

Abstract
Heart activity and long-term function are regulated by the sympathetic and parasympathetic branches of the nervous system. Parasympathetic neurons have received increased attention recently because acetylcholine (ACh) has been shown to play protective roles in heart disease. However, parasympathetic innervation is sparse in the heart, raising the question of how cholinergic signaling regulates cardiomyocytes. We hypothesized that non-neuronal secretion of ACh from cardiomyocytes plays a role in cholinergic regulation of cardiac activity. To test this possibility, we eliminated secretion of ACh exclusively from cardiomyocytes by targeting the vesicular acetylcholine transporter (VAChT). We find that lack of cardiomyocyte-secreted ACh disturbs the regulation of cardiac activity and causes cardiomyocyte remodeling. Mutant mice present normal hemodynamic parameters under nonstressful conditions; however, following exercise, their heart rate response is increased. Moreover, hearts from mutant mice present increased oxidative stress, altered calcium signaling, remodeling, and hypertrophy. Hence, without cardiomyocyte-derived ACh secretion, hearts from mutant mice show signs of imbalanced autonomic activity consistent with decreased cholinergic drive. These unexpected results suggest that cardiomyocyte-derived ACh is required for maintenance of cardiac homeostasis and regulates critical signaling pathways necessary to maintain normal heart activity. We propose that this non-neuronal source of ACh boosts parasympathetic cholinergic signaling to counterbalance sympathetic activity regulating multiple aspects of heart physiology.—Roy, A., Fields, W. C., Rocha-Resende, C., Resende, R. R., Guatimosim, S., Prado, V. F., Gros, R., Prado, M. A. M. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart.
Keywords: cardiac remodeling, VAChT, choline acetyltransferase, parasympathetic activity, cardiac hypertrophy, autonomic function
Acetylcholine (ACh) released by parasympathetic nerves regulates the minute-to-minute changes in heart rate and contractility required for proper cardiovascular function via muscarinic receptors, opposing the activity of the sympathetic nervous system (1). In addition to regulating atrial activity, ACh plays multiple roles in ventricular function (2, 3), notwithstanding limited parasympathetic innervation in ventricular regions (4, 5). The exact mechanisms by which ACh can have such widespread effects, despite somewhat limited parasympathetic innervation in regions other than the atria, are not fully understood. Interestingly, in the pancreas (6) as well as the immune system (7, 8), non-neuronal sources of ACh secretion have recently been shown to play a role in regulating insulin secretion (6) and the cholinergic antiinflammatory system (9, 10), respectively. However, information is limited regarding the contribution of non-neuronal ACh for autonomic regulation of other bodily functions in vivo.
Recent experiments have indicated that cultured cardiomyocytes, similar to pancreatic alpha cells (6) and lymphocytes (7), can express the enzymatic machinery required for ACh synthesis (11, 12). Moreover, secretion of ACh from cultured cardiomyocytes depends on the activity of the vesicular acetylcholine transporter (VAChT; ref. 12). However, whether this non-neuronal source of ACh plays a physiological role in the regulation of heart activity or function is unknown. Here, we used the Cre-loxP system to eliminate VAChT exclusively in cardiomyocytes and test for potential roles for cardiomyocyte-secreted ACh in heart activity. We found that this novel form of cellular communication is required for physiological regulation of heart size and stress levels, likely by maintaining high levels of ACh at synaptic junctions. We also discovered that the recovery of heart rate following exercise or stress is disturbed in the absence of this non-neuronal source of ACh, which suggests the widespread effects of non-neuronal cardiomyocyte-derived ACh. Our study provides a novel mechanism for autocrine/paracrine regulation of cardiomyocytes by non-neuronal ACh secretion.
MATERIALS AND METHODS
Animals
Transgenic mice expressing Cre under the control of the cardiac-specific murine α-myosin-heavy chain promoter (Myh6-Cre) were obtained from Jackson Laboratory [Bar Harbor, ME, USA; B6.FVB-Tg(Myh6-cre)2182Mds/J; stock no. 011038] and bred to VAChT-floxed (flanked by loxP) mice (VAChTflox/flox) (13). VAChTflox/flox mice have been backcrossed 5 times to C57BL6/j mice as described previously (13). VAChTflox/flox mice are not different from wild-type (WT) mice (14). Littermates (VAChTWT/flox,Myh6-cre+ × VAChTWT/flox,Myh6-cre−) were crossed to generate VAChTMyh6-Cre-flox/flox and VAChTflox/flox−. F2 littermates were then bred to generate mice used for this study. VAChTflox/flox littermates were used as controls.
To generate the cardiomyocyte-specific choline acetyltransferase (ChAT)-knockout (KO) mice, Myh6-cre mice were bred to ChAT-floxed mice (15) obtained from Jackson Laboratory (B6.129-Chattm1Jrs/J; ChATflox/flox) (13). Littermates (ChATWT/flox,Myh6-cre+ × ChATWT/flox,Myh6-cre−) were crossed to generate ChATMyh6-Cre-flox/flox and ChATflox/flox.
The Rosa-EYFP strain [B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J; stock no. 006148; Jackson Laboratory) was used as a Cre reporter. Animals were maintained and cared for according to an approved animal protocol at the University of Western Ontario (2008-127) and following the Canadian Council on Animal Care. Only male mice were used for adult cardiomyocyte isolation and all in vivo experiments. Neonatal cardiomyocytes were isolated and cultured from mice of both genders.
Neonatal cardiomyocyte culture
Neonatal cardiomyocytes were isolated as described previously (16). Briefly, cardiac cells were plated in dishes containing M199 medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 10% FBS, and 2 mM l-glutamine. Cytosine-D-arabinofuranoside (ARA-c; 20 μg/ml) was used to prevent growth of fibroblasts. For hypertrophy studies, cardiac cells at d 4 in culture were incubated with vesamicol (VES; 5 μM) or hemicholinium-3 (HC-3; 10 μM) for 48 h and then used for immunofluorescence or qPCR analyses.
qPCR/RT-PCR
RNA was extracted from isolated cardiomyocytes, and cDNA was synthesized as described previously (17). A brain sample was used as a positive control for VAChT, and a nontemplate reaction was used as a negative control. qPCR for atrial natriuretic peptide (ANP) and G-protein-coupled receptor kinases 2 and 5 (GRK2 and GRK5) was performed as described previously (17). For primer sequences, see Supplemental Table S1.
Immunoblotting
Isolated adult cardiomyocytes were lysed using ice-cold modified RIPA buffer. Protein (80 μg) was separated using SDS-PAGE, and PVDF membranes were probed with anti-VAChT antibody (1:200; Synaptic Systems, Goettingen, Germany). α-Actinin (1:2000; Sigma-Aldrich, St. Louis, MO, USA) was used as a loading control.
Immunostaining
Adult cardiomyocytes were subjected to immunofluorescence protocol as described previously (12). Cells were incubated with one of the following antibodies: anti-VAChT (1:50; Synaptic Systems), anti-ChAT (1:100; Abcam, Cambridge, MA, USA), anti-CHT1 (1:500; kindly supplied by R. Jane Rylett; University of Western Ontario, London, ON, Canada; ref. 18), anti-ANP (1:200; Abcam), or anti-GRK5 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The cells were colabeled with α-actinin (1:200; Sigma-Aldrich). α-Actinin labeled cells were used to measure cardiomyocyte cell surface area. Images were acquired using either the Leica SP5 II (Leica Microsystems, Wetzlar, Germany) or Zeiss LSM 510 Meta (Carl Zeiss, Oberkochen, Germany) confocal system (×63 objective, 488-nm Ar laser and 546-nm HeNe laser were used for excitation of fluorophores).
Measurement of ACh secretion (fluorometric assay)
Transmitter release was measured using the Choline/ACh Quantification Kit (Biovision, Milpitas, CA, USA) as described previously (19). Briefly, cultured medium was collected from VAChTflox/flox and VAChTMyh6-Cre-flox/flox cardiomyocytes and incubated with either 100 μM pyridostigmine bromide (Sigma-Aldrich; P9797) or 100 μM pyridostigmine bromide and 1 μM VES HCl (Sigma-Aldrich; V100) at 37°C for 4 h. Cultured medium was collected and centrifuged at 10,000 rpm for 5 min at 4°C. The resulting supernatant was collected and filtered using a 0.2-μm Acrodisc Syringe Filter (Pall Life Sciences, Pensacola, FL, USA) and placed on ice. ACh concentration was determined using the fluorometric Choline/ACh Quantification Kit (λex=35 nm, λem=590 nm). Each sample was assayed in duplicate and experiments were conducted ≥4 times using separate cultures.
Measurement of ACh secretion (HPLC-electrochemical detection)
Cultured neonatal cardiomyocytes from VAChTflox/flox and VAChTMyh6-Cre-flox/flox mice were treated with 100 μM pyridostigmine bromide (Sigma Aldrich; P9797) and incubated for 4 h at 37°C. It has been reported previously that both subacute treatment with pyridostigmine (20) as well as acute exposure to stress (21) can increase the transcription of acetylcholinesterase (AChE) in the brain. However, in vitro studies have confirmed that pyridostigmine is a very potent AChE inhibitor (IC50=0.33 μM for inhibition of erythrocyte AChE activity; ref. 22). As such, 100 μM pyridostigmine was used in our experiments to ensure the inhibition of virtually all AChE activity throughout the experiment. Cultured media was collected and centrifuged at 13,200 RPM for 5 min at 4°C. The supernatant was filtered through a 0.2 μM Acrodisc Syringe Filter (Pall Life Sciences, Port Washington, NY, USA; PN 4602) and injected into an ESA UltiMATE 3000 system with a Choulochem electrochemical detector (Thermo Scientific, Waltham, MA, USA). The sequence was run using the following components and parameters: flow rate, 0.300 ml/min; injection volume, 20 μl; cell potential, 275 mV; column, MGII Capcell Pak C18 column (Shiseido, Tokyo, Japan; 92461); column temperature, 40°C; and ACh postcolumn solid-phase reactor (Thermo Scientific; 70-0640A).
Nitric oxide (NO) measurement
Neonatal cardiomyocytes from control and cardiomyocyte-specific VAChT-KO (cVAChT; VAChTMyh6-Cre-flox/flox) mice were used to measure NO production, as described previously (12). Cells were incubated with either carbachol (10 μM) or pyridostigmine (100 μM). Images were acquired using the Leica SP5 II confocal system and analyzed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry (IHC)
Hearts were excised and fixed using 4% PFA. IHC was performed on slices as described previously (23). Slices were incubated with either anti-VAChT (1:200; Synaptic Systems) or anti-CHT1 (1:200) (18) to visualize intracardiac ganglia and terminals, respectively.
Heart rate and blood pressure recording
Both heart rate and blood pressure were recorded from conscious animals using the CODA tail-cuff blood pressure system (Kent Scientific, Torrington, CT, USA), as described previously (24).
Electrocardiography (ECG)
Electrocardiograms were recorded using radiotelemeters, as described previously (3). HR was recorded continuously over 24 h to obtain baseline recordings. In addition, heart rates were recorded in the home cages immediately following i.p. injection of saline or following an acute exercise routine (ramp up from 5 m/min to 15 m/min over 60 s, followed by 180 s at 15 m/min).
Cardiomyocyte morphometry
In situ cardiomyocyte cell surface area was measured as described previously (3).
Reactive oxygen species (ROS) measurement
ROS levels were measured using the MitoSOX Red superoxide indicator (Invitrogen, Carlsbad, CA, USA), as described previously (17).
Protein oxidation measurement
Protein was isolated from whole hearts using ice-cold modified RIPA buffer, separated using SDS-PAGE, and transferred onto PVDF membrane. The levels of oxidatively modified proteins were analyzed in control and cVAChT hearts using the OxyBlot Protein Oxidation Detection Kit (Millipore. Bedford, MA, USA) following the manufacturer's directions.
Indirect calorimetry, activity, and inactivity
These experiments were performed using the Comprehensive Laboratory Animal Monitoring System (CLAMS) metabolic chamber (Columbus Instruments, Columbus, OH, USA) as described previously (25, 26). Vo2, Vco2, food and water intake, and activity/inactivity were measured. Respiratory exchange ratio and energy expenditure/heat were calculated within the Oxymax software (Columbus Instruments). Periods of inactivity (sleep) were obtained using the sleep detection algorithms available within the Oxymax software.
Preparation of siRNA
Potential target sites within the VAChT gene were selected and then searched with the U.S. National Center for Biotechology Information (NCBI; Bethesda, MD, USA) Basic Local Alignment Search Tool (BLAST; http://blast.ncbi.nlm.nih.gov) was used to confirm specificity for the transporter. The siRNA for VAChT was prepared as described previously by our group (27). The sense and antisense oligonucleotides of siRNA were 5′-GGAGCAGGGAGGCAGAAGAAGCTGT-3′ and 5′-ACAGCTTCTTCTGCCTCCCTGCTCCAT-3′, respectively. For siRNA studies, neonatal cardiomyocyte cultures were transfected at d 4 with 100 nM of siRNA 48 h prior to measurements. The cells were then used for immunofluorescence, immunobloting, or qPCR analyses.
Cardiomyocyte isolation and Ca2+ recording
Adult cardiomyocytes were isolated and calcium transients were recorded as described previously (3, 17) using line-scan imaging on a Zeiss LSM 510 Meta confocal microscope. Images were processed and Ca2+ recordings were analyzed using ImageJ software.
Hemodynamic measurements
Invasive left ventricular (LV) hemodynamic measurements were obtained under baseline as well as following administration of isoproterenol (ISO; 0.5 μg i.p.) using a Millar Mikro-tip pressure transducer (Millar Instruments, Houston, TX, USA), as described previously (3). All the LV parameters were obtained using the PowerLab Chart Analysis software (AD Instruments, Colorado Springs, CO, USA).
Statistical analyses
Results for experiments are reported as means ± sem. Student's t test, 1-way ANOVA with a Tukey's post hoc test, or 2-way ANOVA was used to assess statistical differences between experimental groups as required, using GraphPad (San Diego, CA, USA) or SigmaStat (Systat Software Inc., San Jose, CA, USA). Values of P < 0.05 were considered statistically significant.
RESULTS
Generation of cardiomyocyte-specific VAChT null mice
Previous experiments indicated that, in cultured neonatal cardiomyocytes, ACh secretion is dependent on the activity of VAChT (12), a transporter that is critical for ACh storage in nerve endings (28, 29). To selectively interfere with secretion of ACh from cardiomyocytes in vivo, we generated the VAChTMyh6-Cre-flox/flox (cVAChT henceforth) mice by crossing Myh6-Cre mice [B6.FVB-Tg(Myh6-cre)2182Mds/J; Jackson Laboratory] with VAChT floxed mice (ref. 13 and Supplemental Fig. S1A, B). The α-myosin heavy chain (α-MHC; Myh6) promoter drives Cre expression exclusively in cardiomyocytes (30). Although previous reports indicated that Cre is expressed mainly in ventricles in Myh6-Cre mice, Cre expression in the atria has been observed during development (31). To confirm elimination of VAChT in cVAChT mice, we isolated adult cardiomyocytes and performed RT-PCR to detect VAChT transcripts (Fig. 1A). A band of 167 bp was detected in VAChTflox/flox but not in cVAChT mice. Sequencing analysis confirmed that this band represented the VAChT sequence. Moreover, immunoblot (Fig. 1B) and immunofluorescence analysis (Fig. 1C) confirmed elimination of VAChT in isolated ventricular cardiomyocytes from cVAChT mice. Moreover, whole-mount fluorescent IHC in the atria of control mice revealed that VAChT is also expressed in sinoatrial (SA) nodal cells, as colocalization was observed between VAChT and HCN4, a marker for the SA node (Fig. 1D). Notably, VAChT staining in SA nodal cells was absent in cVAChT tissue, indicating that VAChT was also deleted in these cells in cVAChT mice (Fig. 1D). Furthermore, VAChT expression in parasympathetic ganglia and in nerve terminals was not altered in cVAChT mice (Fig. 1D, E), indicating that we specifically eliminated non-neuronal VAChT in the heart. In support of these data, we used a reporter mouse line to demonstrate that Cre expression is absent in parasympathetic nerve terminals stained with an antibody against the high-affinity choline transporter (CHT1; Supplemental Fig. S1C). In addition, expression of CHT1 was not affected in isolated cardiomyocytes from cVAChT mice (Supplemental Fig. S1D). However, cVAChT cardiomyocytes appeared to have greater levels of ChAT expression (Supplemental Fig. S1E), an observation similar to that previously reported in VAChT-KO mice, which reflects, in part, a rearrangement of the cholinergic gene locus following Cre-mediated recombination (23).
Figure 1.
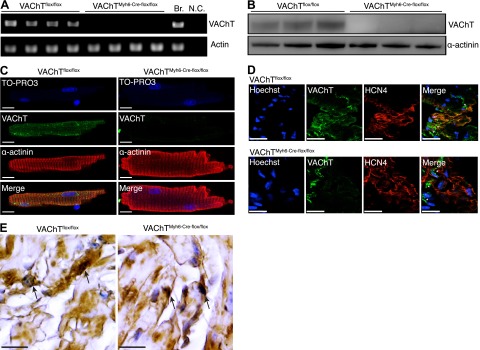
Selective elimination of VAChT in cardiomyocytes of cVAChT mice. A–C) VAChT expression was determined by PCR (A; Br., brain, N.C., negative control), immunoblotting (B), and immunofluorescence (C) in adult cardiomyocytes. Scale bars = 25 μm. D) Colabeling for VAChT and HCN4 in whole-mount atrial tissue from control and cVAChT mice. Asterisks indicate VAChT staining in parasympathetic nerve terminals. Scale bars = 25 μm. E) VAChT immunoreactivity in intracardiac parasympathetic ganglia from cVAChT mice. Arrows indicate positive staining for VAChT. Scale bars = 25 μm.
Genetic removal of VAChT from cardiomyocytes inhibits ACh release
Secretion of ACh from cultured neonatal cardiomyocytes was analyzed using a fluorometric assay to detect choline/ACh levels in culture media. The assay was performed in neonatal cardiomyocytes after 4 d in culture, to avoid potential contribution of parasympathetic neurons that could contaminate adult cardiomyocyte preparations. Using this method, we could confidently detect as low as 10 pmols/well for ACh (Supplemental Fig. S2A). ACh release from control myocytes could be easily detected in the presence of pyridostigmine, a cholinesterase inhibitor (Supplemental Fig. S2B) used to preserve secreted neurotransmitter. In the presence of VES, a specific inhibitor of the VAChT, ACh release from these cells was significantly diminished (Supplemental Fig. S2B). In agreement with the pharmacological data, cVAChT cardiomyocytes showed no detectable ACh release (Fig. 2A).
Figure 2.

A) ACh release from neonatal cardiomyocytes isolated and cultured from control and cVAChT mice. n = number of separate cell isolations for each genotype. ***P < 0.001 vs. control. B) ACh release from control and cVAChT neonatal cardiomyocytes as detected through HPLC with electrochemical detection. n = number of separate cell isolations for each genotype. **P < 0.01 vs. control. C) Bioassay to measure ACh release in cultured neonatal cardiomyocytes using DAF fluorescence. Cells were treated with either carbachol (Carb.) to activate muscarinic receptors and increase NO levels or pyridostigmine (PYR), which preserves secreted ACh, which can then activate muscarinic receptors and increase the production of NO (12). n = number of cells examined from 5 separate cell isolations/genotype. Scale bars = 25 μm. Data are represented as means ± sem. *P < 0.05 vs. control.
We confirmed these findings by performing HPLC with electrochemical detection of ACh in media from cultured cardiomyocytes. We could confidently detect up to 125 fmol of ACh on the column (Supplemental Fig. S2C). ACh secretion was detectable from WT cardiomyocytes in the presence of pyridostigmine (Supplemental Fig. S2D); however, ACh secretion from cells treated with VES was significantly reduced (Supplemental Fig. S2D). Furthermore, no ACh was detected in media from cVAChT cardiomyocytes using HPLC with electrochemical detection, whereas ACh could be detected in media from WT cardiomyocytes (Fig. 2B and Supplemental Fig. S2E).
We further confirmed that secretion of ACh was inhibited in cVAChT neonatal myocyte cultures through the use of a bioassay with the fluorescent dye DAF-FM, as described previously (12). DAF becomes fluorescent in the presence of NO, which is produced following activation of muscarinic receptors by ACh secreted from cardiomyocytes. To validate this bioassay, we used neonatal cardiomyocytes from C57BL/6 WT mice. We showed that DAF fluorescence was significantly increased in WT cardiomyocytes following treatment with carbachol (a muscarinic agonist; Supplemental Fig. S2F). Pyridostigmine also augmented the fluorescent signal (Supplemental Fig. S2F). Conversely, no fluorescence increase was observed in cardiomyocytes cotreated with pyridostigmine and either HC-3 (to inhibit the production of ACh) or VES (Supplemental Fig. S2F). These results indicate that this bioassay has enough sensitivity to detect changes in ACh release from WT cardiomyocytes. When cardiomyocytes from cVAChT mice were tested, DAF fluorescence was significantly increased in response to carbachol; however, no response was observed following treatment with the cholinesterase inhibitor pyridostigmine. In contrast, cardiomyocytes from littermate controls presented a robust response to carbachol and cholinesterase inhibition (Fig. 2C). Together, these results clearly establish using multiple methodologies that secretion of ACh from cardiomyocytes is impaired in cVAChT mice.
Inhibition of ACh secretion from cardiomyocytes alters heart activity
cVAChT mice did not present any gross abnormalities in appearance or body weight (29.1±4.1 vs. 30.8±4.9 g in control vs. cVAChT mice). Furthermore, these mice did not present alterations in several metabolic parameters, as measured using metabolic cages (Supplemental Table S2), which suggests that the cardiomyocyte changes observed were not deleterious to mutant mice under control conditions. Accordingly, we did not observe changes in blood pressure, as determined by tail cuff (Fig. 3A), nor in basal heart rate, as measured by radiotelemetry (ECG, Fig. 3B). In contrast, cVAChT mice showed an increase in heart rate when analyzed using the noninvasive tail-cuff system (Fig. 3A). This increase in heart rate was not due to the presence of Cre recombinase, as it was not observed in Myh6-cre mice when compared to WT mice under the same conditions (Supplemental Fig. S3A). These results suggest a possible role for cardiomyocyte-derived ACh in the regulation of heart rate.
Figure 3.

Analysis of heart rate in cVAChT mice. A) Blood pressure and heart rate analysis in VAChTflox/flox and cVAChT mice using the CODA tail-cuff system. B) Heart rate over 24 h in awake, freely moving VAChTflox/flox and cVAChT mice in their home cage (n≥4 mice/genotype). C) Heart rate response following gentle restraint and i.p. saline injection (n≥4 mice/genotype). D) Heart rate recovery following acute, brief exercise in VAChTflox/flox and cVAChT mice (n≥4 mice/genotype). Data are represented as means ± sem. *P < 0.05 vs. control mice.
To further confirm that the increased heart rate observed in cVAChT mice via tail cuff was due to inhibition of the non-neuronal cholinergic system in cardiomyocytes, we generated cardiomyocyte-specific ChAT-KO (ChATMyh6-Cre-flox/flox) mice. This ChAT-floxed line has been previously characterized; when crossed with Cre lines, ChAT expression is abolished, which parallels elimination of ACh (15, 32). Tail-cuff analysis in these mice also revealed increased heart rate in mice lacking ACh production (Supplemental Fig. S3B).
Tail-cuff measurements require physical restraint, which is stressful for mice, while ECG measurements are obtained under conditions similar to the home cage environment. Hence, we hypothesized that the difference in heart rate observed using these two techniques might reflect an imbalance in cardiac regulation in mutant mice. Specifically, that cardiomyocyte-derived ACh could play a physiological role in boosting the parasympathetic signaling required for heart rate recovery in response to stress. To test this hypothesis, we used ECG telemetry to record heart rates immediately after mice were stressed by an intraperitoneal injection of saline. We observed that heart rate recovery of cVAChT mice to baseline levels was slower when compared to control VAChTflox/flox mice (Fig. 3C). These data suggest that, following increased sympathetic demand due to stress, regulation of heart function by the parasympathetic system relies, at least in part, on ACh secreted from cardiomyocytes. To further test this possibility, cVAChT and littermate controls implanted with ECG telemeters were submitted to an acute, low-intensity, treadmill exercise test. Immediately following this mild exercise routine, heart rates in control and cVAChT mice were recorded in their home cage. The results demonstrate that this exercise routine led to a significantly greater increase in heart rate in cVAChT mice as compared to control mice (Fig. 3D). Moreover, heart rate recovery to preexercise levels took significantly longer in cVAChT mice when compared to control mice (Fig. 3D). These results suggest that cardiomyocyte-derived ACh may normally offset sympathetic activity, helping the heart to respond to increased sympathetic demand.
cVAChT mice display cardiac hypertrophy and molecular remodeling
The results above suggest that control of heart rate, particularly under stress, is affected in cVAChT mice potentially due to decreased cholinergic signaling. Previous experiments indicated that in vitro inhibition of either VAChT activity or ACh synthesis increased hypertrophy in cultured neonatal cardiomyocytes treated with sympathetic agonists (12). To determine whether cardiomyocyte-secreted ACh plays a role in cardiac remodeling in vivo, we analyzed heart weight in 3-mo-old mutant mice. As shown in Fig. 4A, cVAChT mice showed increased heart size. Cardiac hypertrophy was due to remodeling of cVAChT myocytes, which exhibited a significant increase in surface area when measured both in vitro (Fig. 4B) and in situ (Fig. 4C). Notably, the presence of Cre recombinase alone did not lead to either cardiac or cardiomyocyte hypertrophy (Supplemental Fig. S4A, B).
Figure 4.
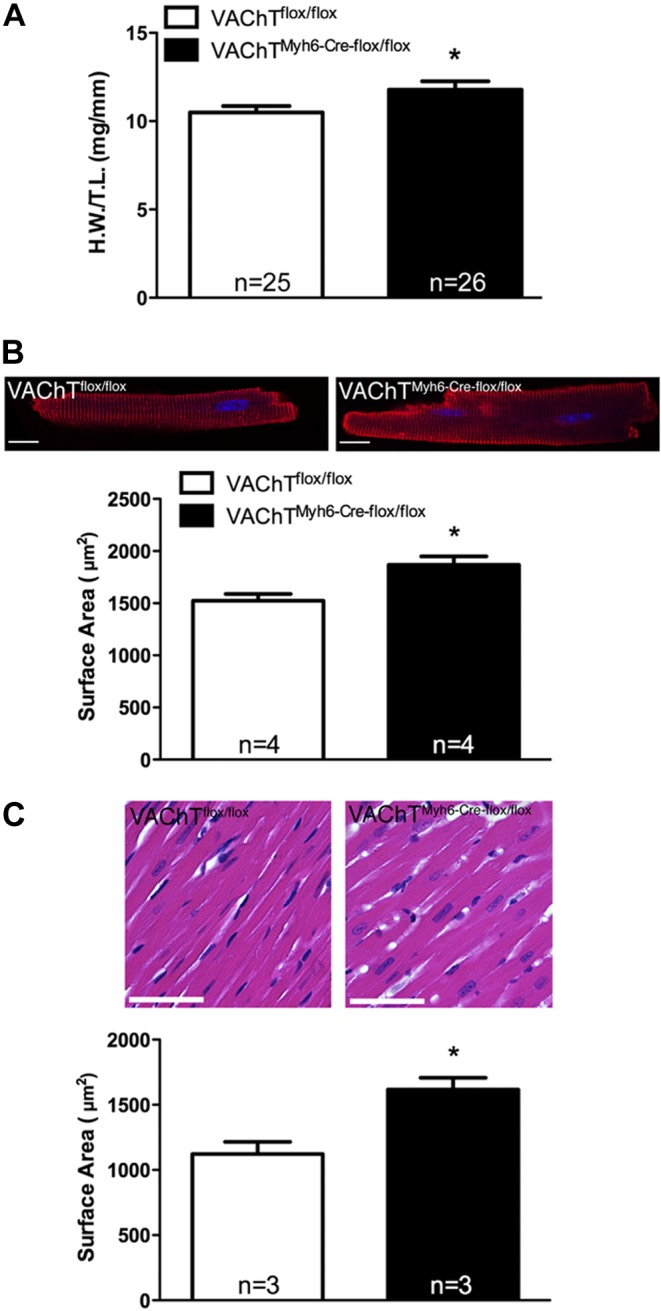
Cardiac hypertrophy in cVAChT mice. A) Heart weight normalized to tibia length in VAChTflox/flox and cVAChT mice (n=number of mice). B) Surface area of isolated cardiomyocytes (n=number of mice/genotype; ≥75 cells/genotype were analyzed). C) Surface area of cardiomyocytes in situ (n=number of mice/genotype, ≥70 cells/genotype were analyzed). Scale bars = 25 μm. Data are represented as means ± sem. *P < 0.05 vs. control.
To further test whether the observed hypertrophic response was indeed dependent on VAChT expression and activity, we cultured WT neonatal cardiomyocytes and treated them with a pharmacological inhibitor of VAChT activity (VES), or with siRNA against VAChT (Supplemental Fig. S4C). VAChT knockdown was confirmed through qPCR and immunoblotting, which revealed an 80 and 60% decrease in VAChT mRNA and protein levels, respectively. Both of these treatments led to a significant increase in cardiomyocyte size after 48 h in culture. Furthermore, cardiomyocytes treated with an inhibitor of CHT1, HC-3, which blocks ACh synthesis, presented similar hypertrophy (Supplementary Fig. S4C), thus confirming that the hypertrophic response was due to inhibition of cholinergic signaling at the level of the myocytes.
Cardiac hypertrophy is associated with increased expression of fetal program genes, which contribute to cardiac remodeling (33). Expression of the markers of cardiac remodeling and stress, β-myosin heavy chain (β-MHC) and ANP, was increased severalfold in cardiomyocytes from cVAChT mice (Fig. 5A). Immunostaining in isolated cardiomyocytes also revealed increased ANP protein levels (Fig. 5B, C), compatible with cardiomyocyte remodeling. Cardiomyocyte remodeling and reactivation of the fetal gene program are usually associated with increased cellular stress (34). Furthermore, hypertrophy induced through hyperadrenergic signaling has been coupled to increased ROS levels (35). If removal of VAChT from cardiomyocytes leads to imbalanced autonomic control of the heart, it is possible that mutant cardiomyocytes are under considerably more stress than control cardiomyocytes. We tested this possibility and found that cardiomyocytes isolated from cVAChT mice showed increased levels of ROS (as determined via mitochondrial superoxide levels; Fig. 5D), suggesting that myocytes lacking the intrinsic cholinergic system display increased levels of stress. Furthermore, we determined that increased ROS levels in myocytes led to an increase in overall protein oxidation in the whole heart in cVAChT mice (Fig. 5E), as measured using the OxyBlot protein oxidation kit (Millipore), which can detect the levels of oxidized proteins.
Figure 5.

Cellular stress in cVAChT cardiomyocytes. A) Expression of the cardiac stress markers, β-MHC and ANP, in control and cVAChT cardiomyocytes (n=number of mice). B, C) Immunostaining for ANP in adult cardiomyocytes from control and cVAChT mice. D) Measurement of ROS levels in isolated cardiomyocytes loaded with the MitoSOX superoxide indicator (n=number of cells; ≥3 mice/genotype). E) Measurement of oxidized protein levels in whole hearts from control and cVAChT mice (n=number of mice). F, G) Assessment of calcium transients in isolated ventricular myocytes from VAChTflox/flox and cVAChT mice. F) Representative recordings of line-scan profile of Ca2+ transients in control and cVAChT myocytes. G) Summary of peak Ca2+ (n=number of cardiomyocytes; cells isolated from ≥3 mice/genotype). Scale bars = 25 μm. Data are represented as means ± sem. *P < 0.05 vs. control.
Increased levels of mitochondrial ROS have previously been shown to increase [Ca2+]i in arterial myocytes, especially following induction of hypoxic stress (36). Therefore, we sought to determine whether the increased levels of ROS observed in cVAChT myocytes were accompanied with changes in calcium transients in ventricular cardiomyocytes. In agreement with this possibility, we detected an alteration in calcium handling in cardiomyocytes obtained from mutant mice with an increase in peak calcium (Ca2+) transients (Fig. 5F, G). These results suggest that elimination of VAChT in cardiomyocytes leads to remodeling of cardiomyocytes, with overactivation of Ca2+ transients potentially altering cardiac function.
It is likely that inhibition of the non-neuronal cholinergic system leads to imbalanced regulation of cardiac activity. As such, we analyzed whether components involved in sympathetic signaling, and thus, regulation of cardiomyocyte function, were altered in cVAChT mice. GRKs have been shown to play important roles in cardiac hypertrophy in response to increased sympathetic activation (37, 38). Therefore, we examined the expression of GRK2 and GRK5, the two most predominantly expressed GRKs in cardiac tissue. Although GRK2 did not show any changes in expression, mRNA expression of GRK5 was significantly increased in cVAChT myocytes (Fig. 6A). Moreover, immunostaining analysis showed that subcellular localization of GRK5 was altered in cVAChT cardiomyocytes, as mutated cells showed increased nuclear localization for GRK5 as compared to control cardiomyocytes (Fig. 6B, C).
Figure 6.
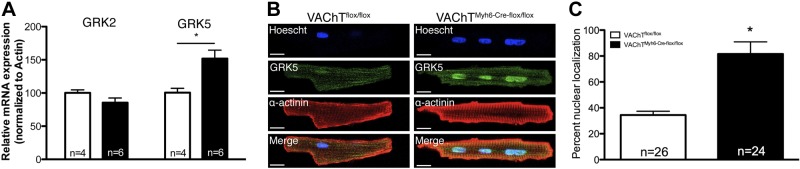
Cardiac remodelling in cVAChT cardiomyocytes. A) mRNA expression of GRK5 in isolated cells from control and cVAChT mice (n=number of mice). B, C) Immunostaining for GRK5 in isolated myocytes. Scale bars = 25 μm. Data are represented as means ± sem. *P < 0.05 vs. control.
Cardiomocyte remodelling in cVAChT mice affects LV function
Ventricular cardiomyocytes from cVAChT mice display remodeling and hypertrophy; therefore, we tested whether these alterations affected LV contractility. Invasive hemodynamic assessments were performed on anesthetized control and cVAChT mice. As shown in Table 1, the hemodynamic parameters were similar in both genotypes under baseline conditions; however, the LV hemodynamics were altered in cVAChT mice following a bolus dose of ISO, used to increase cardiac contractility. Interestingly, although heart rate was similar in both genotypes under baseline, the cVAChT mice showed a significant decrease in heart rate following ISO treatment. Furthermore, the maximum rate of LV pressure rise (peak +dP/dtmax) was significantly decreased, and the maximum rate of LV pressure fall (peak −dP/dtmin) showed a tendency for decrease in cVAChT mice (P=0.0508). In addition, the contractility index of cVAChT mice was not different from control counterparts under baseline; however, a significant reduction was found in the contractility index following ISO treatment in cVAChT mice. These results suggest compromised LV function in the mutant mice, especially under increased cardiac demand, as induced through treatment with ISO.
Table 1.
Hemodynamic parameters for control (n=9) and cVAChT (n=9) mice under baseline and following isoproterenol stimulation
| Parameter | Baseline |
Isoproterenol |
||
|---|---|---|---|---|
| VAChTflox/flox | VAChTMyh6-cre-flox/flox | VAChTflox/flox | VAChTMyh6-cre-flox/flox | |
| HR (bpm) | 289.9 ± 24.7 | 267.4 ± 16.7 | 629.2 ± 11.2 | 566.1 ± 15.7* |
| LVSP (mmHg) | 103.3 ± 6.5 | 101.8 ± 4.0 | 97.2 ± 7.4 | 92.1 ± 5.6 |
| LVEDP (mmHg) | 7.0 ± 2.4 | 12.5 ± 3.3 | −0.3 ± 1.5 | 2.3 ± 2.0 |
| +dP/dTmax (mmHg/s) | 7699 ± 809 | 6999 ± 720 | 16819 ± 1766 | 12432 ± 861* |
| −dP/dTmin (mmHg/s) | −7370 ± 395 | −6752 ± 518 | −9770 ± 1019 | −7413 ± 212# |
| Contractility index (s−1) | 154.8 ± 8.1 | 148.9 ± 13.9 | 347.5 ± 14.1 | 280.1 ± 15.6* |
+dP/dTmax, maximum first derivative of the change in left ventricle pressure; dP/dTmin, minimum first derivative of the change in left ventricle pressure HR, heart rate; LVEDP, left ventricular end diastolic pressure; LVSP, left ventricular systolic pressure. Values are represented as means ± se.
P < 0.05,
P = 0.0508.
DISCUSSION
Our experiments reveal a novel and unexpected way by which cardiovascular function is regulated. These data indicate that cardiomyocytes can secrete significant amounts of ACh in vivo via a VAChT-dependent mechanism, similar to that observed in nerve terminals (39). cVAChT mice seem to lack the capacity to sustain normal levels of cholinergic signaling, implicating non-neuronal ACh as part of a physiological system that controls cardiac function.
Lack of cardiomyocyte-secreted ACh leads to altered heart rate regulation under stress. The SA node controls heart rate; therefore, the alteration in heart rate we observed in cVAChT mice is likely related to the altered expression of VAChT in SA node myocytes observed in these mutants. Notably, measurements of heart rate in the absence of stress indicate that regulation of basal activity does not seem to depend on this cardiomyocyte cholinergic system. Hence, contribution of cardiomyocyte-derived ACh is revealed only with increased demand on the parasympathetic system, such as following exercise. Notably, an inverse relationship between decreased serum AChE levels and delayed heart rate recovery following exercise has previously been reported in healthy human subjects (40). This previous work provides further support for the importance of peripheral ACh levels, including cardiomyocyte-derived ACh, in regulating cardiac function.
It is likely that non-neuronal ACh, secreted from cardiomyocytes, functions through similar second messenger systems as those activated when neuronal ACh binds to muscarinic receptors. Future electrophysiological studies will determine the mechanisms leading to alterations in chronotropic responses observed following inhibition of non-neuronal cholinergic signaling in the specialized cardiomyocytes of the SA node.
It is well established that increased sympathetic signaling can lead to cardiac remodelling and increased cardiomyocyte stress in the working myocardium (41). Indeed, even though cVAChT mice presented normal regulation of heart rate under nonstressful conditions, changes in heart rate, due to lack of cardiomyocyte-derived ACh, may cause long-term changes in heart function. It appears that lack of the intrinsic cholinergic system in cardiomyocytes leads to an increase in basal stress. This may play an important role in the regulation of cardiac function because it has previously been reported that increased stress can lead to an up-regulation of AChE transcription in the brain (42). It will be important to determine whether cVAChT mice display altered AChE transcription in the heart, which may then serve to further exacerbate the stress response by increasing degradation of neuronal ACh.
Lack of cardiomyocyte-derived ACh was associated with ventricular cardiomyocyte hypertrophy, molecular remodelling, and increased oxidative stress, which argues for a widespread effect of cardiomyocyte-secreted ACh. These molecular changes were accompanied by alterations in left ventricular function and cardiac contractility under increased demand due to treatment with ISO. The observed hypertrophy may be related to the increased expression, as well as altered subcellular localization, of GRK5 observed in cVAChT cardiomyocytes. It has been reported that GRK5 overexpression leads to hypertrophy, which is dependent on its nuclear function (37). Conversely, GRK5 null mice display a delay in hypertrophy following transverse aortic constriction (43). Further studies will be required to elucidate potential GRK5-specific pathways affected in cVAChT myocytes that can lead to remodeling and hypertrophy. In addition, it will be important to determine whether the transcriptional activation of hypertrophic genes, including GRK5, are regulated by miRNAs as it has previously been shown that overexpression of the miR-212/132 family of miRNAs leads to pathological cardiac hypertrophy (44). It will be interesting to determine whether lack of cardiomyocyte-derived ACh leads to hypertrophy in a miRNA dependent mechanism.
Although the detailed mechanisms that trigger the release of ACh from cardiomyocytes are not clear at the moment, a clear possibility might involve sympathetic signaling. Previous experiments in vitro suggest that adrenergic stimulation can induce the expression of cholinergic genes in cardiomyocytes (12). Whether reciprocal interactions occur between sympathetic activation and cardiomyocyte-derived ACh is unknown. Interestingly, previous experiments have demonstrated that proteins involved with exocytic release of neurotransmitters, or their homologues, are expressed in vesicular compartments in cardiomyocytes (45). Hence, secretory vesicle compartments in cardiomyocytes are likely to be the source of ACh secretion (Fig. 7). It should be noted we have previously established that VAChT is present in recycling vesicles in cardiomyocytes (12). Our observation that the cholinergic cardiomyocyte machinery seems to control heart rate after stress and exercise suggests that cardiomyocyte-derived ACh secretion may be regulated by sympathetic activity.
Figure 7.

Non-neuronal release of Ach from cardiomyocytes. WT cardiomyocytes secrete ACh in response to increased physiological stress (e.g., exercise) in a VAChT-dependent manner to regulate heart rate. This response is blunted in mice lacking VAChT specifically in cardiomyocytes due to the lack of non-neuronal ACh release.
Interestingly, activation of the cholinergic anti-inflammatory pathway has been shown to play an important role in preventing the release of proinflammatory cytokines from circulating macrophages and thereby attenuating the effects of infection or injury (46). Immune cells can serve as a non-neuronal source of ACh, and its levels appear to be altered in a miR-132 dependent mechanism involving targeting of AChE mRNA (47). Although the activity of the cholinergic anti-inflammatory reflex has not been measured here, it is expected that its function will be unaltered due to the specificity of the Cre line used.
Recently, non-neuronal secretion of ACh in a VAChT-dependent manner was demonstrated from pancreatic α-cells, a process that regulates insulin secretion in humans (6). Here, we show that secretion of ACh from cardiomyocytes also has physiological relevance as this non-neuronal cholinergic system plays a previously unsuspected role in regulating heart rate as well as remodeling. Based on these results, it is necessary to reevaluate how the sympathetic-parasympathetic systems control heart activity to include this novel form of autocrine/paracrine communication from cardiomyocytes. We propose that secretion of ACh from cardiomyocytes enhances cholinergic signaling from parasympathetic neurons required to regulate cardiomyocyte activity (Fig. 7). Cardiomyocyte-derived ACh augments extracellular levels of this neurotransmitter in synaptic junctions between parasympathetic terminals and cardiomyocytes during high demand; for example, in response to increased sympathetic activation due to physiological stress. This novel mode of communication may serve to offset constant sympathetic signaling avoiding increased cardiomyocyte stress and hypertrophic responses.
Supplementary Material
Acknowledgments
The authors thank Dr. R. Jane Rylett (University of Western Ontario, London, ON, Canada) for kindly providing the CHT1 antibody and Drs. Dale Laird and Qingping Feng (University of Western Ontario) for their insightful comments regarding the manuscript.
This work was supported by the Heart and Stroke Foundation of Ontario (grants NA6656 and G-13-0002843), the Canadian Institutes for Health Research (grants MOP-82756 and MOP-89919), the Canadian Foundation for Innovation, the Ontario Research Fund, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; Brazil), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG; Brazil), and the U.S. National Institutes of Health (grant R03TW008425 from the Fogarty International Center to S.G.). A.R. is supported by an Ontario Graduate Scholarship. R.G. was supported by a New Investigator Award from the Heart and Stroke Foundation of Canada.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ACh
- acetylcholine
- AChE
- acetylcholinesterase
- ANP
- atrial natriuretic peptide
- α-MHC
- α-myosin heavy chain (Myh6)
- β-MHC
- β-myosin heavy chain
- ChAT
- choline acetyltransferase
- CHT1
- high-affinity choline transporter
- cVAChT
- cardiomyocyte-specific vesicular acetylcholine transporter-knockout (VAChTMyh6-Cre-flox/flox)
- ECG
- electrocardiography
- floxed
- flanked by loxP
- GRK
- G-protein-coupled receptor kinase
- HC-3
- hemicholinium-3
- IHC
- immunohistochemistry
- ISO
- isoproterenol
- KO
- knockout
- LV
- left ventricular
- Myh6
- α-myosin heavy chain (α-MHC)
- NO
- nitric oxide
- ROS
- reactive oxygen species
- SA
- sinoatrial
- VAChT
- vesicular acetylcholine transporter
- VES
- vesamicol
- WT
- wild type
REFERENCES
- 1. Levy M. N. (1997) Neural control of cardiac function. Baillieres Clin. Neurol. 6, 227–244 [PubMed] [Google Scholar]
- 2. Kanazawa H., Ieda M., Kimura K., Arai T., Kawaguchi-Manabe H., Matsuhashi T., Endo J., Sano M., Kawakami T., Kimura T., Monkawa T., Hayashi M., Iwanami A., Okano H., Okada Y., Ishibashi-Ueda H., Ogawa S., Fukuda K. (2010) Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J. Clin. Invest. 120, 408–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lara A., Damasceno D. D., Pires R., Gros R., Gomes E. R., Gavioli M., Lima R. F., Guimaraes D., Lima P., Bueno C. R., Jr., Vasconcelos A., Roman-Campos D., Menezes C. A., Sirvente R. A., Salemi V. M., Mady C., Caron M. G., Ferreira A. J., Brum P. C., Resende R. R., Cruz J. S., Gomez M. V., Prado V. F., de Almeida A. P., Prado M. A., Guatimosim S. (2010) Dysautonomia due to reduced cholinergic neurotransmission causes cardiac remodeling and heart failure. Mol. Cell. Biol. 30, 1746–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoover D. B., Ganote C. E., Ferguson S. M., Blakely R. D., Parsons R. L. (2004) Localization of cholinergic innervation in guinea pig heart by immunohistochemistry for high-affinity choline transporters. Cardiovasc. Res. 62, 112–121 [DOI] [PubMed] [Google Scholar]
- 5. Kawano H., Okada R., Yano K. (2003) Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels 18, 32–39 [DOI] [PubMed] [Google Scholar]
- 6. Rodriguez-Diaz R., Dando R., Jacques-Silva M. C., Fachado A., Molina J., Abdulreda M. H., Ricordi C., Roper S. D., Berggren P. O., Caicedo A. (2011) Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat. Med. 17, 888–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fujii T., Kawashima K. (2001) An independent non-neuronal cholinergic system in lymphocytes. Jpn. J. Pharmacol. 85, 11–15 [DOI] [PubMed] [Google Scholar]
- 8. Rosas-Ballina M., Olofsson P. S., Ochani M., Valdes-Ferrer S. I., Levine Y. A., Reardon C., Tusche M. W., Pavlov V. A., Andersson U., Chavan S., Mak T. W., Tracey K. J. (2011) Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wessler I., Kirkpatrick C. J., Racke K. (1998) Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol. Ther. 77, 59–79 [DOI] [PubMed] [Google Scholar]
- 10. Kawashima K., Fujii T. (2000) Extraneuronal cholinergic system in lymphocytes. Pharmacol. Ther. 86, 29–48 [DOI] [PubMed] [Google Scholar]
- 11. Rana O. R., Schauerte P., Kluttig R., Schroder J. W., Koenen R. R., Weber C., Nolte K. W., Weis J., Hoffmann R., Marx N., Saygili E. (2010) Acetylcholine as an age-dependent non-neuronal source in the heart. Auton. Neurosci. 156, 82–89 [DOI] [PubMed] [Google Scholar]
- 12. Rocha-Resende C., Roy A., Resende R., Ladeira M. S., Lara A., de Morais Gomes E. R., Prado V. F., Gros R., Guatimosim C., Prado M. A., Guatimosim S. (2012) Non-neuronal cholinergic machinery present in cardiomyocytes offsets hypertrophic signals. J. Mol. Cell. Cardiol. 53, 206–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guzman M. S., De Jaeger X., Raulic S., Souza I. A., Li A. X., Schmid S., Menon R. S., Gainetdinov R. R., Caron M. G., Bartha R., Prado V. F., Prado M. A. (2011) Elimination of the vesicular acetylcholine transporter in the striatum reveals regulation of behaviour by cholinergic-glutamatergic co-transmission. PLoS Biol. 9, e1001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martins-Silva C., De Jaeger X., Guzman M. S., Lima R. D., Santos M. S., Kushmerick C., Gomez M. V., Caron M. G., Prado M. A., Prado V. F. (2011) Novel strains of mice deficient for the vesicular acetylcholine transporter: insights on transcriptional regulation and control of locomotor behavior. PLoS One 6, e17611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Misgeld T., Burgess R. W., Lewis R. M., Cunningham J. M., Lichtman J. W., Sanes J. R. (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36, 635–648 [DOI] [PubMed] [Google Scholar]
- 16. Guatimosim S., Amaya M. J., Guerra M. T., Aguiar C. J., Goes A. M., Gomez-Viquez N. L., Rodrigues M. A., Gomes D. A., Martins-Cruz J., Lederer W. J., Leite M. F. (2008) Nuclear Ca2+ regulates cardiomyocyte function. Cell Calcium 44, 230–242 [DOI] [PubMed] [Google Scholar]
- 17. Roy A., Lara A., Guimaraes D., Pires R., Gomes E. R., Carter D. E., Gomez M. V., Guatimosim S., Prado V. F., Prado M. A., Gros R. (2012) An analysis of the myocardial transcriptome in a mouse model of cardiac dysfunction with decreased cholinergic neurotransmission. PLoS One 7, e39997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ribeiro F. M., Alves-Silva J., Volknandt W., Martins-Silva C., Mahmud H., Wilhelm A., Gomez M. V., Rylett R. J., Ferguson S. S., Prado V. F., Prado M. A. (2003) The hemicholinium-3 sensitive high affinity choline transporter is internalized by clathrin-mediated endocytosis and is present in endosomes and synaptic vesicles. J. Neurochem. 87, 136–146 [DOI] [PubMed] [Google Scholar]
- 19. Lau J. K., Brown K. C., Thornhill B. A., Crabtree C. M., Dom A. M., Witte T. R., Hardman W. E., McNees C. A., Stover C. A., Carpenter A. B., Luo H., Chen Y. C., Shiflett B. S., Dasgupta P. (2013) Inhibition of cholinergic signaling causes apoptosis in human bronchioalveolar carcinoma. Cancer Res. 73, 1328–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friedman A., Kaufer D., Shemer J., Hendler I., Soreq H., Tur-Kaspa I. (1996) Pyridostigmine brain penetration under stress enhances neuronal excitability and induces early immediate transcriptional response. Nat. Med. 2, 1382–1385 [DOI] [PubMed] [Google Scholar]
- 21. Kaufer D., Friedman A., Seidman S., Soreq H. (1998) Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 393, 373–377 [DOI] [PubMed] [Google Scholar]
- 22. Lorke D. E., Hasan M. Y., Nurulain S. M., Shafiullah M., Kuca K., Petroianu G. A. (2011) Pretreatment for acute exposure to diisopropylfluorophosphate: in vivo efficacy of various acetylcholinesterase inhibitors. J. Appl. Toxicol. 31, 515–523 [DOI] [PubMed] [Google Scholar]
- 23. de Castro B. M., De Jaeger X., Martins-Silva C., Lima R. D., Amaral E., Menezes C., Lima P., Neves C. M., Pires R. G., Gould T. W., Welch I., Kushmerick C., Guatimosim C., Izquierdo I., Cammarota M., Rylett R. J., Gomez M. V., Caron M. G., Oppenheim R. W., Prado M. A., Prado V. F. (2009) The vesicular acetylcholine transporter is required for neuromuscular development and function. Mol. Cell. Biol. 29, 5238–5250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beraldo F. H., Soares I. N., Goncalves D. F., Fan J., Thomas A. A., Santos T. G., Mohammad A. H., Roffe M., Calder M. D., Nikolova S., Hajj G. N., Guimaraes A. L., Massensini A. R., Welch I., Betts D. H., Gros R., Drangova M., Watson A. J., Bartha R., Prado V. F., Martins V. R., Prado M. A. (2013) Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. [E-pub ahead of print] FASEB J. 27, 10.1096/fj.13-232280 [DOI] [PubMed] [Google Scholar]
- 25. Guzman M. S., De Jaeger X., Drangova M., Prado M. A., Gros R., Prado V. F. (2013) Mice with selective elimination of striatal acetylcholine release are lean, show altered energy homeostasis and changed sleep/wake cycle. J. Neurochem. 124, 658–669 [DOI] [PubMed] [Google Scholar]
- 26. Assini J. M., Mulvihill E. E., Sutherland B. G., Telford D. E., Sawyez C. G., Felder S. L., Chhoker S., Edwards J. Y., Gros R., Huff M. W. (2013) Naringenin prevents cholesterol-induced systemic inflammation, metabolic dysregulation, and atherosclerosis in Ldlr(-)/(-) mice. J. Lipid Res. 54, 711–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aguiar C. J., Andrade V. L., Gomes E. R., Alves M. N., Ladeira M. S., Pinheiro A. C., Gomes D. A., Almeida A. P., Goes A. M., Resende R. R., Guatimosim S., Leite M. F. (2010) Succinate modulates Ca(2+) transient and cardiomyocyte viability through PKA-dependent pathway. Cell Calcium 47, 37–46 [DOI] [PubMed] [Google Scholar]
- 28. Prado M. A., Reis R. A., Prado V. F., de Mello M. C., Gomez M. V., de Mello F. G. (2002) Regulation of acetylcholine synthesis and storage. Neurochem. Int. 41, 291–299 [DOI] [PubMed] [Google Scholar]
- 29. Prado V. F., Roy A., Kolisnyk B., Gros R., Prado M. A. (2013) Regulation of cholinergic activity by the vesicular acetylcholine transporter. Biochem. J. 450, 265–274 [DOI] [PubMed] [Google Scholar]
- 30. Agah R., Frenkel P. A., French B. A., Michael L. H., Overbeek P. A., Schneider M. D. (1997) Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 100, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lyons G. E., Schiaffino S., Sassoon D., Barton P., Buckingham M. (1990) Developmental regulation of myosin gene expression in mouse cardiac muscle. J. Cell Biol. 111, 2427–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stacy R. C., Demas J., Burgess R. W., Sanes J. R., Wong R. O. (2005) Disruption and recovery of patterned retinal activity in the absence of acetylcholine. J. Neurosci. 25, 9347–9357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frey N., Olson E. N. (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65, 45–79 [DOI] [PubMed] [Google Scholar]
- 34. Thum T., Galuppo P., Wolf C., Fiedler J., Kneitz S., van Laake L. W., Doevendans P. A., Mummery C. L., Borlak J., Haverich A., Gross C., Engelhardt S., Ertl G., Bauersachs J. (2007) MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116, 258–267 [DOI] [PubMed] [Google Scholar]
- 35. Amin J. K., Xiao L., Pimental D. R., Pagano P. J., Singh K., Sawyer D. B., Colucci W. S. (2001) Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell. Cardiol. 33, 131–139 [DOI] [PubMed] [Google Scholar]
- 36. Waypa G. B., Marks J. D., Mack M. M., Boriboun C., Mungai P. T., Schumacker P. T. (2002) Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ. Res. 91, 719–726 [DOI] [PubMed] [Google Scholar]
- 37. Martini J. S., Raake P., Vinge L. E., DeGeorge B. R., Jr., Chuprun J. K., Harris D. M., Gao E., Eckhart A. D., Pitcher J. A., Koch W. J. (2008) Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 105, 12457–12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choi D. J., Koch W. J., Hunter J. J., Rockman H. A. (1997) Mechanism of beta-adrenergic receptor desensitization in cardiac hypertrophy is increased beta-adrenergic receptor kinase. J. Biol. Chem. 272, 17223–17229 [DOI] [PubMed] [Google Scholar]
- 39. Parsons S. M. (2000) Transport mechanisms in acetylcholine and monoamine storage. FASEB J. 14, 2423–2434 [DOI] [PubMed] [Google Scholar]
- 40. Canaani J., Shenhar-Tsarfaty S., Weiskopf N., Yakobi R., Assayag E. B., Berliner S., Soreq H. (2010) Serum AChE activities predict exercise heart rate parameters of asymptomatic individuals. Neurosci. Med. 1, 43–49 [Google Scholar]
- 41. Mann D. L., Kent R. L., Parsons B., Cooper G. t. (1992) Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 85, 790–804 [DOI] [PubMed] [Google Scholar]
- 42. Sailaja B. S., Cohen-Carmon D., Zimmerman G., Soreq H., Meshorer E. (2012) Stress-induced epigenetic transcriptional memory of acetylcholinesterase by HDAC4. Proc. Natl. Acad. Sci. U. S. A. 109, E3687–E3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gold J. I., Gao E., Shang X., Premont R. T., Koch W. J. (2012) Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ. Res. 111, 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ucar A., Gupta S. K., Fiedler J., Erikci E., Kardasinski M., Batkai S., Dangwal S., Kumarswamy R., Bang C., Holzmann A., Remke J., Caprio M., Jentzsch C., Engelhardt S., Geisendorf S., Glas C., Hofmann T. G., Nessling M., Richter K., Schiffer M., Carrier L., Napp L. C., Bauersachs J., Chowdhury K., Thum T. (2012) The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 3, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ferlito M., Fulton W. B., Zauher M. A., Marban E., Steenbergen C., Lowenstein C. J. (2010) VAMP-1, VAMP-2, and syntaxin-4 regulate ANP release from cardiac myocytes. J. Mol. Cell. Cardiol. 49, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Borovikova L. V., Ivanova S., Zhang M., Yang H., Botchkina G. I., Watkins L. R., Wang H., Abumrad N., Eaton J. W., Tracey K. J. (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462 [DOI] [PubMed] [Google Scholar]
- 47. Shaked I., Meerson A., Wolf Y., Avni R., Greenberg D., Gilboa-Geffen A., Soreq H. (2009) MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 31, 965–973 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.