. J., Parker I. (2003) Video-rate confocal microscopy. Methods Enzymol. 360, 447–481 [DOI] [PubMed] [Google Scholar]
- 20. Wang J., Homer R. J., Chen Q., Elias J. A. (2000) Endogenous and exogenous IL-6 inhibit aeroallergen-induced Th2 inflammation. J. Immunol. 165, 4051–4061 [DOI] [PubMed] [Google Scholar]
- 21. Neuhaus-Steinmetz U., Glaab T., Daser A., Braun A., Lommatzsch M., Herz U., Kips J., Alarie Y., Renz H. (2000) Sequential development of airway hyperresponsiveness and acute airway obstruction in a mouse model of allergic inflammation. Int. Arch. Allergy Immunol. 121, 57–67 [DOI] [PubMed] [Google Scholar]
- 22. Mehra D., Sternberg D. I., Jia Y., Canfield S., Lemaitre V., Nkyimbeng T., Wilder J., Sonett J., D'Armiento J. (2010) Altered lymphocyte trafficking and diminished airway reactivity in transgenic mice expressing human MMP-9 in a mouse model of asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L189–L196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cho S. H., Woo C. H., Yoon S. B., Kim J. H. (2004) Protein kinase Cdelta functions downstream of Ca2+ mobilization in FcepsilonRI signaling to degranulation in mast cells. J. Allergy Clin. Immunol. 114, 1085–1092 [DOI] [PubMed] [Google Scholar]
- 24. Blacquiere M. J., Hylkema M. N., Postma D. S., Geerlings M., Timens W., Melgert B. N. (2010) Airway inflammation and remodeling in two mouse models of asthma: comparison of males and females. Int. Arch. Allergy Immunol. 153, 173–181 [DOI] [PubMed] [Google Scholar]
- 25. Taylor A., Verhagen J., Blaser K., Akdis M., Akdis C. A. (2006) Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology 117, 433–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaffar Z., Sivakuru T., Roberts K. (2004) CD4+CD25+ T cells regulate airway eosinophilic inflammation by modulating the Th2 cell phenotype. J. Immunol. 172, 3842–3849 [DOI] [PubMed] [Google Scholar]
- 27. Fattouh R., Al-Garawi A., Fattouh M., Arias K., Walker T. D., Goncharova S., Coyle A. J., Humbles A. A., Jordana M. (2011) Eosinophils are dispensable for allergic remodeling and immunity in a model of house dust mite-induced airway disease. Am. J. Respir. Crit. Care Med. 183, 179–188 [DOI] [PubMed] [Google Scholar]
- 28. Naureckas E. T., Ndukwu I. M., Halayko A. J., Maxwell C., Hershenson M. B., Solway J. (1999) Bronchoalveolar lavage fluid from asthmatic subjects is mitogenic for human airway smooth muscle. Am. J. Respir. Crit. Care Med. 160, 2062–2066 [DOI] [PubMed] [Google Scholar]
- 29. Lee J. H., Johnson P. R., Roth M., Hunt N. H., Black J. L. (2001) ERK activation and mitogenesis in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L1019–L1029 [DOI] [PubMed] [Google Scholar]
- 30. Eynott P. R., Nath P., Leung S. Y., Adcock I. M., Bennett B. L., Chung K. F. (2003) Allergen-induced inflammation and airway epithelial and smooth muscle cell proliferation: role of Jun N-terminal kinase. Brit. J. Pharmacol. 140, 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou X. B., Wulfsen I., Lutz S., Utku E., Sausbier U., Ruth P., Wieland T., Korth M. (2008) M2 muscarinic receptors induce airway smooth muscle activation via a dual, Gbetagamma-mediated inhibition of large conductance Ca2+-activated K+ channel activity. J. Biol. Chem. 283, 21036–21044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kume H., Mikawa K., Takagi K., Kotlikoff M. I. (1995) Role of G proteins and KCa channels in the muscarinic and beta-adrenergic regulation of airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 268, L221–L229 [DOI] [PubMed] [Google Scholar]
- 33. Semenov I., Wang B., Herlihy J. T., Brenner R. (2011) BK channel beta1 subunits regulate airway contraction secondary to M2 muscarinic acetylcholine receptor mediated depolarization. J. Physiol. 589, 1803–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barnes P. J. (1985) Clinical studies with calcium antagonists in asthma. Br. J. Clin. Pharmacol. 20(Suppl. 2), 289S–298S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Janssen L. J. (1997) T-type and L-type Ca2+ currents in canine bronchial smooth muscle: characterization and physiological roles. Am. J. Physiol. Cell Physiol. 272, C1757–C1765 [DOI] [PubMed] [Google Scholar]
- 36. Gschwendt M., Muller H. J., Kielbassa K., Zang R., Kittstein W., Rincke G., Marks F. (1994) Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun. 199, 93–98 [DOI] [PubMed] [Google Scholar]
- 37. Soltoff S. P. (2007) Rottlerin: an inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol. Sci. 28, 453–458 [DOI] [PubMed] [Google Scholar]
- 38. Davies S. P., Reddy H., Caivano M., Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soltoff S. P. (2001) Rottlerin is a mitochondrial uncoupler that decreases cellular ATP levels and indirectly blocks protein kinase Cdelta tyrosine phosphorylation. J. Biol. Chem. 276, 37986–37992 [DOI] [PubMed] [Google Scholar]
- 40. Langlois A., Chouinard F., Flamand N., Ferland C., Rola-Pleszczynski M., Laviolette M. (2009) Crucial implication of protein kinase C (PKC)-delta, PKC-zeta, ERK-1/2, and p38 MAPK in migration of human asthmatic eosinophils. J. Leukoc. Biol. 85, 656–663 [DOI] [PubMed] [Google Scholar]
- 41. Schubl S., Tsai S., Ryer E. J., Wang C., Hu J., Kent K. C., Liu B. (2009) Upregulation of protein kinase cdelta in vascular smooth muscle cells promotes inflammation in abdominal aortic aneurysm. J. Surg. Res. 153, 181–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roose J. P., Mollenauer M., Gupta V. A., Stone J., Weiss A. (2005) A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol. Cell. Biol. 25, 4426–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J. S., Kim I. S., Ryu J. S., Yun C. Y. (2008) House dust mite, Dermatophagoides pteronissinus increases expression of MCP-1, IL-6, and IL-8 in human monocytic THP-1 cells. Cytokine 42, 365–371 [DOI] [PubMed] [Google Scholar]
- 44. Liu W., Liang Q., Balzar S., Wenzel S., Gorska M., Alam R. (2008) Cell-specific activation profile of extracellular signal-regulated kinase 1/2, Jun N-terminal kinase, and p38 mitogen-activated protein kinases in asthmatic airways. J. Allergy Clin. Immunol. 121, 893–902 e892 [DOI] [PubMed] [Google Scholar]
- 45. Grunstein M. M., Veler H., Shan X., Larson J., Grunstein J. S., Chuang S. (2005) Proasthmatic effects and mechanisms of action of the dust mite allergen, Der p 1, in airway smooth muscle. J. Allergy Clin. Immunol. 116, 94–101 [DOI] [PubMed] [Google Scholar]
- 46. Miura K., Schroeder J. T., Hubbard W. C., MacGlashan D. W., Jr. (1999) Extracellular signal-regulated kinases regulate leukotriene C4 generation, but not histamine release or IL-4 production from human basophils. J. Immunol. 162, 4198–4206 [PubMed] [Google Scholar]
- 47. Bates M. E., Green V. L., Bertics P. J. (2000) ERK1 and ERK2 activation by chemotactic factors in human eosinophils is interleukin 5-dependent and contributes to leukotriene C(4) biosynthesis. J. Biol. Chem. 275, 10968–10975 [DOI] [PubMed] [Google Scholar]
- 48. Moore P. E., Church T. L., Chism D. D., Panettieri R. A., Jr., Shore S. A. (2002) IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: a role for ERK. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L847–L853 [DOI] [PubMed] [Google Scholar]
- 49. Kampen G. T., Stafford S., Adachi T., Jinquan T., Quan S., Grant J. A., Skov P. S., Poulsen L. K., Alam R. (2000) Eotaxin induces degranulation and chemotaxis of eosinophils through the activation of ERK2 and p38 mitogen-activated protein kinases. Blood 95, 1911–1917 [PubMed] [Google Scholar]
- 50. Hallsworth M. P., Moir L. M., Lai D., Hirst S. J. (2001) Inhibitors of mitogen-activated protein kinases differentially regulate eosinophil-activating cytokine release from human airway smooth muscle. Am. J. Respir. Crit. Care Med. 164, 688–697 [DOI] [PubMed] [Google Scholar]
- 51. Duan W., Chan J. H., Wong C. H., Leung B. P., Wong W. S. (2004) Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J. Immunol. 172, 7053–7059 [DOI] [PubMed] [Google Scholar]
- 52. Goplen N., Karim Z., Guo L., Zhuang Y., Huang H., Gorska M. M., Gelfand E., Pages G., Pouyssegur J., Alam R. (2012) ERK1 is important for Th2 differentiation and development of experimental asthma. FASEB J. 26, 1934–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alrashdan Y. A., Alkhouri H., Chen E., Lalor D. J., Poniris M., Henness S., Brightling C. E., Burgess J. K., Armour C. L., Ammit A. J., Hughes J. M. (2012) Asthmatic airway smooth muscle CXCL10 production: mitogen-activated protein kinase JNK involvement. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L1118–L1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tan X., Alrashdan Y. A., Alkhouri H., Oliver B. G., Armour C. L., Hughes J. M. (2013) Airway smooth muscle CXCR3 ligand production: regulation by JAK-STAT1 and intracellular calcium. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L790–L802 [DOI] [PubMed] [Google Scholar]
- 55. Gujral M. L., Varma D. R., Sareen K. N., Roy A. K. (1960) Oral contraceptives. Part II. Antifertility effect of Mallotus philippinensis Mueller-Argoviensis. Indian J. Med. Res. 48, 52–58 [PubMed] [Google Scholar]
- 56. Zhang D., Anantharam V., Kanthasamy A., Kanthasamy A. G. (2007) Neuroprotective effect of protein kinase C delta inhibitor rottlerin in cell culture and animal models of Parkinson's disease. J. Pharmacol. Exp. Ther. 322, 913–922 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.