

Abstract
Summary: Studies of the human microbiome, and microbial community ecology in general, have blossomed of late and are now a burgeoning source of exciting research findings. Along with the advent of next-generation sequencing platforms, which have dramatically increased the scope of microbiome-related projects, several high-performance sequence analysis pipelines (e.g. QIIME, MOTHUR, VAMPS) are now available to investigators for microbiome analysis. The subject of our manuscript, the graphical user interface-based Explicet software package, fills a previously unmet need for a robust, yet intuitive means of integrating the outputs of the software pipelines with user-specified metadata and then visualizing the combined data.
Availability and Implementation: Explicet is implemented in C++ via the Qt framework and supported in native code on all major operating systems (Windows, Macintosh, Linux). The source code, documents and tutorials are freely available under an open-source license at www.explicet.org.
Contact: daniel.frank@ucdenver.edu
1 INTRODUCTION
The scope of microbial ecology studies has increased significantly over the past decade, driven in part by the realization that microbial communities are critical mediators of human and ecosystem health. Furthermore, a 10 000-fold decrease in the cost of DNA sequencing (www.genome.gov/sequencingcosts) has resulted in a commensurate increase in the volume of data to analyze. In response, many software systems (e.g. 16S pipelines) have been created to deal with the resulting glut of data (Frank 2008, 2009; Frank and Robertson, 2011; Giongo et al., 2010; Hartman et al., 2010; Kuczynski et al., 2012; Schloss et al., 2009; vamps.mbl.edu). At present, software tools for microbiome analysis often require some sophistication in computer skills to perform even basic analyses. Because tools using graphical user interfaces provide more facile interactivity for data exploration, we have developed Explicet (Latin for ‘explanation’), which consumes the output from existing sequence analysis pipelines and greatly expedites metadata-driven management, analysis and visualization of sequence classification results.
2 OVERVIEW OF EXPLICET
Explicet is a scalable (laptop to server) open-source software package implemented in C++ using the Qt cross platform application framework (qt-project.org) and the plotting methods from Qwt (qwt.sourceforge.net). As such, the software is provided as native code on multiple operating systems (Windows, Macintosh, Linux). Performance of the primary display window and file size are O [number of samples X number of operational taxonomic units (OTUs)], yielding good performance with modest compute capability.
Explicet is compatible with any upstream sequence analysis pipeline that produces OTU table data, which Explicet displays in a spreadsheet-like window (Fig. 1A). Metadata, for instance environmental or clinical attributes, are imported through tab-delimited or comma separated value formatted flat files. OTU data and metadata are managed within a single file as an Explicet Project. Projects may contain one or more user-defined Workspaces, which allow bioinformatic experiments to be performed on metadata-derived subsets of Project samples or OTUs (or both), (e.g. samples from males aged >40 years and only the OTUs belonging to the phylum Firmicutes). Workspaces store user-generated Figures displaying descriptive data or Explicet computations. Workspaces and Figures are persistent and available for user reference or revision during the analysis life cycle (Fig. 1B).
Fig. 1.
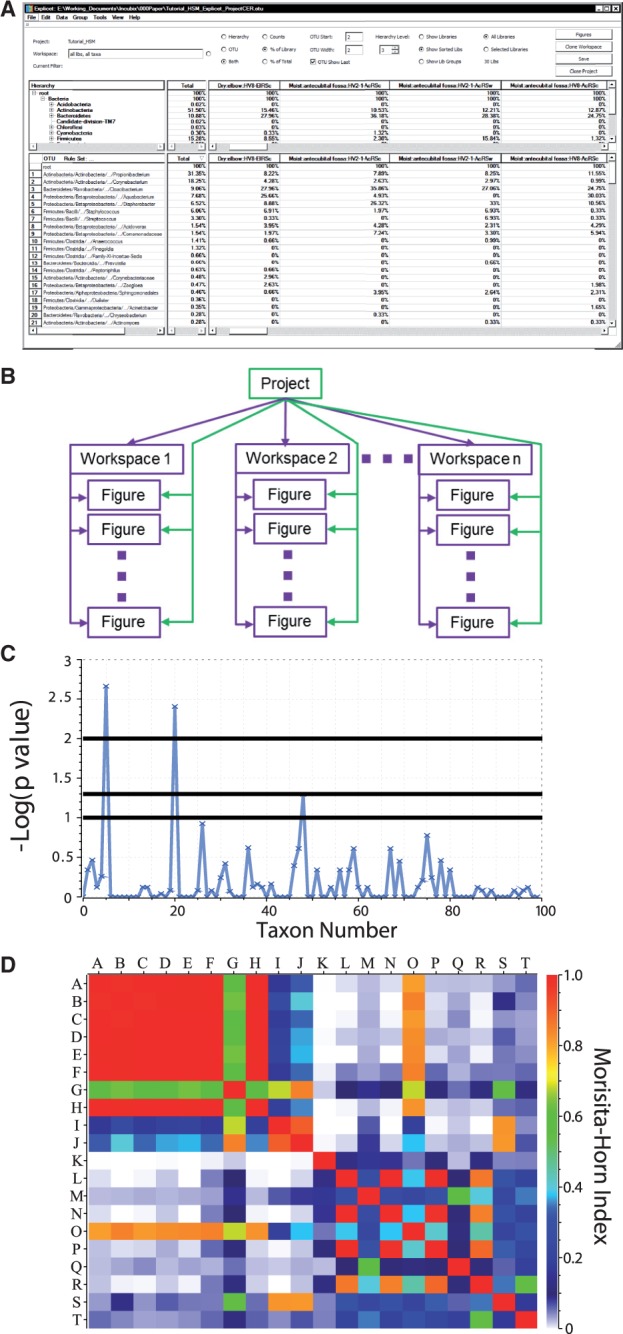
Explicet windows displaying a portion of a human skin microbiome dataset (Grice et al., 2009). (A) Explicet workspace window showing the row/column data displays characteristics of an OTU file. The upper pane displays the taxonomic hierarchy of the dataset and individual samples. The lower pane shows the full OTU table perspective on the data. (B) Relationships between data management entities in Explicet. (C) Manhattan plot of a Two-Part (Wagner et al., 2011) non-parametric statistical comparison of samples, indicating that two taxa differ significantly between sample types. (D) Heatmap of Morisita–Horn similarity between samples
Explicet provides several tools for metadata-driven analysis of microbiome datasets. Basic distributions of OTUs in samples or categories of samples (defined by user-supplied metadata) can be displayed via stacked bar plots, pie charts or heatmaps. OTUs that differ in abundance and/or prevalence between user-defined groups of samples can be identified by Wilcoxon, two-proportion and two-part statistical tests (Wagner et al., 2011) and graphically portrayed as Manhattan plots (Fig. 1C). Ecological alpha and beta diversity indices (Schloss and Handelsman, 2007) can be calculated through resampling and rarefaction; results are presented as line plots, heatmaps and data tables (Fig. 1D). All tabulated data are exportable as delimited text files for import into other tools (e.g. R or SAS), whereas Figures are exported as editable PDFs.
3 CONCLUSIONS
Explicet fills a critical need for a robust, yet intuitive means of integrating the outputs of sequence processing and classification pipelines with user-specified metadata and then visualizing the combined data. Although designed initially for 16S analysis, Explicet can be applied to any dataset organized through a hierarchical classification scheme (e.g. other genes or metagenomes). For bioinformaticians, Explicet offers a powerful means of rapidly evaluating and segmenting large microbial ecology datasets. For non-bioinformaticians, a familiar easy-to-use mouse-driven software application softens the training focus away from software logistics and toward the microbiology and statistical methods that underpin microbial ecology. Practical use of Explicet in several laboratory settings has been found to reduce the requirements for personnel with bioinformatics/computational expertise (Hara et al., 2013; Markle et al., 2013; Robertson et al., 2013). Consequently, the analysis of complex microbiome datasets is now much more accessible to the growing number of investigators who wish to bring a microbial ecological perspective to their fields of interest, especially in medicine and the environmental sciences.
ACKNOWLEDGEMENT
The authors thank Mr. Bruce Holland of Incubix Inc. (Boulder, CO, USA) for his generous support for this project.
Funding: National Institutes of Health (HG005964 to D.N.F.) and the Alfred P. Sloan Foundation (Microbiology of the Built Environment to N.R.P.).
Conflict of Interest: none declared.
REFERENCES
- Frank DN. XplorSeq: a software environment for integrated management and phylogenetic analysis of metagenomic sequence data. BMC Bioinformatics. 2008;9:420. doi: 10.1186/1471-2105-9-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN. BARCRAWL and BARTAB: software tools for the design and implementation of barcoded primers for highly multiplexed DNA sequencing. BMC Bioinformatics. 2009;10:362. doi: 10.1186/1471-2105-10-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, Robertson CE. The Phyloware Project: a software framework for phylogenomic virtue. In: de Bruijn FJ, editor. Handbook of Molecular Microbial Ecology I: Metagenomics and Complementary Approaches. Hoboken, New Jersey, USA: John Wiley & Sons, Inc; 2011. [Google Scholar]
- Giongo A, et al. PANGEA: pipeline for analysis of next generation amplicons. ISME J. 2010;4:852–861. doi: 10.1038/ismej.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara N, et al. The role of the intestinal microbiota in type 1 diabetes. Clin. Immunol. 2013;146:112–119. doi: 10.1016/j.clim.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Hartman AL, et al. Introducing W.A.T.E.R.S.: a workflow for the alignment, taxonomy, and ecology of ribosomal sequences. BMC Bioinformatics. 2010;11:317. doi: 10.1186/1471-2105-11-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J, et al. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Microbiol. 2012 doi: 10.1002/9780471729259.mc01e05s27. Chapter 1, Unit 1E 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markle JG, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339:1084–1088. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- Robertson CE, et al. Culture-independent analysis of aerosol microbiology in a metropolitan subway system. Appl. Environ. Microbiol. 2013;79:3485–3493. doi: 10.1128/AEM.00331-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J. The last word: books as a statistical metaphor for microbial communities. Annu. Rev. Microbiol. 2007;61:23–34. doi: 10.1146/annurev.micro.61.011507.151712. [DOI] [PubMed] [Google Scholar]
- Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communitie. Appl. Environ. Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BD, et al. Application of two-part statistics for comparison of sequence variant counts. PLoS One. 2011;6:e20296. doi: 10.1371/journal.pone.0020296. [DOI] [PMC free article] [PubMed] [Google Scholar]