

Abstract
Lipoxygenases (LOXs), which are essential in eukaryotes, have no confirmed function in prokaryotes that are devoid of polyunsaturated fatty acids. The structure of a secretable LOX from Pseudomonas aeruginosa (Pa_LOX), the first available from a prokaryote, presents significant differences with respect to eukaryotic LOXs, including a cluster of helices acting as a lid to the active center. The mobility of the lid and the structural variability of the N-terminal region of Pa_LOX was confirmed by comparing 2 crystal forms. The binding pocket contains a phosphatidylethanolamine phospholipid with branches of 18 (sn-1) and 14/16 (sn-2) carbon atoms in length. Carbon atoms from the sn-1 chain approach the catalytic iron in a manner that sheds light on how the enzymatic reaction might proceed. The findings in these studies suggest that Pa_LOX has the capacity to extract and modify unsaturated phospholipids from eukaryotic membranes, allowing this LOX to play a role in the interaction of P. aeruginosa with host cells.—Garreta, A., Val-Moraes, S. P., García-Fernández, Q., Montserrat Busquets, C. J., Oliver, A., Ortiz, A., Gaffney, B. J., Fita, I., Manresa, A., Carpena, X. Structure and interaction with phospholipids of a prokaryotic lipoxygenase from Pseudomonas aeruginosa.
Keywords: lipid peroxidation, protein–phospholipid complexes, enzyme mechanisms, membrane interaction, host–pathogen interactions
Lipoxygenases (LOXS) are nonheme iron dioxygenases that are responsible for a wide range of essential functions in eukaryotes (1–5), including the formation of the fatty acid hydroperoxides necessary for the production of many biological mediators and signaling molecules (6, 7), the mobilization of lipids (8, 9), and the modification of membrane structures (10). LOXs are monomeric, with molecular masses of 75–80 kDa in animals, 94–104 kDa in plants and fungi, and 49–75 kDa in bacteria. LOX-1 from soybeans, the first LOX crystal structure determined (11, 12), has an N-terminal domain with a characteristic β-barrel topology—the PLAT (polycystin-1, lipoxygenase, α-toxin) domain (13), which is followed by a larger domain formed mainly by helices and containing a catalytic iron. This organization appears to be fully conserved among eukaryotic LOXs (4, 11, 14–20), although some fungi enzymes contain manganese instead of iron (21).
LOXs are also found in some prokaryotes, where their possible biological roles remain unclear, as prokaryotes lack or produce few polyunsaturated fatty acids. The opportunistic pathogen Pseudomonas aeruginosa carries a secretable LOX (Pa_LOX) with the capacity to convert arachidonic acid into 15-hydroxyeicosatetraenoic acid (22). In this regard, it has been proposed that Pa_LOX modulates host defense and inflammation by altering the synthesis of anti-inflammatory molecules. We present the crystal structure of Pa_LOX, the first LOX solved from a prokaryote. Although sequence analysis had predicted an organization of Pa_LOX similar to that of eukaryotic LOXs (22), the structure determined showed significant differences. Even more unexpected was the finding of a large substrate binding pocket containing a complete phospholipid. Despite the presence of the bound phospholipid, the iron was oxidized by linoleate 13S-hydroperoxyoctadecadienoic acid (13S-HPODE), and Pa_LOX was characterized further by electron paramagnetic resonance (EPR) spectroscopy. Finally, we examined the possible biological functions of Pa_LOX by biophysical studies of the interaction of Pa_LOX with lipid membranes and by in vivo analysis of the cell invasiveness and cytotoxicity of P. aeruginosa variants, with and without this enzyme. The results indicate that some prokaryotic LOXs may be capable of acting autonomously on the membrane phospholipids of their eukaryotic cell hosts.
MATERIALS AND METHODS
See Supplemental Data for complementary details.
Protein expression and purification
The gene for Pa_LOX (AF479686.2), corresponding to the Pseudomonas aeruginosa strain 42A2 originally isolated from soil, was cloned in pET28a and expressed as described previously (23). A His-tag version beginning at Ala19 was expressed in Escherichia coli BL21 (DE3) and subjected to an Ni-chelating column. Pa_LOX was then further purified by passage through a Superdex 200 10/300 GL column (Amersham Pharmacia Biotech, Munich, Germany) and concentrated up to 75 mg/ml in 50 mM potassium phosphate buffer (pH 7.0) and 150 mM NaCl.
Crystallization and structure determination
The His-tag–labeled Pa_LOX was crystallized in 2 crystal forms at 20°C by the vapor diffusion, hanging-drop method at a protein concentration of 15mg/ml. The first crystal form was obtained over a reservoir containing 10% polyethylene glycol (PEG) 3350, 50 mM MgCl2, and 0.1 M HEPES (pH 7.5). These crystals belonged to space group P21212 with unit cell parameters of a = 132.7 Å, b = 116.0 Å, and c = 42.6 Å. A diffraction dataset (1.7 Å resolution) from a cryocooled crystal with an extra 20% of glycerol applied to the original mother liquor was collected by using a fixed 0.9334-Å wavelength at beam line ID14eh1 [European Synchrotron Radiation Facility (ESRF), Grenoble, France]. A second crystal form was obtained with a mother liquor (12% PEG 3350, 0.2 M MgCl2, and 0.1 M Tris, pH 7.0) very similar to the one used with the first crystal form. The new crystals belonged to space group C2221 with unit cell parameters of a = 84.8 Å, b = 97.3 Å, and c = 157.2 Å. For these new crystals, a diffraction dataset at 2.0 Å resolution was collected at ID14eh2. Datasets were integrated and scaled by the program DENZO/SCALEPACK (24). Structure determination was performed with the program MOLREP (25), by using as a searching model the truncated structure of LOX from Plexaura homomalla [Protein Data Bank (PDB) entry 2FNQ], which included residues 498–1066. Model building was performed with COOT (26) and was refined with REFMAC (27) and BUSTER (28) (Table 1). After noting the presence of the ligand in the active center, we took special care to avoid introducing any sort of model bias. All figures were prepared with the program PyMOL (29). Coordinates and structure factors have been deposited in the PDB with accession codes 4G32 and 4G33 for the first and second crystal forms, respectively.
Table 1.
Data collection and refinement statistics
| Parameter | Value | |
|---|---|---|
| Data collection | ||
| Space group | P21212 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 132.7, 116.0, 42.6 | 84.8, 97.3, 157.2 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 20–1.75 (1.81–1.75) | 20–2.0 (2.08–2.03) |
| Rsym | 9.1 (63.0) | 10.9 (64.8) |
| I/σ(I) | 10.7 (2.5) | 9.0 (2.2) |
| Multiplicity | 3.9 (3.5) | 4.7 (4.1) |
| Completeness (%) | 97.9 (97.0) | 99.2 (94.9) |
| Refinement | ||
| Resolution (Å) | 20–1.75 (1.79–1.75) | 20–2.0 (2.08–2.03) |
| Reflections (n) | 65,882 (4,720) | 41,570 (2,888) |
| Rwork/Rfree | 18.1/22.1 (21.3/25.3) | 18.7/22.5 (22.5/25.8) |
| Atoms (n) | ||
| Protein | 5384 | 5117 |
| Phospholipid | 47 | 47 |
| Glycerol | 66 | 36 |
| Water | 598 | 222 |
| B factors (Å2) | ||
| Protein | 22.6 | 42.6 |
| Phospholipid | 28.1 | 36.7 |
| Glycerol | 48.7 | 52.1 |
| Water | 31.9 | 35.9 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.010 | 0.008 |
| Bond angles (deg) | 1.01 | 0.98 |
Values in parentheses are for the highest resolution shell. Rsym = Σhkl Σi |Ii(hkl) – 〈I(hkl)〉|/Σhkl Σi Ii(hkl), where Ii(hkl) is the intensity of an observation, and 〈I(hkl)〉 is the mean value of observations for a unique reflection. Rwork = Σh |Fo(h) – Fc(h)|/Σh |Fo(h), where Fo and Fc are the observed and calculated structure-factor amplitudes, respectively. Rfree was calculated with 5% of the data, which was excluded from the refinement. I/σ(I) = 〈〈I(hkl)〉/σ[〈I(hkl)〉]〉, where I〈(hkl)〉 is the weighted mean of all measurements for reflection hkl, and σ[〈I(hkl)〉] is the standard deviation of the weighted mean.
EPR spectroscopy
EPR spectra were obtained on an E109 spectrometer (Varian Associates, Palo Alto, CA, USA; ref. 12) operating at 9.2 GHz (X band) and equipped with a pumped ESR9/10 liquid helium-flow cryostat set at 3.5 K (Oxford Instruments, Concord, MA, USA). Temperature was measured with a Cernox sensor (Lake Shore Cryotronics, Westerville, OH, USA) just under the 4-mm quartz EPR sample tube. The X-band frequency was measured at the spectrometer bridge. Standard instrument settings for recorded spectra were as follows: microwave power of 5 mW, scan time 30 min/200 G, and time constant 0.5 s. Protein concentrations of EPR samples were 0.4-0.47 mM in 0.2 M potassium phosphate without cryoprotectant. To favor the formation of single-component ferric EPR signals, we oxidized dilute (0.02 mM) Pa_LOX samples by treatment with linoleate 13S-HPODE and washed and concentrated them in a centrifugal concentrator. Complete oxidation of iron was confirmed by comparison with soybean LOX-1 spectra (30). Direct addition of 1 equivalent of the Pa_LOX product 13S-HPODE to 0.47 mM Pa_LOX also gives high-spin ferric EPR signals, but oxidation is relatively slow and a 10-fold excess of 13S-HPODE is necessary for complete oxidation of iron. Although slightly different spectra are obtained with different buffers, a striking change is observed with ethanol addition (to 1%).
The assignments of the EPR signals are to two overlapping spin 5/2 ferric species, each from a different subpopulation of the enzyme, with one dominating when ethanol is present. This dominant component has a sharp peak at effective gy = 7.23, or at 91 mT, and a broad feature, with maxima and minima extending from ∼136 to 160 mT, centered at effective gx = 4.2. The spectrum when ethanol is added results from two iron species, one approximately as already discussed, and another with effective gy and gx features appearing at ∼103 and 106 mT. The interpretation of these data is that the iron center in Pa_LOX is roughly axially symmetric, with various degrees of rhombic distortion being influenced by weak affinity sites on the protein for solvent components. The zero-field splitting ratio, E/D, used to simulate the major component of iron EPR spectra is 0.06, and it is 0.013 for the minor component.
Samples of ferric Pa_LOX were prepared by addition of 13S-HPODE (formed by soybean LOX-1), purified on an LC-SI column (5 μm, 25×0.46 cm; Supelcosil; Sigma-Aldrich, St. Louis, MO, USA) eluted at 1 ml/min with hexane/isopropanol/acetic acid (100/2/0.1 by volume). Also, the Pa_LOX linoleic acid major oxidation product was confirmed to be 13S-HPODE by HPLC analysis of the methyl ester (as in ref. 31) by a Chiralpak AD column (0.46×25 cm; Daicel Chemical Industries, Ltd., Osaka, Japan).
Preparation of vesicles and tryptophan fluorescence measurements
Phospholipid small unilamellar vesicles (SUVs) were prepared by probe sonication. Phospholipids (5–10 μmol; Avanti Polar Lipids, Alabaster, AL, USA) were dissolved in chloroform. The solvent was gently evaporated under a stream of dry N2 to obtain a thin film at the bottom of a glass tube. The last traces of solvent were removed by a further 3 h of desiccation under high vacuum. Buffer (1 ml) was added to the dry samples, and multilamellar vesicles were formed by vortexing the mixture at a temperature well above the gel-to-liquid crystalline phase transition temperature of the phospholipids. This suspension was sonicated for 10 min in the pulsed mode in an ice-water bath in an ultrasound apparatus (Cole-Parmer Instruments, Park Hanwell, UK) equipped with a titanium probe. For DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine), SUV sonication was performed at 50°C. Finally, SUV suspensions were centrifuged for 30 min in a bench microfuge to remove large vesicles, as well as any titanium particles released from the probe, and phospholipid phosphorous was determined (32).
Measurements of changes in the intrinsic tryptophan fluorescence of the protein on SUV addition were performed in a PTI Quantamaster spectrofluorometer (Photon Technology, Birmingham, NJ, USA) provided with a thermostated cuvette holder and magnetic stirring. A 10- × 10-mm quartz cuvette was used. Temperature was maintained at 25°C. Excitation wavelength was set at 290 nm, and emission spectra were recorded between 300 and 370 nm. Excitation and emission slits were set at 4 nm. Pa_LOX concentration was kept constant in the cuvette (usually 0.2 μM), SUV aliquots were added from a concentrated stock suspension (4–10 mM), samples were incubated for 2 min, and the emission spectrum was collected. Emission spectra were corrected for dilution and light-scattering when SUVs were added (33).
Interaction with membranes by isothermal titration calorimetry (ITC)
High-sensitivity ITC measurements were obtained in a VP-ITC Titration Calorimeter (MicroCal, Commerce, CA, USA). The mixing cell had a volume of 1.442 ml. Data were processed with the software (OriginLab Corp. Northampton, MA, USA) provided with the equipment. The calorimeter cell was filled with a suspension of phospholipid SUVs (usually 0.5 mM) and the syringe with a 0.2 mM Pa_LOX solution, both in 0.2 M borate (pH 9.0) buffer. Usually, 10-μl injections were performed. All measurements were obtained at 25°C. The heat of dilution of Pa_LOX was determined in separate experiments by injecting a 0.2 mM solution into the buffer in the absence of liposomes.
Invasion and cytotoxicity assays
The invasive wild-type strain PAO1 and its knockout mutants (PAO3110 and PAO3111) in PA1169, obtained from the transposon collection by Jacobs et al. (34), were used in the invasion experiments. The wild-type cytotoxic strain PA14 and its knockout mutant in PA1169 (kindly supplied by Dr. J. Blázquez, Hospital Universitari, Palma de Mallorca, Spain) were used for cytotoxicity assays. In all cases, mutants were checked through specific polymerase chain reactions (PCRs) for the inactivated genes, as described previously (34). Results obtained for the knockout mutants were compared with those of their respective wild-type strains in the invasion and cytotoxicity experiments.
The invasion assay was performed on A549 human lung cells, with slight modification to the procedure described by Harrison et al. (35). Monolayers were seeded in 24-well tissue culture plates in Roswell Park Memorial Institute (RPMI) 1640 culture medium supplemented with glutamine, 10% fetal calf serum (FCS), and 10 μg/ml gentamicin. Semiconfluent cell monolayers were washed with warm phosphate-buffered saline (PBS). Fresh medium (500 μl) containing ∼5 × 106 bacteria was added per well [multiplicity of infection (MOI) of ∼50] and incubated for 1 and 3 h at 37°C and 5% CO2. To remove extracellular bacteria, 1 ml of medium containing 400 μg/ml amikacin was added in the last hour of incubation. Finally, the A549 cells were washed with PBS and lysed in 0.25% Triton X-100. After cell lysis, the bacteria were resuspended in saline solution and quantified by plating serial dilutions. Assays were performed in quadruplicate, and the results from ≥3 independent experiments are expressed as the total number of internalized bacteria per well (means±sd) (36).
The A549 cells and P. aeruginosa cultures were prepared as described previously for the invasion assay (35, 36). After 3 h of incubation, cytotoxicity toward A549 cells was quantified by measuring the amount of lactate dehydrogenase (LDH) released into the culture supernatant with the Cytotoxicity Detection Kit Plus LDH (Roche Applied Science, Indianapolis, IN, USA) in the same wells used for the invasion experiments. The assays were conducted in quadruplicate in 3 independent experiments for each strain. The percentage of cytotoxicity was calculated relative to the value for the maximum LDH release control, according to the manufacturer's instructions.
In both invasion and cytotoxicity assays, Student's t test was performed with Prism 5 software (GraphPad, San Diego, CA) to ascertain whether differences between means were significant (P<0.05).
RESULTS
Overall structure of Pa_LOX
The structure from a construct of Pa_LOX (GenBank AF479686.2), which started at Ala19 to skip the signal peptide, was determined at 1.75 and 2.0 Å resolution (Fig. 1A, B) using two crystal forms (Table 1). Pa_LOXs are monomers containing a single domain structurally related to the catalytic domain of eukaryotic LOXs (Figs. 1C and 2). Therefore, the N-terminal PLAT domain found in all the eukaryotic enzymes (13) is absent in Pa_LOX, despite having a molecular size (∼70 kDa) similar to that of animal LOXs (23). Instead, Pa_LOX contains an insertion of ∼100 residues before helix α3, which corresponds mainly to a longer and more regular helix α2 and to a pair of long, antiparallel new helices (α2A and α2B) of 36 and 38 residues, respectively (Figs. 1A, B and 3A, B). The orientations of α2A and α2B present a ∼7° rigid-body rotation between the Pa_LOX structures from the 2 crystal forms solved (Fig. 3C). This rotation may be facilitated by the flexibility of the 6 residues linking α2B and α3 (Thr201-Gln202-Gly203-Gly204-Gln205-Gly206), which are disordered in the 2 Pa_LOX structures. Helices α2A and α2B interact exclusively with each other, with the remodeled helix α2, and with a phospholipid found in the substrate binding pocket (see below and Figs. 1A, B and 4). Besides the rotation of helices α2A and α2B, the 2 Pa_LOX structures available differ mainly in the N-terminal region, with models starting at residues Ile50 (Fig. 1A) and Ala19 (Fig. 1B). The longer N-terminal tail of the structure starting at Ala19 protrudes toward a neighboring molecule, where it is stabilized by a large number of interactions, in particular with the polar head of the phospholipid (Fig. 4C).
Figure 1.
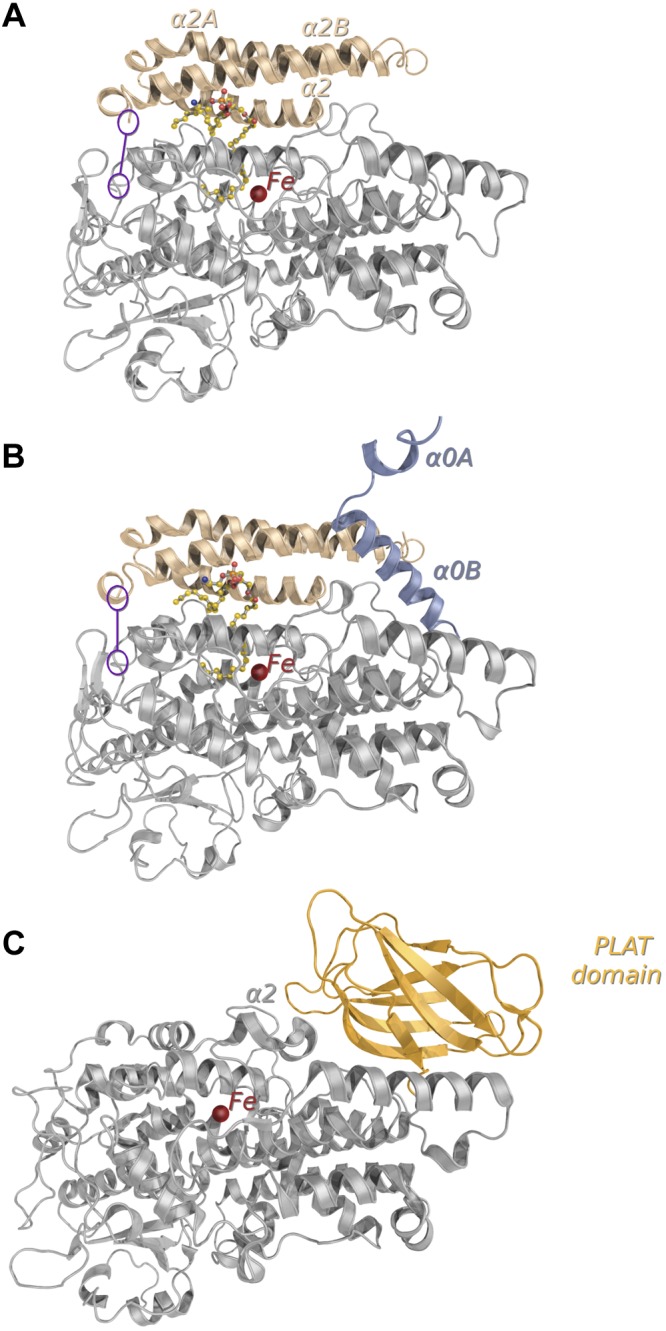
Comparison of prokaryotic and eukaryotic LOX structures. A, B) Overall structures of the prokaryotic Pa_LOX determined for crystal forms (PDB codes: 4G32, 4G33) at resolutions of 1.75 Å (A), and 2.0 Å (B). In the 2 structures, the phospholipid found in the substrate binding pocket is shown as balls and sticks and the missing loop linking α2B and α3 is indicated in purple. C) Structure of a representative eukaryotic LOX from P. homomalla (PDB: 2FNQ) showing the N-terminal PLAT domain (light orange), which is absent in Pa_LOX. In the 3 structures, the catalytic iron is shown as a red sphere. The insertion of ∼100 residues in Pa_LOX with respect to eukaryotic LOXs (A, B, beige) corresponds mainly to a remodeled helix α2 and to the new helices α2A and α2B. The extension of the N-terminal region (B, bluish gray), with 2 new helices labeled α0A and α0B, was visible only in the Pa_LOX structure corresponding to the 2.0 Å resolution crystal form.
Figure 2.

Structural alignment of plant, mammalian, and bacterial LOXs. ESPript output (45) is shown of the structural alignment of Pa_LOX (4G32) with LOXs from coral P. homomalla (2FNQ), rabbit reticulocyte Oryctolagus cuniculus (1LOX), and soybean Glycine max (1YGE). The secondary structure as determined for Pa_LOX is also shown. Residues missing in the corresponding coordinate models are orange. The absence of the PLAT domain and the long insertion after helix α2 (blue) are evident in Pa_LOX.
Figure 3.
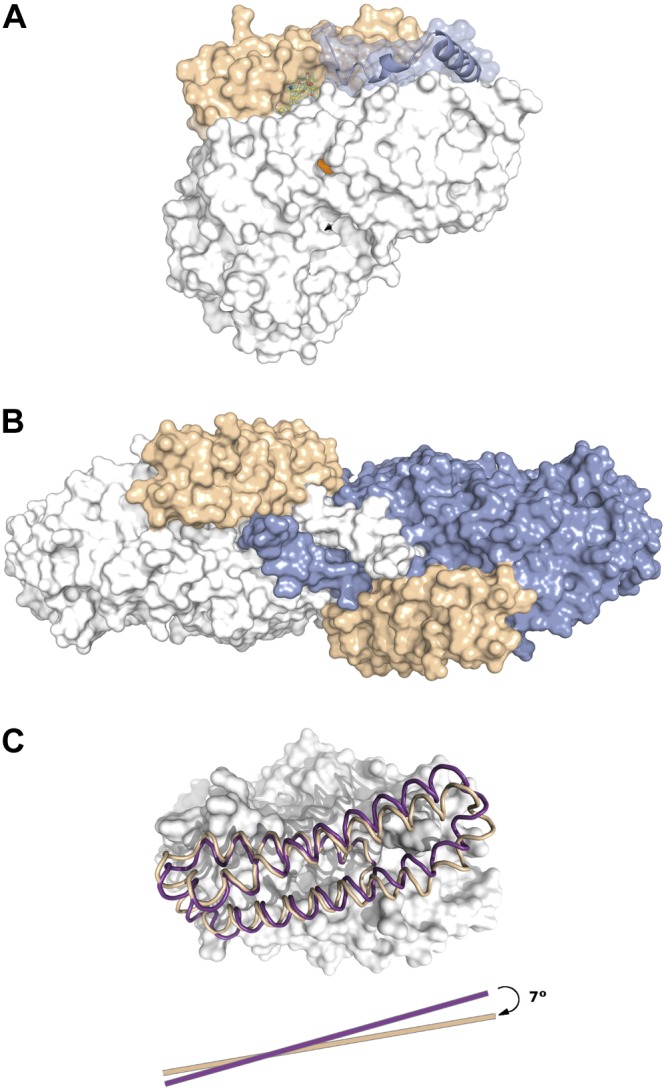
Surface representation of Pa_LOX. A) Surface representation of Pa_LOX, as determined in the crystal form at 2.0 Å resolution. The surface corresponding to the new helices α2A and α2B is indicated (beige). The polar head of the bound PE molecule (solid sticks) remains visible from outside the protein. The N tail of a crystal neighbor molecule interacting with the polar head of PE is also shown (bluish gray). The entrance to the putative oxygen channel corresponds to the orange spot on the surface of Pa_LOX (see also Fig. 5C). B) A 90° rotated view showing the 2 neighboring subunits exchanging their N tails. C) Displacement of helices α2A and α2B in the Pa_LOX structures corresponding to the crystal forms at 1.75 Å (beige) and 2.0 Å (purple), which suggests that these helices act as a removable lid covering the entrance to the binding pocket.
Figure 4.
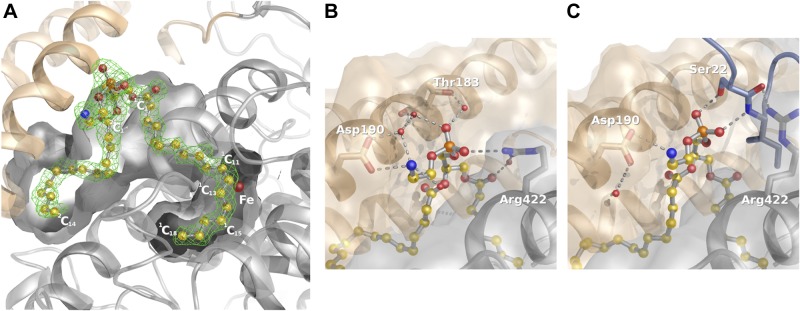
Phospholipid binding to Pa_LOX. A) Section of the binding-pocket showing a 1.75 Å resolution omit electron density map at 2.5 σ (green) of the molecule found in the substrate binding pocket of Pa_LOX. The density, which allows positioning of all the nonhydrogen atoms, corresponds to a PE molecule (balls and sticks). B) The presence of 2 ionic bonds between Pa_LOX and the PE head in the crystal form determined at 1.75 Å resolution suggests specificity toward small polar or charged head phospholipids. C) However, interactions of the PE head are altered in the crystal form determined at 2.0 Å resolution because of the presence of the N-terminal tail of a neighboring molecule (bluish gray).
Active center and bound phospholipid
Given the high conservation of active center residues (Fig. 2), the observation of an enlarged substrate binding pocket with respect to the one in eukaryotic LOXs was unexpected. Moreover, this pocket contains a complete and well-defined phospholipid in the 2 Pa_LOX structures (Figs. 1 and 4A). The new helices, α2A and α2B, found in Pa_LOX act as a lid above the entrance to the large binding pocket (Fig. 3A, B). The different orientation of these helices in the 2 Pa_LOX structures suggests mobility of the lid, a feature that may be necessitated by the flux of substrates and products in and out of the active center.
The phospholipid found inside the binding pocket of Pa_LOX was spontaneously incorporated during the recombinant overexpression of the protein and then was retained throughout the purification procedure with essentially full occupancy, as shown by the low values of the temperature factors. Given the quality of the electron density map, the phospholipid was unambiguously identified as a phosphatidylethanolamine (PE) with fatty acid chains in positions sn-1 (1C) and sn-2 (2C) of 18 and 14 (or even possibly 16 in the second crystal form) carbon atoms in length, respectively (Fig. 4A). The catalytic center in Pa_LOX retains the essential features described for other LOX enzymes (4, 8, 11, 14, 16–20): a nonheme iron atom with a slightly distorted octahedral coordination (Fig. 5A). The 6 iron ligands correspond to the Oδ atom from Asn559, to an oxygen atom from the carboxylate group of the terminal residue Ile658, to a water molecule (W1, Fig. 5A), and to the 3 Nε atoms from His377, His382, and His555. Water molecule W1 also makes a strong (short: 2.46 Å) bond with the second oxygen atom of the terminal carboxylate, thereby completing the characteristic geometry of the iron first coordination sphere in LOXs. Although both ferric and ferrous iron forms are intermediates in LOX catalysis, samples of the isolated Pa_LOX enzyme have no EPR signal, thus supporting the ferrous assignment.
Figure 5.
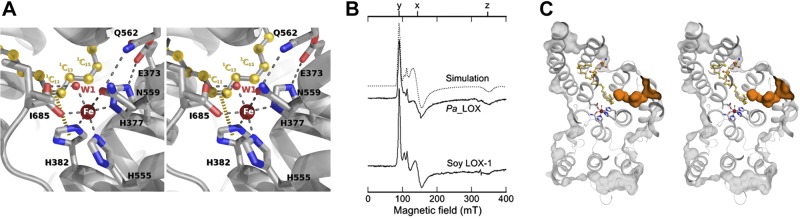
The active center of Pa_LOX. A) Stereo view of the catalytic center of Pa_LOX with the iron atom and the residues from the first and second coordination spheres represented. Five carbon atoms of the phospholipid 1C chain approach the iron with a conformation suggesting that a pentadiene, when present, would stack on 2 of the histidine ligands, as further depicted by a π-interaction between H382 and the suggested C11–C12 double bond (yellow dashes). B) The catalytic center of Pa_LOX is well conserved with respect to the eukaryotic enzymes, as confirmed by the comparison of the ferric EPR spectra from Pa_LOX and soybean LOX-1. To obtain equivalent EPR signals, a longer period (∼20 min) was necessary to oxidize Pa_LOX than for soybean LOX-1 (1 min). Simulation of a ferric EPR spectrum is shown for comparison (dotted line). The y, x, z features, corresponding to transitions in the lower Kramers doublet, are indicated on the top axis. C) Representation of the Pa_LOX solvent accessible surface (light gray) showing the presence of a channel (orange) spanning from the protein surface to the substrate pocket occupied by the phospholipid 1C chain. The surface was computed, including the experimental solvent molecules, to emphasize the strong hydrophobic character of the channel, where no water molecules were detected.
Inherent to the accepted catalytic mechanism of LOXs is that iron cycles ferric–ferrous–ferric in 1 turnover and that the native ferrous enzyme is preactivated by consuming 1 equivalent of a hydroperoxide product (1). Remarkably, for the lipid-loaded Pa_LOX, the ferric EPR spectra closely resemble those found for the soybean enzyme LOX-1 (30)—a more rhombic component (as in Fig. 5B) and additional axial components when ethanol is present (Supplemental Fig. S1A, curve d). Pa_LOX reacts more slowly with substrate or hydroperoxide than does soybean LOX-1, and so the maximum ferric EPR signal from Pa_LOX was achieved more slowly (as described in Materials and Methods). Other spectroscopy studies (37) have compared ferric soybean LOX-1 with model iron compounds and concluded that the iron form with the largest rhombic distortion corresponds to a 5-coordinated iron. One Asn-Fe(II) bond, corresponding to Asn559 in Pa_LOX, ranges from ∼2.4 Å in Pa_LOX to 2.8-3.3 Å in various soybean LOX-1 structures (11, 14, 15). These observations suggest that the rhombic EPR spectrum arises because the Fe-Asn bond in ferric Pa_LOX, or in LOX-1, is considerably longer than observed in the resting ferrous structure.
The 1C chain of the phospholipid approaches the catalytic iron using subpocket 1 of Pa_LOX, which corresponds to the substrate binding pocket described for eukaryotic 12/15-LOX structures (14, 15, 20). The geometry determined for the 1C chain suggests that binding into subpocket 1 favors a cis conformation, generally associated with unsaturated bonds in fatty acids, between carbon atoms 11 and 12 with a deviation from planarity of only ∼5° (Figs. 4A and 5A). The cis conformation may also be favored between carbon atoms 14 and 15, which would complete the 2 double bonds of the pentadiene moiety needed for a substrate to be catalytically suitable. However, the value observed (∼50°) for this 14–15 torsional angle appears too high to correspond to a double bond in the trapped phospholipid. Three other torsional angles at the beginning of the 1C chain (between carbon atoms 2–3, 5–6, and 6–7 with torsional angles of approximately 55, 75, and 55°, respectively) could also favor the presence of double bonds for some of these positions. A channel, strongly hydrophobic in character and with no well-ordered water molecules inside, starts at the protein surface and reaches subpocket 1 near carbon atom 15 (1C15) (Figs. 4A and 5C). This channel could allow oxygen entry into the active center in a way similar to that proposed in other LOXs (15, 20). The geometry of the 2C chain, which occupies subpocket-2 and is without equivalence in any other LOX structure, suggests that a cis conformation may be favored between carbon atoms 5 and 6, or 10 and 11, or both, with observed deviations from planarity of ∼15 and ∼35°, respectively. Therefore, the phospholipid found is most likely O-(sn-1-cis-vaccenyl-sn-2-myristoyl-sn-glycero-3-phosphoryl) ethanolamine, although a sn-1-stearoyl chain is also possible. cis-Vaccenic acid is a fatty acid found in E. coli that is particularly abundant in cultures grown at low temperature, although a chain as short as 14 carbon atoms in length occupying the 2C position of glycerolipids is not commonly observed (38, 39). In fact, the relatively loose packing seen in the 2C aliphatic chain end suggests that this binding subpocket can accommodate aliphatic chains that are 2 or even 4 carbon atoms longer. The 1C-chain is bent, with the methyl end curved back toward the lipid headgroup (C1–C18 distance of 13.6 Å) and 5 potentially reactive carbon atoms in a pentadiene-like conformation approach the iron–water center. The 4 of these carbons closest to the reactive center have slightly longer distances to the iron (average, 5.1 Å) than to W1 (average, 3.7 Å) (Fig. 5A and Supplemental Fig. S1B). The putative double bond between carbon atoms 11 and 12 is the closest to the iron, and the associated hydrogen atoms would be pointing away from the metal. Carbon atoms 11 and 14 lay over the imidazole rings of the iron ligands His382 and His377 at ∼3.9 Å from the corresponding δ-nitrogen atoms. The binding pocket is almost completely hydrophobic except for the presence of the iron ligand water (W1) and the polar atoms from the side chains of residues Tyr609 and Asn607 at the bottom of subpockets 1 and 2, respectively.
In the Pa_LOX structure determined at 1.75 Å resolution, the phospholipid head is partially exposed to the solvent and forms 2 ionic bonds with the protein (Fig. 4B). One ionic bond is between the phosphate moiety and the guanidinium group of Arg422. The second is between the terminal amino group of the phospholipid and the carboxylic group of Asp190 of helix α2B. The presence of Asp190 and Arg422 indicates that Pa_LOX is suited to binding zwitterionic phospholipids—in particular, those with small positively charged terminal groups, such as PE. The side chain of Arg422 presents flexibility (reflected in high atomic temperature factor values), a feature that may offer versatility when binding to the acidic groups of various substrates. However, in the Pa_LOX structure determined at 2.0 Å resolution, Arg422 does not interact with the phospholipid phosphate but forms hydrogen bonds with the N-terminal extension from a neighboring molecule (Figs. 3B and 4C). These observations suggest that the flexible N-terminal tail of Pa_LOX participates initially in “fishing” for the phospholipid by interacting with its polar head, contributing later to the stabilization of the complex once the phospholipid is placed within the binding pocket.
Interaction with membranes
To study whether the bound phospholipid reflects the capacity of Pa_LOX to interact directly with membranes, SUVs of various phospholipid compositions were analyzed in the presence of Pa_LOX by intrinsic fluorescence. Titration with the 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC) SUVs of a Pa_LOX solution resulted in a progressive decrease of fluorescence intensity (Fig. 6A). This reduction could be due either to a decrease in the intrinsic tryptophan fluorescence of the protein, possibly resulting from the interaction with the membranes, or to quenching by neighboring amino acid residues, an effect that may result from conformational changes in the protein (40, 41). In both cases the decrease observed can be used as a measure of the interaction of Pa_LOX with membranes (33). Experiments were then conducted with SUVs of various compositions: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC); PAPC/1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine (PAPE) (1:1); DPPC at 25°C (i.e., in the gel phase); and brain phosphatidylserine (PS). POPC mimics mammalian phospholipid composition well and it is a model lipid often used for biophysical studies. PAPC incorporates arachidonic acid instead of oleic acid, and PAPE was used because of its PE polar head. Finally, brain PS was assayed to study the effect of a negatively charged membrane on Pa_LOX binding. From the spectra obtained for each of the compositions, relative fluorescence intensities at excitation/emission wavelengths of 290 and 330 nm were taken and plotted as a function of the phospholipid concentration added (Fig. 6B). The partition coefficient (Kx) and fluorescence decreases on maximum binding (F∞) show a sharp contrast between brain PS (with the highestF∞ 122.2 and the lowest of 0.16) and the rest of the compositions tested (with Kx and F∞ values in the 15–35 and 0.64-0.84 range, respectively). Assuming that F∞ can be taken as a measure of conformational changes, PS membranes appear to trigger large changes in Pa_LOX. Of note, Pa_LOX also interacted with DPPC membranes in the gel phase, with an affinity similar to that observed for the fluid membranes of POPC and PAPC.
Figure 6.

In vitro binding of Pa_LOX to membranes. A) Intrinsic tryptophan fluorescence decay, collected at an excitation wavelength of 290 nm, with aliquots of PAPC SUVs added progressively to a solution of Pa_LOX. B) After the spectra for dilution and scattering were corrected, the relative fluorescence (taking the protein fluorescence without liposomes as initial fluorescence) was plotted for vesicles composed of DPPC (△) and PAPC (□) (spectra shown in A), as well as POPC (○), PAPC/PAPE (1:1) (●), and brain PS (■), as a function of phospholipid concentration. Solid lines represent the best fit.
ITC studies also provided support for the interaction between Pa_LOX and SUVs, as well as for the enzyme activity when membranes contain polyunsaturated phospholipids (Supplemental Fig. S2). Sequential addition of the enzyme into a thermally equilibrated suspension of POPC SUVs resulted in a series of identical endothermic peaks (Supplemental Fig. S2, trace a). These peaks were only a little larger than those corresponding to the heat of dilution of the protein, thereby indicating that the heat produced by the binding was endothermic, but almost negligible with respect to that caused by dilution. The addition of Pa_LOX into a suspension of PAPC SUVs gave very different results. On injection of this enzyme, an endothermic peak corresponding to the heat of dilution was observed, as above, but the baseline did not return to its initial value (i.e., there was a decrease in the instrument thermal power (dQ1/dt) necessary to maintain isothermal conditions (Supplemental Fig. S2, trace b). This remaining power can be explained only by an enzymatic reaction, in such a way that the total thermal power remained constant (33, 42). Successive injections of enzyme resulted in additional decreases in thermal power, but of progressively lower magnitude, because the substrate/enzyme ratio was dropping gradually. These results indicate that Pa_LOX presents enzymatic activity toward phospholipid membranes containing a polyunsaturated fatty acid, such as PAPC, but not toward POPC, which contains the monounsaturated oleic acid.
In vivo analysis of P. aeruginosa strains lacking Pa_LOX
To study whether the capacity of Pa_LOX to interact directly with membranes has a biological function, we performed in vivo invasiveness and cytotoxicity analysis with reference wild-type invasive (PA01) and cytotoxic (PA14) P. aeruginosa strains and their corresponding knockout mutants in the gene coding for Pa_LOX, PA1169 (Fig. 7). Invasion capacity was reduced (∼50%) in 2 independent PA1169 knockout mutants at 2 exposure times (Fig. 7A, B). This finding confirms that the interaction of Pa_LOX with eukaryotic membranes participates in altering the properties of host cell membranes. However, no significant differences were found between PA14 and its PA1169 knockout mutant regarding LDH release (Fig. 7C), thus ruling out a role of Pa_LOX in the activity of the type 3 secretion system (TTSS) machinery, which is the basis for the virulence of cytotoxic strains such as PA14.
Figure 7.

In vivo assays of Pa_LOX. Comparative invasion at 1 h (A) and 3 h (B) of postinfection exposure and cytotoxicity (C) assays of P. aeruginosa wild-type strains and Pa_LOX (PA1169 gene) knockout mutants. A, B) Number of invasive CFUs per well of A549 cells (means±sd of 3 independent experiments performed in quadruplicate). Wild-type reference strain PAO1 and its Pa_LOX-knockout mutants PAO3110 and PAO3111. *P < 0.05 vs. PAO1. C) Cytotoxicity assay. Percentage of maximum release of LDH per well (when all the A549 cells were lysed with lysis reagent) after 3 h of infection (means±sd of 3 independent experiments performed in quadruplicate). Wild-type reference strain PA14 and its knockout mutant in the PA1169 gene: PA14Δ1169. N.S., not significant.
DISCUSSION
The structure of a secretable LOX from the opportunistic pathogen P. aeruginosa, the first determined from a prokaryote, displays significant differences from the structures of eukaryotic LOX. First, Pa_LOX lacks the membrane-binding N-terminal PLAT domain (13) found in eukaryotic LOXs. Instead Pa_LOX has an insertion, of ∼100 residues, mostly corresponding to a pair of long, antiparallel helices. This insertion forms a lid over the entrance to a large binding pocket. Second, the binding pocket is bifurcated and contains a complete phospholipid with a 1C chain 18 carbons in length that occupies the catalytic site and a 2C chain of 14 (or more) carbons bound below the insertion. Sequence comparisons indicate that the distinctive features found in Pa_LOX would also be present in other prokaryotic LOXs (Fig. 8).
Figure 8.
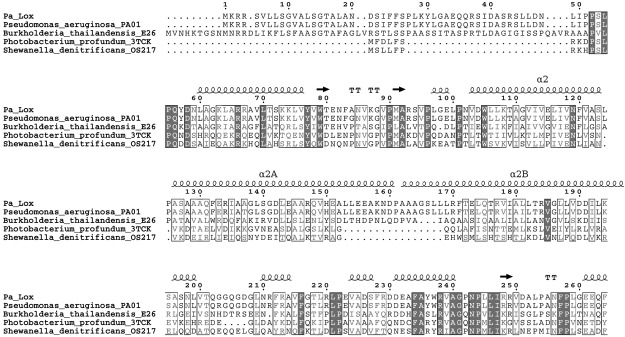
Sequence alignment of the N-terminal region of prokaryotic LOXs. The high sequence homology found in the N-terminal region suggests that prokaryotic LOXs may lack the PLAT domain and contain an insertion similar to that in Pa_LOX.
LOXs produce diverse and highly specific additions of oxygen into polyunsaturated chains. Considerable progress in elucidating important aspects of LOX catalysis has been made through modeling, kinetics, and mutational studies. Although other LOX complexes have been reported (19, 20), a structure with a complete and nonoxidized acyl chain occupying the active site cavity has not been available until the present study, affording an unprecedented opportunity to evaluate mechanistic steps. The 18-carbon acyl chain has a single double bond between carbon atoms 11 and 12. The penultimate double bond of arachidonic acid, a known Pa_LOX substrate (22), could occupy the same position, and its C13 would be well positioned for pro-S hydrogen atom abstraction by the water bound to iron (Supplemental Fig. S1B). The C13 carbon in the Pa_ LOX structure is 4 Å from W1 in this ferrous complex. The corresponding distance in a ferric complex is 3.1 Å, based on a recent computational study of the LOX mechanism (43). The geometry of the lipid 1C chain observed in Pa_LOX suggests that a true substrate with bis-allelic carbon is poised to react with an activated Fe3+-OH center to form the delocalized 5-carbon pentadienyl radical intermediate. The proximity of the double bond carbons to the rings of the histidine-Fe ligands found in the Pa_LOX structure (Fig. 5A) may contribute favorably to stabilizing the intermediate through a π-interaction. Carbon 15 in the Pa_LOX structure is more distant than the other 4 reactive carbons (5.2 Å from W1). This element is an important one in the calculated mechanism (43) because, in the final regeneration of the ferric enzyme, a rotation of the C14–C15 bond allows the proton-coupled electron transfer to proceed. For a linoleic acid substrate, the acyl chain would have to shift further into the Pa_LOX cavity to have the double bond between carbon atoms 9 and 10 in the correct position. It appears that additions to the methyl end (arachidonic) or small shifts of the carboxyl end (linoleic) would be readily accommodated in the large binding cavity.
The observation that iron in Pa_LOX can be oxidized by 13S-HPODE (Fig. 5B), poses an interesting question about the possible entry of substrates into the pocket already occupied by a lipid chain. The low water solubility of phospholipids makes it unlikely that the phospholipid in Pa_LOX is completely removed from the protein when ferric enzyme is prepared. There are long-standing debates on substrate entry into unoccupied binding pockets via the tail (methyl)-first or head (carboxyl)-first approach. However, the reactivity of lipid-bound Pa_LOX with 13S-HPODE and the shape of the binding pocket in LOXs, which approach iron and then bend back toward the protein surface (15–17, 20), suggest that the acyl chain is directly internalized. Direct internalization of substrate is consistent with the structures of the 2 Pa_LOX crystal forms reported here. Comparison of the 2 forms indicates that the active site lid, composed of helices α2A and α2B, can move as a unit with a hinge involving residues 201–206 (Fig. 1). Helix α2 involvement in guiding access to the active site is a consistent recurrent theme in discussions of LOXs that have different positional specificities. Suggestions for the function of this helix focus on rotation of one, or a few, side chains (4, 14) or, alternatively, on rigid body displacements (ref. 16 and this report). Another helix, adjacent to the lid in Pa_LOX and designated helix-11 in most LOX structures, shows rearrangements of interactions of Arg422 with the headgroup of the bound phospholipid in 1 case or with a second Pa_LOX in the other. This helix contains a residue that regulates the S- or R- stereochemical preference of LOXs by being either an alanine or a glycine, respectively (31). In Pa_LOX, the corresponding residue is Ala420, in agreement with the established 15S-product (22). The picture emerging from the Pa_LOX structures presented here and from other organisms is that details of how the polar end of a substrate interacts with helices 2 and 11, the presence of a bend in the substrate cavity to dock a specific substrate double bond, and an appropriately placed oxygen access channel all contribute to the details of how the substrate interacts specifically with the metal center of catalysis.
Tryptophan fluorescence and ITC analysis indicated that, as suggested by the presence of the bound phospholipid and despite the absence of the PLAT domain, Pa_LOX can interact with phospholipid membranes in a composition-dependent manner. Extracting and modifying unsaturated phospholipids, which are commonly found in eukaryotic organisms but are rarely present in bacteria, without cooperation from any other protein, offers a new framework for the elucidation of the biological roles of prokaryotic LOXs. A possible participation of Pa_LOX in the interaction with host cells could explain the reduced invasiveness—about halved—of Pa_LOX knockout variants of P. aeruginosa. Whether this effect on bacterial virulence results directly from the modification of host cell membranes by Pa_LOX or from its potential role in modulating the inflammatory response through interference with arachidonic acid metabolism (22) remains to be elucidated. Secretable prokaryotic LOXs can provide an essential complement to the activities of several specialized microbial toxin enzymes that exert their action at the level of host cell membranes (44).
Supplementary Material
Acknowledgments
Special thanks go to S. Rodríguez-Puente, J. Linacero, J. Pous, and A. Guasch [Plataforma Automatitzada de Cristal·lografia (PAC), Parc Científic de Barcelona, Barcelona, Spain] for valuable help with crystal preparation. The authors thank T. Bates for carefully revising the manuscript.
This work was supported by grants CTQ2010-21283-C02/01/PPQ, HBP2006-0027 from the Ministerio de Ciencia e Innovacion (MICINN) and 2009SGR819 from the Generilitat de Catalunya to A.M.; grants BFU2012-36827 from MICINN and SGR2009-00327 from the Generalitat de Catalunya to I.F.; grant R01GM065268 from the U.S. National Institutes of Health to B.J.G.; and grants SAF2012-38539, CP12/03324, and RD12/0015 from the Ministerio de Economia y Competitividad-Instituto de Salud Carlos III of Spain to C.J. and A.Ol.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 13S-HPODE
- 13S-hydroperoxyoctadecadienoic acid
- DPPC
- 1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- EPR
- electron paramagnetic resonance
- ITC
- isothermal titration calorimetry
- LDH
- lactate dehydrogenase
- LOX
- lipoxygenase
- MOI
- multiplicity of infection
- Pa_LOX
- Pseudomonas aeruginosa LOX
- PAPC
- 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- PAPE
- 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine
- PBS
- phosphate-buffered saline
- PDB
- Protein Data Bank
- PE
- phosphatidylethanolamine
- PEG
- polyethylene glycol
- PLAT
- polycystin-1, lipoxygenase, α-toxin
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PS
- phosphatidylserine
- SUV
- small unilamellar vesicle
- W1
- water molecule 1
REFERENCES
- 1. Kulkarni A. P. (2001) Lipoxygenase: a versatile biocatalyst for biotransformation of endobiotics and xenobiotics. Cell. Mol. Life Sci. 58, 1805–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kessler A., Halitschke R., Baldwin I. T. (2004) Silencing the jasmonate cascade: induced plant defenses and insect populations. Science 305, 665–668 [DOI] [PubMed] [Google Scholar]
- 3. Pidgeon G. P., Lysaght J., Krishnamoorthy S., Reynolds J. V., O'Byrne K., Nie D., Honn K. V. (2007) Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev. 26, 503–524 [DOI] [PubMed] [Google Scholar]
- 4. Gilbert N. C., Bartlett S. G., Waight M. T., Neau D. B., Boeglin W. E., Brash A. R., Newcomer M. E. (2011) The structure of human 5-lipoxygenase. Science 331, 217–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haeggström J. Z., Funk C. D. (2011) Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 111, 5866–5898 [DOI] [PubMed] [Google Scholar]
- 6. Feussner I., Kühn H., Wasternack C. (1997) Do specific linoleate 13-lipoxygenases initiate beta-oxidation? FEBS Lett. 406, 1–5 [DOI] [PubMed] [Google Scholar]
- 7. Funk C. D. (2001) Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875 [DOI] [PubMed] [Google Scholar]
- 8. Brash A. R. (1999) Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 274, 23679–23682 [DOI] [PubMed] [Google Scholar]
- 9. Schneider C., Pratt D. A., Porter N. A., Brash A. R. (2007) Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem. Biol. 14, 473–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rapoport S. M., Schewe T. (1986) The maturational breakdown of mitochondria in reticulocytes. Biochim. Biophys. Acta 864, 471–495 [DOI] [PubMed] [Google Scholar]
- 11. Boyington J. C., Gaffney B. J., Amzel L. M. (1993) The three-dimensional structure of an arachidonic acid 15-lipoxygenase. Science 260, 1482–1486 [DOI] [PubMed] [Google Scholar]
- 12. Gaffney B. J. (1996) Lipoxygenases: structural principles and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 25, 431–459 [DOI] [PubMed] [Google Scholar]
- 13. Bateman A., Sandford R. (1999) The PLAT domain: a new piece in the PKD1 puzzle. Curr. Biol. 9, R588–R590 [DOI] [PubMed] [Google Scholar]
- 14. Minor W., Steczko J., Stec B., Otwinowski Z., Bolin J. T., Walter R., Axelrod B. Crystal structure of soybean lipoxygenase L-1 at 1.4 A resolution. Biochemistry 35:10687–10701, 1996 [DOI] [PubMed] [Google Scholar]
- 15. Youn B., Sellhorn G. E., Mirchel R. J., Gaffney B. J., Grimes H. D., Kang C. (2006) Crystal structures of vegetative soybean lipoxygenase VLX-B and VLX-D, and comparisons with seed isoforms LOX-1 and LOX-3. Proteins 65, 1008–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choi J., Chon J. K., Kim S., Shin W. (2008) Conformational flexibility in mammalian 15S-lipoxygenase: reinterpretation of the crystallographic data. Proteins 70, 1023–1032 [DOI] [PubMed] [Google Scholar]
- 17. Neau D. B., Gilbert N. C., Bartlett S. G., Boeglin W., Brash A. R., Newcomer M. E. (2009) The 1.85 A structure of an 8R-lipoxygenase suggests a general model for lipoxygenase product specificity. Biochemistry 48:7906–7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eek P., Järving R., Järving I., Gilbert N. C., Newcomer M. E., Samel N. (2012) Structure of a calcium-dependent 11R-lipoxygenase suggests a mechanism for Ca2+ regulation. J. Biol. Chem. 287, 22377–22386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilbert N. C., Rui Z., Neau D. B., Waight M. T., Bartlett S. G., Boeglin W. E., Brash A. R., Newcomer M. E. (2012) Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J. 26, 3222–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu S., Mueser T. C., Marnett L. J., Funk M. O., Jr. (2012) Crystal structure of 12-lipoxygenase catalytic-domain-inhibitor complex identifies a substrate-binding channel for catalysis. Structure 20, 1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Su C., Oliw E. H. (1998) Manganese lipoxygenase: purification and characterization. J. Biol. Chem. 273, 13072–13079 [DOI] [PubMed] [Google Scholar]
- 22. Vance R. E., Hong S., Gronert K., Serhan C. N., Mekalanos J. J. (2004) The opportunistic pathogen Pseudomonas aeruginosa carries a secretable arachidonate 15-lipoxygenase. Proc. Natl. Acad. Sci. U. S. A. 101, 2135–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vidal-Mas J., Busquets M., Manresa A. (2005) Cloning and expression of a lipoxygenase from Pseudomonas aeruginosa 42A2. Antonie van Leeuwenhoek 87, 245–251 [DOI] [PubMed] [Google Scholar]
- 24. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 25. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. Allogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. Allogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Allogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 28. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O., Vonrhein C., Womack T. (2011) Buster 2.10.0. Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 29. DeLano W. (2002) The PyMOL Molecular Graphics System, Version 1.5.0.3 Schroedinger, LLC, Portland, OR, USA [Google Scholar]
- 30. Gaffney B. J., Mavrophilipos D. V., Doctor K. S. (1993) Access of ligands to the ferric center in lipoxygenase-1. Biophys. J. 64, 773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coffa G., Brash A. R. (2004) A single active site residue directs oxygenation stereospecificity in lipoxygenases: stereocontrol is linked to the position of oxygenation. Proc. Natl. Acad. Sci. U. S. A. 101, 15579–15584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bottcher C. J. F., Van Gent C. P. C. (1961) A rapid and sensitive submicro phosphorous determination. Anal. Chim. Acta 24, 203–204 [Google Scholar]
- 33. Ladokhin A. S., Jayasinghe S., White S. H. (2000) How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal. Biochem. 285, 235–245 [DOI] [PubMed] [Google Scholar]
- 34. Jacobs M. A., Alwood A., Thaipisuttikul I., Spencer D., Haugen E., Ernst S., Will O., Kaul R., Raymond C., Levy R., Chun-Rong L., Guenthner D., Bovee D., Olson M. V., Manoil C. (2003) Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100, 14339–14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harrison E. M., Carter M. E. K., Luck S., Ou H.-Y., He X., Deng Z., O'Callaghan C., Kadioglu A., Rajakumar K. (2010) Pathogenicity islands PAPI-1 and PAPI-2 contribute individually and synergistically to the virulence of Pseudomonas aeruginosa strain PA14. Infect. Immun. 78, 1437–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shaver C. M., Hauser A. R. (2006) Interactions between effector proteins of the Pseudomonas aeruginosa type III secretion system do not significantly affect several measures of disease severity in mammals. Microbiology 152, 143–152 [DOI] [PubMed] [Google Scholar]
- 37. Solomon E. I., Brunold T. C., Davis M. I., Kemsley J. N., Lee S. K., Lehnert N., Neese F., Skulan A. J., Yang Y. S., Zhou J. (2000) Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 100, 235–350 [DOI] [PubMed] [Google Scholar]
- 38. Oursel D., Loutelier-Bourhis C., Orange N., Chevalier S., Norris V., Lange C. M. (2007) Identification and relative quantification of fatty acids in Escherichia coli membranes by gas chromatography/mass spectrometry. Rapid Commun. Mass. Spectrom. 21, 3229–3233 [DOI] [PubMed] [Google Scholar]
- 39. Oursel D., Loutelier-Bourhis C., Orange N., Chevalier S., Norris V., Lange C. M. (2007) Lipid composition of membranes of Escherichia coli by liquid chromatography/tandem mass spectrometry using negative electrospray ionization. Rapid Commun. Mass. Spectrom. 21, 1721–1728 [DOI] [PubMed] [Google Scholar]
- 40. Callis P., Liu T. (2004) Quantitative prediction of fluorescence quantum yields for tryptophan in proteins. J. Phys. Chem. B 108, 4248–4259 [Google Scholar]
- 41. Pande A. H., Qin S., Tatulian S. A. (2005) Membrane fluidity is a key modulator of membrane binding, insertion, and activity of 5-lipoxygenase. Biophys. J. 88, 4084–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bianconi M. L. (2007) Calorimetry of enzyme-catalyzed reactions. Biophys. Chem. 126, 59–64 [DOI] [PubMed] [Google Scholar]
- 43. Bushnell E. A. C., Jamil R., Gauld J. W. (2013) Gaining insight into the chemistry of lipoxygenases: a computational investigation into the catalytic mechanism of (8R)-lipoxygenase. J. Biol. Inorg. Chem. 18, 343–355 [DOI] [PubMed] [Google Scholar]
- 44. Montes L.-R., Ibarguren M., Goñi F. M., Stonehouse M., Vasil M. L., Alonso A. (2007) Leakage-free membrane fusion induced by the hydrolytic activity of PlcHR(2), a novel phospholipase C/sphingomyelinase from Pseudomonas aeruginosa. Biochim. Biophys. Acta 1768, 2365–2372 [DOI] [PubMed] [Google Scholar]
- 45. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.