

Abstract
The yeast Pichia pastoris is a common host for the recombinant production of biopharmaceuticals, capable of performing posttranslational modifications like glycosylation of secreted proteins. However, the activity of the OCH1 encoded α-1,6-mannosyltransferase triggers hypermannosylation of secreted proteins at great heterogeneity, considerably hampering downstream processing and reproducibility. Horseradish peroxidases are versatile enzymes with applications in diagnostics, bioremediation and cancer treatment. Despite the importance of these enzymes, they are still isolated from plant at low yields with different biochemical properties. Here we show the production of homogeneous glycoprotein species of recombinant horseradish peroxidase by using a P. pastoris platform strain in which OCH1 was deleted. This och1 knockout strain showed a growth impaired phenotype and considerable rearrangements of cell wall components, but nevertheless secreted more homogeneously glycosylated protein carrying mainly Man8 instead of Man10 N-glycans as a dominant core glycan structure at a volumetric productivity of 70% of the wildtype strain.
The methylotrophic yeast Pichia pastoris has long been used for the production of recombinant proteins at high titers. Up to 22 g·L−1 have been reported for intracellularly produced recombinant hydroxynitrile lyase1 and approximately 15 g·L−1 for secreted recombinant gelatin2, demonstrating the high production capacity of this microbial host while being able to grow on comparatively simple and inexpensive media. Not only can P. pastoris be grown to cell densities as high as 160 g·L−1 dry cell weight3, it is also capable of performing posttranslational modifications, including the formation of correct disulfide bridges and the glycosylation of secretory proteins, rendering P. pastoris specifically suitable for the production of complex eukaryotic proteins4.
Glycosylation has long been known to affect various protein properties such as solubility, stability and enzymatic activity (e.g.5,6), which need to be evaluated on a case-by-case basis. Whereas only little is known about O-linked glycosylation, the biosynthesis of N-glycans is well understood. N-glycans are linked to the amido groups of asparagine residues that are recognized by glycotransferases in the sequence motif N-X-S/T, where X is any amino acid but proline. Initially, the biosynthesis steps of N-glycans in yeast and mammals are identical. Dolichol phosphate-linked N-acetylglucosamine (DolP-GlcNAc) is synthesized by the transfer of GlcNAc from uridine diphosphate (UDP) onto DolP on the cytoplasmic side of the ER. After extension to DolP-linked Man5GlcNAc2, this structure is enzymatically flipped to the ER lumen, where further glucose (Glc) and Man residues are added to form a core glycan, Glc3Man9GlcNAc2, which is transferred to an asparagine within the N-X-S/T sequence motif of a nascent protein chain. Subsequently, the three terminal Glc residues and one Man residue are trimmed by glucosidases I and II and an ER-residing α-1,2-mannosidase to form Man8GlcNAc2. At this point, the newly formed glycoprotein is transported to the Golgi apparatus, which is where the yeast and mammalian N-glycosylation pathways diverge7,8,9. In the mammalian Golgi apparatus, α-1,2-mannosidases trim the core glycan further to form Man5GlcNAc2. Ultimately, addition of GlcNAc by a β-N-acetylglucosaminyltransferase I (GnTI), trimming of two further Man residues by a mannosidase II and yet further addition of GlcNAc, galactose (Gal) and sialic acid (Sia) residues by the respective transferases result in the complex N-glycan structures of mammalian proteins10. In the yeast Golgi, on the other hand, the Man8GlcNAc2 glycan is not subjected to further trimming reactions but is substantially extended. In Saccharomyces cerevisiae, more than 100 Man residues may account for hypermannosyl N-glycans on secretory proteins. However, the extent of hypermannosylation varies considerably and seems to depend on so far unknown influences, causing vast heterogeneity in the N-glycan pattern of secreted glycoproteins. In S. cerevisiae as well as in P. pastoris and other yeasts, the first reaction in hypermannosylation is catalyzed by an α-1,6-mannosyltransferase (Och1p) that is encoded by the gene Outer CHain elongation 1 (OCH1), which was first discovered and characterized in S. cerevisiae11,12. Och1p uses Man from guanosin diphosphate and links it to the core glycan by an α-1,6-glycosidic bond, forming a substrate that triggers additional mannosylation (Figure 1). Whereas S. cerevisiae holds a repertoire of Golgi-resident α-1,2-, α-1,3 and α-1,6-mannosyl and mannosylphosphate transferases, P. pastoris seems to lack the Golgi-resident α-1,3-mannosyltransferase, but to possess four additional β-mannosyltransferases instead7,13,14.
Figure 1. Och1p in N-glycan biosynthesis.

In the Golgi, the α-1,6-mannosyltransferase activity (α-1,6-MnT) of Och1p extends the N-linked Man8GlcNAc2 core glycan, which is then heterogeneously hyperglycosylated by several additional (phospho-) mannosyltransferases (MnTs). Galanthus nivalis lectin (GNL) and Lens culinaris lectin (LCA) bind to the different glycan structures either with high (thick arrow) or low (thin arrow) specificity.
Although not as extensive as those of S. cerevisiae, the N-glycans of P. pastoris are also of the high mannose type and the humanization of the N-glycosylation machinery of P. pastoris has been the subject of several studies (Table 1).
Table 1. Humanization of N-glycans in P. pastoris. Selected studies focusing on the humanization of the N-glycans on P. pastoris derived glycoproteins. Bmt, β-mannosyltransferase; Mns, mannosidase; GnT, β-N-acetylglucosaminyltransferase; UDP-GlcNAc, uridine diphosphate-N-acetylglucosamine; OCH1, outer chain elongation gene 1.
| content | references |
|---|---|
| introduction of α-1,2-Mns, GnTI and an UDP-GlcNAc transporter via a combinatorial genetic library approach in a Δoch1::URA3 strain | 13,50 |
| introduction of an UDP-GlcNAc transporter, α-1,2-MnsIA, MnsII, GntI, GntII in a Δoch1::URA3 strain | 51 |
| introduction of α-1,2-Mns and GnTI, OCH1 inactivation via a knockin plasmid | 10 |
| GlycoSwitch plasmids for OCH1 inactivation and introduction of glycosidase and glycosyltransferase activities to produce complex terminally galactosylated glycoproteins | 52 |
| introduction of sialic acid biosynthesis pathway and corresponding transporter and transferase activities to produce complex terminally sialylated glycoproteins | 53 |
| elimination of α-Mns resistant glycan structures by inactivation of the activities of Bmt1p, Bmt2p and Bmt3p | 54 |
Here, we report the deletion of the OCH1 gene from the P. pastoris genome in an irreversible and straight forward approach. Thereby, we generated a new P. pastoris platform strain that allows the production of recombinant proteins with shorter glycan structures of considerably increased homogeneity compared to proteins produced in a wildtype strain. In contrast to previous glycoengineering studies, which required several time- and labor-intensive steps of strain engineering, we achieved more homogeneously glycosylated protein with a single gene knockout step. Horseradish peroxidase (HRP) is a versatile enzyme with applications in diagnostics and histochemistry, bioremediation and cancer treatment. However, due to the lack of an appropriate recombinant production process, HRP preparations are still derived from horseradish roots as mixtures of different isoenzymes15. In the present study, we produced recombinant HRP in an och1 knockout strain in the controlled environment of a bioreactor, purified and characterized the enzyme, thus demonstrating the general applicability of this new platform strain by the example of this industrially and medically relevant enzyme.
Results
Knockout of OCH1 from Ppku70- and PpMutS
In yeast, the OCH1 gene encodes an α-1,6-mannosyltransferase whose activity triggers the subsequent transfer of further mannose and phosphomannose residues onto the N-glycans of secreted proteins in the Golgi apparatus, resulting in heterogeneously hyperglycosylated protein species that appear as a smear on SDS gels, e.g.16,17. This hyperglycosylation not only limits the use of yeast derived proteins as biopharmaceuticals but also greatly impedes traditional downstream processing. Hence, a P. pastoris strain that allows the production of less heterogeneously glycosylated proteins would considerably relieve protein production processes with P. pastoris.
A flipper cassette targeting to the OCH1 locus was transformed to a Ppku70- strain to replace the OCH1 open reading frame. This Ppku70- strain has to rely on homologous recombination for gene integration events, in contrast to a wildtype strain18. The cassette construct and the knockout workflow are schematically shown in Fig. 2.
Figure 2. Schematic workflow of the knockout of OCH1 using a flipper cassette.

The regions 5′ OCH1 and 3′ OCH1 represent sequences upstream and downstream of the OCH1 ORF, respectively. The 34 bp flipper recombinase target (FRT) sequences flank the AOX1 promoter (P AOX1), the FLP recombinase ORF, the CYC1 transcription terminator (CYC TT), a constitutive eukaryotic and a prokaryotic promotor (P ILV and P EM72, respectively), a ble ORF mediating Zeocin™ resistance and an AOD transcription terminator (AOD TT). A double homologous recombination event replaced the OCH1 ORF in the genome with the flipper cassette. Growth of recombinant cells on methanol induced the production of the FLP recombinase which recognized the two FRT sites and excised the inner sequence, leaving only one FRT site in the genome. Single fragments are not drawn to scale.
Transformation of the flipper cassette to the Ppku70- strain resulted in only few Zeocin™ resistant clones. However, Sanger sequencing of a PCR amplified fragment of the OCH1 locus from genomic DNA showed that the majority of the tested transformants had correct integration of the transformed cassette. The transformants grew slowly and formed colonies of abnormal shape. This phenotype was preserved when the strains were grown on minimal methanol agar plates to induce the production of the FLP recombinase and subsequent excision of the inner part of the flipper cassette containing the expression cassettes for the FLP recombinase and the Zeocin™ resistance enzyme. Reconstituted sensitivity to Zeocin™, PCR and Sanger sequencing of the former OCH1 locus confirmed the successful excision of the inner part of the flipper cassette and the efficient replacement of the former OCH1 ORF with a single FRT site of 34 bp.
Transformation of the same OCH1 flipper cassette to PpMutS resulted in hundreds of clones resistant to Zeocin™. Initial PCR based screenings of over 100 randomly chosen clones did not give any positive hits, analogously to what has been described by Vervecken et al.10. However, after having identified the corresponding phenotype of positive transformants in the Ppku70- based och1 knockout strain, designated PpFWK1, also PpMutS based transformants with correct integration of the flipper cassette could be spotted easily on the agar plates since they showed the same unusual colony phenotype as colonies of PpFWK1 (Figure 3).
Figure 3. Colony phenotypes.

(a), PpMutS; (b), PpFWK3. Both strains were grown on YPD agar.
Increasing the incubation time of the transformed cells on the agar plates to at least four days allowed growth of the och1 knockout colonies to a size at which their abnormal shape was an obvious hint to their genotype (Figure 3). Again, Zeocin™ sensitivity was reconstituted by induction of the FLP recombinase on minimal methanol agar plates and the replacement of the OCH1 ORF by a single FRT site was shown by PCR and Sanger sequencing. The observed phenotype of the generated och1 knockout strain PpFWK3 included slow growth, abnormal colony shape and temperature sensitivity at 37°C (data not shown). Upon transformation of the wildtype OCH1 promoter and OCH1 ORF to PpFWK3, the detrimental phenotype was found to be complemented. PCR analyses confirmed the unaltered replacement of the former OCH1 ORF by a FRT site, but the presence of the complementing OCH1 ORF somewhere else in the genome due to ectopic integration of the transformed plasmid pPpT4_BamHI_OCH1rescue.
Strain morphology
Cell morphology and cell division of an och1 knockout strain
Consistent with previous reports on Och1p deficient S. cerevisiae strains19,20, we found the N-glycosylation mutant to be characterized by an altered phenotype and growth profile (Table 2). In contrast to the wildtype based strain PpMutS, the och1 knockout strain PpFWK3 grew in the form of large cell clusters with clumpy appearance and multibudded cells (Figure 4). Daughter cells within these clusters displayed clearly segregated vacuoles, but remained stably attached to the wall of mother cells (Supplementary Figure 1).
Table 2. Strain specific parameters of the different P. pastoris strains. Parameters are defined in the footnotex.
| PpMutS | PpMutSHRP | PpFWK3 | PpFWK3HRP | |
|---|---|---|---|---|
| max. μGly (h−1) | 0.30 | 0.31 | 0.20 | 0.20 |
| qGly (mmol·g−1·h−1) | 2.90 | 3.10 | 1.90 | 1.90 |
| YX/Gly (Cmol·Cmol−1) | 0.63 | 0.41 | 0.61 | 0.54 |
| YCO2/Gly (Cmol·Cmol−1) | 0.33 | 0.64 | 0.37 | 0.44 |
| Δtimeadapt (h) | 15.7 | 19.9 | 6.60 | 8.90 |
| qMeOH (mmol·g−1·h−1) | 0.62 | 0.70 | 0.52 | 0.43 |
| max. qMeOH-(mmol·g−1·h−1) | 0.67 | 0.78 | 0.69 | 0.53 |
| YX/MeOH (Cmol·Cmol−1) | 0.39 | 0.07 | 0.05 | 0.04 |
| YCO2/MeOH (Cmol·Cmol−1) | 0.57 | 1.02 | constantly decreasing | constantly decreasing |
| C-balance | 0.97 | 1.04 | constantly decreasing | constantly decreasing |
| qp (U·g−1·h−1) | - | 0.77 | - | 0.50 |
| vol. productivity (U·L−1·h−1) | - | 2.60 | - | 1.80 |
| efficiency factor (η) (U·mmol−1 26) | - | 1.10 | - | 1.20 |
Xmax. μGly, maximum specific growth rate on glycerol; qGly, specific uptake rate of glycerol during the batch; YX/Gly, biomass yield on glycerol; YCO2/Gly, CO2 yield on glycerol; Δtimeadapt, time from first addition of methanol to a maximum in offgas activity; qMeOH, average specific uptake rate of methanol during consecutive methanol pulses; max. qMeOH, maximum specific uptake rate of methanol during consecutive methanol pulses; YX/MeOH biomass yield on methanol; YCO2/MeOH, CO2 yield on methanol; C-balance, sum of biomass and CO2 yields; qp, specific productivity of HRP; vol. productivity, volumetric productivity of HRP; efficiency factor; efficiency of the conversion of substrate methanol into product HRP.
Figure 4. Phenotypic change in P. pastoris upon OCH1 knockout.
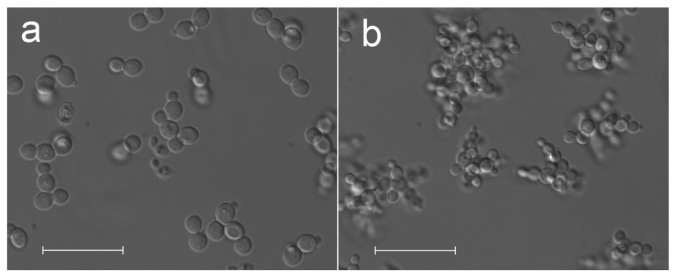
Representative DIC micrographs of PpMuts and PpFWK3. (a), PpMutS cells in batch culture. (b), covalently linked clusters of multibudded cells in PpFWK3 during the same cultivation phase. Scale bars represent 25 μm.
Knockout induced stress response is reflected by a spatial rearrangement of WGA/STL binding sites
The direct functional implication of the altered N-glycosylation in PpFWK3 for cell morphology and cytokinesis was further illustrated by a striking difference in the chitin deposition after OCH1 knockout (Supplementary Figure 1). In lectin based glycoprofiling studies, we observed substantially altered binding patterns for the GlcNAc-specific lectin WGA, with the reactive carbohydrate motifs being homogeneously distributed across the entire cell surface in the och1 knockout strain, instead of remaining confined to the bud scars as in PpMutS (Figure 5 and Supplementary Figure 1).
Figure 5. Lectin based glycoprofiling of surface carbohydrate motifs in PpMutS and PpFWK3.

Live cells harvested in the exponential growth phase of batch cultivation were incubated with fluorescein labeled lectins. Micrographs show the isolated channel for FITC detection and merged images with DIC (overlay). Scale bars represent 10 μm.
In order to assess whether a mere alteration in the steric accessibility of chitin chains in the lateral wall was responsible for the difference in the WGA staining behavior or whether chitin was actually specifically localized at bud scars, PpMutS cells were subjected to the same fluorescence microscopic analysis after treatment with concentrated methanol, leading to denaturation of mannoproteins and a substantial increase in cell wall permeability21. The efficiency of the cell wall permeabilization protocol was validated by concomitant incubation with a usually non-membrane penetrating DAPI dye, which could readily access the nuclear space after methanol treatment. Still, methanol permeabilized PpMutS cells displayed only the conventional, bud scar selective staining pattern, contrasting to the generalized binding of GNL, which served as a control (Supplementary Figure 2).
Quantitatively, comparative overall ratios of WGA/FM® 4–64 were found for PpMutS and the och1 knockout strain PpFWK3 (Figure 6), indicating that mainly a spatial redistribution of chitin may be induced by altering the glycosylation machinery, but without general enhancement of the total cellular chitin level. In other words, the rather high chitin concentration at the bud scars in PpMutS seemed to be reduced in favor of an increased chitin deposition in the lateral cell walls of PpFWK3. These results were also confirmed via the chitin binding lectin STL, which has a similar carbohydrate specificity profile as WGA but only limited affinity to isolated GlcNAc residues (Figure 6).
Figure 6. Quantitative determination of lectin binding on PpMutS and PpFWK3 cells.

Cell suspensions adjusted to the same concentration level were incubated with FITC labeled lectins. After thorough washing, cells were lysed and the fluorescence intensity of the lysis buffer recorded. Binding data was normalized to the average cellular content of FM® 4–64, which was shown to be similar in both strains via FACS analysis. Values represent mean ± SD of three independent experiments.
Inactivation of the Och1p activity shifted the interaction capacity of mannose specific lectins
The och1 knockout cells of the PpFWK3 strain displayed a lower binding capacity for the lectin GNL than PpMutS cells (Figure 6). GNL interacts with high mannose N-glycans and preferably reacts with α-1,3-Man residues, but also binds to α-1,6 linked Man residues22,23. In the current study, the GNL/FM® 4–64 ratio was reduced by 50% in PpFWK3 as compared to PpMutS, but still remained the lectin with highest binding capacity for this strain (Figure 6). In striking contrast, the binding levels of the glycan core-binding lectin LCA were increased upon OCH1 knockout (Figure 6). Both, GNL and LCA showed an equal distribution across the entire cell wall (Figure 5), corresponding to the established localization of their putative targets, high-mannose N-glycans and core glycans, respectively.
Production of HRP in the strains PpMutSHRP and PpFWK3HRP
To show the applicability of the generated och1 knockout strain for the production of recombinant proteins, a vector harboring a gene coding for an acidic HRP isoenzyme was transformed into either PpMutS or PpFWK3. Transformation of the linearized constructs into PpFWK3 resulted in fewer Zeocin™ resistant clones than for PpMutS, but sufficient to allow screening for HRP activity after cultivation in a 96-deep well plate in minimal media. Despite the apparent growth defect of PpFWK3, the volumetric yields in HRP activity in these micro-scale cultivations were comparable to those of PpMutS based transformants. Prior to cultivation of PpMutSHRP or PpFWK3HRP in the bioreactor, both strains were analyzed in terms of copy number of the transformed HRP gene to ensure comparability on this level. Both strains were found to have a single copy integration of the HRP encoding gene and were thus considered suitable for comparative bioreactor cultivations.
Strain characterization in bioreactors
We characterized four P. pastoris strains (Table 2) with a recently published method of conducting dynamic experiments during batch cultivations in the controlled environment of a bioreactor16,24,25. After depletion of glucose, a first methanol adaption pulse with a final concentration of 0.5% (v/v) was applied. The adaptation times to the new substrate methanol (Δtimeadapt), defined as the maximum in offgas activity, were determined for all four P. pastoris strains and are shown in Table 2.
The calculated carbon dioxide evolution rate (CER), illustrating the metabolic activity of the different strains, the specific substrate uptake rate (qs) and, where appropriate, the specific productivity (qp), during the methanol pulses are shown in Supplementary Fig. 3–6. As shown in Supplementary Fig. 3 and 4, the CER profiles for the strains PpMutS and PpMutSHRP showed a similar pattern during the consecutive methanol pulses and qs values stayed constant over time. In contrast, the CER profiles for the strains PpFWK3 and PpFWK3HRP substantially changed over time (Supplementary Figures 5 and 6). After each methanol pulse, less CO2 was produced per time and volume, indicating that the P. pastoris cells became metabolically less active. Thus, the consumption of 1% (v/v) methanol took longer after each consecutive pulse (compare Supplementary Figures 3 and 4 with Supplementary Figures 5 and 6). The altered metabolic activity of the och1 knockout strains was also depicted in the calculated yields (YX/S and YCO2/S), which are shown in Fig. 7.
Figure 7. Calculated yields for the different P. pastoris strains during batch cultivations with methanol pulses.

(a), PpMutS; (b), PpMutSHRP; (c), PpFWK3; (d), PpFWK3HRP. Black square, carbon dioxide yield (YCO2/S); black dot, biomass yield (YX/S).
For the strains PpMutS and PpMutSHRP both the carbon dioxide yield (YCO2/S) and the biomass yield (YX/S) stayed constant during the six conducted consecutive methanol pulses (Figure 7a and b). Evidently, the insertion of the HRP gene into strain PpMutS affected its physiology as YX/S decreased, whereas YCO2/S increased (compare Figure 7a and b). Hence, PpMutSHRP mainly used the substrate methanol for protein production and dissimilation than for biomass growth.
Interestingly, the calculated yields for PpFWK3 and PpFWK3HRP strains showed a very different behaviour. Although YX/S was again rather constant, YCO2/S decreased dramatically in the course of the six to seven consecutive methanol pulses (Figure 7c and d), indicating that these two strains became more and more metabolically inactive. Since HPLC analysis revealed that no undesired metabolites were produced in substantial amounts during any of the four cultivations, the C-balances for strains PpFWK3 and PpFWK3HRP were determined close to 1.0 only at the beginning of the cultivation but rapidly decreased over time, whereas the C-balances for strains PpMutS and PpMutSHRP were always determined to be close to 1.0 (Supplementary Figure 7). A summary of the determined strain specific parameters of the four different P. pastoris strains is given in Table 2.
As shown in Table 2, the maximum specific growth rates on glycerol (max. μGly) for PpFWK3 strains were approximately 1.5-fold lower than for PpMutS strains. The yields on glycerol showed a similar pattern for PpMutS and PpFWK3 strains, as the yields were shifted towards production of carbon dioxide rather than biomass when the strains were hosting the gene for recombinant HRP. Both PpFWK3 strains needed less than half of the adaptation time to methanol (Δtimeadapt) compared to the PpMutS strains. The altered glycosylation machinery in the PpFWK3 strains seems to allow a faster adaption to methanol, which could be of great significance for industrial applications where fast and efficient bioprocesses are required. As expected, the adaptation times of the strains hosting the recombinant enzyme were longer compared to the strains not carrying this additional gene. The average specific substrate uptake rates for both substrates (qs) glycerol and methanol were lower for the PpFWK3 strains than for PpMutS strains. In respect to the maximum specific uptake rate for methanol (max. qMeOH) the values for the different strains were quite similar. However, these values were determined during the 3rd and the 5th methanol pulse for PpMutS and PpMutSHRP, respectively. The values during the other pulses were alike. For PpFWK3 strains on the other hand, these maxima could only be determined during the 1st methanol pulse. After that, the values for qs constantly decreased, indicating a progressional reduction of the metabolic activity of the PpFWK3 strains during consecutive methanol pulses. This was also underlined by constantly decreasing YCO2/S and C-balances. However, despite this seemingly negative impact, PpFWK3HRP produced the HRP isoenzyme at specific and volumetric productivities which were only reduced by 35% and 30%, respectively, compared to PpMutSHRP. In a previous study, we introduced the efficiency factor η, which puts the productivity of the strains in direct relation to the consumed substrate26. In this respect, the och1 knockout strain PpFWK3HRP even showed a higher ratio than PpMutSHRP and thus proved to be of justifiable interest for the production of recombinant proteins.
Enzyme purification and characterization
In order to demonstrate the successful decrease of heterogeneity in the glycosylation of HRP when produced in an och1 knockout strain, size exclusion chromatography (SEC) was performed with cell free cultivation broth (Figure 8). A SEC elution profile has higher specificity and sensitivity than an image of an SDS polyacrylamide gel and was therefore preferably used for this purpose. HRP produced in PpMutSHRP eluted with higher heterogeneity than HRP produced in PpFWK3HRP.
Figure 8. Size exclusion chromatogram of HRP glycovariants.

Solid line, HRP produced in PpMutSHRP; dashed line, HRP produced in PpFWK3HRP. The run was performed at a flow of 9 cm·h−1 and fractions of 1.2 mL were collected. The measured HRP activities per fraction are shown as relative activities with the respective maximum activities set to 100% for better comparability. The unnormalized maximum activities were 2.6 and 15.1 U·mL−1 for HRP from PpMutSHRP and from PpFWK3HRP, respectively. The loaded volume was approximately 200 μL for HRP from either PpMutSHRP or PpFWK3HRP.
Judging by the size exclusion chromatogram (Figure 8), HRP produced in a PpMutSHRP strain substantially differed in its surface glycosylation pattern compared to HRP produced in a PpFWK3HRP strain, which showed higher homogeneity. To analyze this phenomenon in detail, we enzymatically released the glycans from the produced recombinant HRPs and analyzed them via liquid chromatography-mass spectrometry (LC-MS). Reducing glycans were observed mainly as doubly charged [M + H + NH4]2+ ions (Figure 9). Analysis of enzymatically released glycans from HRP produced in either PpMutSHRP or PpFWK3HRP confirmed the expected decrease in both N-glycan size and heterogeneity. The dominant core glycan structure shifted from Man10 to Man8 in strain PpFWK3HRP (Table 3). As shown in Fig. 9, HRP produced in the strain PpMutSHRP carried a greater variety of different glycan chains consisting of up to 17 mannoses and a higher amount of phosphorylated sugars than HRP produced in PpFWK3HRP.
Figure 9. Chromatogram of liquid chromatography-mass spectrometry of glycans released from HRP produced in either PpMutSHRP or in PpFWK3HRP.

HRP [M + 2H]2+ and [M + 2NH4]2+ ions are marked with ° and *, respectively. “Man10-P” or just “12-P” indicate phosphorylated glycans. lock, lock mass.
Table 3. Relative peak areas of identified glycan structures cleaved off from HRP recombinantly produced in either PpMutSHRP or in PpFWK3HRP.
| PpMutSHRP | PpFWK3HRP | |
|---|---|---|
| glycan structure | relative peak area [%] | |
| Man7 | 0.0 | 7.9 |
| Man8 | 0.0 | 57.3 |
| Man9 | 15.7 | 16.8 |
| Man10 | 31.4 | 6.1 |
| Man10-P | 5.6 | 0.0 |
| Man11 | 18.7 | 6.8 |
| Man11-P | 6.7 | 0.0 |
| Man12 | 6.7 | 3.2 |
| Man12-P | 4.4 | 0.0 |
| Man13 | 3.7 | 1.8 |
| Man13-P | 1.8 | 0.0 |
| Man14 | 2.1 | 0.0 |
| Man14-P | 1.1 | 0.0 |
| Man15 | 1.2 | 0.0 |
| Man16 | 0.5 | 0.0 |
| Man17 | 0.4 | 0.0 |
| total | 100.0 | 100.0 |
The evaluation of the relative peak areas underlined this observation (Table 3), as around 60% of the identified glycan structures cleaved off from HRP produced in the och1 knockout strain PpFWK3HRP were of the Man8 type, whereas no structure of that type was identified for HRP from PpMutSHRP. As shown in Table 3 there were also much more different glycanchains identified on HRP from PpMutSHRP and interestingly no phosphorylated mannose structures were found on HRP from PpFWK3HRP.
To check whether the kinetic constants or the stability of the enzyme were affected by the altered glycosylation pattern, we characterized purified preparations of HRP. HRP preparations produced by either PpMutSHRP or PpFWK3HRP in bioreactor cultivations were purified by using a recently described strategy for HRP isoenzyme C1A27. Both HRP preparations did not bind to the mixed mode HCIC resin but were found in the flowthrough, i.e. 93% of HRP produced in PpMutSHRP and 87% of HRP produced in PpFWK3HRP. Contaminating proteins were retained on the resin, leading to a partial purification at a factor of approximately 2.5 for both enzyme solutions. A subsequent size exclusion step gave an additional purification factor of approximately 2.0. After purification, the fractions with the highest purification factor were pooled and ultrafiltrated. The enzyme HRP produced in PpMutSHRP was concentrated to around 3.0 mg·mL−1, whereas HRP produced in PpFWK3HRP could not be concentrated due to the immediate formation of precipitates during ultrafiltration and the resulting clogging of the membrane, indicating a reduced solubility of the extracellular proteins in this preparation. We determined the kinetic constants for both enzyme preparations with H2O2 as electron donor at saturating concentration and ABTS as electron acceptor in varying concentrations (Table 4; Supplementary Figure 8).
Table 4. Enzymatic characterization of homogeneously glycosylated HRP. Kinetic constants of HRP produced in either PpMutSHRP or PpFWK3HRP with H2O2 as electron donor at saturating concentration and ABTS as electron acceptor in varying concentrations were determined at 420 nm and 30°C.
| production strain | KM_ABTS [mM] | Vmax [μmol·s−1·μg−1] |
|---|---|---|
| PpMutSHRP | 2.40 | 3.07 |
| PpFWK3HRP | 2.03 | 2.46 |
As shown in Table 4, the affinity of HRP towards the substrate ABTS was increased, as the KM was found to be decreased by approximately 15% by the altered surface glycosylation. However, Vmax was decreased by nearly 20%. Also, the thermal stability of the produced HRP glycovariants at 60°C was studied (Supplementary Figure 9) and the half life times (τ1/2) were determined28. The τ1/2 of HRP produced in PpMutSHRP was determined with 384 s, whereas HRP from PpFWK3HRP showed a reduction in its thermal half life time of around 50% with 198 s.
Discussion
Despite the numerous advantages of using P. pastoris as a host organism for recombinant protein production, its inherent heterogeneous yeast type hyperglycosylation of secreted proteins has to be addressed by extensive and elaborated strain modifications. Here, we present a straight forward approach for the generation of a wildtype based P. pastoris platform strain that allows the production of more homogeneously glycosylated recombinant proteins due to an irreversible deletion of the OCH1 gene.
Yeast hypermannosylation largely depends on the initial activity of an α-1,6-mannosyltransferase in the Golgi apparatus. Elimination of this activity was achieved by replacement of the OCH1 ORF with a single 34 bp FRT site by using a flipper cassette. However, this approach required double homologous recombination at the correct locus in the genome. Unfortunately, homologous integration events only play a minor part in P. pastoris, as recently demonstrated by Näätsaari et al.18 and which was found to be especially true for the OCH1 locus by Vervecken et al.10. A new P. pastoris strain with inactivated non-homologous end joining pathway, designated Ppku70-, proved to be a particularly convenient tool to identify the phenotype of the specific knockout strain in this study. Since homologous integration was the sole possibility for recombination events in the Ppku70- strain, the total number of positive transformants was predominantly made up by transformants with homologous integration. The fact that particularly few colonies were obtained by targeting of the transformed flipper cassette to the OCH1 locus when using the Ppku70- strain also supported the hypothesis of increased difficulty of homologous recombination in that locus.
An och1 knockout strain of S. cerevisiae was described to show several defects such as impaired budding and increased temperature sensitivity11. Choi et al. mentioned temperature sensitivity and increased flocculation for their P. pastoris och1 knockout strain13, but neither they nor Vervecken et al.10 described any further severe growth defects. However, the och1 knockout strain in the present study was found to show not only formation of cell clusters and temperature sensitivity, but also decreased growth which might be due to an impaired cell wall structure and thus complicated bud formation. Similarly, a recently generated och1 knockout strain based on the his4 mutant strain P. pastoris GS115 was described with slower growth and rough colony surface29. Explanations for these divergent findings remain speculative, but might be due to single nucleotide polymorphisms and hence different strain backgrounds. Also, a secondary integration event of the transformed flipper cassette cannot be completely excluded. However, considering that as little as 100 ng of the cassette were transformed and that all clones that exposed the described phenotype had the correct integration of the cassette in the OCH1 locus indicates that the observed phenotype can be ascribed rather to the knockout of OCH1 than to any additional genomic rearrangement. Also, the similarity of the observed phenotype in the present P. pastoris och1 knockout strain to the phenotype described for a S. cerevisiae och1 knockout strain11 very much suggests that the deletion of the OCH1 gene is actually responsible for the phenotype observed in this study. Most strikingly, reintroduction of the wildtype OCH1 gene to the och1 knockout strain PpFWK3 restored its phenotype, thus conclusively linking the observed phenotype of PpFWK3 to the deletion of the OCH1 gene. Transformation of the och1 knockout strain with a linearized vector harboring an expression cassette for the production of an HRP isoenzyme via electroporation was found to result in lower transformation efficiency than electroporation of a P. pastoris wildtype strain. This increased sensitivity to electroporation might be another reflection of the altered cell wall composition that could be shown by lectin based glycoprofiling.
The highly specific interaction of the lectin WGA with the exposed chitin ring of bud scars30 has previously been reported for other yeasts and can be used for the precise determination of the number of cell divisions performed31,32. To the best of our knowledge, this is the first confirmation of this structure-related specificity for P. pastoris. However, in the och1 knockout strain, we noticed an almost complete loss of bud scar selectivity. The reasons for the regionally diverse distribution of chitin in PpFWK3 may either lie in an increased accessibility of previously cryptic chitin chains in the lateral cell wall (i.e. when the protective polymannan layer is missing), or in an actively increased chitin synthesis and deposition at the cell wall, i.e. as a compensatory response to the cell wall stress caused by the impaired barrier function. Such stimulation of counter-regulatory pathways upon impairment of cell wall integrity has been observed in several yeast species33,34 and involves diverse mechanisms and signaling cascades35, which are believed to be directly or indirectly connected to the deletion of Och1p activity20. Especially the osmotic stress exerted on the cell as a result of the impaired cell wall integrity is known to present an important factor for the induction of counter-regulatory pathways36. Due to the persistence of bud scar specific binding of WGA in methanol treated PpMutS cells, we concluded that the strong affinity of WGA to the overall cell wall of PpFWK3 cells traced back to a de novo deposition of carbohydrate epitopes (most probably chitin) in this och1 knockout strain, and not an increased exposure of constitutive cell wall glycans that are invariably present but usually shielded by an outer chain hypermannan structure in the wildtype strain. The somewhat reduced STL/FM® 4–64 ratio compared to the WGA/FM® 4–64 ratio may be connected to the minor differences regarding the preferentially binding ligand. Based on the similarity between STL and WGA staining patterns, it is unlikely that any GlcNAc motifs other than chitin (e.g. in the core glycan of mannoproteins) were the primary binding epitopes detected by either of the two lectins. As a side aspect of the current work, we were able to demonstrate the use of fluorescence labeled lectins as convenient and versatile probes for visualizing stress responses derived from impaired cell wall integrity in yeast. The decreased GNL/FM® 4–64 ratio of PpFWK3 compared to PpMutS could be explained by the minimized amount of high mannose N-glycans and the therefore inherently lower amount of potential GNL target ligands (i.e. α-1,3- and α-1,6-Man residues) of PpFWK3 compared to PpMutS. Nevertheless, the remaining core glycan provided sufficient GNL targets for a distinct signal. The increased LCA/FM® 4–64 signal of PpFWK3 on the other hand, may be explained by the preference of this lectin for short chain X-α-1,2-Man–Man motifs, with X representing either α-Man or β-GlcNAc37. In mannoproteins, such short motifs may be found in the core glycans, the accessibility of which may be enhanced in absence of the usually highly branched polymannan structures of a P. pastoris wildtype strain33,34.
The detailed characterization of the different P. pastoris strains in the controlled environment of a bioreactor revealed that the och1 knockout strains were physiologically impaired compared to their wildtype equivalents. During the consecutive pulses, the carbon dioxide yield YCO2/S and the C-balances constantly decreased, indicating a loss in metabolic activity. This was also apparent in the CER signals during the single methanol pulses (Supplementary Figure 5 and 6). At the beginning of the cultivation, methanol was metabolized much faster than during later methanol pulses. We followed the morphology of the P. pastoris cells during cultivations via microscopy and identified formation of cell clusters by the och1 knockout strains. Obviously, the altered surface glycosylation of och1 knockout cells also affected the budding process. Instead of budding off, the daughter cells stayed attached to the mother cell. The microscopically observed increased tendency for cluster formation may be regarded as an effect which is intrinsically linked to the loss of the polymannan layer upon Och1p inactivation. Cell disruption experiments showed that neither treatment with Triton X-100, 0.5% EDTA or 5 M urea, nor extensive mechanic shearing via sonication allowed breaking the clusters to single cells. Thus, we conclude that deficiencies in the constitutive cell division machinery upon Och1p inactivation led to a strong, covalent linkage between the cell walls, which remained intact even after the end of the normal budding process. The exact nature of this linkage remains speculative at the current point, but may be associated to aberrant glycosylation steps in the glycan backbone, occurring as a compensatory adaptation to the lack of Och1p activity20,38. Due to these very dense and compact formations we hypothesize that the cells in the center of these clusters became limited in oxygen and nutrients and showed no more metabolic activity. Since these cell clusters increased in size over time, the overall metabolic activity of the total amount of cells in the bioreactor decreased, which we observed in decreasing YCO2/S and C-balances. However, these cell clusters were still able to produce recombinant protein. In fact, both the specific productivity and the volumetric productivity, the main focus of industrial bioprocesses, were only reduced by 35% and 30%, respectively, compared to the P. pastoris strain with an intact OCH1 gene. However, due to the constant decrease in metabolic activity over time, use of the och1 knockout strain in industrial processes will require considerable modifications to current standard protocols to optimize recombinant protein production in this strain.
When we analyzed the HRP produced by either a PpMutSHRP strain or the och1 knockout strain PpFWK3HRP, we found the enzyme preparation from PpFWK3HRP to be considerably more homogenously glycosylated. More detailed analyses of the surface glycan chains of HRP by mass spectrometry revealed striking differences in the glycosylation pattern between HRP produced in PpMutSHRP and PpFWK3HRP. HRP produced in PpMutSHRP carried a more heterogeneous glycopattern with several high-mannose structures and a great amount of phosphorylated sugars. The most dominant glycan was found to be a Man10 structure. In contrast, the most dominant glycan of HRP produced in PpFWK3HRP was a Man8 core glycan structure. This reduction is in agreement with our expectations since no Och1p could act on Man8GlcNAc2 core glycan structures in the Golgi of the PpFWK3HRP strain as it could in PpMutSHRP. Due to the missing Och1p activity, other glycosyltransferases, especially mannosylphosphate transferases, were reduced in their activity resulting in a more homogeneous glycopattern on the surface of recombinantly produced HRP from PpFWK3HRP (Table 3).
Enzymatic characterizations of HRP revealed an increase in the affinity to the substrate ABTS, but a decrease of Vmax for HRP with a more homogeneous glycopattern. Also, a decreased stability at 60°C for the homogeneously glycosylated HRP compared to the heterogeneously hyperglycosylated glycovariant was observed.
In conclusion, we irreversibly eliminated the OCH1 encoded α-1,6-mannosyltransferase activity of P. pastoris. The phenotype of the generated och1 knockout platform strain resembled the phenotype described for the same knockout in S. cerevisiae. Nevertheless, the strain was successfully employed for the production of recombinant HRP as a reporter enzyme. Strain specific parameters were determined in comparative bioreactor cultivations. Recombinant HRP from either an unaltered P. pastoris strain or the och1 knockout strain was purified and characterized. The main findings of this study can be summarized as:
The och1 knockout strains were characterized by slow growth, increased temperature sensitivity and formation of cell clusters. The altered N-glycosylation pathway and resultant structural impacts in the PpFWK3 strain appears to have triggered the dynamic reorganization of surface mannose residues and other glycan structures. The cellular response seemed to be more diverse than just a simple lack of an outer chain hypermannan structure, and may also involve secondary counter-regulatory mechanisms on the metabolic or structural level20,38. Our results in this regard are in direct agreement with previous work on och1 deletion strains of S. cerevisiae19,39. Lectin based glycoprofiling represented a rapid and reliable method to provide functional proof for the successful deletion of Och1p activity in P. pastoris.
In the course of consecutive methanol pulses, the och1 knockout strains lost their metabolic activity due to the formation of cell clusters, thus making the adaption of current production processes necessary.
As shown by detailed LC-MS data, the produced recombinant enzyme exhibited a more homogeneous surface glycosylation, which is beneficial for subsequent downstream processing and applications.
Vmax with ABTS as substrate and the thermal stability at 60°C were reduced for the homogeneously glycosylated HRP, whereas its affinity for ABTS was increased, rendering the enzyme suitable for most applications.
Here, we report the thorough biotechnological characterization of a P. pastoris platform strain that allows the production of recombinant proteins with considerably increased homogeneity in their glycosylation pattern due to an irreversible knockout of the OCH1 gene. Currently, efforts are driven forward to elucidate the potential benefits of the cell morphological changes in glycoengineered P. pastoris strains for the expression of recombinant proteins40. Also, future studies will focus on rescuing the growth impaired phenotype to generate a strain that shares the favorable growth phenotype of a wildtype strain but still allows the production of homogeneously glycosylated secreted proteins.
Methods
Chemicals
Enzymes and deoxynucleotide triphosphates were obtained from Thermo Scientific (formerly Fermentas, Germany). Phusion™ High-Fidelity DNA-polymerase was from Finnzymes (Finland). 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt (ABTS) was purchased from Sigma-Aldrich (Austria). Difco™ yeast nitrogen base w/o amino acids (YNB), Difco™ yeast nitrogen base w/o amino acids and ammonia sulfate (YNB2), Bacto™ tryptone and Bacto™ yeast extract were purchased from Becton Dickinson (Austria). Zeocin™ was purchased from InvivoGen (France) via Eubio (Austria). Fluorescein isothiocyanate (FITC) labeled lectins for microscopic analysis were obtained from Vector Laboratories (USA), comprising wheat germ agglutinin (WGA) from Triticum vulgaris, Lens culinaris agglutinin (LCA), Galanthus nivalis lectin (GNL) and Solanum tuberosum lectin (STL). FM® 4–64 membrane stain and Hoechst 33342 or 4′,6-diamidino-2-phenylindole (DAPI) nucleic acid stains were purchased from Life Technologies (USA). Other chemicals were obtained from Carl Roth (Germany).
Microorganisms
DNA manipulations were performed in accordance to standard protocols41 in E. coli Top10F' (Life Technologies, formerly Invitrogen, Austria). All P. pastoris strains in this study were based on the wildtype strain CBS 7435 (identical to NRRL Y-11430 or ATCC 76273). Initial OCH1 knockout studies were performed in a ku70 deletion strain, previously described by Näätsaari et al.18, hereafter called Ppku70-. Since P. pastoris strains with MutS phenotype have been repeatedly shown to be superior over strains with Mut+ phenotype for the production of recombinant proteins (e.g.26), the ultimate och1 knockout strain was based on a MutS strain described in18, hereafter called PpMutS.
Deletion of the OCH1 gene
Based on the genome sequence of the P. pastoris wildtype strain CBS 743542, the primers OCH1-5int-fw1 and OCH1-5int-rv1 were designed to amplify a DNA fragment upstream the OCH1 open reading frame from genomic DNA isolated according to43. Primers OCH1-3int-fw1b and OCH1-3int-rv1b were designed to amplify a fragment downstream of the OCH1 ORF. OCH1-5int-rv1 and OCH1-3int-fw1b were designed to add sequences that overlap with the FRT flanked inner part of a flipper cassette. All primer sequences are listed in Table 5.
Table 5. Oligonucleotide primer list. Primers used for the amplification of upstream and downstream sequences of the OCH1 locus from genomic DNA, for amplification of the whole OCH1 locus for Sanger sequencing, for amplification of fragments to assemble pPpT4_BamHI_OCH1rescue and to amplify the OCH1 ORF.
| primer name | sequence (5′ - 3′) |
|---|---|
| OCH1-5int-fw1 | GAACTGTGTAACCTTTTAAATGACGGGATCTAAATACGTCATG |
| OCH1-5int-rv1 | CTATTCTCTAGAAAGTATAGGAACTTCGGCTGATGATATTTGCTACGAACACTG |
| OCH1-3int-fw1b | GTTCCTATACTTTCTAGAGAATAGGAACTTCGCGAGATTAGAGAATGAATACCTTCTTCTAAGCGATCG |
| OCH1-3int-rv1b | GAAGTATTAGGAGCTGAAGAAGCAGAGGCAGAG |
| OCH1check-fw1 | CACACATATAAAGGCAATCTACG |
| OCH1check-rv2 | CAATAACTTCTGCAATAGACTGC |
| OCH1rescue-fw1 | TTCATAGGCTTGGGGTAATAG |
| OCH1rescue-rv1 | CTTGAGCGGCCGCTTAGTCCTTCCAACTTCCTTC |
| OCH1rescue_T4fw | GCATACATTTGAAGGAAGTTGGAAGGACTAAGCGGCCGCTCAAGAGGAT |
| OCH1rescue_T4rv | CTATTTCTCTGTCATCTATCTATTACCCCAAGCCTATGAAGGATCTGGGTACCGCAGG |
| OCH1-ORF-fw | ATGGCGAAGGCAGATGGC |
| OCH1-ORF-rv | TTAGTCCTTCCAACTTCCTTCAAATG |
The two OCH1 targeting fragments of approximately 1.5 kb each were used to assemble a flipper cassette via overlap extension PCR18. Transformation of 100 ng of the assembled flipper cassette into either Ppku70- or PpMutS was performed as described by Lin-Cereghino et al.44. Transformants were identified on yeast extract-peptone-dextrose (YPD) agar plates containing 100 mg·L−1 Zeocin™. Double homologous recombination of the flipper cassette in the OCH1 locus was verified by PCR using the primers OCH1check-fw1 and OCH1check-rv2 (Table 5) and Sanger sequencing, using isolated genomic DNA as template. Expression of the FLP recombinase gene was induced by growing positive transformants on minimal methanol agar plates. The FLP recombinase mediated excision of the FRT flanked inner part of the flipper cassette was shown by restored sensitivity of the cells towards Zeocin™ and again by PCR and Sanger sequencing. The resulting Ppku70- och1 knockout strain was designated PpFWK1, the PpMutS och1 knockout strain was designated PpFWK3.
Complementation of the observed phenotype of the PpFWK3 strain was performed by transforming a plasmid that was constructed by assembly45 of two fragments, which were generated by PCR using the primers OCH1rescue-fw1 and OCH1rescue-rv1 using genomic DNA from PpMutS as template, and OCH1rescue_T4fw and OCH1rescue_T4rv using the plasmid pPpT4_S18 as template (Table 5). The resulting plasmid contained the wildtype OCH1 ORF plus 698 bp of upstream sequence, putatively harboring the natural OCH1 promoter, and was designated pPpT4_BamHI_OCH1rescue. Approximately 500 ng of BamHI linearized plasmid were transformed to PpFWK3, aliquots were plated on YPD Zeocin™ agar plates and incubated at 28°C for two days. PCR with the primers OCH1check-fw1 and OCH1check-rv2 from isolated genomic DNA of transformant strains was performed to confirm the unaltered replacement of the former OCH1 ORF by a single FRT site. A second PCR with the primers OCH1-ORF-fw and OCH1-ORF-rv from the same genomic DNA was performed to confirm the presence of a plasmid transmitted OCH1 ORF somewhere else in the genome. A resulting strain with restored wildtype phenotype was designated PpFWK3R.
Phenotypic strain characterization
Lectin based glycoprofiling via fluorescence microscopy
Qualitative analysis of lectin binding was performed by incubating 500 μL of PpMutS or PpFWK3 cell suspensions in 20 mM HEPES buffer, pH 7.4, at an OD600 of 0.3 with 500 μL of the respective lectin solution (250 pmol·mL−1 in 20 mM HEPES, pH 7.4) for 30 min at 4°C. If appropriate, 5 μg·mL−1 HOECHST 33342 nucleic acid stain or 0.5 μg·mL−1 FM® 4–64 membrane stain were included in the incubation mix. After thorough washing by repeated centrifugation (1700 × g, 5 min) and resuspension, cells were diluted in 1.0 mL of particle free phosphate buffered saline (PBS; 50 mM, pH 7.4) and mounted in FlexiPERM® coverslip 12-well plates for microscopic analysis. Images were acquired on a Zeiss Epifluorescence Axio Observer.Z1 deconvolution microscopy system (Carl Zeiss, Germany) equipped with LD Plan-Neofluar objectives and the LED illumination system Colibri®. Exposure wavelengths and filter sets of the individual channels were chosen according to the respective fluorophore(s) (DAPI/Hoechst 33342: ex/em 365/450 nm; FITC: ex/em 485/525 nm; FM® 4–64 ex/em 485/>620 nm), and combined with differential interference contrast (DIC) images for ease of orientation. Exposure time and illumination parameters were adjusted individually for optimal visibility. For bud scar visualization, Z-stack image series of representative spots were recorded and processed via moderate iterative deconvolution. Lectin cytoadhesion was quantified by incubating the cells with the respective lectin solutions and FM® 4–64 as described above (4°C, 30 min), followed by extensive washing. Cells were then lysed by treatment with Triton X-100/SDS (1.0/1.0%) for 24 h under vivid agitation. The FITC fluorescence intensity in the lysis buffer was assessed in a microplate reader (TECAN, Austria) at ex/em 485/525 nm and normalized to the content of FM® 4–64 for direct comparison between the individual samples. Lectin solutions without cells were subjected to the same treatment and analyzed in order to exclude potential degradation of the fluorophore. Control experiments via fluorescence-activated cell sorting (FACS) were performed to verify similar uptake of the membrane stain in both strains.
Membrane permeabilization experiments
To gain information on the steric accessibility of cell wall-embedded chitin and other carbohydrates, cell permeability was enhanced by treatment with concentrated methanol at −20°C for 20 min, followed by lectin staining. The cells contained in 1.0 mL of precooled suspension (OD600 of 0.3) were harvested by centrifugation, resuspended in 100 μL PBS buffer and added dropwise to 1.0 mL of icecold methanol under vivid agitation. After 20 min, cells were pelleted again and excessive solvent was removed via repeated washing and centrifugation with fresh PBS buffer. After rehydration in 1.0 mL of 20 mM HEPES buffer, pH 7.4, cells were subjected to the same lectin staining protocol as described above and analyzed via fluorescence microscopy. Efficient membrane permeabilization was verified via successful counterstaining of the nuclear DNA with a normally non-membrane permeable DAPI dye.
Production of the reporter enzyme horseradish peroxidase in shake flask experiments
Aliquots of approximately 2 μg of SmiI linearized plasmid pPpT4_S18, harboring a HRP gene containing nine potential N-glycosylation sites were transformed into either PpMutS or PpFWK3. The transformed HRP gene encodes for a new acidic HRP isoenzyme. A detailed description on the identification of new HRP isoenzymes will be given elsewhere (Näätsaari et al., manuscript in preparation). The HRP gene was codon optimized for expression in P. pastoris based on a codon table described in46. Expression of the gene was regulated by the AOX1 promoter. Efficient secretion of HRP to the supernatant was facilitated by fusion of the prepro signal sequence of the S. cerevisiae mating factor alpha to the N-terminus of the mature HRP. Transformations were performed according to44 with the following modification: Whereas an overnight culture of PpMutS was diluted to an OD600 of 0.2 to grow to an OD600 of 0.8–1.0 in approximately 5 h prior to preparation of the cells for electroporation, an overnight culture of PpFWK3 was diluted to a starting OD600 of 0.7 to account for its decreased growth rate. Transformants were grown on YPD Zeocin™ agar plates and randomly chosen for screening in micro scale cultivations in 96-deep well plates, similarly to47. The cells were cultivated in 250 μL iron-supplemented BMD1% (11 g·L−1 α-D(+)-glucose monohydrate, 13.4 g·L−1 YNB, 0.4 mg·L−1 D(+)-biotin, 278 mg·L−1 FeSO4 7H2O, 0.1 M potassium phosphate buffer, pH 6.0) for approximately 60 h, then induced once with 250 μL BMM2 (1% (v/v) methanol, 13.4 g·L−1 YNB, 0.4 mg·L−1 D(+)-biotin, 0.1 M potassium phosphate buffer, pH 6.0) and three times with 50 μL BMM10 (5% (v/v) methanol, 13.4 g·L−1 YNB, 0.4 mg·L−1 D(+)-biotin, 0.1 M potassium phosphate buffer, pH 6.0) per well 12 h, 24 h and 36 h after the first addition of BMM2. Induction with the methanol containing media BMM2 and BMM10 induced the production of HRP which was under control of the AOX1 promoter. The respective HRP production strains were designated PpMutSHRP and PpFWK3HRP.
Small scale cultivations were performed in 0.5 L Ultra Yield Flasks (BioSilta, Finland) in 45 mL iron-supplemented BMD1%. After approximately 60 h, 5 mL BMM10 were added. Twelve hours and 36 h after the first induction pulse, 0.5 mL pure methanol were added. Twentyfour hours and 48 h after the first induction pulse, 0.25 mL pure methanol were added. HRP activity in the supernatant was determined by mixing 15 μL of culture supernatant with 140 μL of assay solution (1 mM ABTS, 0.8 mM H2O2, 50 mM NaOAc buffer, pH 4.5) and following the increase in absorbance at 405 nm in a Spectramax Plus 384 platereader (Molecular Devices, Germany) at room temperature for 3 min. Promising clones were streaked to single colonies and cultivated again in quadruplicates for rescreening. The copy number of the HRP gene in selected PpMutS and PpFWK3 transformant strains was determined via quantitiative real-time PCR according to a protocol of Abad et al.48 and as described previously in26.
Bioreactor cultivations
Four different P. pastoris strains (Table 6) were characterized in terms of physiology, biomass growth and productivity by a novel, dynamic strategy of conducting methanol pulses during batch cultivations in the controlled environment of a bioreactor, which we have described recently16,24,26.
Table 6. P. pastoris strains used for biotechnological characterisation during bioreactor cultivations.
| strain | name |
|---|---|
| P. pastoris MutS | PpMutS |
| P. pastoris MutS HRP | PpMutSHRP |
| P. pastoris MutS och1 | PpFWK3 |
| P. pastoris MutS och1 HRP | PpFWK3HRP |
Culture media
Yeast nitrogen base medium (YNBM): 20 g·L−1 α-D(+)-glucose monohydrate, 3.4 g·L−1 YNB2, 10 g·L−1 (NH4)2SO4, 0.4 g·L−1 D(+)-biotin, 0.1 M potassium phosphate buffer, pH 6.0.
Trace element solution (PTM1): 6 g·L−1 CuSO4.5H2O, 0.08 g·L−1 NaI, 3 g·L−1 MnSO4·H2O, 0.2 g.L−1 Na2MoO4·2H2O, 0.02 g·L−1 H3BO3, 0.5 g·L−1 CoCl2, 20 g·L−1 ZnCl2, 65 g·L−1 FeSO4·7H2O, 0.2 g·L−1 D(+)-biotin, 5 mL·L−1 95–98% H2SO4.
Basal salt medium (BSM): 44 g·L−1 α-D(+)-glucose monohydrate, 1.17 g·L−1 CaSO4·2H2O, 18.2 g·L−1 K2SO4, 14.9 g·L−1 MgSO4.7H2O, 4.13 g·L−1 KOH, 26.7 mL·L−1 85% (v/v) o-phosphoric acid, 0.2 mL·L−1 Antifoam Struktol J650, 4.35 mL·L−1 PTM1, NH4OH as N-source (see experimental procedure).
Base: NH4OH, concentration was determined by titration with 0.25 M potassium hydrogen phthalate.
Preculture
Frozen stocks (−80°C) of either PpMutSHRP and PpFWK3HRP were precultivated in 100 mL of YNBM in 1 L shake flasks at 30°C and 230 rpm for max. 24 h. The preculture was transferred aseptically to the respective culture vessel. The inoculation volume was 10% of the final starting volume.
Batch cultivation
Batch cultivations were carried out in a 3 L working volume Labfors glass bioreactor (Infors, Switzerland). BSM was sterilized in the bioreactor and pH was adjusted to pH 5.0 by using concentrated ammonia solution after autoclaving. Sterile filtered PTM1 was transferred to the reactor aseptically. Dissolved oxygen (dO2) was measured with a sterilizable polarographic dissolved oxygen electrode (Mettler Toledo, Switzerland). The pH was measured with a sterilizable electrode (Mettler Toledo, Switzerland) and maintained constant with a step controller using 2.5 M ammonia solution. Base consumption was determined gravimetrically. Cultivation temperature was set to 30°C and agitation was fixed to 1495 rpm. The culture was aerated with 2.0 vvm dried air and offgas of the culture was measured by using an infrared cell for CO2 and a paramagnetic cell for O2 concentration (Servomax, Switzerland). Temperature, pH, dO2, agitation as well as CO2 and O2 in the offgas were measured online and logged in a process information management system (PIMS Lucullus; Biospectra, Switzerland).
After the complete consumption of the substrate glucose, which was indicated by an increase of dO2 and a drop in offgas activity, the first methanol pulse (adaptation pulse) of a final concentration of 0.5% (v/v) was conducted with methanol supplemented with PTM1 (12 mL PTM1 per 1 L of methanol). Subsequently, between five and seven pulses were performed with 1% or 2% (v/v) methanol for each strain. For each pulse, at least two samples were taken to determine the concentrations of the substrate methanol and product as well as dry cell weight (DCW) and OD600 to calculate the strain specific parameters. The induction period for PpMutSHRP and PpFWK3HRP was carried out in the presence of 1 mM of the heme precursor δ-aminolevulinic acid.
Analysis of growth- and expression-parameters
DCW was determined by centrifugation of 5 mL culture broth (4,000 × g, 10 min, 4°C), washing the pellet with 5 mL deionized water and subsequent drying at 105°C to a constant weight in an oven. OD600 of the culture broth was measured using a spectrophotometer (Genesys 20; Thermo Scientific, Austria). The activity of HRP was determined using a CuBiAn XC enzymatic robot (Innovatis, Germany). Cell free samples (10 μL) were added to 140 μL of 1 mM ABTS in 50 mM potassium phosphate buffer, pH 6.5. The reaction mixture was incubated at 37°C and was started by the addition of 20 μL of 0.075% H2O2. Changes of absorbance at 415 nm were measured for 80 s and rates were calculated. Calibration was done using commercially available horseradish peroxidase (Type VI-A, Sigma-Aldrich, Austria, P6782, Lot# 118K76734) as standard at six different concentrations (0.02; 0.05; 0.1; 0.25; 0.5 and 1.0 U·mL−1). Protein concentrations were determined at 595 nm using the Bradford Protein Assay Kit (Bio-Rad Laboratories GmbH, Austria) with bovine serum albumin as standard.
Substrate concentrations
Concentration of methanol was determined in cell free samples by HPLC (Agilent Technologies, USA) equipped with a Supelcoguard column, a Supelcogel C-610H ion-exchange column (Sigma-Aldrich, Austria) and a refractive index detector (Agilent Technologies, USA). The mobile phase was 0.1% H3PO4 with a constant flow rate of 0.5 mL·min−1 and the system was run isocratically. Calibration was done by measuring standard points in the range of 0.1 to 10 g·L−1 methanol.
Data analysis
Strain characteristic parameters were determined at a carbon dioxide evolution rate (CER) above 2.5 mmol·L−1·h−1 during each methanol pulse. Measurements of biomass, product and substrate concentration were executed in duplicates. Along the observed standard deviation for the single measurement, the error was propagated to the specific rates qs and qp as well as to the yield coefficients. The error of determination of the specific rates and the yields was therefore set to 10% and 5%, respectively16,24.
Enzyme purification
Size exclusion chromatography
The supernatants from PpMutSHRP and PpFWK3HRP produced in small scale cultures in 0.5 L Ultra Yield Flasks were concentrated to approximately 500 μL each using Vivaspin 20 tubes (Sartorius Stedim Biotech, Germany) with 10 kDa MWCO and recovered from the tubes resulting in a volume of max. 1500 μL, prior to size exclusion chromatography (SEC) on a HiLoad™ 16/60 Superdex 200 prep grade column (GE Healthcare Europe, Austria). SEC was performed at a flow rate of approximately 9 cm·h−1, fractions of 1.2 mL were collected and assayed for HRP activity using ABTS as substrate.
2-step purification protocol
To purify the secreted HRP produced in bioreactor cultivations, the fermentation broths were harvested and centrifuged (4,000 × g, 20 min) and the cell free supernatants were subjected to diafiltration with buffer (500 mM NaCl, 20 mM NaOAc, pH 6.0) for a subsequent purification step via a mixed mode resin (hydrophobic charge induction chromatography, HCIC) followed by a size exclusion step (SEC). We have recently described this 2-step flowthrough based strategy for the HRP isoenzyme C1A27. The catalytic activity and the protein content in all fractions were determined, active fractions were pooled and concentrated via ultrafiltration to approximately 3 mg·mL−1 for HRP produced in PpMutsHRP and 0.3 mg·mL−1 for HRP produced in PpFWK3HRP for subsequent enzyme characterization.
Enzyme characterization
The two HRP preparations produced in either PpMutsHRP or in PpFWK3HRP were characterized to determine differences between the hypermannosylated HRP from PpMutSHRP and its glycovariant produced in PpFWK3HRP.
Liquid chromatography-mass spectrometry (LC-MS) analysis
Protein N-glycosylation was analyzed by releasing the N-glycans with peptide:N-glycosidase F (Roche, Mannheim). The released N-glycans were desalted and analyzed using a porous graphitic carbon capillary column (ThermoScientific) coupled to a mass spectrometer (Maxis 4 G, Bruker, Bremen). Deviating from previous work49, glycans were not reduced and a steep gradient was applied leading to the elution of all glycans within approximately 2 min.
Kinetic constants with ABTS
Protein concentrations of the HRP preparations were determined at 595 nm using the Bradford Protein Assay Kit (Bio-Rad Laboratories GmbH, Austria) with bovine serum albumin as standard. The kinetic constants for ABTS were determined for both HRP glycovariants. The reaction was started by adding 10 μL enzyme solution (3 mg·mL−1 HRP from PpMutSHRP and 0.3 mg·mL−1 HRP from PpFWK3HRP) to 990 μL reaction buffer containing ABTS in varying concentrations (0.01–10 mM), 1 mM H2O2 and 50 mM potassium phosphate, pH 6.5. The change in absorbance at 420 nm was recorded in a spectrophotometer UV-1601 (Shimadzu, Japan) at 30°C controlled with a temperature controller (CPS controller 240 A; Shimadzu, Japan). Absorption curves were recorded with a software program (UVPC Optional Kinetics; Shimadzu, Japan). Measurements were performed in triplicates.
Thermal stability
Both enzyme solutions were incubated at 60°C for 1 h. At different time points, aliquots were withdrawn, the solutions were immediately cooled and centrifuged (20,000 × g, 15 min) to pellet precipitated proteins and the remaining catalytic activity in the supernatants was measured28.
Author Contributions
F.W.K. and O.S. conceived of and planned the study. F.W.K., C.G., L.N., M.W., R.P. and O.S. conducted the different experiments. C.H., A.G. and F.A. supervised parts of the research. F.W.K. and O.S. wrote the paper.
Supplementary Material
Supplementary Info
Acknowledgments
The authors are very grateful to the Austrian Science Fund (FWF): project P24861-B19 and FWF W901 DK Molecular Enzymology for financial support. We would also like to thank Michaela Gerstmann, Astrid Weninger and Karl Metzger for excellent technical assistance in the lab and Daniel Maresch for excellent assistance with LC-MS.
References
- Hasslacher M. et al. High-level intracellular expression of hydroxynitrile lyase from the tropical rubber tree Hevea brasiliensis in microbial hosts. Protein Expr. Purif. 11, 61–71 (1997). [DOI] [PubMed] [Google Scholar]
- Werten M. W., van den Bosch T. J., Wind R. D., Mooibroek H. & de Wolf F. A. High-yield secretion of recombinant gelatins by Pichia pastoris. Yeast 15, 1087–1096 (1999). [DOI] [PubMed] [Google Scholar]
- Jahic M., Rotticci-Mulder J. C., Martinelle M., Hult K. & Enfors S.-O. Modeling of growth and energy metabolism of Pichia pastoris producing a fusion protein. Bioprocess Biosyst. Eng. 24, 385–393 (2002). [Google Scholar]
- Lin-Cereghino G. P., Lin-Cereghino J., Ilgen C. & Cregg J. M. Production of recombinant proteins in fermenter cultures of the yeast Pichia pastoris. Curr. Opin. Biotechnol. 13, 329–332 (2002). [DOI] [PubMed] [Google Scholar]
- Parekh R. B. Effects of glycosylation on protein function. Curr. Opin. Struct. Biol. 1, 750–754 (1991). [Google Scholar]
- Ryckaert S., Martens V., De Vusser K. & Contreras R. Development of a S. cerevisiae whole cell biocatalyst for in vitro sialylation of oligosaccharides. J. Biotechnol. 119, 379–388 (2005). [DOI] [PubMed] [Google Scholar]
- Wildt S. & Gerngross T. U. The humanization of N-glycosylation pathways in yeast. Nat. Rev. Microbiol. 3, 119–128 (2005). [DOI] [PubMed] [Google Scholar]
- Hamilton S. R. & Gerngross T. U. Glycosylation engineering in yeast: the advent of fully humanized yeast. Curr. Opin. Biotechnol. 18, 387–392 (2007). [DOI] [PubMed] [Google Scholar]
- De Pourcq K., De Schutter K. & Callewaert N. Engineering of glycosylation in yeast and other fungi: current state and perspectives. Appl. Microbiol. Biotechnol. 87, 1617–1631 (2010). [DOI] [PubMed] [Google Scholar]
- Vervecken W. et al. In vivo synthesis of mammalian-like, hybrid-type N-glycans in Pichia pastoris. Appl. Environ. Microbiol. 70, 2639–2646 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasu T. et al. Isolation of new temperature-sensitive mutants of Saccharomyces cerevisiae deficient in mannose outer chain elongation. Yeast 8, 535–547 (1992). [DOI] [PubMed] [Google Scholar]
- Nakayama K., Nagasu T., Shimma Y., Kuromitsu J. & Jigami Y. OCH1 encodes a novel membrane bound mannosyltransferase: outer chain elongation of asparagine-linked oligosaccharides. EMBO J. 11, 2511–2519 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi B.-K. et al. Use of combinatorial genetic libraries to humanize N-linked glycosylation in the yeast Pichia pastoris. Proc. Natl. Acad. Sci. U.S.A. 100, 5022–5027 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mille C. et al. Identification of a new family of genes involved in beta-1,2-mannosylation of glycans in Pichia pastoris and Candida albicans. J. Biol. Chem. 283, 9724–9736 (2008). [DOI] [PubMed] [Google Scholar]
- Veitch N. C. Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry 65, 249–259 (2004). [DOI] [PubMed] [Google Scholar]
- Dietzsch C., Spadiut O. & Herwig C. A dynamic method based on the specific substrate uptake rate to set up a feeding strategy for Pichia pastoris. Microb. Cell Fact. 10, 14–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawski B. et al. Functional expression of horseradish peroxidase in Saccharomyces cerevisiae and Pichia pastoris. Protein Eng. 13, 377–384 (2000). [DOI] [PubMed] [Google Scholar]
- Näätsaari L. et al. Deletion of the Pichia pastoris KU70 homologue facilitates platform strain generation for gene expression and synthetic biology. PLoS ONE 7, e39720 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Zhang H., Liu X., Wang P. G. & Qi Q. Influence of N-glycosylation on Saccharomyces cerevisiae morphology: a golgi glycosylation mutant shows cell division defects. Curr. Microbiol. 55, 198–204 (2007). [DOI] [PubMed] [Google Scholar]
- Lee B. N. & Elion E. A. The MAPKKK Ste11 regulates vegetative growth through a kinase cascade of shared signaling components. Proc. Natl. Acad. Sci. U.S.A. 96, 12679–12684 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Non-Conventional Yeasts In Genetics, Biochemistry And Biotechnology. (Springer Berlin Heidelberg, 2003). 10.1007/978-3-642-55758-3. [Google Scholar]
- Shibuya N., Goldstein I. J., Van Damme E. J. & Peumans W. J. Binding properties of a mannose-specific lectin from the snowdrop (Galanthus nivalis) bulb. J. Biol. Chem. 263, 728–734 (1988). [PubMed] [Google Scholar]
- Fouquaert E. et al. Related lectins from snowdrop and maize differ in their carbohydrate-binding specificity. Biochem. Biophys. Res. Commun. 380, 260–265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzsch C., Spadiut O. & Herwig C. A fast approach to determine a fed batch feeding profile for recombinant Pichia pastoris strains. Microb. Cell Fact. 10, 85–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalai D., Dietzsch C., Herwig C. & Spadiut O. A dynamic fed batch strategy for a Pichia pastoris mixed feed system to increase process understanding. Biotechnol. Prog. 28, 878–886 (2012). [DOI] [PubMed] [Google Scholar]
- Krainer F. W. et al. Recombinant protein expression in Pichia pastoris strains with an engineered methanol utilization pathway. Microb. Cell Fact. 11, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadiut O., Rossetti L., Dietzsch C. & Herwig C. Purification of a recombinant plant peroxidase produced in Pichia pastoris by a simple 2-step strategy. Protein Expr. Purif. 86, 89–97 (2012). [DOI] [PubMed] [Google Scholar]
- Spadiut O. et al. Improving thermostability and catalytic activity of pyranose 2-oxidase from Trametes multicolor by rational and semi-rational design. FEBS J. 276, 776–792 (2009). [DOI] [PubMed] [Google Scholar]
- Chen Z. et al. Enhancement of the gene targeting efficiency of non-conventional yeasts by increasing genetic redundancy. PLoS ONE 8, e57952 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabib E. & Bowers B. Chitin and yeast budding. Localization of chitin in yeast bud scars. J. Biol. Chem. 246, 152–159 (1971). [PubMed] [Google Scholar]
- Chen C. et al. A high-throughput screening system for genes extending life-span. Exp. Gerontol. 38, 1051–1063 (2003). [DOI] [PubMed] [Google Scholar]
- Chaudhari R. D., Stenson J. D., Overton T. W. & Thomas C. R. Effect of bud scars on the mechanical properties of Saccharomyces cerevisiae cell walls. Chem. Eng. Sci. 84, 188–196 (2012). [Google Scholar]
- Lesage G. & Bussey H. Cell wall assembly in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 70, 317–343 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot P. W. J., Ram A. F. & Klis F. M. Features and functions of covalently linked proteins in fungal cell walls. Fungal Genet. Biol. 42, 657–675 (2005). [DOI] [PubMed] [Google Scholar]
- Klis F. M., Boorsma A. & De Groot P. W. J. Cell wall construction in Saccharomyces cerevisiae. Yeast 23, 185–202 (2006). [DOI] [PubMed] [Google Scholar]
- Kapteyn J. C. et al. Altered extent of cross-linking of beta1,6-glucosylated mannoproteins to chitin in Saccharomyces cerevisiae mutants with reduced cell wall beta1,3-glucan content. J. Bacteriol. 179, 6279–6284 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupin K. A., Liden D. & Haab B. B. The fine specificity of mannose-binding and galactose-binding lectins revealed using outlier motif analysis of glycan array data. Glycobiology 22, 160–169 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama H. & Suzuki T. Metabolism of free oligosaccharides is facilitated in the och1D mutant of Saccharomyces cerevisiae. Glycobiology 21, 1341–1348 (2011). [DOI] [PubMed] [Google Scholar]
- Nakanishi-Shindo Y., Nakayama K., Tanaka A., Toda Y. & Jigami Y. Structure of the N-linked oligosaccharides that show the complete loss of alpha-1,6-polymannose outer chain from och1, och1 mnn1, and och1 mnn1 alg3 mutants of Saccharomyces cerevisiae. J. Biol. Chem. 268, 26338–26345 (1993). [PubMed] [Google Scholar]
- Jacobs P. P. et al. Pichia surface display: display of proteins on the surface of glycoengineered Pichia pastoris strains. Biotechnol. Lett. 30, 2173–2181 (2008). [DOI] [PubMed] [Google Scholar]
- Current Protocols In Molecular Biology. (John Wiley & Sons, Inc., 2001). 10.1002/0471142727. [Google Scholar]
- Küberl A. et al. High-quality genome sequence of Pichia pastoris CBS7435. J. Biotechnol. 154, 312–320 (2011). [DOI] [PubMed] [Google Scholar]
- Harju S., Fedosyuk H. & Peterson K. R. Rapid isolation of yeast genomic DNA: Bust n′ Grab. BMC Biotechnol. 4, 8–13 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Cereghino J. et al. Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. BioTechniques 38, 44–48 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- Abad S. et al. Stepwise engineering of a Pichia pastoris D-amino acid oxidase whole cell catalyst. Microb. Cell Fact. 9, 24–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis R. et al. Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res. 5, 179–89 (2004). [DOI] [PubMed] [Google Scholar]
- Abad S. et al. Real-time PCR-based determination of gene copy numbers in Pichia pastoris. Biotechnol. J. 5, 413–420 (2010). [DOI] [PubMed] [Google Scholar]
- Pabst M. et al. Isomeric analysis of oligomannosidic N-glycans and their dolichol-linked precursors. Glycobiology 22, 389–399 (2012). [DOI] [PubMed] [Google Scholar]
- Nett J. H. et al. A combinatorial genetic library approach to target heterologous glycosylation enzymes to the endoplasmic reticulum or the Golgi apparatus of Pichia pastoris. Yeast 28, 237–252 (2011). [DOI] [PubMed] [Google Scholar]
- Hamilton S. R. et al. Production of complex human glycoproteins in yeast. Science 301, 1244–1246 (2003). [DOI] [PubMed] [Google Scholar]
- Jacobs P. P., Geysens S., Vervecken W., Contreras R. & Callewaert N. Engineering complex-type N-glycosylation in Pichia pastoris using GlycoSwitch technology. Nat. Protoc. 4, 58–70 (2009). [DOI] [PubMed] [Google Scholar]
- Hamilton S. R. et al. Humanization of yeast to produce complex terminally sialylated glycoproteins. Science 313, 1441–1443 (2006). [DOI] [PubMed] [Google Scholar]
- Hopkins D. et al. Elimination of β-mannose glycan structures in Pichia pastoris. Glycobiology 21, 1616–1626 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Info