

Abstract
The stability of fluorotelomer alcohols under basic conditions was studied. HF elimination across the CF2-CH2 junction is shown to be facilitated by an intramolecular hydrogen bond, while solvation is the key determinant in the stability of alcohols of various perfluoroalkyl lengths. Finally, fluorotelomer alcohols can be rendered kinetically stable if either the alcohol or the base has low solubility in the reaction medium.
Keywords: Fluorotelomer alcohols, HF-elimination
1. Introduction
Fluorous synthesis [1,2] and separation [3–6] have proven to be extraordinarily useful, but are dependent on efficient synthetic methods to introduce fluorous moieties. Many fluorous synthons have been developed [7], yet fluorotelomer alcohols are uniquely absent. Fluorotelomer alcohols, F-(CF2)x(CH2)2-OH generally designated FXH2-OH, are rarely used in the literature and are cited as giving abnormal results [8–10]. The anomalies associated with fluorotelomer alcohols have been generally attributed to hydrogen-fluoride elimination across the CF2-CH2 junction [11]. Yet, this proposed elimination of HF fails to explain why fluorotelomer alcohols decompose more readily than other fluorous synthons, such as the fluorous analogs of ether protecting groups used to great effect by Curran et al [4].
Similarly, CF2-CH2 junction instability has been implicated in the conversion of fluorotelomer alcohols to perfluorinated acids as observed in in vivo studies of mice. In this case, HF elimination has been proposed to follow metabolic oxidation of the hydroxyl group [12].
Here we report detailed studies on the mechanism of fluorotelomer alcohol decomposition in synthetically relevant conditions. Specifically, we show the fundamental importance of solvation – the better solvated an alcohol, the more rapid its decomposition. These results also explain the peculiar instability of perfluorohexyl-telomer alcohols, a case well documented in the literature but never completely explained [8–10]. As such, the elucidated mechanism should be taken into account when devising schema for introducing fluorinated chains in organic molecules.
2. Results and Discussion
Fluorotelomer alcohol decomposition came to our attention when a straightforward Williamson ether synthesis under basic conditions in THF yielded no product. Visually, the solution went from clear to brown-black. Further in the presence of a poly(ethylene glycol) mesylate no substitution was observed. To provide for reproducible and comparable results, a standard decomposition reaction system was developed: 12 mM perfluorotelomer alcohol was dissolved in THF and heated to reflux with three equivalents of base for 24 hours. Sodium hydride or potassium tert-butoxide were used as the base. Stability was then defined as a lack of color change and lack of new signals by 1H- and 19F-NMR. NMR confirmed the correlation between instability – the appearance of new signals by 1H- and 19F-NMR – and the empirical observation of color change.
Alcohols F4H2-OH, F6H2-OH, F8H2-OH and F10H2-OH were all subjected to the standard stability test along with decan-1-ol and 1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluorooctane pentyl ether (F6H2-O-H5) as controls. The controls showed no color change. Decan-1-ol showed no change by NMR, while F6H2-O-H5 showed the appearance of new, low intensity, signals at 5.84, 5.75, 4.22, 3.75, 1.85 and 1.26 ppm. These new signals correspond to those seen in the decomposition of fluorotelomer alcohols (Fig. 1);1 however, the extent of decomposition was dramatically less than that seen for any alcohol except F10H2-OH. In contrast, the fluorotelomer alcohols showed variable rates of decomposition ranging from immediate color change (F4H2-OH) to only mild color change after 24 hours (F10H2-OH). The 1H-NMR spectra for the four, stability-tested alcohols are shown in Fig. 1. As can be seen (Fig. 1), only F10H2-OH shows little sign of decomposition by NMR.
Fig. 1.

1H-NMR spectra from top to bottom of neat F6H2-OH (all starting fluorotelomer alcohol 1H-NMR are identical) and F4H2-OH, F6H2-OH, F8H2-OH and F10H2-OH after stability tests in NaH/THF.
As seen above, F6H2-OH is not uniquely unstable, but rather exists along a continuum of instability, with increased stability coming with larger fluorocarbon-chain lengths. The more interesting data come from the control tests. The lack of decomposition of decan-1-ol confirms that a CF2-CH2 junction is necessary for decomposition to occur. Furthermore, as expected, F6H2-O-H5 does show decomposition – it has a CF2-CH2 junction that can eliminate HF – though much less than comparable alcohol (F6H2-OH). This suggests that the hydroxyl group plays a role in enhancing the decomposition of fluorotelomer alcohols over other species that have a CF2-CH2 junction. Ellis et al. has suggested that the unique chemical ionization fragmentations observed by mass spectrometry for fluorotelomer alcohols was the result of the intramolecular hydrogen bond shown in Fig. 2 [13].
Fig. 2.
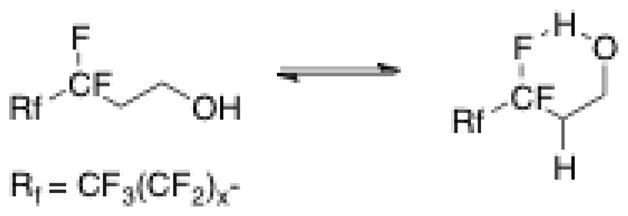
Intramolecular hydrogen bond structure proposed by Ellis et al [13].
Our hypothesis is that such an intramolecular hydrogen bond (Fig. 2) catalyzes the elimination of HF, starting the chain of events that leads to further decomposition of the fluorinated alcohols. Other heteroatoms usually outcompete aliphatic fluorine as hydrogen-bond acceptors, but aliphatic fluorines have been demonstrated to form hydrogen bonds [14]. To further test this hypothesis, fluorinated alcohols with a single methylene spacer, F3H1-OH, F6H1-OH and F8H1-OH, were tested and showed no decomposition. This behavior can be explained by considering that alcohols with a single methylene between the first CF2 and the hydroxyl group are unable to form the intramolecular hydrogen bond that catalyzes HF elimination. This is supported by recent a theoretical study by Cormanich et al., where calculations at the B3LYP/aug-cc-pVDZ level of theory and basis set showed that an intramolecular H-bond cannot form between fluorine and hydroxyl groups when involved in five-membered rings [15]. Altogether, hydroxyl group-catalyzed HF elimination appears to be the best explanation for the uniquely enhanced instability of fluorotelomer alcohols.
The mechanism of HF elimination and subsequent alcohol decomposition – through reaction of the intermediate alkene – was investigated via timed stability tests of F6H2-OH. The reaction was quenched after 15 minutes, 30 minutes, 1 hour, 2 hours and 24 hours. All reactions were worked up by simple aqueous extraction using D2O. Free fluoride was observed by 19F-NMR in the aqueous washings from all stability tests, while no starting alcohol was observed in the D2O washings. An overlay of the 1H-NMR spectra for each time point between 4.5 and 6.5 ppm is shown in Fig. 3. A double of triplets at 5.9 ppm with coupling constants of 33 and 6.5 Hz is seen to increase in intensity up to 1 hour and then decrease to a low, stead-state level between 2 and 24 hours. The NMR chemical shifts and coupling constants are comparable to those observed from the literature for semi-fluorinated alkenes [16,17]. Together, these results suggest that the semi-fluorinated alkene is indeed an intermediate in the decomposition pathway. It should be noted that the results do not rule out the possibility of a second elimination to form a highly polarized alkene, but there is currently no evidence to support this pathway.
Fig. 3.

Appearance and disappearance of vinyl proton signal during stability test of F6H2-OH, times from top to bottom, 0 min, 15 min, 30 min, 1 hour, 2 hour, 24 hour.
By Bent’s rule, the semi-fluorinated alkene produced by HF elimination is expected to be unstable. Furthermore, it is highly polarized and susceptible to nucleophilic attack and oligomerization via an anionic mechanism (Scheme 1). This explains the myriad of new signals observed in the product NMR (Fig. 1) and why the product mixture proved intractable to purification, possessed a low Rf by TLC and was un-analyzable by mass spectrometry.
Scheme 1.
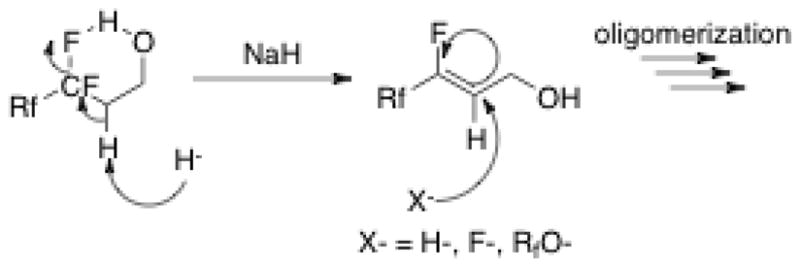
Proposed decomposition path of fluorotelomer alcohols using sodium hydride as the base. Other bases with lower pKa showed the same results.
The hydrogen-bond-facilitated elimination mechanism can explain the unique instability of fluorotelomer alcohols but not the differences in the rate and extent of decomposition observed among the alcohols. The change from a non-hydrogen-bonded conformer to a hydrogen-bonded conformer (Fig. 2) necessarily involves a change in the dipole moment of the fluorotelomer alcohols and this offered potential insight into the role that polarity might play in stability. Using Gaussian 09 [18] through geometry optimization at B3LYP/6-31G** level of theory and basis set, the dipole moments of each conformer for all alcohols were calculated. The non-hydrogen-bond conformers have an average dipole moment of 2.14 ± 0.16 Debye. The hydrogen-bond conformers have an average dipole moment of 1.04 ± 0.03 Debye. Given the large change in dipole moment upon forming the intramolecular hydrogen bond, solvent polarity was investigated as a potential factor in favoring the hydrogen-bond conformer over the linear conformer. Polar solvents were expected to favor the non-hydrogen-bond conformer and hence increase stability.
To test the effect of solvent polarity on alcohol stability, F6H2-OH was concomitantly tested in hexanes, THF and DMF using NaH as the base. The time to it took each reaction to turn brown was 24 hours in hexanes, 50 minutes in THF and 12 minutes in DMF. The trend is the exact opposite of that expected: polar solvents decrease stability instead of increasing it. This unexpected trend can be attributed to two factors. First, HF elimination proceeds through a very polar transition state (formation of a carbanion and subsequent elimination of fluoride). A polar solvent is better able to stabilize the developing charges, thus lowering the barrier to decomposition. Secondly, different solvents affect the solubility of the base. The more poorly soluble the base, the more selective that base becomes for the more acidic – and more likely to react with the insoluble base – hydroxyl proton over the less acidic methylene proton.
To further test the role of base solubility, potassium tert-butoxide was chosen as a more soluble base. The overall trend remains the same as that observed with NaH, though the rate of decomposition is dramatically accelerated: 16 minutes to brown color in hexanes, 12 minutes in THF and 7 minutes in DMF. The ability of potassium tert-butoxide to initiate decomposition also implies that any fluorotelomer alcohol – deprotonated by potassium tert-butoxide or NaH – could also act as a secondary base to eliminate HF. Thus, as the solubility of the base increased, the stability of the alcohol diminished. The most dramatic example of this base solubility was obtained when F6H2-OH was tested in BTF (α,α,α-trifluorotoluene) with NaH. Sodium hydride is completely insoluble in BTF and the results of the test show as little decomposition as F10H2-OH in THF with NaH, the least amount of decomposition for any FxH2-OH system tested. Overall, solvent polarity affects the rate of decomposition by two means: increased polarity lowers the activation barrier to HF elimination and increases the base solubility thus allowing the base to abstract the less acidic methylene proton.
Base solubility was shown to play a key role in determining alcohol stability. As such, the solubility of the alcohols was investigated as a means of explaining the differences in fluorocarbon chain length. The solubility of all fluorotelomer alcohols in THF was determined by addition of 100 mg of each alcohol to 5 mL of THF, vortexing the solution and then letting it sit for 10 minutes. If there was no phase separation or crystallization of the alcohol, 100 mg more was added and the process repeated. The results of the solubility tests indicate that F4H2-OH and F6H2-OH are both fully miscible in THF; however, F8H2-OH and F10H2-OH were soluble up to 1.1 M and 4.4 mM, respectively. F10H2-OH, which showed the least decomposition, also has the lowest solubility in THF. This suggests a correlation between the solvation of the alcohol, vis-à-vis solubility, and stability of the alcohol under basic conditions.
Given the vastly different miscibility of F6H2-OH and F10H2-OH in THF, both were retested in DMF, as a more solubilizing solvent. F6H2-OH turned brown in 12 minutes, while F10H2-OH did the same in14 minutes. These results show very similar times for color changes to occur between alcohols that possessed dramatic differences in THF. This suggests that the solvation of the alcohol, in terms of solubility, is also key to the degradation of fluorotelomer alcohols under basic conditions. When the alcohol is not very soluble the CF2-CH2 junction is less accessible to the base. Hence, base-catalyzed HF elimination is afforded kinetic protection. The result is increased stability in THF for the less soluble F8H2-OH and F10H2-OH.
The possibility that the slow rate of decomposition observed with the larger alcohols could be due to the spontaneous formation of supramolecular fluorous aggregates, which would prevent the intramolecular elimination of HF, was also considered. To probe the viability of aggregation by fluorotelomer alcohols, solutions of 12 mM fluorotelomer alcohol in THF were prepared and analyzed by dynamic light scattering (DLS). No aggregates were observed for any alcohol at the concentrations used to monitor decomposition behavior.
A subsequent systematic study of aggregation behavior of F8H2-OH in THF was carried out by 19F-NMR studies of solutions ranging from 1 mM to 1 M. Using 0.1 mM BTF as an internal reference, 19F-NMR spectra were collected and the change in chemical shift, in Hz, of the trifluoromethyl groups was plotted against the log of concentration (Fig. 4). By NMR, F8H2-OH was only observed to aggregate at concentrations at or above 1 M (Fig. 4), concentrations that approach the solubility limit of F8H2-OH. Therefore, while possible, aggregation seems unlikely as an explanatory factor for alcohol stability. Aggregation can further be ruled out for two reasons. First, F4H2-OH and F6H2-OH are both fully soluble in THF and show no signs of aggregation by DLS, yet F6H2-OH decomposes more slowly than F4H2-OH. Second, F8H2-OH is more stable still and yet shows no signs of aggregating at the concentrations used – either by DLS or by NMR.
Fig. 4.
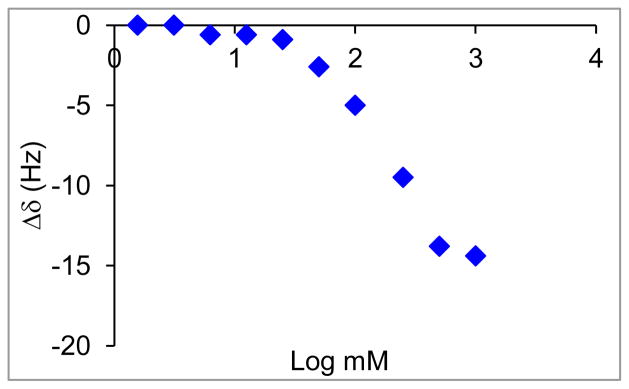
Change in chemical shift of F8H2-OH’s –CF3 as compared to changes in concentration
3. Conclusion
The unique instability of fluorotelomer alcohols was investigated. Fluorotelomer alcohols are uniquely unstable due to the formation of an intramolecular hydrogen bond, which facilitates HF elimination. It has been demonstrated, spectroscopically, that HF elimination is indeed the mechanism by which decomposition initiates. The resulting, unstable alkene then oligomerizes to give an intractable mixture of unidentifiable products. Solvation is the best mechanism to explain the differences in the decomposition with changes in solvent, base and fluorous-segment length. In contrast to the mechanism elucidated in in vivo degradation, these results demonstrate an HF-elimination pathway that does not first depend on oxidation of the alcohol. The data also suggest a means of circumventing the instability problem for synthetic ends: the use of solvents in which either the base or alcohol is not very soluble. The effectiveness of this type of methodology has been used in a recent publication by Zaggia et al. to yield semi-fluorinated ethers from fluorotelomer alcohols and bromoalkanes in aqueous KOH in good to excellent yields (60 – 97 %) [19].
4. Materials and Methods
4.1 Materials
Fluorous chemicals were purchased from SynQuest Laboratories Inc. (Alachua, FL). 1H- and 19F-NMR spectra were obtained on 400 and 500 spectrometers using CDCl3 as solvent and CFCl3 as internal reference. Particle sizes were determined by dynamic light scattering (DLS).
4.2 Stability protocol
To a dry 25 mL roundbottom flask on ice, under argon were added 10 mL of dry solvent (THF, BTF, hexane or DMF) and 0.36 mmol of base (NaH or potassium tert-butoxide). To this were added 0.12 mmol of FxH2OH. The reaction was then slowly warmed from 0 °C to reflux and allowed to react for 24 hours. The reaction was then cooled, diluted with 50 mL DCM and washed with 50 mL water. The organic layer was then dried over magnesium sulfate and condensed under reduced pressure.
Supplementary Material
Highlights.
The instability of fluorotelomer alcohols – a problem widely acknowledged yet not elucidated in the literature – is investigated.
The mechanism of decomposition relies on formation of an intramolecular hydrogen bond to facilitate HF-elimination.
The effects of solvent, base and fluorocarbon-chain length are thoroughly investigated and rationalized through solubility.
Acknowledgments
The work described in this article was supported by NIH grant NIGMS 079375 to S.M.
Footnotes
Full sized NMR spectra for each starting alcohol before and after the stability tests and all NMR spectra not shown herein can be found in Supplementary Information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Curran DP. Angew Chem Int Ed. 1998;37:1174–1196. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1174::AID-ANIE1174>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 2.Zhang W. Chem Rev. 2009;109:749–795. doi: 10.1021/cr800412s. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida J-i, Itami K. Chem Rev. 2002;102:3693–3716. doi: 10.1021/cr0103524. [DOI] [PubMed] [Google Scholar]
- 4.Curran DP, Sinha MK, Zhang KZ, Sabatini JJ, Cho DH. Nat Chem. 2012;4:124–129. doi: 10.1038/nchem.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tzschucke CC, Markert C, Bannwarth W, Roller S, Hebel A, Haag R. Angew Chem Int Ed. 2002;41:3964–4000. doi: 10.1002/1521-3773(20021104)41:21<3964::AID-ANIE3964>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 6.Zhang W, Curran DP. Tetrahedron. 2006;62:11837–11865. doi: 10.1016/j.tet.2006.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rábai J. In: Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horváth IT, editors. Wiley-VCH GmbH; Weinheim, Germany: 2004. pp. 156–174. [Google Scholar]
- 8.Xu W, Osei-Prempeh G, Lema C, Oldham ED, Aguilera RJ, Parkin S, Rankin SE, Knutson BL, Lehmler HJ. Carbohydr Res. 2012;349:12–23. doi: 10.1016/j.carres.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meinert H, Geister U. J Fluor Chem. 1994;68:221–226. [Google Scholar]
- 10.Harada N, Nishikata T, Nagashima H. Tetrahedron. 2012;68:3243–3252. [Google Scholar]
- 11.Rocaboy C, Rutherford D, Bennett BL, Gladysz JA. J Phys Org Chem. 2000;13:596–603. [Google Scholar]
- 12.Zhang XJ, Lai TB, Kong RYC. Top Curr Chem. 2012;308:365–404. doi: 10.1007/128_2011_270. [DOI] [PubMed] [Google Scholar]
- 13.Ellis DA, Mabury SA. J Am Soc Mass Spectrom. 2003;14:1177–1191. doi: 10.1016/S1044-0305(03)00450-1. [DOI] [PubMed] [Google Scholar]
- 14.Caminati W, MElandri S, Maris A, Ottaviani P. Angew Chem Int Ed. 2006;45:2438–2442. doi: 10.1002/anie.200504486. [DOI] [PubMed] [Google Scholar]
- 15.Cormanich RA, Freitas MP, Tormena CF, Rittner R. RSC Adv. 2012;2:4169–4174. [Google Scholar]
- 16.Petrov VA. J Org Chem. 1995;60:3423–3426. [Google Scholar]
- 17.Banks RE, Barlow MG, Nikkho-Amiry MJ. J Fluor Chem. 1997;82:171–174. [Google Scholar]
- 18.Frisch MJT, GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralata JE, Ogliao F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Corss JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision B.1. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 19.Zaggia A, Conte L, Ceretta PF. J Fluor Chem. 2010;131:844–851. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.