

Abstract
Oxidation of retinol via retinaldehyde results in the formation of the essential morphogen all-trans-retinoic acid (ATRA). Previous studies have identified critical roles in the regulation of embryonic ATRA levels for retinol, retinaldehyde, and ATRA-oxidizing enzymes; however, the contribution of retinaldehyde reductases to ATRA metabolism is not completely understood. Herein, we investigate the role of the retinaldehyde reductase Dhrs3 in embryonic retinoid metabolism using a Dhrs3-deficient mouse. Lack of DHRS3 leads to a 40% increase in the levels of ATRA and a 60% and 55% decrease in the levels of retinol and retinyl esters, respectively, in Dhrs3−/− embryos compared to wild-type littermates. Furthermore, accumulation of excess ATRA is accompanied by a compensatory 30–50% reduction in the expression of ATRA synthetic genes and a 120% increase in the expression of the ATRA catabolic enzyme Cyp26a1 in Dhrs3−/− embryos vs. controls. Excess ATRA also leads to alterations (40–80%) in the expression of several developmentally important ATRA target genes. Consequently, Dhrs3−/− embryos die late in gestation and display defects in cardiac outflow tract formation, atrial and ventricular septation, skeletal development, and palatogenesis. These data demonstrate that the reduction of retinaldehyde by DHRS3 is critical for preventing formation of excess ATRA during embryonic development.—Billings, S. E., Pierzchalski, K., Butler Tjaden, N. E., Pang, X.-Y., Trainor, P. A., Kane, M. A., Moise, A. R. The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development.
Keywords: development, metabolism, nuclear receptors, retinoid, vitamin A
All-trans-retinoic acid, (ATRA) a metabolite of vitamin A (retinol), carries out essential roles in embryonic development as well as in postnatal life (1, 2). ATRA binds and activates heterodimers of the retinoic acid receptor (RAR)/retinoid X receptor (RXR) (3–5) which activates or represses target genes (6). Since either excess or deficiency of ATRA can have severe teratogenic effects, its levels need to be exquisitely well regulated. This regulation is imparted by a complex interaction of enzymes, transporters, and binding proteins involved in ATRA metabolism. In the model currently accepted in the field, the extent of ATRA signaling is limited primarily by the activity of the cytochrome P450 enzymes CYP26A1, B1, and C1 (7, 8).
Oxidation of all-trans-retinol to all-trans-retinaldehyde, considered to be a critical, rate-limiting step in the synthesis of ATRA (9, 10), can be carried out by either microsomal short-chain dehydrogenase/reductase (SDR) enzymes or by cytosolic medium-chain alcohol dehydrogenases (ADHs) (9–11). Of these enzymes, the only one whose role in embryonic retinoid metabolism is supported by genetic studies is the SDR retinol dehydrogenase 10 (RDH10). Loss of RDH10 results in a reduced capacity to oxidize retinol, which leads to reduced ATRA levels, alterations in the expression of ATRA targets and defects in embryonic development (12–16). As a result, RDH10-null mice typically die by embryonic day 10.5 (E10.5; ref. 17).
Reduction of all-trans-retinaldehyde to all-trans-retinol is proposed to be carried out by both SDR enzymes and by several members of the aldo-keto-reductase (AKR) family (11, 18). This biotransformation is essential for the conversion of provitamin A carotenoids, like β-carotene, to all-trans-retinol and for the recycling of the visual chromophore. Several SDRs have been genetically demonstrated to play a role in the reduction of all-trans-retinaldehyde in the eye (19). However, as of yet, we do not know which retinaldehyde reductases are physiologically relevant for retinoid metabolism during embryogenesis or in adult nonvisual tissues, nor whether this reaction is important in the regulation of retinoid metabolism outside the eye.
Dehydrogenase/reductase superfamily member 3 (DHRS3), first referred to as retinal-specific SDR1 (retSDR1), carries out the NADPH-dependent reduction of retinaldehyde in vitro (20). In addition to the retina, Dhrs3 was shown to be expressed in many other embryonic and adult tissues (20–24) and the expression of Dhrs3 is up-regulated by ATRA and down-regulated in response to inflammation (21, 22, 24–27). The expression of Dhrs3 can also be induced by p53, p63 (28–30), RXR (31) and PPARγ (32). Notably, knockdown of the zebrafish homologue dhrs3a using antisense morpholinos (MOs) leads to up-regulation of ATRA target genes. Conversely, forced overexpression of dhrs3a leads to reduced ATRA signaling in zebrafish embryos (22). Taken together, these results suggest that all-trans-retinaldehyde reduction by Dhrs3a is an important regulatory process in retinoid metabolism during zebrafish development. However, it is not clear whether this role is conserved in mammals, which, unlike zebrafish, do not rely on all-trans-retinaldehyde as a storage form of vitamin A during embryogenesis (33–35). Therefore, in our studies, we further expand on this model of homeostasis by demonstrating that DHRS3 plays an indispensable and evolutionarily conserved role in the maintenance of ATRA homeostasis.
MATERIALS AND METHODS
Mouse strains, husbandry, and genotyping of Dhrs3+/− mice
Dhrs3+/− founder mice were produced by Lexicon Pharmaceuticals (The Woodlands, TX, USA) and Genentech (South San Francisco, CA, USA) (36) and were obtained through the U.S. National Institutes of Health Mutant Mouse Regional Resource Center (MMRRC) Program. Dhrs3 was targeted with a selection cassette through homologous recombination in 129S5/SvEvBrd embryonic stem (ES) cell clones (Fig. 1A). The selection cassette is composed of a bicistronic neomycin resistance and lacZ; however, the expression of the LacZ gene cannot be detected in the founder mice (F. J. de Sauvage, Genentech; personal communication, July 19, 2013) or succeeding generations (results not shown). The selection cassette replaces Dhrs3 exon 1 of the Dhrs3 NM_011303.6 transcript, which codes for the translation start codon and for residues comprising the essential Rossmann-fold motif, which is involved in binding dinucleotide cofactors (37) (Fig. 1A). Thus, the disrupted Dhrs3 allele is predicted to be nonfunctional. Correctly targeted clones were microinjected into C57BL/6J blastocysts.
Figure 1.

Generation of Dhrs3-deficient mice. A) Genomic map of Dhrs3 on mouse chromosome 4 indicating replacement of exon 1 with a selection cassette (red). Arrows indicate positions of hybridization for primers used for genotyping. B) Analysis of the products of the genotyping reaction of genomic DNA of Dhrs3+/+ (WT), Dhrs3+/− (Het), and Dhrs3−/− (KO) embryos. C) Analysis of the expression of Dhrs3 mRNA by RT-PCR indicates that Dhrs3 mRNA cannot be detected in Dhrs3−/− embryos. D) Analysis of the expression of DHRS3 protein by immunohistochemistry of limb sections indicates that expression of DHRS3 protein cannot be detected in the Dhrs3−/− embryos. Table 2 shows statistics of the frequency of embryos of the WT, Het, and KO genotypes resulting from Dhrs3+/− crosses recovered at E14.5 or E17.5 or observed at birth (P0).
The wild-type (WT) allele of Dhrs3 was detected by PCR of genomic DNA using the primers 5′-CTGAGGGTAAAGGGACTCTGG-3′ and 5′ AATAGCCAGCGAGATACCAATC-3′, which generate a 242-bp amplification product. The knockout allele was detected using the primers 5′-GCAGCGCATCGCCTTCTATC-3′ and 5′ AATAGCCAGCGAGATACCAATC-3′, which generate a 308-bp amplification product.
The RARE-LacZ mice generated and described by Rossant et al. (38) were obtained from the Jackson Laboratories (Bar Harbor, ME, USA) and maintained on a CD1 background. The RARE-LacZ transgenic mice express a β-galactosidase (LacZ) reporter gene under the control of a retinoic acid response element (RARE) derived from the promoter of the Rarβ gene (39). The presence of the RARE-lacZ allele was detected with allele-specific primers: 5′-CGTCGTCCCCTCAAACTGGCAGA-3′ and 5′-TTCGGCGCTCCACAGTTTCGGGTTTTC-3′. The conditions of PCR for all genotyping reactions were: 94°C for 1 min; 40 cycles of 94°C for 30 s, 68°C for 30 s with a 0.2°C drop every cycle, 68°C for 1 min; and 68°C for 10 min.
Animal husbandry and dietary treatment
Dhrs3+/− founder mice generated on a mixed 129/SvEvBrd x C57BL/6 background were backcrossed to C57BL/6 (Charles River, Wilmington, MA, USA) for three generations and inbred to generate Dhrs3−/− homozygotes. Mice were maintained in a controlled climate with a 12-h light-dark cycle and fed a vitamin A-sufficient (VAS) diet with defined vitamin A content (Research Diets, New Brunswick, NJ, USA) from weaning and through gestation. VAS diet is derived from AIN93G (40) and contains 4 IU preformed vitamin A as retinyl esters per gram diet, and soybean oil instead of cottonseed oil to minimize provitamin A carotenoid content. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Kansas.
RNA isolation and quantitative RT-PCR (qRT-PCR)
RNA was extracted from E14.5 embryos using Trizol (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's protocol. Complementary first-strand DNA was synthesized from 1 μg total RNA using SuperScript III reverse transcriptase (Life Technologies) and oligo-dT primers. qRT-PCR reactions were performed in a 10-μl volume using 25 ng cDNA, 250 nM of each primer, and 5 μl of QuantiFast SYBR Green PCR Master Mix (Qiagen, Germantown, MD, USA) on the Applied Biosystems StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA). The conditions of qPCR amplification were: 95°C for 10 min, 95°C for 10 s, and 60°C for 30 s for 40 cycles. The primer sets for the various target genes are listed in Table 1. The relative levels of cDNA were quantified by the comparative threshold cycle method using β-actin as an internal standard. Each sample corresponds to one distinct embryo from each genotype.
Table 1.
Primers for quantitative real-time PCR analysis
| Gene | Accession number | Sequence | |
|---|---|---|---|
| Actin-β | NM_007393 | Forward | 5′-TATTGGCAACGAGCGGTTCC-3′ |
| Reverse | 5′-GGCATAGAGGTCTTTACGGATGTC-3′ | ||
| CRABP1 | NM_013496 | Forward | 5′-CAGCAGCGAGAATTTCGACGA-3′ |
| Reverse | 5′-CGCACAGTAGTGGATGTCTTGA-3′ | ||
| CRBP-I | NM_011254 | Forward | 5′-AATGAGAGCTGAGGGTG-3′ |
| Reverse | 5′-GGTATGCGTTTCGGTCC-3′ | ||
| CRBP-II | NM_009034.4 | Forward | 5′-ATAAAGGGGAGAAGGAGAA-3′ |
| Reverse | 5′-CCATCACTTCTTTTTGAACA-3′ | ||
| CRBP-III | NM_022020.2 | Forward | 5′-CTTGAGGCAACTACTCAATG-3′ |
| Reverse | 5′-TTCATCAACCCAAAACAGAC-3′ | ||
| Cyp26a1 | NM_007811 | Forward | 5′-GAACATTCGCGCCAAGATCC-3′ |
| Reverse | 5′-TTAGTGCCTGCATATCCAGCC-3′ | ||
| Cyp26B1 | NM_001177713 | Forward | 5′-CCGTGAGAAGCTGCAGTGTA-3′ |
| Reverse | 5′-GGGTTCCATCCTTCAGCTCC-3′ | ||
| Cyp26C1 | NM_001105201 | Forward | 5′-CTGGCCCAACAACTCTGGAC-3′ |
| Reverse | 5′-AACGAGAGCCCTGTACCAAC-3′ | ||
| Hoxa1 | NM_010449 | Forward | 5′-CAAGAAGCCTGTCGTTCCCC-3′ |
| Reverse | 5′-ACTCTCCAACTTTCCCTGTTTTG-3′ | ||
| HoxA13 | NM_008264 | Forward | 5′-CGAACGGGAATACGCTACGA-3′ |
| Reverse | 5′-GAACCAGATTGTGACCTGCC-3′ | ||
| HoxD13 | NM_008275 | Forward | 5′-TGATTTAGGACCGCTGGGAG-3′ |
| Reverse | 5′-AGCAAAGGAGACTGCCAAAGA-3′ | ||
| Lrat | NM_023624 | Forward | 5′-AGCAAGGGTGGTGACTAGAC-3′ |
| Reverse | 5′-ATCCCCCAACTCTCTACTGC-3′ | ||
| Raldh1a1 | NM_013467 | Forward | 5′-ATACTTGTCGGATTTAGGAGGCT-3′ |
| Reverse | 5′-GGGCCTATCTTCCAAATGAACA-3′ | ||
| Raldh1a2 | NM_009022 | Forward | 5′-CAGAGAGTGGGAGAGTGTTCC-3′ |
| Reverse | 5′-CACACAGAACCAAGAGAGAAGG-3′ | ||
| Raldh1a3 | NM_053080 | Forward | 5′-AGGTCTACGGGGAGTTTGTG-3′ |
| Reverse | 5′-CTGCTTTTGGTCGATCTGAGG-3′ | ||
| Rarb | NM_011243 | Forward | 5′-CAAGCTCCAAGAACCACTGC-3′ |
| Reverse | 5′-ATTACACGTTCGGCACCTTTC-3′ | ||
| Rdh10 | NM_133832 | Forward | 5′-CTGGAGTTGAGGATTACTGTGC-3′ |
| Reverse | 5′-TCTGAACATGCCCGTGTCTA-3′ | ||
| Stra6 | NM_001162476 | Forward | 5′-GATACCCAGAGCTGACCAACC-3′ |
| Reverse | 5′-ATGTGTGGTAGCCTGGATCCA-3′ |
Immunohistochemistry
Embryos were fixed in 10% formalin for 90 min at room temperature, washed in PBS, and equilibrated in 20% sucrose/PBS overnight at 4°C. Embryos were then embedded in optimum cutting temperature (OCT) compound (cat. no. 4583; Sakura FineTek USA Inc., Torrance, CA, USA) and frozen on dry ice. Cryosections (14 μm) were blocked for 1 h in blocking buffer [2% bovine serum albumin (BSA), 2% normal goat serum, and 0.05% Tween 20 in PBS, pH 7.4]. Primary antibody (anti-DHRS3, 15393-1AP 1:200; ProteinTech, Chicago, IL, USA) was diluted in blocking buffer, applied to sections, and incubated overnight at 4°C. Sections were washed with PBS 3 times for 10 min each, then secondary antibody (diluted 1:1000 in blocking buffer; anti-rabbit AlexaFluor 568, Life Technologies) and 1 μM DAPI (Life Technologies) were applied. Sections were washed again and mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA). Images were acquired with a ×10 objective (Nikon Plan Fluor 0.30 DIC infinity/0.17 WD 16.0; Nikon, Tokyo, Japan) on an epifluorescent microscope (Nikon Eclipse TE 2000-U) using Metamorph software (Nikon). Alternatively, images spanning the entire embryo were acquired in montage capture mode using the ×10 objective (Olympus/3I spinning disk confocal/epifluorescence/TIRF inverted microscope; Olympus, Tokyo, Japan) and assembled in a composite image using SlideBook 5.0 (Olympus).
Retinoid analysis by high performance liquid chromatography with UV detection (HPLC-UV) and liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Embryos were harvested under yellow light, frozen in liquid nitrogen and kept at −80°C until extraction. Retinoids were quantified in extracted samples by LC-MS/MS for ATRA isomers or by HPLC-UV for neutral retinoids (retinol, retinaldehyde, and retinyl esters) using previously described methodologies (41–47). Retinoid concentrations were determined from calibration curves (for each analyte) constructed from authentic standards. Retinoid levels were normalized per gram of tissue. Results are expressed as means ± sd based on the averaged values for ≥4 embryos/genotype. Statistical significance was assessed with a 2-tailed, unpaired Student's t test.
Assays of LacZ reporter gene expression
Dhrs3−/−; RARE-LacZ and Dhrs3+/+; RARE-LacZ embryos were harvested at E14.5, and the activity of LacZ was assayed using established X-gal staining procedures (48). The staining was performed in parallel for the same length of time under identical conditions, and the embryos were imaged using a dissecting microscope under identical conditions.
Skeletal staining
E14.5 or E17.5 embryos were stained with Alcian blue or Alizarin red for cartilage and bone, respectively, using established procedures (48) with the following modifications. The embryos were dissected and rinsed in PBS and fixed in ice-cold 95% ethanol. Skin and organs were removed for E17.5 embryos. Embryos were transferred to stain base solution (70% ethanol, 5% acetic acid), and then stained overnight in fresh staining solution (70% ethanol, 5% acetic acid, 0.02% Alcian blue, 0.005% Alizarin red). After staining, the embryos were briefly rinsed in stain base solution, then in water, followed by potassium hydroxide (KOH). E14.5 embryos received 1% KOH treatments for 30 min, while E17.5 embryos received 2% KOH treatments for 6 h. The embryos were rinsed in 0.25% KOH for 30 min and then cleared in a 0.25% KOH-glycerol series.
Serial section histology
E14.5 embryos were fixed in neutral buffered 10% formalin, embedded in paraffin, and processed by serial sectioning at 7 μm. The slides were stained using hematoxylin and eosin using established procedures (49) and imaged with a dissecting microscope.
RESULTS
Dhrs3−/− mice exhibit late embryonic and perinatal lethality
We derived Dhrs3−/− mice by crossing Dhrs3+/− mice that were maintained on a VAS diet from weaning and through gestation. VAS diet contains 4 IU/g of preformed vitamin A (as retinyl palmitate) and was chosen based on cumulative evidence that chow diet varies in content and exceeds the required recommended values of vitamin A (50, 51). The VAS diet represents a moderate, yet, adequate amount of vitamin A compared to chow (15 IU/g; ref. 50).
To verify that the Dhrs3−/− mice are indeed null for Dhrs3, we assayed Dhrs3 transcript expression by RT-PCR (Fig. 1C) by amplifying a region outside the disrupted region of the Dhrs3 gene. We also assayed for the expression of DHRS3 protein by immunohistochemistry of E14.5 sections from Dhrs3−/− embryos and WT littermates. Neither Dhrs3 transcript nor DHRS3 protein can be detected in Dhrs3−/− embryos (Fig. 1D), indicating that Dhrs3−/− animals lack both Dhrs3 transcript and translation product.
Very few Dhrs3−/− mice were recovered at birth (frequency of 2 vs. 25% expected; Table 2). The few Dhrs3−/− pups obtained were smaller than heterozygous or WT littermates, lacked a milk spot, and were dead soon after birth. Gross examination of postnatal day 0 (P0) Dhrs3−/− pups revealed they had open eyes, and a hypoplastic mandible (micrognathia; Supplemental Fig. S1).
Table 2.
Frequency of embryos of different genotypes resulting from Dhrs3+/− crosses recovered at E14.5 or E17.5 or observed at birth (P0)
| Age | Genotype (%) |
n | ||
|---|---|---|---|---|
| WT | Het | KO | ||
| E14.5 | 26 | 54 | 20 | 163 |
| E17.5 | 31 | 56 | 13 | 39 |
| P0 | 42 | 56 | 2* | 43 |
Differences between observed and expected percentages of WT, heterozygous (Het), and knockout (KO; Dhrs3−/−) mice were evaluated using the χ2 test.
P < 0.001.
To identify the stage of development during which ablation of Dhrs3 leads to reduced survival of embryos, we evaluated the genotypic ratios of Dhrs3−/− embryos obtained from heterozygous matings and collected at E14.5, E17.5, and at birth (P0). While E14.5 and E17.5 Dhrs3−/− embryos can be recovered in close to expected ratios (25%), the percentage of Dhrs3−/− embryos recovered at birth is significantly lower than expected (Dhrs3−/− frequency of 2%; P < 0.001; Table 2). Therefore, Dhrs3 deficiency results in late gestation/perinatal lethality.
Ablation of Dhrs3 results in changes in retinoid metabolism
We propose that the lack of DHRS3 results in decreased reduction of all-trans-retinaldehyde, which leads to changes in the levels of ATRA in Dhrs3−/− embryos. Therefore, we evaluated the role of Dhrs3 in retinoid metabolism and ATRA signaling at E14.5.
We quantified retinoid metabolites in E14.5 WT and Dhrs3−/− littermates obtained from Dhrs3+/− dams maintained on VAS diet. Endogenous ATRA was quantified by LC-MS/MS, while the more abundant nonpolar retinoids were quantified by HPLC-UV (47). Global levels of ATRA are increased in Dhrs3−/− embryos (Fig. 2). However, the levels of retinaldehyde were not significantly altered in Dhrs3−/− vs. Dhrs3+/− or WT littermates. This data is consistent with the proposed function of DHRS3, because the substrate of DHRS3, all-trans-retinaldehyde, is quickly and irreversibly oxidized to ATRA by the cytosolic retinaldehyde dehydrogenases (RALDHs) 1, 2, and 3 (52–54). Furthermore, we demonstrate that the product of DHRS3, all-trans-retinol, and its ester forms (retinyl esters) are decreased (Fig. 2). These data argue strongly that DHRS3 has a physiologically important role as a retinaldehyde reductase.
Figure 2.

Alterations in the levels of retinoid metabolites as a result of Dhrs3-deficiency. Global levels of all-trans-retinol, retinyl esters, and ATRA were analyzed in WT, Dhrs3+/− (Het), or Dhrs3−/− (KO) mice. E14.5 embryos were derived from dams maintained on a VAS diet. Retinoids were extracted from whole embryos and analyzed using LC-UV in the case of all-trans-retinol, retinaloximes, and retinyl esters and LC-MS/MS in the case of ATRA. Amount of each retinoid was normalized based on the weight of embryonic tissue. Data represent means ± sd of retinoid levels from 4–5 embryos/genotype; *P < 0.05, ***P < 0.001 vs. WT; ##P < 0.01, ###P < 0.001 vs. Het.
The level of enzymes involved in the synthesis and breakdown of ATRA are very tightly regulated in vivo (7, 9, 10). The excess formation of ATRA seen in Dhrs3−/− embryos could cause the ATRA regulatory system to seek to counteract the excess ATRA levels by either reduced synthesis or by increased degradation of ATRA. To assay the levels of expression of enzymes known to be involved in ATRA metabolism, we performed qRT-PCR using mRNA derived from whole WT and Dhrs3−/− embryos. Consistent with the accumulation of excess ATRA, there is a general down-regulation of enzymes involved in ATRA synthesis; i.e., Rdh10, Raldh1, Raldh2, and Raldh3, and an increase in the expression of Cyp26A1, an ATRA catabolic enzyme, in Dhrs3−/− embryos vs. their WT littermates (Fig. 3A). The up-regulation of Cyp26A1 in response to excess ATRA is in agreement with the established direct regulation of this gene by RAR/RXR (55, 56). Ablation of Dhrs3 did not result in changes in the expression levels of Cyp26B1 and Cyp26C1, also involved in ATRA catabolism (57–59). We also found no evidence of changes in the expression levels of other genes involved in retinoid metabolism, such as lecithin retinol:acyltransferase (Lrat), cellular retinol binding protein (CRBP)-I, CRBP-II, CRBP-III, cellular retinoic acid binding protein 1 (CRABP1), CRABP2, Isx, or carotenoid-15,15′-monooxygenase (Bcmo1) in Dhrs3−/− vs. WT embryos (results not shown). Nevertheless, the changes observed in the expression levels of several ATRA synthetic and catabolic enzymes in Dhrs3−/− vs. WT embryos are consistent with an attempt to restore normal ATRA levels through a metabolic compensatory response. The role and feedback regulation of the studied enzymes in the ATRA biosynthetic pathway is schematically depicted in Fig. 3C.
Figure 3.
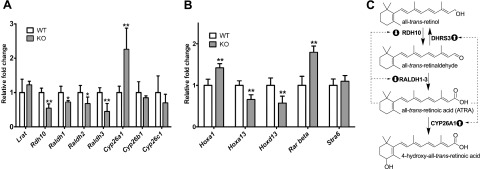
Alterations in the expression levels of genes involved in ATRA synthesis and signaling in Dhrs3−/− mice. Expression of candidate genes was measured by qRT-PCR using mRNA isolated from whole E14.5 WT or Dhrs3−/− (KO) embryos from dams maintained on VAS. A) Data represent means ± sd; n = 4 KO and 5 WT embryos. *P < 0.05, **P < 0.01. B) Alterations in the expression levels of known ATRA target genes in Dhrs3−/− mice vs. WT littermates. For each gene, WT expression level is normalized to 1. Data represent means ± sd of expression levels from 4 KO and 5 WT. **P < 0.01. C) Diagram of ATRA metabolism and feedback-regulation mechanisms operating during embryogenesis. Accumulation of ATRA in Dhrs3−/− embryos leads to feedback mechanisms, resulting in increased expression of Cyp26a1 (up-arrow bullet) and, conversely, decreased expression of Rdh10, Raldh1-3 (down-arrow bullet). The expression of Dhrs3 is itself upregulated by ATRA (refs. 21, 22, 25–27). Despite compensatory responses, Dhrs3−/− embryos have decreased levels of all-trans-retinol and retinyl esters, accumulate excess ATRA and display altered ATRA signaling.
Ablation of Dhrs3 results in changes in ATRA-mediated signaling
To establish whether ablation of Dhrs3 can alter ATRA-dependent signaling, we studied the expression of known ATRA targets and the activity of an RAR reporter gene in Dhrs3−/− and WT embryos.
We assayed the expression of several genes known to be controlled by ATRA via RARs by qRT-PCR using mRNA derived from whole Dhrs3−/− and WT embryos. Our results indicate that the expression of Rarb and the homeobox (Hox) gene Hoxa1 are significantly increased in Dhrs3−/− vs. WT embryos (Fig. 3B), consistent with the reported direct activation of these genes by ATRA (39, 61). In contrast, we observed significantly decreased expression of the distal (5′-located) Hox genes, Hoxa13 and Hoxd13. This observation is consistent with an increased level of ATRA in the limb bud, causing the expansion of the expression domain of more proximal Hox genes at the expense of the more distal ones (62). Other known ATRA target genes, including Rara, Hoxb1, Nrip1, Otx2, stimulated by retinoic acid 6 (Stra6), and Stra8, did not display a statistically significant change in expression as a result of Dhrs3 ablation (data not shown). These results indicate that ATRA signaling is altered in Dhrs3−/− embryos, in agreement with the increase in global levels of ATRA.
Next, we employed a RARE-LacZ reporter mouse model which carries a LacZ transgene (38) driven by a RARE element derived from the Rarβ gene (63). The extent of LacZ activity reflects RAR signaling in situ (38), thus allowing for the visualization of RAR signaling in a spatial manner.
We compared the pattern of RAR-mediated ATRA signaling in E14.5 Dhrs3−/−; RARE-LacZ vs. Dhrs3+/+; RARE-LacZ mice. Most tissues of Dhrs3−/−; RARE-LacZ display higher levels of RARE-LacZ transgene activity in comparison with corresponding tissues of Dhrs3+/+; RARE-LacZ embryos; this was particularly noticeable in the tails and frontonasal regions as seen in all four E14.5 Dhrs3−/−, RARE-LacZ embryos examined (Fig. 4). In general, we observed that the Dhrs3−/−; RARE-LacZ embryos that display the most severe developmental abnormalities also exhibit the largest increases in RARE-LacZ transgene activity, suggesting that the developmental defects and the levels of ATRA signaling are correlated. In summary, the absence of Dhrs3 is consistently associated with expanded domains of ATRA signaling.
Figure 4.

Alterations in ATRA-responsive transgene activity in Dhrs3−/− mice. Dhrs3−/− (A–C) and WT (D–F) embryos harboring a RARE-LacZ transgene were collected at E14.5 and stained for β-galactosidase (β-gal) reporter gene activity. Scale bars = 5 mm. Magnifications of the head (B, E) and tail (C, F) regions of Dhrs3−/−; RARE-LacZ (B, C) and Dhrs3+/+; RARE-LacZ (E, F) embryos are shown. Arrows indicate areas of expanded RARE-LacZ activity in the Dhrs3−/−; RARE-LacZ embryos. The staining pattern is representative of that seen in ≥4 embryos/genotype.
Cumulatively, our results indicate that the pattern of expression of the RARE-LacZ transgene and the levels of expression of several known ATRA-regulated target genes are altered as a result of Dhrs3 ablation. These results are consistent with the hypothesis that the increased levels of ATRA cause disruptions in ATRA signaling in Dhrs3−/− embryos.
Dhrs3 is expressed in tissues involved in embryonic retinoid metabolism
The pattern of expression of Dhrs3 in mouse embryos has not been described. Therefore, we determined the tissue localization of Dhrs3 in embryonic mouse tissues.
E14.5 Dhrs3−/− embryos can be recovered in expected genotypic ratios; therefore, we used immunohistochemistry to examine the expression of DHRS3 protein at E14.5. The specificity of the polyclonal anti-DHRS3 antisera was verified using Dhrs3−/− littermates, as we did not observe significant staining in any of the tissues, except a faint signal in the liver of Dhrs3−/− embryos which is likely to be autofluorescence (Fig. 5A, B).
Figure 5.

Expression pattern of DHRS3 protein in E14.5 embryo sections. Sagittal sections from Dhrs3−/− mice and WT littermates were stained using anti-DHRS3 polyclonal antiserum (red) and DAPI (blue) for nuclear staining. Ssections were examined by epifluorescent microscopy, and the individual images were assembled to obtain a montage of the whole section. A) DHRS3 is expressed at high levels in fetal liver, kidney (k), nasal epithelium (n), along the sternum (st) and the spinal cord (sc), and the limb (arrow) of WT embryos. Moderate immunoreactivity staining is observed in the cerebellum (Cb), the developing pituitary (pit), and the telencephalic choroid plexus (TCh). Weaker staining is observed in the lung (L) and a distinct population of cells in the heart (h). B) Dhrs3−/− embryo stained and examined under the same conditions as the WT littermate shows minimal staining with the anti-DHRS3 antiserum. C–J) Expression of DHRS3 in selected tissues, including the developing lung (C), the heart (D), nasal epithelium (E), neural retina (F), intestinal epithelium (G), kidney (H), interdigital area of developing limb (I, arrow) and cerebellum (J). Scale bars = 1 mm. Images are representative of the staining pattern observed using sections obtained from 4 WT and 3 Dhrs3−/− littermates.
At E14.5, DHRS3 is found at high levels in fetal liver, kidney, nasal epithelium, and the interdigital zones of limbs (Fig. 5). Particularly high expression of DHRS3 was detected in the pericardial mesothelium and in the roof and floor plates along the dorsal and ventral midline of the neural tube, respectively (Fig. 5A). Moderate expression of DHRS3 was detected in the fetal brain in the developing pituitary, cerebellum, and choroid plexus. Lower levels of expression of DHRS3 were detected in the developing heart, lung, intestine, thyroid (not shown), and retina. This pattern of expression is in agreement with that observed for the Dhrs3 homologs from zebrafish and Xenopus (22, 23). Thus, DHRS3 is found in many embryonic tissues where retinoid metabolism and retinoid signaling play important roles (1, 2, 64).
Ablation of Dhrs3 results in altered developmental processes
Dhrs3−/− embryos exhibit late gestation/perinatal lethality and have altered levels of retinoids and ATRA-target genes and an altered pattern of expression of the RAR-reporter gene. Late gestation and perinatal lethality in embryos can often be attributed to a limited number of primary causes of death, which include cardiovascular defects, hematopoietic defects, developmental delay of the lungs, cranial nerve defects, or skeletal defects (65). Next, we examined the development of the heart and the axial and craniofacial skeleton in E14.5 Dhrs3−/− embryos.
The heart morphology of E14.5 Dhrs3−/− embryos was compared to that of WT littermates and published mouse models that display cardiac malformations (66). All of the 5 E14.5 Dhrs3−/− embryos examined displayed cardiac malformations, including a ventricular septation defect (VSD) present in 4 of 5 embryos examined (Fig. 6). Ventricle septation of WT mice is usually complete by E13.5. In contrast, the ventricles of these E14.5 Dhrs3−/− embryos communicate via a large opening due to an incomplete interventricular septum (Fig. 6E, F vs. I, J); the VSD being confined to the membranous part of the septum. Examination of the development of the outflow tract of the hearts of E14.5 Dhrs3−/− embryos revealed that in 3 of 5 embryos the aorta is positioned in the right ventricle adjacent to the pulmonary trunk, resulting in a double-outlet right ventricle (DORV; Fig. 6E). Thus, the only exit for blood from the left ventricle of these Dhrs3−/− embryos is through the incomplete septum and subsequently the right ventricle. In addition to VSD and DORV, these 3 E14.5 Dhrs3−/− embryos examined also display a complete lack of atrial septation (ASD) (Fig. 6F, J). A more severely affected Dhrs3−/− embryo displayed additional developmental delays and cardiac and skeletal malformations.
Figure 6.

Dhrs3−/− mice display abnormalities in heart and craniofacial development. Sagittal sections of E14.5 Dhrs3−/− (A) and WT (B) embryos stained with hematoxylin and eosin (H&E) indicates a hypoplastic jaw and cleft palate in the Dhrs3−/− embryos. The palatal shelf seen in WT embryos (B, leader) is absent in all sections of Dhrs3−/− embryos (A, asterisk). Cleft palate seen in the Dhrs3−/− embryos could be attributed to the failure of the palatal shelves to elevate in Dhrs3−/− embryos (C) vs. WT (G). Transversal sections of the heart of Dhrs3−/− embryos (proceeding caudally D→F) vs. WT embryos (proceeding caudally H→J) reveal several heart abnormalities. A membranous ventricular septal defect (VSD) caused by an incomplete interventricular septum (IVS) between the left ventricle (LV) and right ventricle (RV) can be observed in sections of the hearts of Dhrs3−/− embryos (E, F) in comparison with corresponding sections of hearts of WT embryos (I, J). Double outlet right ventricle (DORV) is evidenced by the observation that both the pulmonary trunk (Pt) and aortic trunk (Ao) connect to the RV of Dhrs3−/− embryos (E, misaligned aorta indicated by red arrow and DORV). In contrast, the Pt and Ao of WT embryos connect to the RV and LV, respectively (D, E vs. H, I). In the case of Dhrs3−/− embryos, DORV is associated with a repositioning of the atrial semilunar valve to the RV [E, I; aortic valve leaflets (AVL)]. Dhrs3−/− embryos also display an atrial septal defect, as indicated by an incomplete septation between the left atrium (LA) and right atrium (RA) (F, red accolade) in comparison with WT atria (J). E, esophagus; MVL, mitral valve leaflet; NS, nasal septum; PVL, pulmonary valve leaflet; SP, septum primum; TVL, tricuspid valve leaflet. Scale bars = 2 mm (A, B); 0.5 mm (C–J). Sections are representative of morphology of ≥3 WT and 3 Dhrs3−/− littermates.
The E14.5 and E17.5 Dhrs3−/− embryos exhibit defects in axial and craniofacial development. The palatal shelves of Dhrs3−/− embryos fail to elevate by E14.5 in contrast to WT littermates, as seen in all five Dhrs3−/− embryos examined (Fig. 6C, G). Instead, the palatal shelves of Dhrs3−/− embryos continue to grow downward toward the tongue. Both E14.5 and E17.5 Dhrs3−/− embryos also display abnormalities in axial skeleton development, manifested as vertebral fusions and delayed ossification. Specifically, analysis of the axial skeleton of Dhrs3−/− embryos revealed a fusion of the posterior arches of the first two cervical vertebrae, the atlas and axis (C1 and C2, respectively) seen in all three embryos examined (Fig. 7A, C vs. B, D). The E17.5 Dhrs3−/− embryos display delayed ossification, particularly noticeable in the parietal bone of the skull and the mandible (Supplemental Fig. S2).
Figure 7.
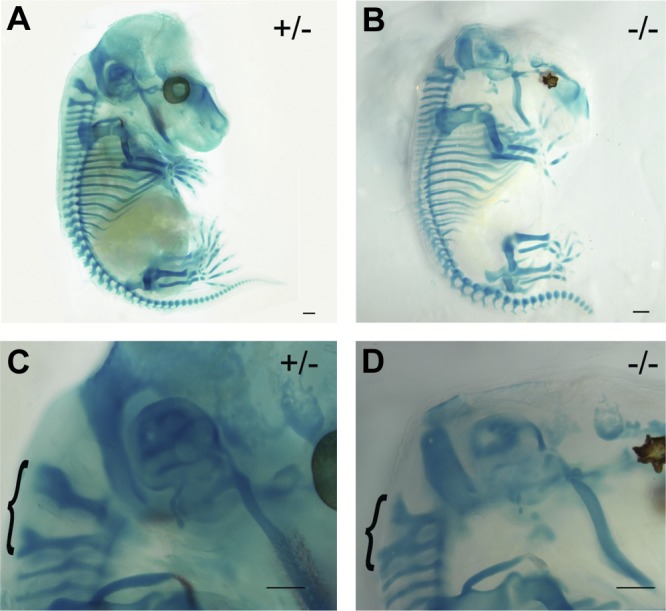
Dhrs3−/− mice display skeletal abnormalities. Skeletal preparations of E14.5 heterozygous (A, C) and Dhrs3−/− (B, D) mice stained with Alcian blue (cartilage) and Alizarin red (bone). Dhrs3−/− embryos display a fusion between the posterior arch of the axis (C2) and atlas (C1) cervical vertebrae, indicated by an accolade (D vs. C). Scale bars = 1 mm. Morphologies are representative of those seen in ≥3 embryos/genotype.
In summary, Dhrs3-deficient mice have disrupted retinoid homeostasis, resulting in alterations in RAR/RXR-based signaling. Consequently, Dhrs3 ablation results in heart septation defects, both VSD and ASD, and defects in the outflow tract formation, DORV, and are nonviable. Dhrs3−/− embryos display additional defects that have also been associated with ATRA embryopathy, such as defects in palatogenesis and vertebral fusions, and delayed ossification. Cumulatively, our findings indicate a critical role for DHRS3 in the metabolism of vitamin A in vivo and demonstrate that DHRS3 is required for proper embryonic development.
DISCUSSION
In the currently accepted model, accumulation of excess ATRA is averted primarily through the actions of CYP26 enzymes (2, 7, 8). We present evidence that retinaldehyde reduction is also an important physiological mechanism to prevent excess ATRA formation. We report that DHRS3 is indispensable for preventing formation of excess levels of ATRA, thereby ensuring proper ATRA-based signaling during development.
Based on its known enzymatic activity, we propose that DHRS3 is responsible for the majority of the retinaldehyde reductase activity during embryogenesis. As a result, the levels of the product of DHRS3, retinol, and its storage form, retinyl esters, are diminished in Dhrs3−/− embryos compared to WT and heterozygous littermates. Conversely, the levels of ATRA are increased in Dhrs3−/− embryos compared to WT littermates. We did not detect any increases in the global levels of retinaldehyde in Dhrs3−/− embryos compared to WT littermates. However, we cannot exclude that retinaldehyde may accumulate transiently in Dhrs3−/− embryos and, thus, play a role in some of the effects of Dhrs3 ablation in an ATRA-independent manner (67, 68). Nevertheless, the majority of the developmental phenotypes observed in Dhrs3−/− embryos are consistent with ATRA embryopathy.
The changes seen in ATRA levels in Dhrs3−/− embryos are similar in magnitude to those seen in other animal models with impaired ATRA synthesis and which also exhibit developmental defects (16, 69). Notably, tissues of Dhrs3−/− embryos show reduced expression of enzymes that contribute to ATRA synthesis, such as Rdh10 and Raldh1-3, and increased expression of ATRA catabolic enzymes, such as Cyp26a1. Thus, despite compensation by enzymes involved in ATRA metabolism and the presence of other potential retinaldehyde reductases, the activity of DHRS3 is fundamentally required for retinoid homeostasis in embryos.
Increased levels of ATRA in Dhrs3−/− embryos lead to increased expression of direct targets of RAR/RXR, such as Hoxa1 and Rarb, and decreased expression of Hoxa13 and Hoxd13. Down-regulation of Hoxa13 and Hoxd13 could be caused by an ATRA-induced proximalizing shift in the positional identity of segments along the limb axis (62). This phenomenon has been previously observed in embryos exposed to excess ATRA as a result of reduced catabolism or exogenous treatment (58, 62, 70, 71). The pattern of expression of the RARE-LacZ transgene in the Dhrs3−/− embryos is generally consistent with the global increases in ATRA levels, showing a consistent expansion of the domains of RARE-LacZ transgene activity at E14.5 on a Dhrs3-deficient background.
DHRS3 is expressed in many embryonic tissues involved in retinoid metabolism (1, 2, 64). The changes observed in ATRA signaling and RAR-dependent patterning in Dhrs3−/− embryos are accompanied by defects in developmental programming, which ultimately result in embryonic lethality. Dhrs3−/− embryos exhibit cardiac septal defects, outflow tract malformations (DORV), cleft palate, and defects and delays in craniofacial and axial skeleton development. Based on the significant changes in retinoid metabolism and the defects in development observed in Dhrs3−/− embryos, our data strongly argues against the existence of enzymes that can successfully compensate for the lack of DHRS3 during embryogenesis.
DHRS3 is a physiologically relevant retinaldehyde reductase that plays an important role in ATRA homeostasis
Based on cumulative studies (22, 24) and our results, we propose that Dhrs3 plays an important role in the regulatory network responsible for the prevention of excess ATRA formation in the embryo, as schematically depicted in Fig. 3C. To ensure homeostasis, ATRA synthetic genes are down-regulated in response to excess ATRA, while those that degrade ATRA or which divert retinol toward an inert storage form are increased in response to ATRA (8, 72).. Here we show that, indeed, the synthetic enzymes, Rdh10, Raldh1, 2, and 3, are down-regulated while the catabolic enzyme Cyp26a1 is up-regulated in the presence of high endogenous levels of ATRA in Dhrs3−/− embryos. ATRA also up-regulates the expression of Dhrs3 itself (21, 22, 24–27), which limits the amount of ATRA by increasing the rate of reduction of all-trans-retinaldehyde to all-trans-retinol. In summary, these results demonstrate that DHRS3 carries out an essential and evolutionarily conserved role in the feedback regulation of ATRA formation.
Developmental consequences of Dhrs3 ablation
Ablation of Dhrs3 leads to defects in cardiovascular, craniofacial and axial skeleton development typically associated with exposure to high levels of ATRA during embryogenesis (73–75). The phenotype of Dhrs3−/− embryos also shares similarities with mouse models that accumulate ATRA as a result of impaired ATRA catabolism, such as mice deficient in Cyp26a1, Cyp26b1, Cyp26c1, or cytochrome P450 oxidoreductase (Por) (reviewed in (7, 76)), including ectopic activation of ATRA reporter genes, eyelid closure defects, vertebral transformations, and craniofacial defects (7, 77, 78). Failure of eyelid fusion in Dhrs3−/− embryos has been observed in animals that have excess ATRA, either as a result of genetic mutations such as those seen in Cyp26b1-deficient mice (58, 77), or in embryos treated with ATRA (74, 79, 80). Similar defects have also been seen in transgenic mouse models that overexpress 3′- Hox genes (81–83). The malformation of the spinous process of C1 in Dhrs3−/− embryos is similar to the one seen in Cyp26a1−/− embryos, which also accumulate ATRA (84). Fusions of cervical vertebrae have also been observed in embryos exposed to high levels of exogenous ATRA (85) and in mouse models that have altered activity or spatial expression pattern of Hox genes (81, 86–88).
Both deficiency and excess of retinol or ATRA during gestation can result in the teratogenic cardiovascular defects observed both in experimental animal models and in patients exposed to ATRA-based therapies or toxic levels of retinol supplements (73–75). ATRA signaling via RAR-RXR plays essential roles in heart anteroposterior (AP) patterning, second heart field formation, and cardiomyocyte differentiation (89–92). RXR signaling is also important in heart development (93). Dhrs3−/− embryos exhibit heart septation defects (ASD, VSD) with misalignment of the outflow tract (DORV); these defects are a likely cause of the late gestational/perinatal lethality. Such malformations have been associated with the teratogenic effects of excess retinol or ATRA administration (94–96) and have also been reported in other genetic models with increased ATRA signaling (97). However, Dhrs3−/− embryos do not display other congenital heart defects associated with the exposure of embryos to high levels of exogenous ATRA such transposition of the great arteries (98–100). The difference between the physiological effects of a sustained increase in the endogenous levels of ATRA vs. those of a single exogenous dose of ATRA could be explained by their different tissue distribution and metabolic kinetics. The heart malformations seen in Dhrs3−/− embryos could be a result of defects in the function of cardiac neural crest-derived cells (NCCs; refs. 101, 102). Notably, Dhrs3 was shown to be expressed in murine NCCs (21). Our studies also indicate that Dhrs3 is expressed in a small distinct population in the heart (Fig. 5D), however, further experiments are necessary to establish the expression and role of Dhrs3 in NCC-derived tissues.
ATRA signaling plays an important role in orofacial development. A striking phenotype of the Dhrs3−/− embryos is the cleft palate as a result of a complete failure of the palatal shelves to elevate. Cleft palate is a common feature of retinoic acid embryopathy (103, 104) and is also observed in mice deficient in Cyp26b1 (105). ATRA affects the expression of the proteoglycan decorin and fibroblast growth factor 10 (FGF10), which play important roles in palatogenesis (105, 106). Interestingly, Dhrs3 is induced by the tumor suppressor p63 (28, 29, 107), and palatogenesis defects are frequently seen in p63-associated disorders such as ectrodactyly-ectodermal dysplasia and cleft syndrome (EEC) (108). It was proposed that lack of induction of Dhrs3 could be responsible for the alteration in the levels of ATRA in response to mutations in p63 (29), and thus play a role in the etiology of p63-associated EEC (104). Our studies indicate that DHRS3 does indeed plays a critical role in ATRA metabolism and that ablation of Dhrs3 leads to palatogenesis defects. However, more studies are required to establish whether the expression or activity of Dhrs3 is altered in p63-associated disorders.
Summary
DHRS3 is a critical enzyme that functions to reduce all-trans-retinaldehyde to all-trans-retinol and to prevent the synthesis of excess ATRA. Dhrs3 ablation leads to increased levels of ATRA and altered ATRA-related signaling, causes late gestation/perinatal death, and is associated with a constellation of defects characteristic of ATRA embryopathy. In summary, these data provide new insights into mechanisms that regulate the endogenous levels of ATRA during development. These studies also set the stage for further investigation into the role and regulation of the expression of Dhrs3 in both embryonic and postnatal tissues.
Supplementary Material
Acknowledgments
The authors thank Drs. Joseph Napoli, Rick Dobrowsky and Jeff Staudinger for valuable suggestions. The authors thank Dr. Janet Rossant (University of Toronto, the Hospital for Sick Children, Toronto, ON, Canada) for the gift of the RARE-LacZ reporter mice and Dr. Fred de Sauvage (Genentech, South San Francisco, CA, USA) and Lexicon Pharmaceuticals (The Woodlands, TX, USA) for sharing the Dhrs3+/− strain. The authors also thank Dr. David Moore, Ms. Heather Shinogle and Dr. Jennifer Hueston for help with microscopy and the use of a dissecting microscope.
This work was supported by new faculty startup funds from the University of Kansas School of Pharmacy (A.R.M.); by grants from the U.S. National Center for Research Resources (NCRR) and National Institute of General Medical Sciences (NIGMS; 5P20RR017708-10 and 8 P20 GM103420-10; A.R.M.); by an award from the General Research Fund from the University of Kansas Center for Research (A.R.M.); by new faculty startup funds from the University of Maryland School of Pharmacy (M.A.K.) and U.S. National Instittue of Allergy and Infectious Diseases contract HHSN272202000046C (M.A.K.); and by the Stowers Institute for Medical Research and the National Institute for Dental and Craniofacial Research (DE016082; P.A.T.).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ADH
- alcohol dehydrogenase
- AKR
- aldo-keto-reductase
- APCI
- atmospheric-pressure chemical ionization
- AP
- anteroposterior
- ASD
- atrial septation defect
- ATRA
- all-trans-retinoic acid
- BCMO1
- carotenoid-15,15′-monooxygenase
- CRABP
- cellular retinoic acid binding protein
- CRBP
- cellular retinol binding protein
- DHRS
- dehydrogenase/reductase superfamily
- DORV
- double-outlet right ventricle
- DR
- direct repeat
- E
- embryonic day
- EEC
- ectrodactyly-ectodermal dysplasia and cleft syndrome
- ES
- embryonic stem
- HPLC-UV
- high performance liquid chromatography with UV detection
- LacZ
- β-galactosidase
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- LRAT
- lecithin:retinol acyltransferase
- MS
- mass spectrometry
- MO
- morpholino
- NCC
- neural crest-derived cell
- P
- postnatal day
- qRT-PCR
- quantitative RT-PCR
- RALDH
- retinaldehyde dehydrogenase
- RAR
- retinoic acid receptor
- RARE
- retinoic acid response element
- RDH
- retinol dehydrogenase
- RXR
- retinoid X receptor
- SDR
- short-chain dehydrogenase/reductase
- STRA6
- stimulated by retinoic acid 6
- VAS
- vitamin A-sufficient
- VSD
- ventricular septation defect
- WT
- wild type
REFERENCES
- 1. Clagett-Dame M., Knutson D. (2011) Vitamin A in reproduction and development. Nutrients 3, 385–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rhinn M., Dolle P. (2012) Retinoic acid signalling during development. Development 139, 843–858 [DOI] [PubMed] [Google Scholar]
- 3. Leid M., Kastner P., Lyons R., Nakshatri H., Saunders M., Zacharewski T., Chen J. Y., Staub A., Garnier J. M., Mader S., Chambon P. (1992) Purification, cloning, and RXR identity of the HELA-cell factor with which RAR or TR heterodimerizes to bind target sequences efficiently. Cell 68, 377–395 [DOI] [PubMed] [Google Scholar]
- 4. Kliewer S. A., Umesono K., Mangelsdorf D. J., Evans R. M. (1992) Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature 355, 446–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kastner P., Mark M., Ghyselinck N., Krezel W., Dupe V., Grondona J. M., Chambon P. (1997) Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development 124, 313–326 [DOI] [PubMed] [Google Scholar]
- 6. Rochette-Egly C., Germain P. (2009) Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl. Recept. Signal. 7, e005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pennimpede T., Cameron D. A., MacLean G. A., Li H., Abu-Abed S., Petkovich M. (2010) The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A Clin. Mol. Teratol. 88, 883–894 [DOI] [PubMed] [Google Scholar]
- 8. Ross A. C., Zolfaghari R. (2011) Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 31, 65–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Napoli J. L. (2012) Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta. 1821, 152–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar S., Sandell L. L., Trainor P. A., Koentgen F., Duester G. (2012) Alcohol and aldehyde dehydrogenases: retinoid metabolic effects in mouse knockout models. Biochim. Biophys. Acta 1821, 198–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kedishvili N. Y. (2013) Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 54, 1744–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sandell L. L., Sanderson B. W., Moiseyev G., Johnson T., Mushegian A., Young K., Rey J. P., Ma J. X., Staehling-Hampton K., Trainor P. A. (2007) RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 21, 1113–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cunningham T. J., Chatzi C., Sandell L. L., Trainor P. A., Duester G. (2011) Rdh10 mutants deficient in limb field retinoic acid signaling exhibit normal limb patterning but display interdigital webbing. Dev. Dyn. 240, 1142–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Farjo K. M., Moiseyev G., Nikolaeva O., Sandell L. L., Trainor P. A., Ma J. X. (2011) RDH10 is the primary enzyme responsible for the first step of embryonic vitamin A metabolism and retinoic acid synthesis. Dev. Biol. 357, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rhinn M., Schuhbaur B., Niederreither K., Dolle P. (2011) Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. U. S. A. 108, 16687–16692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ashique A. M., May S. R., Kane M. A., Folias A. E., Phamluong K., Choe Y., Napoli J. L., Peterson A. S. (2012) Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling. Genesis 50, 415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sandell L. L., Lynn M. L., Inman K. E., McDowell W., Trainor P. A. (2012) RDH10 oxidation of vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PloS One 7, e30698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Porte S., Xavier Ruiz F., Gimenez J., Molist I., Alvarez S., Dominguez M., Alvarez R., de Lera A. R., Pares X., Farres J. (2012) Aldo-keto reductases in retinoid metabolism: search for substrate specificity and inhibitor selectivity. Chem. Biol. Interact. 202, 186–194 [DOI] [PubMed] [Google Scholar]
- 19. Kiser P. D., Golczak M., Maeda A., Palczewski K. (2012) Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta 1821, 137–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haeseleer F., Huang J., Lebioda L., Saari J., Palczewski K. (1998) Molecular characterization of a novel short-chain dehydrogenase/reductase that reduces all-trans-retinal. J. Biol. Chem. 273, 21790–21799 [DOI] [PubMed] [Google Scholar]
- 21. Williams S. S., Mear J. P., Liang H.-C., Potter S. S., Aronow B. J., Colbert M. C. (2004) Large-scale reprogramming of cranial neural crest gene expression by retinoic acid exposure. Physiol. Genomics 19, 184–197 [DOI] [PubMed] [Google Scholar]
- 22. Feng L., Hernandez R. E., Waxman J. S., Yelon D., Moens C. B. (2010) Dhrs3a regulates retinoic acid biosynthesis through a feedback inhibition mechanism. Dev. Biol. 338, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kam R. K., Chen Y., Chan S. O., Chan W. Y., Dawid I. B., Zhao H. (2010) Developmental expression of Xenopus short-chain dehydrogenase/reductase 3. Int. J. Dev. Biol. 54, 1355–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zolfaghari R., Chen Q., Ross A. C. (2012) DHRS3, a retinal reductase, is differentially regulated by retinoic acid and lipopolysaccharide-induced inflammation in THP-1 cells and rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G578–G588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cerignoli F., Guo X., Cardinali B., Rinaldi C., Casaletto J., Frati L., Screpanti I., Gudas L. J., Gulino A., Thiele C. J., Giannini G. (2002) retSDR1, a short-chain retinol dehydrogenase/reductase, is retinoic acid-inducible and frequently deleted in human neuroblastoma cell lines. Cancer Res. 62, 1196–1204 [PubMed] [Google Scholar]
- 26. Mou C., Pitel F., Gourichon D., Vignoles F., Tzika A., Tato P., Yu L., Burt D. W., Bed'hom B., Tixier-Boichard M., Painter K. J., Headon D. J. (2011) Cryptic patterning of avian skin confers a developmental facility for loss of neck feathering. PLoS Biol. 9, e1001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wong Y. F., Wilson P. D., Unwin R. J., Norman J. T., Arno M., Hendry B. M., Xu Q. (2012) Retinoic acid receptor-dependent, cell-autonomous, endogenous retinoic acid signaling and its target genes in mouse collecting duct cells. PloS One 7, e45725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smeenk L., van Heeringen S. J., Koeppel M., van Driel M. A., Bartels S. J., Akkers R. C., Denissov S., Stunnenberg H. G., Lohrum M. (2008) Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 36, 3639–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kirschner R. D., Rother K., Muller G. A., Engeland K. (2010) The retinal dehydrogenase/reductase retSDR1/DHRS3 gene is activated by p53 and p63 but not by mutants derived from tumors or EEC/ADULT malformation syndromes. Cell Cycle 9, 2177–2188 [DOI] [PubMed] [Google Scholar]
- 30. Deisenroth C., Itahana Y., Tollini L., Jin A., Zhang Y. (2011) p53-Inducible DHRS3 is an endoplasmic reticulum protein associated with lipid droplet accumulation. J. Biol. Chem. 286, 28343–28356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y., Zhang Y., Hill J., Kim H. T., Shen Q., Bissonnette R. P., Lamph W. W., Brown P. H. (2008) The rexinoid, bexarotene, prevents the development of premalignant lesions in MMTV-erbB2 mice. Br. J. Cancer 98, 1380–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szatmari I., Pap A., Ruhl R., Ma J. X., Illarionov P. A., Besra G. S., Rajnavolgyi E., Dezso B., Nagy L. (2006) PPARgamma controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J. Exp. Med. 203, 2351–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Costaridis P., Horton C., Zeitlinger J., Holder N., Maden M. (1996) Endogenous retinoids in the zebrafish embryo and adult. Dev. Dyn. 205, 41–51 [DOI] [PubMed] [Google Scholar]
- 34. Irie T., Seki T. (2002) Retinoid composition and retinal localization in the eggs of teleost fishes. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 131, 209–219 [DOI] [PubMed] [Google Scholar]
- 35. Levi L., Ziv T., Admon A., Levavi-Sivan B., Lubzens E. (2012) Insight into molecular pathways of retinal metabolism, associated with vitellogenesis in zebrafish. Am. J. Physiol. Endocrinol. Metab. 302, E626–644 [DOI] [PubMed] [Google Scholar]
- 36. Tang T., Li L., Tang J., Li Y., Lin W. Y., Martin F., Grant D., Solloway M., Parker L., Ye W., Forrest W., Ghilardi N., Oravecz T., Platt K. A., Rice D. S., Hansen G. M., Abuin A., Eberhart D. E., Godowski P., Holt K. H., Peterson A., Zambrowicz B. P., de Sauvage F. J. (2010) A mouse knockout library for secreted and transmembrane proteins. Nat. Biotechnol. 28, 749–755 [DOI] [PubMed] [Google Scholar]
- 37. Rao S. T., Rossmann M. G. (1973) Comparison of super-secondary structures in proteins. J. Mol. Biol. 76, 241–256 [DOI] [PubMed] [Google Scholar]
- 38. Rossant J., Zirngibl R., Cado D., Shago M., Giguere V. (1991) Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 5, 1333–1344 [DOI] [PubMed] [Google Scholar]
- 39. De The H., Vivanco-Ruiz M. M., Tiollais P., Stunnenberg H., Dejean A. (1990) Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 343, 177–180 [DOI] [PubMed] [Google Scholar]
- 40. Reeves P. G. (1997) Components of the AIN-93 diets as improvements in the AIN-76A diet. J. Nutr. 127, 838S–841S [DOI] [PubMed] [Google Scholar]
- 41. Moise A. R., Kuksa V., Imanishi Y., Palczewski K. (2004) Identification of all-trans-retinol: all-trans-13,14-dihydroretinol saturase. J. Biol. Chem. 279, 50230–50242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kane M. A., Chen N., Sparks S., Napoli J. L. (2005) Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem. J. 388, 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moise A. R., Kuksa V., Blaner W. S., Baehr W., Palczewski K. (2005) Metabolism and transactivation activity of 13,14-dihydroretinoic acid. J. Biol. Chem. 280, 27815–27825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kane M. A., Folias A. E., Napoli J. L. (2008) HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Anal. Biochem. 378, 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kane M. A., Folias A. E., Wang C., Napoli J. L. (2008) Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal. Chem. 80, 1702–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moise A. R., Lobo G. P., Erokwu B., Wilson D. L., Peck D., Alvarez S., Dominguez M., Alvarez R., Flask C. A., de Lera A. R., von Lintig J., Palczewski K. (2010) Increased adiposity in the retinol saturase-knockout mouse. FASEB J. 24, 1261–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kane M. A., Napoli J. L. (2010) Quantification of endogenous retinoids. Methods Mol. Biol. 652, 1–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nagy A. (2003) Manipulating the Mouse Embryo: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA [Google Scholar]
- 49. Fischer A. H., Jacobson K. A., Rose J., Zeller R. (2008) Hematoxylin and eosin staining of tissue and cell sections. Cold Spring Harb. Protoc. 2008, pdb.prot4986 [DOI] [PubMed] [Google Scholar]
- 50. Zhang M., Hu P., Krois C. R., Kane M. A., Napoli J. L. (2007) Altered vitamin A homeostasis and increased size and adiposity in the rdh1-null mouse. FASEB J. 21, 2886–2896 [DOI] [PubMed] [Google Scholar]
- 51. Ross A. C. (2010) Diet in vitamin A research. Methods Mol. Biol. 652, 295–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Niederreither K., Subbarayan V., Dolle P., Chambon P. (1999) Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 21, 444–448 [DOI] [PubMed] [Google Scholar]
- 53. Fan X., Molotkov A., Manabe S., Donmoyer C. M., Deltour L., Foglio M. H., Cuenca A. E., Blaner W. S., Lipton S. A., Duester G. (2003) Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell. Biol. 23, 4637–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dupe V., Matt N., Garnier J. M., Chambon P., Mark M., Ghyselinck N. B. (2003) A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. U. S. A. 100, 14036–14041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Loudig O., Babichuk C., White J., Abu-Abed S., Mueller C., Petkovich M. (2000) Cytochrome P450RAI(CYP26) promoter: a distinct composite retinoic acid response element underlies the complex regulation of retinoic acid metabolism. Mol. Endocrinol. 14, 1483–1497 [DOI] [PubMed] [Google Scholar]
- 56. Zhang Y., Zolfaghari R., Ross A. C. (2010) Multiple retinoic acid response elements cooperate to enhance the inducibility of CYP26A1 gene expression in liver. Gene 464, 32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tahayato A., Dolle P., Petkovich M. (2003) Cyp26C1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first branchial arch and tooth buds during murine development. Gene Expr. Patterns 3, 449–454 [DOI] [PubMed] [Google Scholar]
- 58. Yashiro K., Zhao X., Uehara M., Yamashita K., Nishijima M., Nishino J., Saijoh Y., Sakai Y., Hamada H. (2004) Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev. Cell 6, 411–422 [DOI] [PubMed] [Google Scholar]
- 59. Uehara M., Yashiro K., Mamiya S., Nishino J., Chambon P., Dolle P., Sakai Y. (2007) CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev. Biol. 302, 399–411 [DOI] [PubMed] [Google Scholar]
- 60. O'Byrne S. M., Blaner W. S. (2013) Retinol and retinyl esters: biochemistry and physiology. J. Lipid Res 54, 1731–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dupe V., Davenne M., Brocard J., Dolle P., Mark M., Dierich A., Chambon P., Rijli F. M. (1997) In vivo functional analysis of the Hoxa-1 3′ retinoic acid response element (3′RARE). Development 124, 399–410 [DOI] [PubMed] [Google Scholar]
- 62. Gardiner D. M., Blumberg B., Komine Y., Bryant S. V. (1995) Regulation of HoxA expression in developing and regenerating axolotl limbs. Development 121, 1731–1741 [DOI] [PubMed] [Google Scholar]
- 63. Sucov H. M., Murakami K. K., Evans R. M. (1990) Characterization of an autoregulated response element in the mouse retinoic acid receptor type beta gene. Proc. Natl. Acad. Sci. U. S. A. 87, 5392–5396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Spiegler E., Kim Y. K., Wassef L., Shete V., Quadro L. (2011) Maternal-fetal transfer and metabolism of vitamin A and its precursor beta-carotene in the developing tissues. Biochim. Biophys. Acta 1821, 88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Papaioannou V. E., Behringer R. (2005) Mouse Phenotypes: A Handbook Of Mutation Analysis, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA [Google Scholar]
- 66. Savolainen S. M., Foley J. F., Elmore S. A. (2009) Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol. Pathol. 37, 395–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ziouzenkova O., Orasanu G., Sharlach M., Akiyama T. E., Berger J. P., Viereck J., Hamilton J. A., Tang G., Dolnikowski G. G., Vogel S., Duester G., Plutzky J. (2007) Retinaldehyde represses adipogenesis and diet-induced obesity. Nat. Med. 13, 695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kiefer F. W., Vernochet C., O'Brien P., Spoerl S., Brown J. D., Nallamshetty S., Zeyda M., Stulnig T. M., Cohen D. E., Kahn C. R., Plutzky J. (2012) Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat. Med. 18, 918–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim Y. K., Wassef L., Chung S., Jiang H., Wyss A., Blaner W. S., Quadro L. (2011) beta-Carotene and its cleavage enzyme beta-carotene-15,15′-oxygenase (CMOI) affect retinoid metabolism in developing tissues. FASEB J. 25, 1641–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ribes V., Otto D. M., Dickmann L., Schmidt K., Schuhbaur B., Henderson C., Blomhoff R., Wolf C. R., Tickle C., Dolle P. (2007) Rescue of cytochrome P450 oxidoreductase (Por) mouse mutants reveals functions in vasculogenesis, brain and limb patterning linked to retinoic acid homeostasis. Dev. Biol. 303, 66–81 [DOI] [PubMed] [Google Scholar]
- 71. Shimizu H., Lee G. S., Beedanagari S. R., Collins M. D. (2007) Altered localization of gene expression in both ectoderm and mesoderm is associated with a murine strain difference in retinoic acid–induced forelimb ectrodactyly. Birth Defects Res. Clin. Mol. Teratol. 79, 465–482 [DOI] [PubMed] [Google Scholar]
- 72. Zolfaghari R., Ross A. C. (2009) An essential set of basic DNA response elements is required for receptor-dependent transcription of the lecithin: retinol acyltransferase (Lrat) gene. Arch. Biochem. Biophys. 489, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lammer E. J., Chen D. T., Hoar R. M., Agnish N. D., Benke P. J., Braun J. T., Curry C. J., Fernhoff P. M., Grix A. W., Jr., Lott I. T., Richard J. M, Sun S. C. (1985) Retinoic acid embryopathy. N. Engl. J. Med. 313, 837–841 [DOI] [PubMed] [Google Scholar]
- 74. Rothman K. J., Moore L. L., Singer M. R., Nguyen U. S., Mannino S., Milunsky A. (1995) Teratogenicity of high vitamin A intake. N. Engl. J. Med. 333, 1369–1373 [DOI] [PubMed] [Google Scholar]
- 75. Collins M. D., Mao G. E. (1999) Teratology of retinoids. Annu. Rev. Pharmacol. Toxicol. 39, 399–430 [DOI] [PubMed] [Google Scholar]
- 76. White R. J., Schilling T. F. (2008) How degrading: Cyp26s in hindbrain development. Dev. Dyn. 237, 2775–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sakai Y., Meno C., Fujii H., Nishino J., Shiratori H., Saijoh Y., Rossant J., Hamada H. (2001) The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 15, 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Maclean G., Dolle P., Petkovich M. (2009) Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning. Dev. Dyn. 238, 732–745 [DOI] [PubMed] [Google Scholar]
- 79. Cohlan S. Q. (1953) Excessive intake of vitamin A as a cause of congenital anomalies in the rat. Science 117, 535–536 [DOI] [PubMed] [Google Scholar]
- 80. Penniston K. L., Tanumihardjo S. A. (2006) The acute and chronic toxic effects of vitamin A. Am. J. Clin. Nutr. 83, 191–201 [DOI] [PubMed] [Google Scholar]
- 81. Kessel M., Balling R., Gruss P. (1990) Variations of cervical vertebrae after expression of a Hox-1.1 transgene in mice. Cell 61, 301–308 [DOI] [PubMed] [Google Scholar]
- 82. Kaur S., Singh G., Stock J. L., Schreiner C. M., Kier A. B., Yager K. L., Mucenski M. L., Scott W. J., Jr., Potter S. S. (1992) Dominant mutation of the murine Hox-2.2 gene results in developmental abnormalities. J. Exp. Zool. 264, 323–336 [DOI] [PubMed] [Google Scholar]
- 83. McLain K., Schreiner C., Yager K. L., Stock J. L., Potter S. S. (1992) Ectopic expression of Hox-2.3 induces craniofacial and skeletal malformations in transgenic mice. Mech. Dev. 39, 3–16 [DOI] [PubMed] [Google Scholar]
- 84. Abu-Abed S., Dolle P., Metzger D., Beckett B., Chambon P., Petkovich M. (2001) The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 15, 226–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kessel M., Gruss P. (1991) Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell 67, 89–104 [DOI] [PubMed] [Google Scholar]
- 86. Condie B. G., Capecchi M. R. (1993) Mice homozygous for a targeted disruption of Hoxd-3 (Hox-4.1) exhibit anterior transformations of the first and second cervical-vertebrae, the atlas and the axis. Development 119, 579–595 [DOI] [PubMed] [Google Scholar]
- 87. Condie B. G., Capecchi M. R. (1994) Mice with targeted disruptions in the paralogous genes Hoxa-3 and Hord-3 reveal synergistic interactions. Nature 370, 304–307 [DOI] [PubMed] [Google Scholar]
- 88. Horan G. S. B., Kovacs E. N., Behringer R. R., Featherstone M. S. (1995) Mutations in paralogous Hox genes result in overlapping homeotic transformations of the axial skeleton - evidence for unique and redundant function. Dev. Biol. 169, 359–372 [DOI] [PubMed] [Google Scholar]
- 89. Niederreither K., Vermot J., Messaddeq N., Schuhbaur B., Chambon P., Dolle P. (2001) Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 128, 1019–1031 [DOI] [PubMed] [Google Scholar]
- 90. Ryckebusch L., Wang Z., Bertrand N., Lin S.-C., Chi X., Schwartz R., Zaffran S. p., Niederreither K. (2008) Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. U. S. A. 105, 2913–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sirbu I. O., Zhao X., Duester G. (2008) Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev. Dyn. 237, 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lin S. C., Dolle P., Ryckebusch L., Noseda M., Zaffran S., Schneider M. D., Niederreither K. (2010) Endogenous retinoic acid regulates cardiac progenitor differentiation. Proc. Natl. Acad. Sci. U. S. A. 107, 9234–9239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sucov H. M., Dyson E., Gumeringer C. L., Price J., Chien K. R., Evans R. M. (1994) RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 8, 1007–1018 [DOI] [PubMed] [Google Scholar]
- 94. Wilson J. G., Roth C. B., Warkany J. (1953) An analysis of the syndrome of malformations induced by maternal vitamin A deficiency - effects of restoration of vitamin-A at various times during gestation. Am. J. Anat. 92, 189–217 [DOI] [PubMed] [Google Scholar]
- 95. Davis L. A., Sadler T. W. (1981) Effects of vitamin A on endocardial cushion development in the mouse heart. Teratology 24, 139–148 [DOI] [PubMed] [Google Scholar]
- 96. Kolodzinska A., Heleniak A., Ratajska A. (2013) Retinoic acid-induced ventricular non-compacted cardiomyopathy in mice. Kardiol. Pol. 71, 447–452 [DOI] [PubMed] [Google Scholar]
- 97. Guris D. L., Duester G., Papaioannou V. E., Imamoto A. (2006) Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev. Cell 10, 81–92 [DOI] [PubMed] [Google Scholar]
- 98. Yasui H., Nakazawa M., Morishima M., Miyagawa-Tomita S., Momma K. (1995) Morphological observations on the pathogenetic process of transposition of the great arteries induced by retinoic acid in mice. Circulation 91, 2478–2486 [DOI] [PubMed] [Google Scholar]
- 99. Nakajima Y., Morishima M., Nakazawa M., Momma K. (1996) Inhibition of outflow cushion mesenchyme formation in retinoic acid-induced complete transposition of the great arteries. Cardiovasc. Res. 31, E77–E85 [PubMed] [Google Scholar]
- 100. Yasui H., Nakazawa M., Morishima M., Ando M., Takao A., Aikawa E. (1997) Cardiac outflow tract septation process in the mouse model of transposition of the great arteries. Teratology 55, 353–363 [DOI] [PubMed] [Google Scholar]
- 101. Inman K. E., Ezin M., Bronner-Fraser M., Trainor P. A. (2010) Cardiac neural crest cells. In Heart Development and Regeneration, Vol. 1 (Rosenthal N., Harvey R. P., eds) pp. 417–434, Elsevier, Amsterdam [Google Scholar]
- 102. Keyte A., Hutson M. R. (2012) The neural crest in cardiac congenital anomalies. Differentiation 84, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Abbott B. D., Harris M. W., Birnbaum L. S. (1989) Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology 40, 533–553 [DOI] [PubMed] [Google Scholar]
- 104. Ackermans M. M. G., Zhou H., Carels C. E. L., Wagener F. A., Von den Hoff J. W. (2011) Vitamin A and clefting: putative biological mechanisms. Nutr. Rev. 69, 613–624 [DOI] [PubMed] [Google Scholar]
- 105. Okano J., Kimura W., Papaionnou V. E., Miura N., Yamada G., Shiota K., Sakai Y. (2012) The regulation of endogenous retinoic acid level through CYP26B1 is required for elevation of palatal shelves. Dev. Dyn. 241, 1744–1756 [DOI] [PubMed] [Google Scholar]
- 106. Zhang Y., Mori T., Iseki K., Hagino S., Takaki H., Takeuchi M., Hikake T., Tase C., Murakawa M., Yokoya S., Wanaka A. (2003) Differential expression of decorin and biglycan genes during palatogenesis in normal and retinoic acid–treated mice. Dev. Dyn. 226, 618–626 [DOI] [PubMed] [Google Scholar]
- 107. Zhou H. Q., van Bokhoven H. (2010) Regulation of vitamin metabolism by p53 and p63 in development and cancer. Cell Cycle 9, 2709–2709 [PubMed] [Google Scholar]
- 108. Rinne T., Brunner H. G., van Bokhoven H. (2007) p63-associated disorders. Cell Cycle 6, 262–268 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.