

Abstract
Electric fields associated with Raman enhancements are typically inferred from changes in the observed scattering intensity. Here we use the vibrational Stark effect from a nitrile reporter to determine the electric field dependent frequency shift of cyanide (CN) on a gold (Au) surface. Electroplated Au surfaces with surface enhanced Raman (SERS) activity exhibit larger Stark shifts near the edge and in areas with large roughness. The Stark shift is observed to correlate with intensity of a co-adsorbed thiophenol molecule. Gap-mode Tip enhanced Raman scattering (TERS), using a Au nanoparticle tip, show dramatic shifts in the CN stretch that correlate to enhancement factors of 1013 in the gap region. The observed peak widths indicate the largest fields are highly localized. Changes in the nitrile stretch frequency provide a direct measurement of the electric fields in SERS and TERS experiments.
Keywords: vibrational stark effect, SERS, TERS, Raman, plasmonics, nitrile
Electric field enhancements associated with plasmonic structures have been used to enable detection and imaging of individual molecules.1, 2 The excitation of a localized surface plasmon resonance in a noble metal nanostructure results in a local electric field that underpins surface enhanced and tip enhanced spectroscopies.3 Direct measurement of the electric field has been complicated to assess; instead intensities associated with the enhanced molecules have been utilized to infer the magnitude of the electric field.4 In surface enhanced Raman spectroscopy, both excitation field and the Raman emission field are enhanced and contribute to the observed signal.5, 6 Recently, the electric field of a nanoparticle dimer was determined using the vibrational Stark effect of a CO molecule coadsorbed in the gap junction.7 In this letter we show that the Stark shift in a nitrile (CN) group adsorbed on a nanostructured gold surface can be used to map the electric fields that are associated with enhanced Raman scattering. Here we use the combination of surface enhanced Raman (SERS) and tip enhanced Raman (TERS) to investigate electric fields derived from the Stark shift of adsorbed cyanide.
The vibrational Stark effect arises from an external electric field perturbation to a chemical bond.8 The effect can result in either an increase or decrease in vibrational frequency dependent upon the orientation of the bond dipole and direction of the applied field; requiring a preferred molecular orientation for an observable frequency shift.9 Nitriles provide a sensitive probe of electric fields in a variety of environments such as proteins,10–13 biomembranes,14 the electrochemical double layer,15–17 and the energy levels of molecules.18–20 Nitriles adsorb to surfaces with preferred orientations and their Stark tuning parameters are well characterized, suggesting an ideal probe to assess the electric fields associated with plasmon enhanced spectroscopies.
A CN covered SERS active surface was prepared by electroplating onto wire embedded in polystyrene with a AuCN plating solution. Figure 1 shows the AFM topography of the surface, which is recessed slightly (~0.5 μm) into the supporting polystyrene block. To assess the SERS activity, the surface was soaked in 1mM thiophenol solution and the Raman map (Fig. 1C) was obtained. The Raman spectrum of thiophenol is observed most prominently at the edge of the Au surface. The AFM image of the Au surface exhibits a RMS roughness in the center of the electrode of 45 nm and of 300 – 400 nm near the outer region. The Raman spectra also show a large contribution of CN around 2250 cm−1. The frequency of the CN stretch is observed to vary across the mapped surface, shifting to higher frequencies near the edge of the Au surface and showing a consistent blue shift in regions expected to show increased SERS activity. All observed CN vibrational frequencies are blue shifted compared to the CN stretch observed from Au(I) cyanide salt [KAu(CN)2], which we observe at 2164 cm−1 in agreement with literature,21 indicating a different environment than observed on the SERS surface. The CN stretch frequency observed most closely matches reports of AuCN.22 The CN stretch in Au salts is observed at significantly lower energies.21, 23
Figure 1.

A) The 3D AFM topography of an electrodeposited Au microelectrode is shown. B) The observed CN stretch frequency is mapped along the electrode. C) The Raman intensity of codeposited thiophenol is mapped along part of the same electrode shown in A. The inset shows the Raman spectrum at the highest intensity pixel in the thiophenol map at 1572 cm−1 (red line, inset).
Further examination of the CN stretching modes shows evidence of an asymmetric frequency distributions on the Au surface. The heterogeneous and asymmetric line broadening of the nitrile stretch suggests different environments. Figure 2 shows that regions with large Stark shifts are also observed to have shoulders at lower energy. Fitting the observed CN stretch to a two-peak model provides a frequency for both the high and low electric field environments. Figure 2 maps the electric field on the Au surface calculated using the Stark tuning rate of 2.9 cm−1 (MV/cm)−1 for CN,24, 25 and 2243 cm−1, the CN stretch frequency in a isotropic field,16, 26 as the reference frequency necessary to determine the electric field.27 In figure 2B, the low field component is only slightly blue shifted from the reference value and is observed fairly consistently across the Au surface. While in figure 2C, the high field component localizes along the edge where SERS from thiophenol was also observed. Interestingly, there are no bands associated with thiophenol observed in regions exhibiting only the low field component. The electric field enhancement in these regions is either insufficient to generate an observable Raman signal given the cross-section of thiophenol, or the thiophenol is not present in sufficient concentration to be observed.28 The consistent CN across the surface may inhibit thiophenol absorption in all but high curvature areas of the hot spots. Of note, the CN frequencies observed are the same in the absence of thiophenol. Intensity based enhancement calculations are dominated by the signal from molecules in hotspots.29 This bimodal shifting indicates the ability to detect both hotspots and lower enhancements contributing to the observed Raman signal simultaneously.
Figure 2.
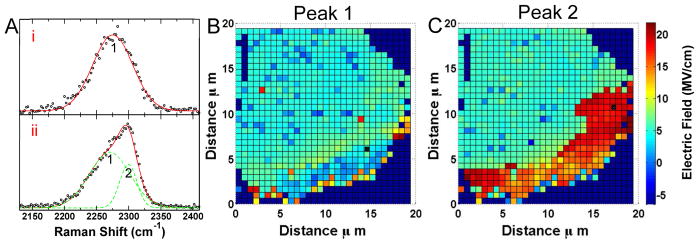
A) Representative SERS spectra of cyanide and the resulting peak fits are shown from flat (i) and edge (ii) regions. The edge region shows an asymmetric peak that can be deconvolved into two peaks, peak 1 and 2. B) A map of the electric field determined from the frequency of peak 1 indicates a low electric field component across the surface. C) The larger CN frequency of peak 2 corresponds to more intense electric fields localized along the edge of the Au deposit. In the absence of an asymmetric lineshape, the single frequency observed is used in both plots. A black cross in (B) and (C) denote where the spectra (i) and (ii) were acquired, respectively.
The magnitude of the fields determined from the Stark shift enable us to calculate an enhancement factor for the surface. SERS enhancement factors (EF) have been shown to arise from the electric field attendant to the excitation field (Eexc) and the Raman emission field (Eemm) as shown:
| Eq. 1 |
The incident electric field (E0) was determined to be 0.025 MV/cm, as calculated from our diffraction limited 632.8 nm laser spot with a measured irradiance of 7.5×108 mW/cm2. Using the field values shown in Figure 2 and our E0 value, Equation 1 suggests enhancement factors ranging from 109 to 1012 on the SERS surface. Since the Raman emission, or re-radiation, is dependent upon the orientation of the vibrational mode relative to the electric field, observed enhancement factors are often lower than predicted by the ideal E4 approximation.30, 31
Considering the SERS activity on the Au surface is non-uniform, the correlation between SERS structure and the molecule that enhanced signal originates from, is ambiguous. TERS provides a controlled means to generate enhancements associated with LSPR excitation. Figure 3 shows the results of a classic, gap-mode configuration that brings our tip into close proximity with our CN covered Au surface. The gap plasmon arises from coupling of the plasmon resonances in the TERS tip and the Au surface.
Figure 3.

i) The TERS spectrum of obtained from forming a gap junction over the CN covered Au surface is shown. ii) Retracting the tip from the surface provides the SERS spectrum lacking the high frequency peak. iii), The spectrum obtained from bringing the same tip into contact with a bare gold surface shows no CN peaks.
For the TERS experiments, a shorter deposition time (60 s to 30 s) provides a low SERS background, but an observable CN signal in a flat area on the Au surface. The TERS tip is comprised of a Au nanoparticle at the apex of a glass fiber. The TERS tip was brought into contact with the CN covered Au surface. Figure 3 shows the change in the CN stretch frequency when the tip is in and out of contact with the surface. The TERS spectrum shows an intense, blue shifted peak at 2381 cm−1. This represents a Stark shift of 129 cm−1 from the CN stretch frequency observed on the Au surface without the tip, and a shift of 138 cm−1 relative to the 2243 cm−1 reference value noted above.
To verify that the peak arises from the CN on the Au surface, the same TERS tip was also brought into contact with a fresh, template stripped Au surface without CN. No peaks were observed in the CN stretch region. This indicates there was no transfer of CN to the TERS probe, as well as the shifted peak not being an artifact of dirt on the probes apex, confirming the shifted peak is indeed due to the gap mode plasmonic field. Using the E4 approximation, the electric field within the gap corresponds to an enhancement factor of 1013.
Pettinger and coworkers reported gap-mode TERS results using an Au scanning-tunneling microscopy (STM) tip on both silver and gold surfaces to investigate the Stark tuning of CN. Their results showed only a 2 cm−1 shift in the CN frequency on the Au surface upon tip approach. With Ag they observed a shift of 43 cm−1. We believe the differences observed arise from differences in plasmon frequency associated with the gap geometry. Coupling between nanostructures, in this case the roughened surface and the TERS tip, results in a red-shifted plasmon.32 The resulting field and enhancement (Eq. 1) in our experiments arises from better overlap between the Raman excitation and emission with the plasmon. It’s been shown that plasmon mode frequencies and intensities, and subsequent Raman enhancement, are greatly affected by the physical properties of the nanostructures.32 In a recent report small changes in the gap resonance dramatically impacted the observed TERS signals, in this case enabling single molecule imaging.2 Similarly, our previous work has shown dramatic signal increases arising from plasmon coupling.33–35 The sharp Au tip used in the work by Petttinger and colleagues may not have resulted in as beneficial overlap with the gap resonance.
The linewidth observed for the TERS peak is also narrower than the original SERS peak observed. A Gaussian fit to the observed signals indicates linewidths of 15 and 25 cm−1 FWHM for the TERS peak at 2381 cm−1 and SERS peak at 2249 cm−1, respectively. The narrower FWHM suggests a more homogeneous electric field in the junction compared to the SERS surface. An analysis of the observed peak areas and enhancement factors determined from the electric fields producing the Stark shift, suggests that the signals observed in gap-mode TERS arise from a spot 6 nm in diameter. Since CN still resides outside this confined region, signal from this CN still contributes to the far field background. This confined enhancement agrees with a recent report of TERS imaging with resolution well below the limit associated with the field in a gap junction.2
Finite element modeling of the tip – surface junction suggests the enhanced field in the gap should be 100× greater than the incident field (E0). The nanoparticle here is approximately 150 nm in diameter and is coupling to a planar gold surface. The observed Stark shift suggests a field 2000× the incident laser excitation. It has been reported that classical electrodynamics, such as that used in our model, break down at distances less than 1 nm from the surface.36 The CN on our surface is well below this limit, which will lead to differences between theory and experimental results. However, the magnitude of the electric field we calculate from the CN Stark shift is consistent with a recent study using CO as a Stark reporter.7
To assess the validity of the electric fields calculated from the Stark shift, Figure 4 plots the observed intensities from the thiophenol (Fig. 4A) and CN (Fig 4B) against the electric field determined from the Stark tuning coefficient. The E4 approximation is plotted over the data and shown to agree within reason when a scaling factor of 0.28 is incorporated. The uncertainty in the electric field, evident in the CN stretch inhomogeneous broadening, is depicted as the shaded region to illustrate the full range of electric fields present.
Figure 4.

A) The CN stretch and B) the thiophenol 1550 cm−1 band intensities observed are plotted against the determined electric fields. The shading is the AE4 model overlaid onto the data. The width of the shading correlates to the FWHM of the CN stretch, and thus the uncertainty in the E field. In both plots A = 0.28.
The coefficient in the AE4 function fit to the data accounts for a number of factors: 1) local field effects, 2) the polarizability or orientation of the molecule, and 3) the incident electric field.
The electric field near the Stark reporter is known to be highly sensitive to the local electric field environment. Local field effects are often included to correct for variance from the observed Stark shift and the known electric field.9 It is common to report shifts relative to the local field (f); however, our factor A clearly contains contributions from other sources.
The plots in figure 4 show the intensities of molecules on the surface vs. electric field. Since the graphs plot Raman intensity against the electric field, there must be dependence on the polarizability of each molecule. The agreement in the A term suggests that either CN and thiophenol have comparable responses to electric fields or polarizability has a minor impact on the observed trend. Additionally differences in the orientation of the molecules are accounted for in the polarizability. There are reports of CN oriented at different angles with respect to the Au surface.22, 23, 37
The E4 model accounts for observed enhancement factors, while we have modeled the data versus absolute electric field. The fact that the model correlates well with our data supports our assertion the Stark shift arises from the plasmonic enhancement of the electric field. A correction for the incident field could be included; however, this would more accurately reflect enhancement factors that would require normalizing the observed intensities against conventional Raman intensities, which was not attempted here.
Other possible sources of error include our E0 value. We have reported our fields based on a literature value for the frequency of CN in an isotropic field that appears to provide a reasonable determination of the observed fields. Another possibility is the Stark tuning rate for CN on Au is different. An early computational study suggested that the Au-C bond is strongly perturbed, which would effect the C-N bonds response to electric fields.8 Additional studies will further refine the tuning rate for use in tracking fields associated with the local electric fields relevant to plasmonic field enhancements.
Our results show that the CN stretch frequency shows a blue shift that correlates with Raman enhancements arising from the excitation of surface plasmon resonances in SERS experiments. Tip enhanced Raman experiments verified that the shift correlates with fields from plasmon excitation. These results indicate a CN Stark shift can be used to investigate the electric fields arising from plasmon excitation on surfaces in diverse problems.
Experimental
AFM/Raman
Atomic force microscopy was performed using two different instruments. A Nanonics MV2000 AFM running NWS at version 1740 b4 control software. A pulled glass pipette with a tip size near 20 nm and length of approximately 150 μm, affixed to a quartz tuning fork, for open air AFM, was used to collect topography.
For combined AFM-Raman maps, a Nanonics MV4000 operated with NWS software version 2.3 was used. A long cantilever (>150 um) air AFM-Raman probe was used to collect topography and Raman spectra simultaneously. Raman spectra were obtained using a 632.8 nm HeNe laser, with a power at the objective of 1.1 mW, for excitation. The AFM was positioned in the sample plane of a custom upright microscope. The objective used for the Raman collection was a 100×, 0.7 NA Nikon L-Plan bright field objective. The microscope objective and AFM were vibrationally isolated from the Raman spectrometer. The Raman signal was collected through the single objective, filtered from the Rayleigh scattering and excitation light using a 633 nm dichroic filter (Semrock) and longpass filter (Semrock), and spectrally resolved using a Horiba Jobin Yvon iHR320 spectrometer with a Horiba Jobin Yvon Synapse CCD. Radial polarization was produced by a liquid crystal mode converter (ArcOptix).
Analysis of the collected Raman maps was preformed using Matlab and an open source peak-fitting package.38 Equal width Gaussian functions were used to fit both the chosen thiophenol band and the cyanide band. Each fit was repeated 15 times to achieve the lowest % RMS error. Any fit with an % RMS error > 15% was discarded from the maps.
TERS
TERS spectra were collected using the same microscope described above. The tip used in these experiments was a CMP Au-ball tip (Nanonics Imaging) appropriate for measurements in air. The diameter of the Au ball tip is approximately 150 nm.
Sample Preparation
A tungsten wire39 was etched to a point with an approximate apex width of 50 nm and embedded in polystyrene. With the etched end pointing down, the wire was held in a brass mold in resistive contact with the polished brass at the bottom of the mold. Polystyrene powder was added around the wire and melted at temperature of 250 °C similar to the method described by Martin.40 The polystyrene was allowed to cool and harden; the polymer disk was then removed from the holder. The polystyrene was polished using alumina embedded paper to expose the tip of the embedded wire. Gold was electrodeposited onto the wire using a commercial plating solution (Technic, Inc; Orotemp 24 RTU rack). The potential for deposition was controlled with a CHI660D potentiostat (CH Instruments) and was set at −1 V. After 170 seconds of deposition, the electrodes were rinsed with water followed by ethanol.
The gold deposits are 10’s of micrometers in diameter with an approximately circular shape. Each electrode was sized using optical microscopy and compared to USAF resolution target (Edmund Optical). Fabricated electrodes ranged from 20–100 micrometers in diameter.
The electrodes were then placed in 0.097 M thiophenol in ethanol for approximately 24 hours to co-adsorb thiophenol onto the gold. After an appropriate soaking time, the solution was removed and the electrode was again rinsed with water followed by ethanol. After fabrication and thiophenol addition, the electrodes were stored covered under ambient conditions.
Acknowledgments
The authors thank Steven Corcelli for helpful discussions. The National Institutes of Health Award RR024367 and the University of Notre Dame supported this work.
References
- 1.Steidtner J, Pettinger B. Tip-Enhanced Raman Spectroscopy and Microscopy on Single Dye Molecules with 15 Nm Resolution. Phys Rev Lett. 2008:100. doi: 10.1103/PhysRevLett.100.236101. [DOI] [PubMed] [Google Scholar]
- 2.Zhang R, Zhang Y, Dong ZC, Jiang S, Zhang C, Chen LG, Zhang L, Liao Y, Aizpurua J, Luo Y, et al. Chemical Mapping of a Single Molecule by Plasmon-Enhanced Raman Scattering. Nature. 2013;498:82–86. doi: 10.1038/nature12151. [DOI] [PubMed] [Google Scholar]
- 3.Moskovits M. Surface-Enhanced Spectroscopy. Reviews of Modern Physics. 1985;57:783–826. [Google Scholar]
- 4.Stiles PL, Dieringer JA, Shah NC, Van Duyne RP. Surface-Enhanced Raman Spectroscopy. Annu Rev Anal Chem. 2008;1:601–626. doi: 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- 5.Wang DS, Kerker M. Enhanced Raman-Scattering by Molecules Adsorbed at the Surface of Colloidal Spheroids. Phys Rev B. 1981;24:1777–1790. [Google Scholar]
- 6.Kerker M, Wang DS, Chew H. Surface Enhanced Raman-Scattering (Sers) by Molecules Adsorbed at Spherical-Particles. Applied Optics. 1980;19:4159–4174. doi: 10.1364/AO.19.004159. [DOI] [PubMed] [Google Scholar]
- 7.Banik M, El-Khoury PZ, Nag A, Rodriguez-Perez A, Guarrottxena N, Bazan GC, Apkarian VA. Surface-Enhanced Raman Trajectories on a Nano-Dumbbell: Transition from Field to Charge Transfer Plasmons as the Spheres Fuse. ACS Nano. 2012;6:10343–10354. doi: 10.1021/nn304277n. [DOI] [PubMed] [Google Scholar]
- 8.Bagus PS, Nelin CJ, Müller W, Philpott MR, Seki H. Field-Induced Vibrational Frequency Shifts of Co and Cn Chemisorbed on Cu (100) Phys Rev Lett. 1987;58:559–562. doi: 10.1103/PhysRevLett.58.559. [DOI] [PubMed] [Google Scholar]
- 9.Bublitz GU, Boxer SG. Stark Spectroscopy: Applications in Chemistry, Biology, and Materials Science. Annu Rev Phys Chem. 1997;48:213–42. doi: 10.1146/annurev.physchem.48.1.213. [DOI] [PubMed] [Google Scholar]
- 10.Stafford AJ, Ensign DL, Webb LJ. Vibrational Stark Effect Spectroscopy at the Interface of Ras and Rap1a Bound to the Ras Binding Domain of Ralgds Reveals an Electrostatic Mechanism for Protein-Protein Interaction. J Phys Chem B. 2010;114:15331–15344. doi: 10.1021/jp106974e. [DOI] [PubMed] [Google Scholar]
- 11.Boxer SG. Stark Realities. J Phys Chem B. 2009;113:2972–2983. doi: 10.1021/jp8067393. [DOI] [PubMed] [Google Scholar]
- 12.Webb LJ, Boxer SG. Electrostatic Fields near the Active Site of Human Aldose Reductase: 1. New Inhibitors and Vibrational Stark Effect Measurements. Biochemistry. 2008;47:1588–1598. doi: 10.1021/bi701708u. [DOI] [PubMed] [Google Scholar]
- 13.Suydam IT, Boxer SG. Vibrational Stark Effects Calibrate the Sensitivity of Vibrational Probes for Electric Fields in Proteins. Biochemistry. 2003;42:12050–12055. doi: 10.1021/bi0352926. [DOI] [PubMed] [Google Scholar]
- 14.Hu WH, Webb LJ. Direct Measurement of the Membrane Dipole Field in Bicelles Using Vibrational Stark Effect Spectroscopy. J Phys Chem Lett. 2011;2:1925–1930. [Google Scholar]
- 15.Oklejas V, Harris JM. Potential-Dependent Surface-Enhanced Raman Scattering from Adsorbed Thiocyanate for Characterizing Silver Surfaces with Improved Reproducibility. Appl Spectrosc. 2004;58:945–951. doi: 10.1366/0003702041655511. [DOI] [PubMed] [Google Scholar]
- 16.Oklejas V, Sjostrom C, Harris JM. Sers Detection of the Vibrational Stark Effect from Nitrile-Terminated Sams to Probe Electric Fields in the Diffuse Double-Layer. J Am Chem Soc. 2002;124:2408–2409. doi: 10.1021/ja017656s. [DOI] [PubMed] [Google Scholar]
- 17.Lambert DK. Vibrational Stark Effect of Adsorbates at Electrochemical Interfaces. Electrochimica Acta. 1996;41:623–630. [Google Scholar]
- 18.Bryan WA, Calvert CR, King RB, Greenwood JB, Newell WR, Williams ID. Controlled Redistribution of Vibrational Population by Few-Cycle Strong-Field Laser Pulses. Faraday Discuss. 2011;153:343–360. doi: 10.1039/c1fd00042j. [DOI] [PubMed] [Google Scholar]
- 19.Zou SY, Ren QH, Balint-Kurti GG, Manby FR. Analytical Control of Molecular Excitations Including Strong Field Polarization Effects. Phys Rev Lett. 2006:96. doi: 10.1103/PhysRevLett.96.243003. [DOI] [PubMed] [Google Scholar]
- 20.Hermansson K, Tepper H. Electric-Field Effects on Vibrating Polar Molecules: From Weak to Strong Fields. Molecular Physics. 1996;89:1291–1299. [Google Scholar]
- 21.Cho K, Jang YS, Gong MS, Kim K, Joo SW. Determination of Cyanide Species in Silver and Gold Plating Solutions by Raman Spectroscopy. Appl Spectrosc. 2002;56:1147–1151. [Google Scholar]
- 22.Murray CA, Bodoff S. Cyanide Adsorption on Silver and Gold Overlayers on Island Films as Determined by Surface Enhanced Raman-Scattering. J Chem Phys. 1986;85:573–584. [Google Scholar]
- 23.Jacobs MB, Jagodzinski PW, Jones TE, Eberhart ME. A Surfaced-Enhanced Raman Spectroscopy and Density Functional Theory Study of [Au(Cn)(2)](−)/[Au(Cn)(4)](−) Adsorbed on Gold Nanoparticles. J Phys Chem C. 2011;115:24115–24122. [Google Scholar]
- 24.Andrews SS, Boxer SG. Vibrational Stark Effects of Nitriles Ii. Physical Origins of Stark Effects from Experiment and Perturbation Models. J Phys Chem A. 2002;106:469–477. [Google Scholar]
- 25.Brewer SH, Franzen S. A Quantitative Theory and Computational Approach for the Vibrational Stark Effect. J Chem Phys. 2003;119:851–858. [Google Scholar]
- 26.Andrews SS, Boxer SG. Vibrational Stark Effects of Nitriles I. Methods and Experimental Results. J Phys Chem A. 2000;104:11853–11863. [Google Scholar]
- 27.Levinson NM, Fried SD, Boxer SG. Solvent-Induced Infrared Frequency Shifts in Aromatic Nitriles Are Quantitatively Described by the Vibrational Stark Effect. J Phys Chem B. 2012;116:10470–10476. doi: 10.1021/jp301054e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Etchegoin PG, Le Ru EC. A Perspective on Single Molecule Sers: Current Status and Future Challenges. Physical Chemistry Chemical Physics. 2008;10:6079–6089. doi: 10.1039/b809196j. [DOI] [PubMed] [Google Scholar]
- 29.Fang Y, Seong NH, Dlott DD. Measurement of the Distribution of Site Enhancements in Surface-Enhanced Raman Scattering. Science. 2008;321:388–392. doi: 10.1126/science.1159499. [DOI] [PubMed] [Google Scholar]
- 30.Le Ru EC, Grand J, Felidj N, Aubard J, Levi G, Hohenau A, Krenn JR, Blackie E, Etchegoin PG. Experimental Verification of the Sers Electromagnetic Model Beyond the Vertical Bar E Vertical Bar (4) Approximation: Polarization Effects. J Phys Chem C. 2008;112:8117–8121. [Google Scholar]
- 31.Ausman LK, Schatz GC. On the Importance of Incorporating Dipole Reradiation in the Modeling of Surface Enhanced Raman Scattering from Spheres. J Chem Phys. 2009;131:10. doi: 10.1063/1.3211969. [DOI] [PubMed] [Google Scholar]
- 32.Halas NJ, Lal S, Chang WS, Link S, Nordlander P. Plasmons in Strongly Coupled Metallic Nanostructures. Chem Rev. 2011;111:3913–3961. doi: 10.1021/cr200061k. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Schultz ZD. The Chemical Origin of Enhanced Signals from Tip-Enhanced Raman Detection of Functionalized Nanoparticles. Analyst. 2013;138:3150–3157. doi: 10.1039/c3an36898j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexander KD, Schultz ZD. Tip-Enhanced Raman Detection of Antibody Conjugated Nanoparticles on Cellular Membranes. Anal Chem. 2012;84:7408–14. doi: 10.1021/ac301739k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrier SL, Kownacki CM, Schultz ZD. Protein-Ligand Binding Investigated by a Single Nanoparticle Ters Approach. Chem Commun. 2011;47:2065–2067. doi: 10.1039/c0cc05059h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esteban R, Borisov AG, Nordlander P, Aizpurua J. Bridging Quantum and Classical Plasmonics with a Quantum-Corrected Model. Nat Commun. 2012:3. doi: 10.1038/ncomms1806. [DOI] [PubMed] [Google Scholar]
- 37.Kunimatsu K, Seki H, Golden WG, Gordon JG, II, Philpott MR. Electrode/Electrolyte Interphase Study Using Polarization Modulated Ftir Reflection-Absorption Spectroscopy. Surf Sci. 1985;158:596–608. [Google Scholar]
- 38.O’Haver T. Mathworks: Matlab File Exchange. 2013. Interactive Peak Fitter 9.2. [Google Scholar]
- 39.Jobbins MM, Raigoza AF, Kandel SA. Note: Circuit Design for Direct Current and Alternating Current Electrochemical Etching of Scanning Probe Microscopy Tips. Review of Scientific Instruments. 2012;83:3. doi: 10.1063/1.3695001. [DOI] [PubMed] [Google Scholar]
- 40.Johnson AS, Anderson KB, Halpin ST, Kirkpatrick DC, Spence DM, Martin RS. Integration of Multiple Components in Polystyrene-Based Microfluidic Devices Part I: Fabrication and Characterization. Analyst. 2013 doi: 10.1039/c2an36168j. [DOI] [PMC free article] [PubMed] [Google Scholar]