


Abstract
WRKY transcription factors constitute a large protein family in plants that is involved in the regulation of developmental processes and responses to biotic or abiotic stimuli. The question arises how stimulus-specific responses are mediated given that the highly conserved WRKY DNA-binding domain (DBD) exclusively recognizes the ‘TTGACY’ W-box consensus. We speculated that the W-box consensus might be more degenerate and yet undetected differences in the W-box consensus of WRKYs of different evolutionary descent exist. The phylogenetic analysis of WRKY DBDs suggests that they evolved from an ancestral group IIc-like WRKY early in the eukaryote lineage. A direct descent of group IIc WRKYs supports a monophyletic origin of all other group II and III WRKYs from group I by loss of an N-terminal DBD. Group I WRKYs are of paraphyletic descent and evolved multiple times independently. By homology modeling, molecular dynamics simulations and in vitro DNA–protein interaction-enzyme-linked immunosorbent assay with AtWRKY50 (IIc), AtWRKY33 (I) and AtWRKY11 (IId) DBDs, we revealed differences in DNA-binding specificities. Our data imply that other components are essentially required besides the W-box-specific binding to DNA to facilitate a stimulus-specific WRKY function.
INTRODUCTION
Changes in the biotic and abiotic environment are sensed and transmitted through various signaling cascades to finally cause gene expression changes in the nucleus. In contrast to animals, sessile organisms such as plants do not have the ability to avoid non-permissive conditions by migration to more favorable sites, but need to specifically respond and adapt by physiological or developmental reprogramming (1). One of the largest classes of transcription factors in plants is the WRKY protein family, which is well known for its engagement in the abiotic and biotic stress response (2–10). Certain WRKY transcription factors also have an important function in the development of seeds or during senescence, in the establishment of cell identities of trichomes or root hairs and in epigenetic processes such as imprinting (11–19).
Since their first discovery in wild oat and sweet potato (20–22), multiple members of WRKY transcription factors have been identified in all existing land plant species (3,7,10,23–31). The number of family members in the different species of land plants varies and increased during the evolution of modern angiosperms and especially in grasses and asterids (10,23,27,30,32–35). It has been proposed that this expansion has co-evolved with the increasing complexity of the body plans and the arms’ race of land plants with pathogens (7,8,25,36,37). Although some WRKY proteins can be large and have a number of additional domains, others consist of only little more than the highly conserved WRKY DNA-binding domain (DBD), which all WRKY transcription factors have in common (3,7,22). All WRKY DBDs are ∼60 amino acids in size and characterized by the conserved heptad WRKYGQK amino acid motif at their N-terminus and a zinc ion chelating finger structure at their C-terminus. The WRKY DBD is responsible for recognition of the cognate ‘TTGACY' DNA-binding motif of the W-box in the promoters of target genes (3,4,22,38–40). Recent analyses have shown that the WRKY DBDs are members of the mammalian GCM (glial cell missing) 1 superfamily of zinc finger transcription factors, which probably evolved from non-catalytic endonuclease DBDs such as transposases (2,25,37,41).
On the basis of the numbers of WRKY DBDs per protein and the type of zinc finger motif consensus, the large family of WRKY transcription factors is divided into three paraphyletic groups and several subgroups (3,7,10). The hallmark of all group I WRKY proteins is the presence of two WRKY DBDs, whereas group II and III WRKY proteins have only a single WRKY DBD (3,7). The N-terminal WRKY DNA-binding domain (nDBD) and the C-terminal WRKY DNA-binding domain (cDBD) display a high degree of structural similarity and must have evolved by a domain duplication event in the early WRKY protein evolution (7,37,42–44). In most cases, both domains share the WRKYGQK amino acid motif and a C-x4–5-C-x22–23-HxH zinc finger consensus. Despite these obvious similarities between the nDBD and the cDBD, the DNA sequences encoding these domains display significant divergence. For example, the coding sequence of group I cDBDs usually harbors an R-type intron between the WRKYGQK motif and the zinc finger, whereas there are no introns in the coding sequences of the nDBDs (3,29). Group I WRKY transcription factors are not restricted to the plant lineage, but are also found in the unicellular eukaryote Giardia lamblia and the slime mold Dictyostelium discoideum (8,10,30). Instead, group II and III proteins appear to be specific for the green plant lineage. Despite the monophyletic nature of all WRKY DBDs, the proteins contained in group II are of noticeable paraphyletic origin and, hence, are more diverse in their DBDs compared with group I DBDs: there are several examples known where the conserved WRKY motif is altered to WRRY, WKRY or WKKY (7,24,25,32). In addition, the R-type intron inside of the WRKY DBD coding sequence differs in its position in some of the group II WRKY clades (3,29). Although Eulgem et al. (3) divided group II WRKY proteins initially into the five subgroups IIa–IIe, phylogenetic analysis of WRKY DBD sequences from Arabidopsis and rice led to reorganization of the group II WRKY proteins and merged four of the clades into only two new sister groups IIa + b and IId + e (7,10). As more accurate genome sequences become available, there is also increasing information on full-length protein sequences of WRKY proteins. Although previous reports could solely focus on the phylogeny of the WRKY DBD, insights gained from full-length WRKY protein evolution analyses describe the descent of other domains besides the WRKY DBD (24,29,33). Subclade-specific functions, differing positions of intron–exon boundaries and divergent domain structures within the group II subgroups are again in favor of the initially defined five WRKY subgroups IIa–IIe (3,24,29,32,45). Almost all group II proteins possess a conserved C-x5-C-x23-HxH zinc finger consensus (3,7). Instead, group III WRKY DBDs differ from group I and II WRKY by the divergent C-x4–7-C-x23-(24–30)-HxC zinc finger motif (3,7). Phylogenetic analyses revealed that group III WRKY DBDs are evolutionary the youngest (3,7,10). Nevertheless, they are present in early land plant species, such as the moss Physcomitrella patens, indicating all WRKY groups evolved prior to the moss lineage ∼500 million years ago.
Despite the differences in their zinc finger motifs, it is accepted that all WRKY DBDs share a high degree in structural similarity (3,7,37). Recently, both a nuclear magnetic resonance (NMR) solution structure of AtWRKY4 cDBD bound to the ‘TTGACY’ W-box consensus and a high-resolution X-ray structure of AtWRKY1 cDBD resolved a β-sheet structure of five (four antiparallel) β-strands that enclose the zinc coordinating finger structure (42–44). For some WRKY proteins, it was shown that the region preceding the second β-strand is important for an interaction with VQ proteins, such as sigma factors (46). The heptad ‘WRKYGQK' sequence forms this second β-strand that contacts the positively charged nucleobases and the negatively charged phosphate backbone (42–44). It protrudes with its rim into the major groove of the DNA (42–44). The nDBD of group I WRKY proteins has never been shown to bind to DNA, although homology modeling of AtWRKY1 nDBD based on the cDBD crystal structure postulated the DNA-binding abilities also for nDBDs (20,39,42,47–49).
Here we discuss a phylogeny of the WRKY DBD that gives evidence for several independent gains and losses of WRKY DBDs throughout evolution that support paraphyletic origins for groups I and II. Furthermore, we have evidence that group IIc WRKY DBDs are direct descendents from an ancestral group IIc-like WRKY DBD that evolved in basal eukaryote lineages. Apart from group IIc, we postulate a monophyletic origin of groups II and III from group I by loss of the nDBD. Interestingly, all WRKYs analyzed so far bound with highest affinity to the W2-box consensus. Therefore, the question arises how stimulus-specific responses are coordinated given that expression patterns and DNA-binding specificities of different WRKY proteins overlap. By DNA–protein interaction (DPI)-enzyme-linked immunosorbent assay (ELISA) screens, we analyzed the variation in WRKY DBD–DNA recognition and were able to assay the binding capabilities of AtWRKY33 c- and nDBD (group I), AtWRKY11 DBD (group IId) and AtWRKY50 DBD (group IIc). These analyses revealed for the first time the postulated group I nDBD DNA-binding ability. Furthermore, we analyzed the WRKY–DNA interaction by homology modeling, molecular dynamics simulations and quantitative DPI-ELISA in detail. AtWRKY11 and AtWRKY50 DBDs significantly differ within the conserved WRKYGQK peptide of the second β-strand of the β-sheet that is protruding into the major groove. We tested the influence of lysine–glutamine exchange on DNA-binding specificities, which led to new conclusions on the WRKY–DNA interface.
MATERIALS AND METHODS
Sequence alignment and phylogeny
Assignment of the WRKY DBDs into different groups and subgroups was performed as was described before (25). To identify putative WRKY proteins from non-plant eukaryotes or basal plant species, we performed the reciprocal-best-hit approach by using the tblastn or blastp at the NCBI (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). Annotated sequence data for 72 Arabidopsis WRKY proteins (AtWRKY) (3), 82 rice WRKY proteins (OsWRKY) (35), 38 WRKY proteins of P. patens (PpWRKY) (26) and 34 WRKY proteins of Selaginella moellendorffii (SmWRKY) (50) were retrieved from NCBI. To achieve a better separation of the different WRKY clades, to counteract possible long-branch attraction effects of possible fast evolving WRKY protein sequences and to generate a robust root for each of the clades as well as the entire phylogenetic tree, we included several basal WRKY DBD sequences and possible out-groups from diverse phyla: G. lamblia (GlambliaWRKY) [GenBank: XM_765980], D. discoideum (DdiscoideumWRKY) [GenBank: XM_638694], Homo sapiens CRAa FLYWCH-type zinc finger 1 [GenBank: EAW85450], Homo sapiens GCMa [GenBank: BAA13651], Aspergillus flavus NRRL3357 [GenBank: XM_002380447], Cyanidioschyzon merolae [GenBank: AP006502], Ostreococcus lucimarinus CCE9901 [GenBank: XP_001420519], Ostreococcus tauri [GenBank: XP_003080785; GenBank: XP_003080527], Micromonas RCC299 (MICPUN_61119) [GenBank: XP_002504180], Micromonas pusilla (MipuWRKY) [GenBank: XP_002509266; GenBank: XP_003061495; GenBank: XP_003054981], Chlamydomonas reinhardtii (ChrhWRKY) [GenBank: BQ821537], Tetrapisispora blattae (TetrapisisporaWRKY) [GenBank: HE806321] and Coccomyxa subellipsoidea C-169 (CocsubWRKY) [GenBank: EIE27409]. The 60 amino acid spanning WRKY core domains of 295 WRKY DBD sequences (Supplementary Table S1) were used to create multiple protein sequence alignments using default settings in ClustalW (http://www.ebi.ac.uk/clustalw/) (51,52). The phylogenetic trees in Figure 1A and Supplementary Figure S1 were strict consensus trees and calculated with three different programs, ClustalW (http://www.ebi.ac.uk/clustalw/) (51), Phylip (http://evolution.genetics.washington.edu/phylip.html) (53,54) and Phylogenetic Analysis Using Parsimony (PAUP) (http://paup.csit.fsu.edu/) (55), which all resulted in identical tree topologies. Phylogenetic trees were drawn using TreeView software version 1.6.6 (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html). The phylogram in Supplementary Figure S1 was rooted with the DBD of H. sapiens CRAa FLYWCH-type zinc finger 1 [GenBank: EAW85450] as an out-group.
Figure 1.

Phylogeny of WRKY DBDs. (A) Unrooted phylogenetic tree of 295 WRKY domain sequences from 16 different species, including all Arabidopsis (AtWRKY) and rice (OsWRKY) members. Basal plant WRKY DBD sequences, e.g. from P. patens and S. moellendorfii, were included to achieve better separation of the different WRKY clades. WRKY groups and subgoups 1–3 are highlighted in different colors. The positions of novel group Ic WRKY proteins and basal group I DBD from G. lamblia and D. discoideum as well as the H. sapiens FLYWCH domain inside the tree are indicated. The tree is drawn to scale, and branch lengths are indicated. A full list of WRKY DBD sequences is provided as Supplementary Table S1. The same data are given as detailed phylogram that shows all labels and names (Supplementary Figure S1). (B) Schematic overview of the evolutionary history of the WRKY DBD. The analysis of ancestral WRKY proteins revealed paraphyletic origin for group I proteins and direct monophyletic descent of group IIc WRKY proteins from an ancestral group IIc-like WRKY domain. Presence of WRKY members in the basal plant species P. patens or S. moellendorfii and in monocot or dicot phyla is indicated by pictographs. Evolutionary relatedness was inferred by the position in the phylogenetic tree, by structural features of the zinc finger and by representative members of the four plant phyla within each of the clades.
Design of the AtWRKY1 cDBD–DNA complex
The only WRKY DBD protein crystal structure available in the Protein Data Bank (PDB) is the structure of the C-terminal DNA-binding domain (cDBD) of AtWRKY1 (PDB id: 2ayd) that lacks the DNA double helix (42,56) (Supplementary Table S2). The search for similar protein structures of AtWRKY1 cDBD with the Dali server retrieved the co-crystal of MmGCM1a bound to DNA (PDB id: 1odh, Z-score > 6.2, status 10/2010), as previously described by Duan et al. (42,57,58). We mapped the AtWRKY1 cDBD protein onto the MmGCM1a protein using root mean square deviation (RMSD)-minimizing superposition (as implemented by the atom bijection method in biochemical algorithms library (BALL)) (59). We defined certain Cα atoms of both proteins and stored them in a paired list. The resulting RMSD value for the optimal aligned β-sheet atoms (26 Cα atoms) was 0.88 Å and for all WRKY DBD atoms (68 Cα atoms) was 3.18 Å. Thereby, a first protein–DNA complex model of AtWRKY1 cDBD bound to DNA, provided by chains B and C of 1odh, can be constructed.
Design of the AtWRKY1 cDBD–W2-box DNA complex
The original 1odh DNA sequence (5′-CGATGCGGGTGCA-3′) was mutated using the DNAmutator method in BALL (57,59). The DNAmutator method builds a new base pair by keeping DNA backbone atoms, deleting atoms of the old nucleobase and inserting atoms of the new nucleobase in the same plane as the old ones. The A·T base pair (bp) at the 3′ position was deleted. The 12-bp-long DNA was mutated according to the parsley PR1-1 W2-box promoter sequence (5′-TCAAAGTTGACCAATAAT-3′) that includes the previously described WRKY target DNA sequence also known as the W2-box (5′-TTGACC-3′) (22). Initially, the first 12 bp of the W2 sequence were tested within the AtWRKY1 cDBD–DNA complex. Subsequently, the 12-bp sequence was shifted relative to the protein domain six times by one base in the 3′ direction; thus, seven complexes were constructed. The AtWRKY1 cDBD was kept at the same relative position to the 12-bp DNA structure and was repeated with the reverse complement DNA strand. That resulted in 14 protein–DNA complexes with different base pair sequences, all containing the W2-box at different positions relative to the protein domain.
AtWRKY1 cDBD–DNA-binding interface
We know from previous studies that conserved WRKYGQK amino acids form contacts with ‘TTGACC’ (42–44). To identify the most probable protein–DNA complex, we performed molecular dynamics (MD)-simulations for all of the 14 different protein–DNA complexes and calculated their binding affinities with the Molecular Mechanic - Poisson-Boltzmann Surface Area (MM-PBSA) approach, provided by Amber 11 (60) (Supplementary Figure S2). The protonation states of the protein’s amino acids were determined with H++ (61), and the residues are renamed according to Amber naming conventions. Because WRKY DBD proteins contain a zinc ion, which is coordinated by two histidines (residues: 361 and 363 in 2ayd) and two cysteines (residues: 332 and 337 in 2ayd), these four residues are protonated and renamed correspondingly. We used the ff99sb force field and loaded specific parameters for nucleic acids (DNA_CI.lib and frcmod.parmbsc0) (62,63). We specified certain values for dihedral angles and improper torsions for the Zn2+ ion in library and frcmod files. Seventeen Na+ ions were added to neutralize the system; additional 33 Na+ and 33 Cl− ions were added to gain a salt concentration of 0.2 M, while the protein–DNA complex was placed in an octahedral water box of ∼9000 TIP3P water molecules with a 15-Å distance to the end of the box (64). This was done using LEaP, which is a preparatory program provided by Amber and creates topology and coordinate files for the MD simulation. The system was minimized in several steps gradually releasing more and more atoms from spatial constraints. Then it is heated up from 100 to 300 K and relaxed at this temperature to an equilibrium state. While heating up, the protein–DNA complex was constrained, and the constraints were released gradually in several steps. In all 100-ps-long relaxation steps, the center-of-mass motion was removed every 1000 steps to avoid energy drains (65–67). When 300 K was reached, the system was kept at this temperature using a Langevin thermostat with a collision frequency of 2.0 ps−1. The particle mesh Ewald method was used to treat long-range electrostatic interactions (68). The SHAKE algorithm was applied to constrain bond lengths involving bonds to hydrogen atoms; therefore, a time step of 2 fs was sufficient (69). After a relaxation period at constant pressure for 100 ps, the system was simulated for 20 ns. During this production run, only the two 3′–5′ bp of the DNA double helix and 23 β-sheet Cα atoms were constrained with a harmonic force constant of 1 kcal/mol A2. During the last 5 ns of the production run, binding free energies and specific contacts between amino acids and base pairs were calculated. The MM-PBSA Perl script of Amber 11 was used to extract 26 complexes evenly spread over the last 5 ns of the production run. Default parameters were used to calculate binding free energies for each complex. Studying contacts in the range between 1 and 2.5 Å over the last 5 ns of the production run, which were lasting for at least 80%, yielded a clear preference for one AtWRKY1 cDBD–DNA complex (Supplementary Figure S2). Thus, we modeled an AtWRKY1 cDBD–DNA complex, which makes specific contacts to the ‘TTGACC’ sequence. Our designed complex is in good agreement with an NMR WRKY4–DNA complex published recently (44).
Homology modeling of four different WRKY DBDs
To study specificity at the binding interface of different WRKY proteins, we modeled the DBD of AtWRKY11, AtWRKY33 c, AtWRKY33 n and AtWRKY50. We used AtWRKY1 cDBD (PDB ID: 2ayd) as template structure and the amino acid sequences described in Supplementary Table S2 for homology modeling in Prime (Version 3.0, Build: 30111) (42,70,71).
AtWRKY11 and AtWRKY50–DNA-binding specificity
The Q29K and K26Q mutations in AtWRKY11 and AtWRKY50, respectively, were created using BALL (59). Then we built four protein–DNA complexes with the atom bijection and RMSDminimizer methods in BALL using the modeled AtWRKY1 cDBD–DNA complex as reference structure. The wild-type and mutated AtWRKY50–DNA complexes were simulated using the same protocol as described for the AtWRKY1 cDBD–DNA complexes. Over the last 5 ns of the production run of the four protein–DNA complexes, snapshots were extracted every 200 ps. Information about proximity sites in these complexes was also extracted (Supplementary Table S3).
AtWRKY33 cDBD and AtWRKY33 nDBD–binding specificity
We simulated AtWRKY33 cDBD and nDBD protein–DNA complexes using the same MD simulation protocol as described for the AtWRKY1 cDBD–DNA complexes. Over the last 5 ns of the production run, snapshots of both protein–DNA complexes were extracted every 200 ps. Binding free energies with the MM-PBSA method using the same parameters as described for the AtWRKY1 cDBD–DNA complexes were estimated. Additionally, proximity sites were extracted every 200 ps over the last 5 ns (Supplementary Table S3).
Molecular cloning
The coding sequences of Arabidopsis thaliana WRKY11 DBD, WRKY33 cDBD, WRKY33 nDBD and WRKY50 DBD were amplified using complementary DNA from A.thaliana flowers as template and gene-specific primers from Biomers.net GmbH, Germany (Supplementary Table S4). The specific polymerase chain reaction products were inserted into the Gateway compatible vector pENTR/D-TOPO (Life Technologies, Germany) and transformed into DH5α Escherichia coli cells (Stratagene, Germany). By site-directed polymerase chain reaction mutagenesis with suitable primers using the respective pENTR/D-TOPO vector as template, the mutated versions AtWRKY11 DBD_Q29K and AtWRKY DBD_K26Q were created (Supplementary Table S4). After sequencing, BP reaction was performed with Gateway pET-DEST42 vector according to the manufacturer’s protocol (Life Technologies, Germany). Owing to the expression vector, a C-terminal His-epitope is translationally fused when expressed in E. coli expression strain BL21 (Stratagene, Germany). As a negative control, we used BL21 cells transformed with pET-DEST42-empty without ccDB cassette (38).
Protein expression and protein extraction
Proteins were expressed and extracted according to Brand et al. (38). After detection of the His epitope-tagged proteins by western blot analyses, the native crude E. coli protein extracts were used for DPI-ELISA.
DPI-ELISA and DPI-ELISA screen
Native crude E. coli protein extracts containing recombinant A. thaliana WRKY11 DBD:His, WRKY33 cDBD:His, WRKY33 nDBD:His, WRKY50 DBD:His, WRKY11 DBD_Q29K:His and WRKY50 DBD_K26Q:His were used for DPI-ELISA and DPI-ELISA screen as described before (38) (Brand,L.H., Henneges,C., Schüssler,A., Kolukisaoglu,H.U., Koch,G., Wallmeroth, N., Hecker, A., Thurow,K., Zell,A., Harter,K. and Wanke,D, submitted) (Supplementary Table S5 and Supplementary Figure S3). The DPI-ELISA screen absorbance data were set relative to the highest signal in each experiment (Supplementary Table S6). The double-stranded DNA probes were valued positive, if the relative absorbance was above the significance threshold. The significance threshold was designated as the 2-fold standard deviation of the average of the relative absorbance of all probes within one experiment (P ≤ 0.05). To deduce a binding consensus for each WRKY DBD, the forward sequences of all positive probes of each individual experiment were analyzed using Multiple Em for Motif Elicitation (MEME) with settings 0/1 per sequence, 4–6 bp and 3 motifs (72). The motif consensus and its position within the sequences were subsequently assessed for each of the quartiles independently. The number of probes that contain the motif displayed as Weblogo is indicated as small numbers (Supplementary Table S7). The Weblogos were created with Weblogo version 3.0 (73). The quantitative DPI-ELISA absorbance data of two technical replicates were normalized by subtraction of the negative BL21 control extract.
RESULTS
Evolution of WRKY DBDs
We aligned the amino acid sequences of the DBD of 295 WRKY proteins from 15 different species to deduce a DBD-specific phylogram (Figure 1A, Supplementary Figure S1). Besides others, the DBD of the HsFLYWCH transcription factor was used as an out-group to resolve the phylogenetic relation of WRKY DBDs in plants. Within the phylogram, the plant-specific clades are highlighted. The previously described three main plant WRKY groups (I–III) can be identified, but groups I and II constitute paraphyla (3,7,10). The monophyletic plant WRKY DBD dates back to an ancestral WRKY protein with one WRKY domain only, presumably found in a basal non-plant eukaryotic organism. For the dissection of the phylogenetic relation of plant WRKY DBDs, we took into account sequence similarities, structures of the zinc finger motif and the existence of WRKY representatives in basal plant species (Figure 1B). To achieve better separation of basal WRKY groups, to counteract long-branch attraction effects and to avoid artifacts due to rapidly evolving sequences, we included several basal WRKY-like DNA-binding zinc finger sequences as out-groups, such as human GCM1a, a human FLYWCH and two aberrant WRKY consensus from the mold Aspergillus and the yeast Tetrapisispora. To dissect the paraphyletic origin of WRKY proteins, we first analyzed group I-like WRKY DBDs from the protozoa G. lamblia and the slime mold Dictyostelium. Both WRKY proteins have an N-terminal (n-) and a C-terminal (c-) DBD and, hence, are by definition group I-like WRKY proteins. Interestingly, the domains of G. lamblia do not cluster with any group I WRKY, despite exhibiting two WRKY DBDs. Both Dictyostelium DBDs cluster with group I cDBDs. The Dictyostelium nDBD is in clade Ib, whereas the cDBD is close to clade Ia. These findings proof the paraphyletic nature of group I WRKY proteins. As a consequence, and in vast contrast to previous assumptions, these ancestral group I-like WRKY proteins with two DBDs cannot be the ancestors of plant WRKY DBDs. Based on our phylogenetic analysis, we can conclude that the ancestral group I-like WRKYs as well as groups Ia and Ib evolved independently due to a duplication of a IIc-like ancestral WRKY DBD. Owing to the proximity of clade IIc to the basal out-groups, such as mouse GCM1a and the DmFLYWCH, we can propose that members of group IIc are most probably direct descendants of a single ancestral IIc-like WRKY DBD. The evolution of group IIc WRKY proteins with only one WRKY DBD from either group Ia or Ib by loss of the DBD requires more evolutionary steps compared with the direct lineage-specific monophyletic offspring and, therefore, is less probable.
In addition, WRKY groups IIc, Ia and Ib form clades that contain representatives from all plant divisions. To achieve better separation of the other clades, we included WRKY DBDs from basal plant species, as mentioned previously, such as moss P. patens and spikemoss Selaginella moellendorfii. Hence, WRKY groups that contain representatives in mosses, ferns, mono- and dicots must have existed already before the evolution of land plants. Those clades that lack basal representatives must have evolved during later phases of land plant speciation. Because of the proximity of group IIa WRKYs with the clades Ia and Ib, it is evident that the IIa WRKY clade evolved from group I WRKY proteins through loss of their nDBD. This is also the case for group IIb WRKY proteins; however, IIb representatives can only be found in mono- and dicotyledonous species and, thus, they must be descendants from group IIa WRKYs. The clade of group IId WRKYs is evolutionary more distant from clade Ia and Ib cDBD and has more changes in the zinc finger compared with clades IIa and IIb. Hence, clade IId WRKYs evolved most likely from group IIa. The same accounts for group IIe WRKYs, but representatives are missing in mosses. Therefore, group IIe WRKY proteins are most probably descendants from clade IId. The evolutionary youngest WRKY domains are found in the clade of group III WRKY proteins. All group III members have an even more diverged C2-HC zinc finger motif. In particular, clade IIIb appears to be evolutionarily young and the descendent from group IIIa because its representatives can be found in angiosperms only. Interestingly, a grass-specific subgroup of clade IIIa WRKY proteins has two WRKY domains and, by definition, must be addressed as novel group Ic WRKY proteins. There is no phylogenetic support for an independent group Ic clade because it evolved recently due to a duplication of single group IIIa WRKY DBDs in the Gramineae. As the amino acid sequences of the Ic cDBD and nDBD diverged only insignificantly, group Ic WRKY proteins belong evidently to clade IIIa, which again supports the paraphyletic origin of group I WRKY proteins in general.
Molecular structure and DNA-binding specificities of WRKY DBDs
The phylogenetic analysis revealed distinct groups of diverged WRKY DBDs, which possibly reflect WRKY group-specific DNA-binding properties. To test this hypothesis, we chose to analyze the DBDs of representatives of three different groups in detail. We chose AtWRKY50 (group IIc) and AtWRKY11 (group IId), which we had used for in vitro DNA-binding studies previouly, as well as the DBDs of the well-characterized AtWRKY33 (group I) (38) (Figure 2A). Although they belong to different groups, homologous proteins can be found in mosses, ferns, mono- and dicotyledonous species. AtWRKY33, a group I member, exhibits two DBDs. Despite the DNA-binding ability of AtWRKY33 cDBD, which was shown before, the DNA-binding ability of AtWRKY33 nDBD or any other group I nDBD has been proposed from sequence comparison, but could never be experimentally validated (20,39,42,47–49).
Figure 2.

Homology models of AtWRKY DBDs. (A) The general protein secondary structure based on the crystal structure of WRKY1 C-terminal DNA-binding domain (cDBD; PDB id: 2ayd) is given above the alignment of AtWRKY cDBDs and N-terminal DBD (nDBD). Black bars highlight the conserved zinc finger; other conserved amino acids are indicated: (*) same amino acid, (:) amino acid with similar chemical properties, (.) majority of amino acids with similar chemical properties. (B) The five conserved β-strands of AtWRKY1 cDBD are colored according to A in the structure shown (PDB id: 2ayd). (C) The overlay of the protein-DNA models of AtWRKY33 cDBD (orange) and WRKY33 nDBD (green) is displayed. The protein structures are homology models and superimposed with respect to their β-sheet Cα atoms.
To analyze the WRKY–DNA complex in detail, we chose a homology modeling-based approach. The tertiary structure of the protein domain was resolved with the crystal structure of AtWRKY1 cDBD (42). In general, the β-sheet consists of five β-strands that are connected by flexible loop regions (Figure 2A). Whereas β2, β3 and β4 are highly similar between the DBDs of interest, the positions β1 and β5 seem to be more variable. The crystal structure model of AtWRKY1 cDBD reveals that β1 and β5 are more distant from the DNA-binding site, which is mainly represented by β2 (Figure 2B). In contrast to other zinc finger DNA-binding proteins, the involvement of the positively charged zinc ion in DNA binding of WRKY proteins can be excluded. The zinc ion is coordinated between β3 and the C-terminal end of the DBD and presumably confers the stable tertiary domain structure. Unfortunately, there is no information from X-ray structures of co-crystalized WRKY proteins bound to a given DNA consensus; to date only NMR data could be found (44). Hence, we decided to map the AtWRKY1 cDBD onto the MmGCM1a-DNA co-crystal structure. The DNA sequence was changed to a well-known WRKY binding site, the W2-box from parsley (22,38,39,48). The homology model of AtWRKY33 cDBD and nDBD with DNA reveals an almost identical co-structure of both domains with DNA, which supported the idea of general group I nDBD DNA-binding abilities (Figure 2C). The molecular dynamic simulation of AtWRKY33 DBDs with DNA even strengthened this hypothesis (Figure 3A): the calculated binding affinity of AtWRKY33 cDBD to the W2-box is even higher compared with AtWRKY1 indicating a strong protein–DNA interaction. The binding affinity of AtWRKY33 nDBD to the ‘TTGACC' motif is slightly lower compared with AtWRKY1 cDBD. This indicates a binding of AtWRKY33 nDBD to DNA, which might be weaker though, compared with the cDBD. We tested the DNA-binding ability of AtWRKY33 n- and cDBD to the known W2-box by DPI-ELISA (Figure 3B). For the first time, we could show the sequence-specific DNA binding of a group I nDBD. The nDBD of AtWRKY33 specifically binds to the W2-box but not to its mutated version W2m. Furthermore, the DNA-binding affinity of AtWRKY33 nDBD seems to be reduced compared with the cDBD, as is consistent with the in silico molecular modeling data. To reveal the WRKY DNA-binding specificities of both DBDs, we performed DPI-ELISA screens (Figure 3C, Supplementary Tables S5–7 and Supplementary Figure S3). In general, both DBDs are binding ‘TTGACY' motifs with highest affinity. Both domains allow certain variability in the 5′ region of the consensus, whereas the ‘GAC' core of the W-box motif is invariant. The AtWRKY33 cDBD, however, allows more changes in the W-box sequences compared with its nDBD (Figure 3C).
Figure 3.
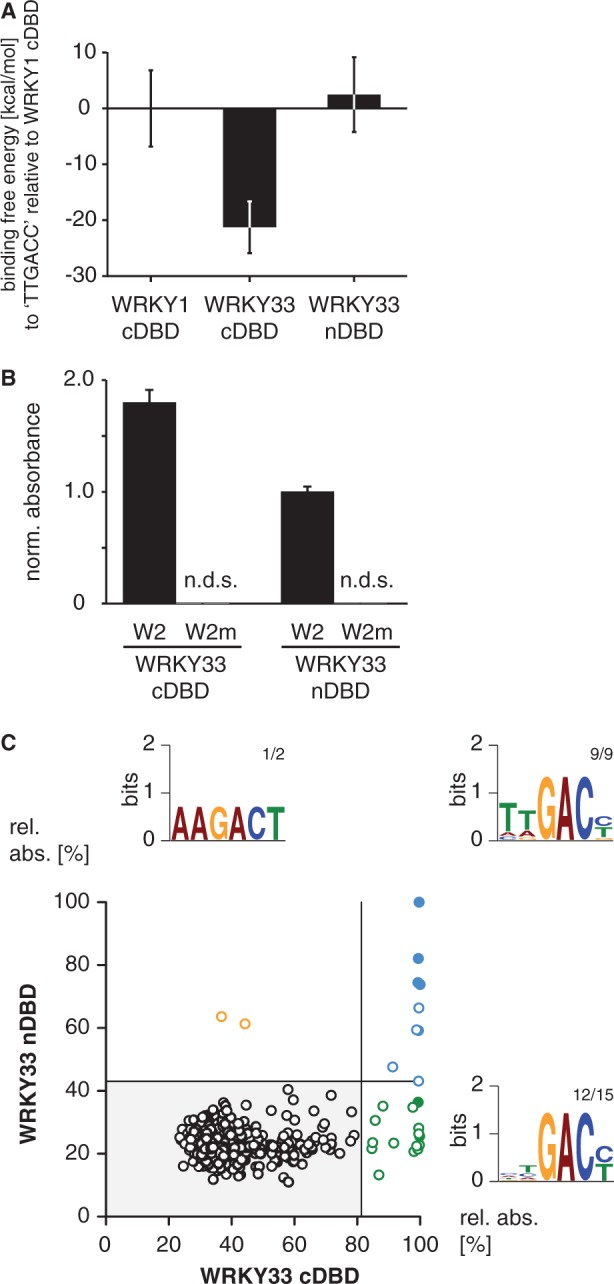
Comparison of the DNA-binding specificities of group I AtWRKY33 N-terminal and C-terminal DNA-binding domain. (A) The relative binding free energies (kcal/mol) of AtWRKY33 C-terminal DNA-binding domain (cDBD) and AtWRKY33 N-terminal DNA-binding domain (nDBD) with respect to WRKY1 cDBD to the DNA sequence (5′-AAAGTTGACCAA-3′) were calculated using the MM-PBSA method in Amber 11. (B) Probing of the W2 or W2m DNA with AtWRKY33 cDBD or nDBD by DPI-ELISA reveals binding ability of both protein domains (n.d.s., no detectable signal; absolute error is shown). Each absorbance value was normalized to the mean of the background control. (C) The XY-plots of relative absorbance values of the DPI-ELISA screens of AtWRKY33 cDBD and nDBD are shown. Those double-stranded DNA probes significantly bound by AtWRKY33 cDBD and nDBD are in blue, those only bound by AtWRKY33cDBD are in green and those only bound by AtWRKY33nDBD are in orange. The filled circles indicate probes exhibiting the known ‘TTGACY’ binding motif. Lines indicate the significance threshold (P ≤ 0.05). The sequences of all positively bound probes were used as MEME input for motif identification. Motif consensus was shown as Weblogos; numbers of probes that contain the motif displayed as Weblogo and number of motifs per quartile are indicated as small numbers.
Hence, by using the DPI-ELISA screen, we can conclude on WRKY DNA-binding specificities in a general view, which means we are able to reveal subtle differences in the DNA recognition spectrum of each DBD. This might help to conclude on WRKY group-specific DNA sequence recognition. Therefore, we tested the DBDs of AtWRKY11 and AtWRKY50 also by DPI-ELISA screen (Figure 4). Both were shown to specifically recognize the W2-box consensus (38). On the basis of our phylogenetic analyses, we propose that the group IId DBD of AtWRKY11 evolved from a group I cDBD (Figure 1B). In contrast, the group IIc DBD of AtWRKY50 is more related to the DBD that was most likely derived directly from an ancestral group IIc-like WRKY. The comparison of AtWRKY11 with the DBDs of AtWRKY33 reveals that all prefer the ‘TTGACY’ consensus as expected (Figure 4A and B, Supplementary Table S7). Despite the fact that AtWRKY11 DBD and AtWRKY33 nDBD share some hits, both bind to certain probes that seem to be DBD-specific. The nDBD of AtWRKY33 seems to bind less specific in the 5′ region of the consensus compared with AtWRKY11 DBD. In contrast, all hits of the AtWRKY11 DBD DPI-ELISA screen were successfully revealed within the AtWRKY33 cDBD experiment. These data underpin the evolutionary relation between group IId DBDs and group I cDBDs. Consistently, AtWRKY50 DBD also prefers to bind to the ‘TTGACY’ consensus (Figure 4C and D, Supplementary Table S7). However, AtWRKY50 DBD shares some hits with the nDBD and the cDBD of AtWRKY33, whereas in both cases DBD-specific probes can be found. Hence, we cannot support the idea that group IIc DBDs might be derived from group I nDBD. Instead, these findings are in line with our hypothesis of a direct descent of group IIc WRKY transcription factors from yet unknown ancestral group IIc-like WRKY proteins. Nevertheless, it should be noted that AtWRKY50 DBD clusters within an angiosperm-specific subgroup of clade IIc, which might have evolved recently and, therefore, lacks homologs in other divisions.
Figure 4.

Comparison of the DNA-binding specificities of four AtWRKY DBDs. The XY-plots of relative absorbance values of the DPI-ELISA screens of the DBDs of AtWRKY11 DBD versus AtWRKY33 nDBD (A), WRKY50 DBD versus WRKY33 nDBD (B), WRKY33 cDBD versus WRKY11 DBD (C) and WRKY33 cDBD versus WRKY50 DBD are graphed. The dsDNA probes significantly bound by both the x- and y-component are in blue, those only bound by the respective x-component are in green and those only bound by the respective y-component are in orange. The filled circles indicate probes exhibiting the known ‘TTGACY’ binding motif. Lines indicate the significance threshold (P ≤ 0.05). The sequences of all positively bound probes were used as MEME input for motif identification. Motif consensus was shown as Weblogos; numbers of probes that contain the motif displayed as Weblogo and number of motifs per quartile are indicated as small numbers.
Identification of residues responsible for the DNA-binding specificity of AtWRKY50 and AtWRKY11
Still the question remains open how the sequence-specific DNA binding of WRKY proteins is mediated. AtWRKY11 DBD and AtWRKY50 DBD are relatively distantly related and exhibit an amino acid difference within the highly conserved second β-strand. Therefore, these WRKYs are favored models to study the sequence-specific DNA recognition of WRKY DBDs in detail (Figure 5). The highly conserved glutamine within β2 that in general favors to contact nucleobases due to its partial negative charge is changed to a lysine within the AtWRKY50 DBD subgroup. Lysine, in contrast, probably favors to contact the negatively charged DNA phosphate backbone because of its partial positive charge. This implies the hypothesis that a reciprocal Q/K change leads to differential DNA-binding specificities of the respective WRKY DBDs. The comparison of the AtWRKY11 and AtWRKY50 DBD by DPI-ELISA screens revealed the high-affinity ‘TTGACY’ DNA-binding motif (Figure 5A). In contrast to AtWRKY11 DBD, AtWRKY50 DBD seems to only require a conserved ‘GAC’ core consensus to interact with high affinity with DNA. The AtWRKY50 DBD permits variations both 5′ and 3′ of the ‘GAC' triplet core. This difference in DNA-binding specificity might be due to the Q/K variance within β2. A close-up of the modeled AtWRKY11 DBD-W2-box co-structure reveals that Gln29 is in proximity to the second thymidine, as was expected, whereas the mutated Lys29 is close to the DNA phosphate backbone, according to what we proposed before (Figure 5B and C). Furthermore, the molecular dynamics simulation of AtWRKY50 DBD and its mutated version AtWRKY50 DBD_K26Q support this statement (Figure 5D). The Lys26 positions close to the DNA phosphate backbone, and Gln26 is close to the thymidine base during molecular dynamics simulations (Figure 5D). Therefore, it can be speculated that the DNA-binding specificity of AtWRKY11 DBD is more similar to the binding specificity of the mutated AtWRKY50 DBD and vice versa.
Figure 5.

Comparison of the DNA-binding specificities of the DBDs of AtWRKY50 versus AtWRKY11. (A) The XY-plots of relative absorbance values of the DPI-ELISA screens of the DBDs of WRKY50 DBD and WRKY11 DBD are shown. The dsDNA probes significantly bound by WRKY50 DBD and WRKY11 DBD are in blue, those only bound by WRKY50 DBD are in green and those only bound by WRKY11 DBD are in orange. The filled circles indicate probes exhibiting the known ‘TTGACY’ binding motif. Lines indicate the significance threshold (P ≤ 0.05). The sequences of all positively bound probes were used as MEME input for motif identification. Motif consensus was shown as Weblogos; numbers of probes that contain the motif displayed as Weblogo and number of motifs per quartile are indicated as small numbers. (B and C) The close-up of the protein-DNA model of WRKY11 DBD (B) and WRKY11 DBD_Q29K (C) is shown (GLN29 - Glutamin - red; LYS29 - Lysin - blue). Both protein structures are homology models and superimposed with respect to their β-sheet Cα atoms. (D) The molecular dynamics simulations (10 ns) of WRKY50 DBD (blue) and WRKY50 DBD_K26Q (red) are shown as close-up.
To quantitatively compare and analyze the respective influences of lysine or glutamine residues on the DNA-binding specificity of WRKY DBDs, we tested the DBD wild-type and mutant versions of AtWRKY11 and AtWRKY50 by quantitative DPI-ELISA (Figure 6). To allow for quantitative analyses, equal amounts of heterologous-expressed protein were applied (Figure 6A). Interestingly, the DNA-binding affinity to the W2-probe of AtWRKY11 DBD_Q29K was drastically decreased compared with the wild-type version (Figure 6B). This indicates that Gln29 is essential for the high affinity of AtWRKY11 DBD to DNA. However, glutamine or lysine on position 26 in the AtWRKY50 DBD does not influence its DNA-binding affinity to the W2-probe. The DNA-binding affinity of AtWRKY50 DBD_K26Q to the other tested DNA probes was drastically decreased compared with the wild-type version, and thus more similar to the AtWRKY11 DBD results. This indicates that the glutamine in the second β-strand is important for sequence-specific DNA recognition and binding. In case of the AtWRKY50, the lysine is necessary for the DNA binding of aberrant W2-box versions.
Figure 6.
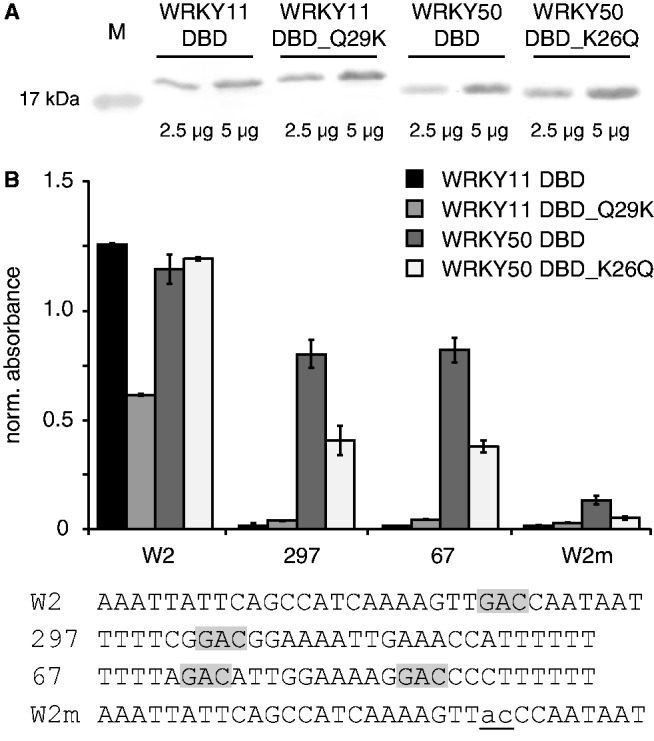
Influence of the glutamine/lysine of the β2-strand of the DBDs of AtWRKY50 DBD and AtWRKY11 DBD on the DNA-binding specificities. (A) Crude E. coli protein extracts of WRKY11 DBD, 11_DBDQ29K, 50 DBD and 50 DBD_Q26K were analyzed by western blot analyses to show comparable protein concentrations and appropriate protein sizes. (B) Equal amounts of protein extract [3 µg/well] were used to probe dsDNA probes [2 pmol/well] with different versions of the W-box by quantitative DPI-ELISA. The forward sequences of the dsDNA probes are given below the graph. The invariable GAC-core is shaded in gray. The absorbance values were normalized to the mean of the background controls. The absolute error is shown.
It is evident from our molecular dynamics simulations and the previously published protein–DNA NMR data that more amino acids are necessary for the specific protein–DNA interaction of the WRKY DBD than the glutamine or lysine within the second β-strand (Figure 7, Supplementary Table S8) (42,44). Our binding interface analyses of molecular dynamics simulations of WRKY DBD–DNA complexes (20 ns) contributed to a refined knowledge of WRKY–DNA interaction sites. Taken together, the simulations of A. thaliana WRKY33 c-/n-, WRKY50 and WRKY11 DBD give evidence that not only the β2-strand is necessary for the specific protein–DNA interaction, but also β3 and β4 are required. This is in accordance with previously published data (Supplementary Figure S4) (42,44). Although the binding interface data extracted from the molecular dynamic simulations should be regarded as possible interaction sites only, many of them overlap with the NMR data of AtWRKY4 with DNA (44). Despite the fact that WRKY DBDs bind to an invariant ‘GAC’ core consensus, the amount of specific amino acid–base interactions within this core is limited. Hence, the mechanism by which the specific WRKY-'GAC' core recognition is mediated remains elusive.
Figure 7.
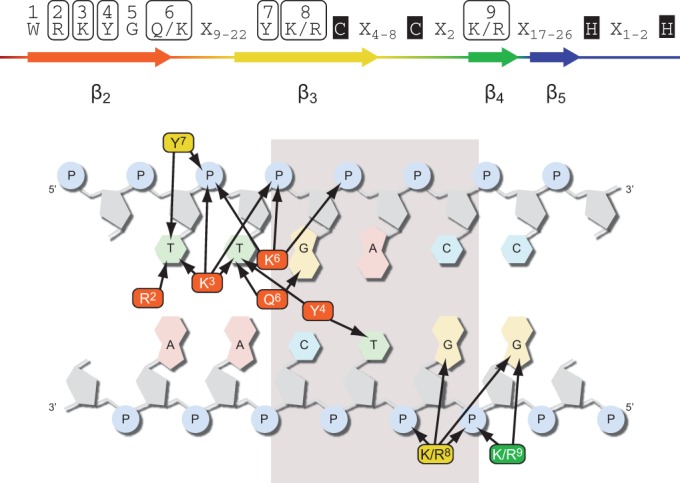
Model of the WRKY–DNA-binding interface. The molecular dynamics simulations of WRKY DBDs led to the identification of amino acids in 1–4Å proximity to either the nucleobases or the phosphate backbone of the DNA (arrow). These amino acids are considered to possibly influence WRKY binding to the W-box. The conserved tryptophan (W1) and the zinc finger are important structural determinants for the WRKY–DNA interaction, but are not in proximity to the DNA (Xn–number of amino acids between conserved proximity sites).
DISCUSSION
Our phylogenetic analysis of the WRKY DBDs from several lineages is coherent with previously published phylogeny and further refines the knowledge on WRKY DBD evolution (3,7,10,45). There were putatively three independent events of DBD duplication resulting in groups I a–c. The DBD duplication in group Ia and Ib proteins is likely not to be of monophyletic origin, but evolved independently at different evolutionary times (10,24,30,74). Nevertheless, these group I proteins were the ancestors of all groups II and III (10), with the exception of group IIc, which we propose derived directly from an ancestral group IIc-like WRKY DBD. It is possible that group IIa and IId proteins evolved independently through loss of the nDBD from group I WRKY proteins (10,29). The hypothesis of independent nDBD losses is strengthened by the comparative analysis of syntenic gene loci in tomato and two Brassicaceae by Rossberg et al. (75). They identified a group I WRKY in tomato that is orthologous to the group II-type WRKY10 in Capsella rubella and A. thaliana (75). WRKY10 experienced a deletion of the nDBD in the Brassicaceae lineage. AtWRKY10 mutation causes the MINISEED3 phenotype, which exhibits severe effects in the formation of seeds (17). Hence, AtWRKY10/Miniseed3 is a lively example of the paraphyletic nature of both group I and II WRKY proteins. Group II seems to have quite recently evolved into groups IIIa and IIIb in the early land plant lineage (10,24). Interestingly, group IIIa includes group I-like proteins. This again underlines the paraphyletic origin of group I and indicates a selective pressure for WRKY transcription factor diversification (24,45). Furthermore, the presence of the novel group Ic and comparably old group I proteins suggests an evolutionary advantageous function for the second DBD. We could show for the first time that both WRKY DBDs of a group Ia WRKY are capable of binding to DNA. One can hypothesize that the two WRKY DBDs of group I proteins bind to two neighboring DNA motifs of the same side of the DNA strand. Consequently, the individual spacing between the two WRKY domains will directly translate into a defined distance of the two consecutively bound W-box motifs. Taking the different evolutionary histories of nDBD and cDBD into account, it is not surprising that both domains displayed different binding specificities. In line with previous speculations, it is likely that the nDBD of group I WRKY proteins also contributes to promoter-specific binding (20,39,47–49). Our DNA-binding assay, along with previously published results, revealed that the majority of tested WRKYs bind to an invariant ‘GAC' core consensus (25,38,39,76). Still, WRKY proteins allow DBD-specific variations in their DNA-binding affinity outside this region (38,39). The variability of their respective DNA-binding consensus supports the proposed phylogeny as well.
Our in silco analyses and DNA-binding assays of AtWRKY11 DBD and AtWRKY50 DBD suggest a new mechanism by which the variability in WRKY DNA-binding specificity is influenced. The aberrant β2 lysine of the AtWRKY50 DBD contacts the phosphate backbone and not the base, and thereby confers the 5′ variability of the AtWRKY50 DNA consensus. The binding mechanism of AtWRKY11 appears to be different compared with AtWRKY50, where other amino acids are necessary for specific protein–DNA interaction. The X-ray structure and NMR data, together with our and previous simulations, helped to identify the amino acids that are most probably in direct contact with the DNA (42,44). This is, however, not sufficient to explain the specific recognition of the ‘GAC' core and suggests the involvement of other binding mechanisms, such as DNA shape read out for specific WRKY-DNA interaction. The analysis of the distantly related mouse GCM1 protein co-crystallized with DNA revealed that the bound DNA is altered in its shape. Instead of the regular B-form of Watson–Crick base pairs, the contact site in the GCM1–DNA crystal complex exhibits a possible Hoogsteen base pairing (57). The WRKY DNA target includes a potential Hoogsteen-dinucleotide step ‘TpG' (‘TTGAC') (77). Therefore, we postulate that not only the DNA base read out is necessary for specific WRKY–DNA interaction, but also the evaluation of the local DNA shape and electrostatic potential, as it was described for other transcription factors previously (78). The influence of adjacent sequences on the WRKY DNA-binding affinity that was reported by Ciolkowski et al. (39) further supports the possible involvement of DNA shape read out mechanisms of the WRKY–W-box interaction. For example, the DNA-binding affinity of E. coli EXTRACYTOPLASMIC FUNCTION σ FACTOR (σE) is influenced by the width of the neighboring minor grove that is not directly bound by σE (79). Recently, the X-ray structure of a NO APICAL MERISTEM/CUPPED SHAPED COTTELYDON (NAC)–DNA complex was published (80). NAC proteins share structural homology with the WRKY/GCM1 proteins, and they possess a WRKY DBD-like exposed β-strand that has similar electrostatic distribution and curvature (81) (Lou,Z.Y., Chen,Q.F., Wang,Q. and Xiong,L.Z, unpublished). The co-structure of Arabidopsis NAC019 (ANAC019) with DNA revealed that this WRKY-like β-strand protrudes into the DNA major groove (81). The DNA bound by the dimeric protein is largely in harmonic B-conformation, and it is proposed that because of the minor flexibility of the ANAC dimer low-affinity DNA motifs are recognized (80). Further analyses by high-resolution co-crystallography are required to disclose whether the DNA bound by WRKY proteins is in harmonic B-conformation or not.
Interestingly, it was shown that VQ-motif-containing proteins SIGMA FACTOR BINDING PROTEIN 1 (SIB1) and SIB2 enhance the DNA-binding affinity of AtWRKY33 to a tandem repeat of the W2-box in vitro (82). This proves that other WRKY interacting proteins influence DNA-binding affinities and render it possible that low-affinity in vitro targets might constitute high-affinity in vivo targets, as it was shown for the viral transcription factor REPLICATION AND TRANSCRIPTION ACTIVATOR (83). Therefore, WRKY protein domains other than the DBD are crucial determinants for complex formation in the nucleus or at the DNA, as well as for promoter-specific functions of WRKY proteins. Recently, Cheng et al. (46) showed that VQ-motif proteins interact with group I cDBD and group IIc DBD but neither with group I nDBD nor with other groups II and III DBDs. This further emphasizes the independent decent of IIc and the group I cDBD from an ancient IIc-like WRKY DBD. Interestingly, the interaction site was mapped to the region of the first β-strand, which we suggest by sequence homology to be different between AtWRKY33 nDBD and cDBD (Figure 2) (46). The retained function of VQ-motif interaction confirms that the descent of group IIc WRKYs from group I WRKY proteins due to a loss of any DBD is almost impossible and can, therefore, be excluded with confidence. VQ-motif protein binding might, therefore, be an evolutionary, rather old, feature, which possibly has already been present in ancestral IIc-like WRKY proteins. Still, the WRKY proteins constitute a transcription factor family that evolves and comprises members that are involved in various developmental programs and stress responses. To answer the question, how stimuli-specific responses are integrated by WRKY proteins, requires further comprehensive research of promoter-specific WRKY complex assembly.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Landesgraduiertenförderung des Landes Baden-Württemberg (to L.H.B.). Funding for open access charge: University of Tübingen.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Joachim Kilian, Friederike Ladwig and Andreas Hecker for statistical analysis and comments on the manuscript.
REFERENCES
- 1.Kilian J, Peschke F, Berendzen KW, Harter K, Wanke D. Prerequisites, performance and profits of transcriptional profiling the abiotic stress response. Biochim. Biophys. Acta. 2012;1819:166–175. doi: 10.1016/j.bbagrm.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Babu MM, Iyer LM, Balaji S, Aravind L. The natural history of the WRKY-GCM1 zinc fingers and the relationship between transcription factors and transposons. Nucleic Acids Res. 2006;34:6505–6520. doi: 10.1093/nar/gkl888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eulgem T, Rushton PJ, Robatzek S, Somssich IE. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5:199–206. doi: 10.1016/s1360-1385(00)01600-9. [DOI] [PubMed] [Google Scholar]
- 4.Pandey SP, Somssich IE. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009;150:1648–1655. doi: 10.1104/pp.109.138990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramirez SR, Basu C. Comparative analyses of plant transcription factor databases. Curr. Genomics. 2009;10:10–17. doi: 10.2174/138920209787581253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riechmann JL, Heard J, Martin G, Reuber L, Jiang C, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, et al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–2110. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
- 7.Rushton PJ, Somssich IE, Ringler P, Shen QJ. WRKY transcription factors. Trends Plant Sci. 2010;15:247–258. doi: 10.1016/j.tplants.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Ulker B, Somssich IE. WRKY transcription factors: from DNA binding towards biological function. Curr. Opin. Plant Biol. 2004;7:491–498. doi: 10.1016/j.pbi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Wenke K, Wanke D, Kilian J, Berendzen K, Harter K, Piechulla B. Volatiles of two growth-inhibiting rhizobacteria commonly engage AtWRKY18 function. Plant J. 2012;70:445–459. doi: 10.1111/j.1365-313X.2011.04891.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol. Biol. 2005;5:1. doi: 10.1186/1471-2148-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ay N, Irmler K, Fischer A, Uhlemann R, Reuter G, Humbeck K. Epigenetic programming via histone methylation at WRKY53 controls leaf senescence in Arabidopsis thaliana. Plant J. 2009;58:333–346. doi: 10.1111/j.1365-313X.2008.03782.x. [DOI] [PubMed] [Google Scholar]
- 12.Besseau S, Li J, Palva ET. WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J. Exp. Bot. 2012;63:2667–2679. doi: 10.1093/jxb/err450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dilkes BP, Spielman M, Weizbauer R, Watson B, Burkart-Waco D, Scott RJ, Comai L. The maternally expressed WRKY transcription factor TTG2 controls lethality in interploidy crosses of Arabidopsis. PLoS Biol. 2008;6:2707–2720. doi: 10.1371/journal.pbio.0060308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia D, Fitz Gerald JN, Berger F. Maternal control of integument cell elongation and zygotic control of endosperm growth are coordinated to determine seed size in Arabidopsis. Plant Cell. 2005;17:52–60. doi: 10.1105/tpc.104.027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishida T, Hattori S, Sano R, Inoue K, Shirano Y, Hayashi H, Shibata D, Sato S, Kato T, Tabata S, et al. Arabidopsis TRANSPARENT TESTA GLABRA2 is directly regulated by R2R3 MYB transcription factors and is involved in regulation of GLABRA2 transcription in epidermal differentiation. Plant Cell. 2007;19:2531–2543. doi: 10.1105/tpc.107.052274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson CS, Kolevski B, Smyth DR. TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell. 2002;14:1359–1375. doi: 10.1105/tpc.001404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo M, Dennis ES, Berger F, Peacock WJ, Chaudhury A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc. Natl Acad. Sci. USA. 2005;102:17531–17536. doi: 10.1073/pnas.0508418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miao Y, Laun T, Zimmermann P, Zentgraf U. Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol. Biol. 2004;55:853–867. doi: 10.1007/s11103-004-2142-6. [DOI] [PubMed] [Google Scholar]
- 19.Ngo QA, Baroux C, Guthorl D, Mozerov P, Collinge MA, Sundaresan V, Grossniklaus U. The Armadillo repeat gene ZAK IXIK promotes Arabidopsis early embryo and endosperm development through a distinctive gametophytic maternal effect. Plant Cell. 2012;24:4026–4043. doi: 10.1105/tpc.112.102384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishiguro S, Nakamura K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and beta-amylase from sweet potato. Mol. Gen. Genet. 1994;244:563–571. doi: 10.1007/BF00282746. [DOI] [PubMed] [Google Scholar]
- 21.Rushton PJ, Macdonald H, Huttly AK, Lazarus CM, Hooley R. Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of alpha-Amy2 genes. Plant Mol. Biol. 1995;29:691–702. doi: 10.1007/BF00041160. [DOI] [PubMed] [Google Scholar]
- 22.Rushton PJ, Torres JT, Parniske M, Wernert P, Hahlbrock K, Somssich IE. Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J. 1996;15:5690–5700. [PMC free article] [PubMed] [Google Scholar]
- 23.He H, Dong Q, Shao Y, Jiang H, Zhu S, Cheng B, Xiang Y. Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 2012;31:1199–1217. doi: 10.1007/s00299-012-1241-0. [DOI] [PubMed] [Google Scholar]
- 24.Ling J, Jiang W, Zhang Y, Yu H, Mao Z, Gu X, Huang S, Xie B. Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genomics. 2011;12:471. doi: 10.1186/1471-2164-12-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangelsen E, Kilian J, Berendzen KW, Kolukisaoglu UH, Harter K, Jansson C, Wanke D. Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genomics. 2008;9:194. doi: 10.1186/1471-2164-9-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud PF, Lindquist EA, Kamisugi Y, et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science. 2008;319:64–69. doi: 10.1126/science.1150646. [DOI] [PubMed] [Google Scholar]
- 27.Rushton PJ, Bokowiec MT, Han S, Zhang H, Brannock JF, Chen X, Laudeman TW, Timko MP. Tobacco transcription factors: novel insights into transcriptional regulation in the Solanaceae. Plant Physiol. 2008;147:280–295. doi: 10.1104/pp.107.114041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryu HS, Han M, Lee SK, Cho JI, Ryoo N, Heu S, Lee YH, Bhoo SH, Wang GL, Hahn TR, et al. A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Rep. 2006;25:836–847. doi: 10.1007/s00299-006-0138-1. [DOI] [PubMed] [Google Scholar]
- 29.Tripathi P, Rabara RC, Langum TJ, Boken AK, Rushton DL, Boomsma DD, Rinerson CI, Rabara J, Reese RN, Chen X, et al. The WRKY transcription factor family in Brachypodium distachyon. BMC Genomics. 2012;13:270. doi: 10.1186/1471-2164-13-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu KL, Guo ZJ, Wang HH, Li J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. 2005;12:9–26. doi: 10.1093/dnares/12.1.9. [DOI] [PubMed] [Google Scholar]
- 31.Zhang ZL, Xie Z, Zou X, Casaretto J, Ho TH, Shen QJ. A rice WRKY gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells. Plant Physiol. 2004;134:1500–1513. doi: 10.1104/pp.103.034967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giacomelli JI, Ribichich KF, Dezar CA, Chan RL. Expression analyses indicate the involvement of sunflower WRKY transcription factors in stress responses, and phylogenetic reconstructions reveal the existence of a novel clade in the Asteraceae. Plant Sci. 2010;178:398–410. [Google Scholar]
- 33.Huang S, Gao Y, Liu J, Peng X, Niu X, Fei Z, Cao S, Liu Y. Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol. Genet. Genomics. 2012;287:495–513. doi: 10.1007/s00438-012-0696-6. [DOI] [PubMed] [Google Scholar]
- 34.Lang D, Weiche B, Timmerhaus G, Richardt S, Riano-Pachon DM, Correa LG, Reski R, Mueller-Roeber B, Rensing SA. Genome-wide phylogenetic comparative analysis of plant transcriptional regulation: a timeline of loss, gain, expansion, and correlation with complexity. Genome Biol. Evol. 2010;2:488–503. doi: 10.1093/gbe/evq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie Z, Zhang ZL, Zou X, Huang J, Ruas P, Thompson D, Shen QJ. Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol. 2005;137:176–189. doi: 10.1104/pp.104.054312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eulgem T, Somssich IE. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007;10:366–371. doi: 10.1016/j.pbi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 37.Yamasaki K, Kigawa T, Seki M, Shinozaki K, Yokoyama S. DNA-binding domains of plant-specific transcription factors: structure, function, and evolution. Trends Plant Sci. 2012;18:267–276. doi: 10.1016/j.tplants.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Brand LH, Kirchler T, Hummel S, Chaban C, Wanke D. DPI-ELISA: a fast and versatile method to specify the binding of plant transcription factors to DNA in vitro. Plant Methods. 2010;6:25. doi: 10.1186/1746-4811-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciolkowski I, Wanke D, Birkenbihl RP, Somssich IE. Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol. Biol. 2008;68:81–92. doi: 10.1007/s11103-008-9353-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turck F, Zhou A, Somssich IE. Stimulus-dependent, promoter-specific binding of transcription factor WRKY1 to its native promoter and the defense-related gene PcPR1-1 in parsley. Plant Cell. 2004;16:2573–2585. doi: 10.1105/tpc.104.024810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joly-Lopez Z, Forczek E, Hoen DR, Juretic N, Bureau TE. A gene family derived from transposable elements during early angiosperm evolution has reproductive fitness benefits in Arabidopsis thaliana. PLoS Genet. 2012;8:e1002931. doi: 10.1371/journal.pgen.1002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duan MR, Nan J, Liang YH, Mao P, Lu L, Li L, Wei C, Lai L, Li Y, Su XD. DNA binding mechanism revealed by high resolution crystal structure of Arabidopsis thaliana WRKY1 protein. Nucleic Acids Res. 2007;35:1145–1154. doi: 10.1093/nar/gkm001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamasaki K, Kigawa T, Inoue M, Tateno M, Yamasaki T, Yabuki T, Aoki M, Seki E, Matsuda T, Tomo Y, et al. Solution structure of an Arabidopsis WRKY DNA binding domain. Plant Cell. 2005;17:944–956. doi: 10.1105/tpc.104.026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamasaki K, Kigawa T, Watanabe S, Inoue M, Yamasaki T, Seki M, Shinozaki K, Yokoyama S. Structural basis for sequence-specific DNA recognition by an Arabidopsis WRKY transcription factor. J. Biol. Chem. 2012;287:7683–7691. doi: 10.1074/jbc.M111.279844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Q, Wang M, Zhang X, Hao B, Kaushik SK, Pan Y. WRKY gene family evolution in Arabidopsis thaliana. Genetica. 2011;139:973–983. doi: 10.1007/s10709-011-9599-4. [DOI] [PubMed] [Google Scholar]
- 46.Cheng Y, Zhou Y, Yang Y, Chi YJ, Zhou J, Chen JY, Wang F, Fan B, Shi K, Zhou YH, et al. Structural and functional analysis of VQ motif-containing proteins in Arabidopsis as interacting proteins of WRKY transcription factors. Plant Physiol. 2012;159:810–825. doi: 10.1104/pp.112.196816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Pater S, Greco V, Pham K, Memelink J, Kijne J. Characterization of a zinc-dependent transcriptional activator from Arabidopsis. Nucleic Acids Res. 1996;24:4624–4631. doi: 10.1093/nar/24.23.4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eulgem T, Rushton PJ, Schmelzer E, Hahlbrock K, Somssich IE. Early nuclear events in plant defence signalling: rapid gene activation by WRKY transcription factors. EMBO J. 1999;18:4689–4699. doi: 10.1093/emboj/18.17.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maeo K, Hayashi S, Kojima-Suzuki H, Morikami A, Nakamura K. Role of conserved residues of the WRKY domain in the DNA-binding of tobacco WRKY family proteins. Biosci. Biotechnol. Biochem. 2001;65:2428–2436. doi: 10.1271/bbb.65.2428. [DOI] [PubMed] [Google Scholar]
- 50.Banks JA, Nishiyama T, Hasebe M, Bowman JL, Gribskov M, dePamphilis C, Albert VA, Aono N, Aoyama T, Ambrose BA, et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science. 2011;332:960–963. doi: 10.1126/science.1203810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Felsenstein J. An alternating least squares approach to inferring phylogenies from pairwise distances. Syst. Biol. 1997;46:101–111. doi: 10.1093/sysbio/46.1.101. [DOI] [PubMed] [Google Scholar]
- 54.Felsenstein J. A comparative method for both discrete and continuous characters using the threshold model. Am. Nat. 2012;179:145–156. doi: 10.1086/663681. [DOI] [PubMed] [Google Scholar]
- 55.Swafford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sunderland, Massachusetts: Sinauer Associates; 2003. http://www.sinauer.com/paup-phylogenetic-analysis-using-parsimony-and-other-methods-4-0-beta.html (9 August 2013, date last accessed) [Google Scholar]
- 56.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen SX, Moulin M, Hashemolhosseini S, Kilian K, Wegner M, Muller CW. Structure of the GCM domain-DNA complex: a DNA-binding domain with a novel fold and mode of target site recognition. EMBO J. 2003;22:1835–1845. doi: 10.1093/emboj/cdg182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hildebrandt A, Dehof AK, Rurainski A, Bertsch A, Schumann M, Toussaint NC, Moll A, Stockel D, Nickels S, Mueller SC, et al. BALL–biochemical algorithms library 1.3. BMC Bioinformatics. 2010;11:531. doi: 10.1186/1471-2105-11-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Case D, Darden TA, Cheatham TE, Simmerling C, Wang J, Duke R, Luo R, Crowley M, Walker R, Zhang W, et al. Amber 11. San Francisco: University of California; 2010. [Google Scholar]
- 61.Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, III, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 65.Cheatham TE, Kollman PA. Molecular dynamics simulations highlight the structural differences among DNA:DNA, RNA:RNA and DNA:RNA hybrid duplexes. J. Amer. Chem. Soc. 1997;119:4805–4825. [Google Scholar]
- 66.Chiu SW, Clark M, Subramaniam S, Jakobsson E. Collective motion artifacts arising in long-duration molecular dynamics simulations. J. Comput. Chem. 2000;21:121–131. [Google Scholar]
- 67.Harvey SC, Tan RKZ, Cheatham TE. The flying ice cube: velocity rescaling in molecular dynamics leads to violation of energy equipartition. J. Comput. Chem. 1998;19:726–740. [Google Scholar]
- 68.Darden T, Pearlman D, Pedersen LG. Ionic charging free energies: spherical versus periodic boundary conditions. J. Chem. Phys. 1998;109:10921. [Google Scholar]
- 69.Ryckaert JP, Ciccotti G, Berendsen H. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 70.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 71.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 72.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramamoorthy R, Jiang SY, Kumar N, Venkatesh PN, Ramachandran S. A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments. Plant Cell Physiol. 2008;49:865–879. doi: 10.1093/pcp/pcn061. [DOI] [PubMed] [Google Scholar]
- 75.Rossberg M, Theres K, Acarkan A, Herrero R, Schmitt T, Schumacher K, Schmitz G, Schmidt R. Comparative sequence analysis reveals extensive microcolinearity in the lateral suppressor regions of the tomato, Arabidopsis, and Capsella genomes. Plant Cell. 2001;13:979–988. doi: 10.1105/tpc.13.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun C, Palmqvist S, Olsson H, Boren M, Ahlandsberg S, Jansson C. A novel WRKY transcription factor, SUSIBA2, participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter. Plant Cell. 2003;15:2076–2092. doi: 10.1105/tpc.014597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Honig B, Rohs R. Biophysics: flipping Watson and Crick. Nature. 2011;470:472–473. doi: 10.1038/470472a. [DOI] [PubMed] [Google Scholar]
- 78.Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010;79:233–269. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lane WJ, Darst SA. The structural basis for promoter -35 element recognition by the group IV sigma factors. PLoS Biol. 2006;4:e269. doi: 10.1371/journal.pbio.0040269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Welner DH, Lindemose S, Grossmann JG, Mollegaard NE, Olsen AN, Helgstrand C, Skriver K, Lo Leggio L. DNA binding by the plant-specific NAC transcription factors in crystal and solution: a firm link to WRKY and GCM transcription factors. Biochem. J. 2012;444:395–404. doi: 10.1042/BJ20111742. [DOI] [PubMed] [Google Scholar]
- 81.Ernst HA, Olsen AN, Larsen S, Lo Leggio L. Structure of the conserved domain of ANAC, a member of the NAC family of transcription factors. EMBO Rep. 2004;5:297–303. doi: 10.1038/sj.embor.7400093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lai Z, Li Y, Wang F, Cheng Y, Fan B, Yu JQ, Chen Z. Arabidopsis sigma factor binding proteins are activators of the WRKY33 transcription factor in plant defense. Plant Cell. 2011;23:3824–3841. doi: 10.1105/tpc.111.090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Song MJ, Hwang S, Wong W, Round J, Martinez-Guzman D, Turpaz Y, Liang J, Wong B, Johnson RC, Carey M, et al. The DNA architectural protein HMGB1 facilitates RTA-mediated viral gene expression in gamma-2 herpesviruses. J. Virol. 2004;78:12940–12950. doi: 10.1128/JVI.78.23.12940-12950.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.