

Abstract
Chronic pressure overload (PO) induces pathological left ventricular hypertrophy (LVH) leading to congestive heart failure (HF). Overexpression of FKBP12.6 (FK506-binding protein [K]) in mice should prevent Ca2+-leak during diastole and may improve overall cardiac function. In order to decipher molecular mechanisms involved in thoracic aortic constriction (TAC)-induced cardiac remodeling and the influence of gender and genotype, we performed a proteomic analysis using two-dimensional differential in-gel electrophoresis (2D-DIGE), mass spectrometry, and bioinformatics techniques to identify alterations in characteristic biological networks. Wild-type (W) and K mice of both genders underwent TAC. Thirty days post-TAC, the altered cardiac remodeling was accompanied with systolic and diastolic dysfunction in all experimental groups. A gender difference in inflammatory protein expression (fibrinogen, α-1-antitrypsin isoforms) and in calreticulin occurred (males > females). Detoxification enzymes and cytoskeletal proteins were noticeably increased in K mice. Both non- and congestive failing mouse heart exhibited down- and upregulation of proteins related to mitochondrial function and purine metabolism, respectively. HF was characterized by a decrease in enzymes related to iron homeostasis, and altered mitochondrial protein expression related to fatty acid metabolism, glycolysis, and redox balance. Moreover, two distinct differential protein profiles characterized TAC-induced pathological LVH and congestive HF in all TAC mice. FKBP12.6 overexpression did not influence TAC-induced deleterious effects. Huntingtin was revealed as a potential mediator for HF. A broad dysregulation of signaling proteins associated with congestive HF suggested that different sets of proteins could be selected as useful biomarkers for HF progression and might predict outcome in PO-induced pathological LVH.
Keywords: 2D-DIGE, cardiac hypertrophy, FKBP12.6, gender, heart failure, pressure overload, Proteomics, transgenic mice
Introduction
Aortic stenosis causes chronic pressure overload (PO) of the left ventricle (LV) that induces myocardial remodeling. In response to the increase in hemodynamic load and neurohormonal stress, the heart initially undergoes an adaptive compensatory left ventricular hypertrophy (LVH) that may progress to maladaptive hypertrophy which, in turn, may eventually lead, with time, to heart failure (HF). Impairment in Ca2+ homeostasis, such as the downregulation of Ca2+-modulating proteins (e.g., sarcoplasmic reticulum [SR] Ca2+-ATPase 2a: SERCA2a), is a common observation in HF. Among the multiple partners mediating the cardiac hypertrophy program and orchestrating Ca2+ homeostasis in the cardiomyocyte, FK506-binding proteins (FKBPs), ubiquitously expressed immunophilins (Schreiber 1991) are involved in the regulation of gene transcription, protein translation, and cell trafficking (Harrar et al. 2001). In particular, the two smallest members of the FKBP family, FKBP12 (calstabin1) and FKBP12.6 (calstabin2), both expressed in cardiomyocytes (Lam et al.1995), play a crucial role in the regulation of intracytoplasmic SR Ca2+ release (Marks 1996; Prestle et al. 2001). The affinity of FKBP12.6 for the Ca2+ release channel (ryanodine receptor [RyR2]) is higher than that of FKBP12 (Jeyakumar et al. 2001) and stabilizes RyR2 in a closed state during diastole, thereby reducing diastolic Ca2+ leak from the SR (Prestle et al. 2001). Disruption of the FKBP12.6 gene in mice results in cardiac hypertrophy in male, but not in female mice (Xin et al. 2002). FKPB12.6-null mice display no structural or functional abnormalities at rest (Wehrens et al. 2003). Indeed, the ablation of FKBP12.6 did not render mice susceptible to stress-induced ventricular arrhythmias (Xiao et al. 2007).
In our transgenic mouse model with specific cardiac overexpression of FKBP12.6 (Gellen et al. 2008), mice of both genders are healthy with no apparent morphologic abnormalities. In male mice we showed that FKBP12.6 overexpression in cardiomyocytes prevents triggered ventricular tachycardia in normal heart in stress conditions, this antiarrhythmic effect resulting likely from increased FKBP12.6 binding to RyR2 (Gellen et al. 2008). More recently, we showed that the cardiac overexpression of FKBP12.6 also protects against catecholamine-promoted burst pacing-induced ventricular tachycardia in the remodeled heart 2 months after thoracic aorta constriction (TAC) (Vinet et al. 2012). This is due at least in part to a decreased sensitivity of cardiomyocytes to catecholamines associated with decreased activation of the Ca2+/Calmodulin-dependent kinase II (decreased phospho-Ser 2814 RyR2) and calcineurin-nuclear factor of activated T cells signaling pathways (Prévilon et al. 2011).
Although much is known about the pathways that promote hypertrophic responses, biological functions can rarely be attributed to individual molecules, but rather arise through complex interactions between numerous cell components. We have shown that FKBP12.6 transgenic mice are protected against TAC-induced mortality, and that female mice develop hypertrophy and HF earlier than males (Prévilon et al. 2011). The latter was associated with temporal changes in Ca2+-dependent and independent cell processes but FKBP12.6 overexpression did not significantly affect the progression of HF following TAC. However, pathological hypertrophy eventually leads to HF via mechanisms that are not yet completely understood and the gender difference in cardiac dysfunction caused by TAC has not yet been clarified. In an effort to elucidate such mechanisms, the aim of this study was to build upon our previous findings to determine whether FKBP12.6 overexpression influences the cardiac remodeling proteome. Using the validated TAC model in mice to induce progressive hypertrophy leading with time to HF, we applied proteomic technology to investigate how FKBP12.6 overexpression could imprint the proteome signature in PO-induced remodeling of mice without (C) or mice with congestive heart failure (H).
Material and Methods
Experimental animals
The transgenic mice overexpressing FKBP12.6 on a B6D2/F1 background were produced in our laboratory and have been described in detail previously (Gellen et al. 2008). Male and female wild type (W) and mice overexpressing FKBP12.6 (K) were used. Mice were housed in a specific pathogen-free facility and handled in accordance with European Union Directives (86/609/EEC) on care and use of laboratory animals. The review and approval of the study was obtained by the local Animal Ethics Committee (No. B 7518 03).
Transverse aortic constriction
Adult female and male mice (5–6 weeks old) of different genotypes (W or K), weighing (18–22 g), underwent TAC using a 27-gauge needle as previously described (Prévilon et al. 2010). Animals were killed 30 days later. Hearts were quickly excised; then chambers (atria, right, and LVs) were dissected, weighed, and immediately frozen in liquid nitrogen and stored at −80°C until use.
Experimental groups for two-dimensional differential in-gel electrophoresis (2D-DIGE) consisted of 12 groups (with four mice per group): female (F) and male (M) mice of the two genotypes were submitted either to TAC (T) or to sham operation (S). TAC groups were further split into two groups according to the presence or absence of lung edema, mice without pulmonary edema (noncongested, C) and mice with congestive heart failure (H). The abbreviations of designated groups are reported in Figure 1.
Figure 1.

Experimental animal groups. The cardiac left ventricle from mice of both genders (n = 24) and genotypes (n = 24) were studied at 30 days postsurgery. Symbols used for genotypes are W (wild type) and K (mice overexpressing FKBP12.6), and for genders F (female) and M (male), sham-operated mice without banding (S) and mice having undergone thoracic aortic constriction (T for TAC). Due to the presence of pulmonary edema, two distinct TAC groups were defined as follows: without [noncongested mice, (C)] or with lung congestion [mice with congestive heart failure, (H)].
Echocardiography
Transthoracic echocardiography was performed with a Toshiba Powervision 6000 (SSA 370A; Toshiba, Tokyo, Japan) device equipped with an 8- to 14-MHz linear and a 6- to 10-MHz sectoriel probe as previously described (Prévilon et al. 2011).
Two-dimensional differential in-gel electrophoresis
To ensure reliability, all samples from the TAC group (C and H) and its respective sham-operated group (S) were processed simultaneously.
Tissue sample preparation
The frozen cardiac LVs from each mouse experimental group (n = 4 per group) were individually pulverized under liquid nitrogen to yield a fine powder using a pestle and mortar. The tissue powder was solubilized in lysis buffer (8 mol/L urea, 2 mol/L thiourea, 4% CHAPS, 60 mmol/L dithiothreitol [DTT]). Then the protein extracts were clarified by ultra-centrifugation at 100,000g for 1 h at 4°C. The supernatants were then treated with the 2D Clean-Up kit (GE Healthcare, Buc, France) according to the manufacturer's instructions. The resulting dry pellets were resuspended in lysis buffer without DTT and adjusted to pH 8.5 with 1 mol/L Tris-base. Protein concentrations of the samples were determined by the Bradford method, and were in the range of 9–12 μg/μL.
Two-dimensional differential in-gel electrophoresis
The LV Samples (50 μg) were labeled with CyDyes™ Fluor minimal dyes (GE Healthcare) Cy3 or Cy5 according to the manufacturer's instructions. The internal standard was prepared by combining equal quantities of all samples used for this study and labeled with Cy2. Fifty micrograms of labeled samples (Cy3 or Cy5) and internal standard (Cy2) were mixed as indicated in Table 1 and each mixed Cy-dye labeled protein extract (150 μg) was added to a rehydration buffer (8 mol/L urea, 2 mol/L thiourea, 2% (w/v) CHAPS, 10 mmol/L DTT, 1,2% (v/v) pH 4–7 IPG buffer (GE Healthcare) and trace of bromophenol blue). Immobiline™ Drystrips (pH 4–7, 18 cm, GE Healthcare) were rehydrated in the dark for 24 h at room temperature under low-viscosity paraffin oil. Isoelectric focusing was performed using an IPGphor system (GE Healthcare) for a total of 52 kVh. IPG strips were then incubated consecutively for 15 min each in equilibration buffer I and II (Buffer I: 50 mmol/L Tris-HCl, pH 8.8, 6 mol/L urea, 2% [w/v] sodium dodecyl sulfate [SDS], 30% [v/v] glycerol and 1% [w/v] DTT; Buffer II: Buffer I with 4.7% [w/v] iodoacetamide and no DTT). Equilibrated strips were placed onto homemade 12% SDS-polyacrylamide gel (SDS-PAGE) and overlaid with agarose solution (0.5% low-melting agarose with a trace of bromophenol blue in running buffer) and electrophoresis was performed in a Ettan-DALT II system (GE Healthcare) at 2.5 W/gel at 12°C until the bromophenol blue dye reached the bottom of the gel. Low fluorescent glass plates were used to minimize background fluorescence during scanning. The gels were scanned using a Typhoon 9400 Trio Variable Mode Imager (GE Healthcare). Gels were scanned using optimal excitation/emission wavelength for each DIGE fluor (Cy2 488/520 nm; Cy3 532/580 nm; Cy5 633/670 nm) and with a resolution set at 100 μm.
Table 1.
Experimental design for 2D-DIGE analysis
| Gel number | Cy3 labeling (50 μg protein) | Cy5 labeling (50 μg protein) | Cy2 labeling (50μg protein) | Number of detected spots | Percentage of matched spot referenced to gel 1 |
|---|---|---|---|---|---|
| 1 | FWH1 | FKS3 | IS | 1795 | 100% |
| 2 | FWH2 | FKC3 | IS | 1634 | 77% |
| 3 | FWC1 | FKH3 | IS | 1630 | 79% |
| 4 | FWC2 | FKS4 | IS | 1546 | 76% |
| 5 | FWS1 | FKC4 | IS | 1524 | 74% |
| 6 | FWS2 | FKH4 | IS | 1543 | 77% |
| 7 | FKH1 | FWH3 | IS | 1650 | 79% |
| 8 | FKH2 | FWS3 | IS | 1505 | 75% |
| 9 | FKC1 | FWC3 | IS | 1627 | 78% |
| 10 | FKC2 | FWH4 | IS | 1541 | 75% |
| 11 | FKS1 | FWC4 | IS | 1589 | 76% |
| 12 | FKS2 | FWS4 | IS | 1519 | 73% |
| 13 | MWH1 | MKS3 | IS | 1502 | 63% |
| 14 | MWH2 | MKC3 | IS | 1741 | 75% |
| 15 | MWC1 | MKH3 | IS | 1702 | 73% |
| 16 | MWC2 | MKS4 | IS | 1572 | 67% |
| 17 | MWS1 | MKC4 | IS | 1754 | 74% |
| 18 | MWS2 | MKH4 | IS | 1733 | 72% |
| 19 | MKH1 | MWH3 | IS | 1662 | 74% |
| 20 | MKH2 | MWS3 | IS | 1562 | 70% |
| 21 | MKC1 | MWC3 | IS | 1606 | 70% |
| 22 | MKC2 | MWH4 | IS | 1490 | 68% |
| 23 | MKS1 | MWC4 | IS | 1487 | 70% |
| 24 | MKS2 | MWS4 | IS | 1672 | 73% |
Fifty micrograms of the proteins extracted from each sample were labeled with Cy3 or Cy5 as indicated. Cy2-labeled Internal Standard (IS) was prepared by combining equal portions of the 48 left ventricular samples. Analytical gels of left ventricular proteins from MWS, male wild-type sham-operated mice (n = 4); MWC, male wild-type noncongested mice (n = 4); MWH, male wild-type mice with congestive heart failure (n = 4); FWS, female wild-type sham-operated mice (n = 4); FWC, female wild-type noncongested mice (n = 4); FWH, female wild-type mice with congestive heart failure (n = 4); MKS, male FKBP12.6 overexpressing sham-operated mice (n = 4); MKC, male FKBP12.6 overexpressing noncongested mice (n = 4); MKH, male FKBP12.6 overexpressing mice with congestive heart failure (n = 4); FKS, female FKBP12.6 overexpressing sham-operated mice (n = 4); FKC, female FKBP12.6 overexpressing noncongested mice (n = 4); FKH, female FKBP12.6 overexpressing mice with congestive heart failure (n = 4).
2D-DIGE differential expression analysis
Image analysis, relative quantification and statistical evaluation and PCA (Principal Component Analysis) were carried out with DeCyder™ 2D software (GE Healthcare, version 7.0). The one-way analysis of variance (ANOVA) test followed by correction for false discovery rate (FDR) (P < 0.05) were used to determine protein spots significantly different between analyses. The fold change (FC) and Student's T-test P-values were calculated across several pairwise comparisons (Male [M] vs. Female [F], FKBP12. 6 [K] vs. Wild type [W], TAC [H, C] vs. Sham [S], congestive failing [H] vs. S, and noncongested mice [C] vs. S) and considered significant for P-values <0.05 and FC >1.2 or <−1.2, taking into account the power of DIGE method to detect a reliable difference in protein abundance down to 15% (Marouga et al. 2005; Viswanathan et al. 2006). FDR correction was applied as a multiple testing correction method to keep the overall error rate as low as possible (Benjamini and Hochberg 2000). Proteins of interest were identified by mass spectrometry.
Protein identification by mass spectrometry
For mass spectrometry analysis, two semi-preparative 2D-gels were prepared as analytical gels. The IPG strips were rehydrated with 400 μg of equal amounts of male LV or female LV samples, respectively. After electrophoresis, 2D-gels were fixed in 30% (v/v) ethanol, 2% (v/v) phosphoric acid (two changes, 30 min each), and then stained for 72 h in 0.01% (w/v) Coomassie Brilliant Blue G-250, 12% (w/v) ammonium sulfate, 18% (v/v) ethanol, and 2% (v/v) phosphoric acid. Spots of interest were manually excised from Coomassie blue-stained semi-preparative gels. Destained and dehydrated gel spots were digested with trypsin (Promega) solution (12.5 ng/μL in 40 mmol/L NH4HCO3-10% acetonitrile) overnight at 40°C. Extracted Peptides were then desalted and concentrated using C18 ZipTips (Millipore, Molsheim, France). Samples were loaded onto a Ziptip by pipetting up and down ten times using a fresh 96-well plate. Samples (in ZipTips) were washed three times with 10 μL of 0.1% trifluoroacetic acid (TFA) and eluted with 4 μL of 50% acetonitrile/0.1% TFA and with 4 μL of 70% acetonitrile/0.1% TFA. Eluted peptides were pooled, dried, and a total of 1.5 μL of eluate was pipetted onto a clean Matrix-Assisted Laser Desorption/Ionisation plate covered with 1.5 μL of α-cyano-4-hydroxycinnamic acid MALDI matrix (LaserBioLabs, Sophia-Antipolis, France).
Mass spectra for each spot were acquired with a MALDI-TOF/TOF mass spectrometer (MALDI-TOF-TOF 4800, ABSciex, les Ulis, France), running version 3.5.28193 of 4000 series explorer software. After filtering tryptic-, keratin-, and matrix-contaminant peaks up to 15 parent ions were selected for subsequent MS/MS fragmentation according to mass range, signal intensity, signal to noise ratio, and absence of neighboring masses in the MS spectrum. Database searching was carried out using Mascot version 2.2 (MatrixScience, London, U.K.) via GPS explorer software (ABSciex) version 3.6 combining MS and MS/MS interrogations on Mus musculus from Swiss-Prot databank 57.13 containing 16271 sequences (January 2010) (http://www.expasy.org). The search parameters were as follows: carbamidomethylation as a variable modification for cysteine and oxidation as a variable modification for methionine residues. Up to 1 missed tryptic cleavage was permitted and mass accuracy tolerance of 30 ppm for precursors and 0.3 Da for fragments were used for all trypsic mass searches. Positive identification was based on a Mascot score above the significance level (i.e., <5%). The reported proteins were always those with the highest number of peptide matches. Under our identification criteria, no result was found to match multiple members of a protein family.
Modeling with ingenuity pathway analysis
In order to gain insights into the biological pathways and networks that were significantly represented in our proteomic datasets (regulated proteins identified by 2D-DIGE and mass spectrometry) we used ingenuity pathway analysis (IPA; Ingenuity Systems, Redwood City, CA). http://www.ingenuity.com). IPA builds hypothetical networks from these focus proteins, and other non–2D DIGE-identified proteins. The networks are displayed graphically as nodes (individual proteins) and edges (the biologic relationships between the nodes). IPA computes a score for each network from the P-value that indicates the likelihood of the focus proteins in a network being found together due to random chance. We selected only networks scoring ≥2, with P < 0.01 of not being generated by chance. Biological functions were assigned to each network by use of annotations from the scientific literature and stored in the Ingenuity Pathways Knowledge Base (IPKB). The Fisher exact test was used to calculate the P-value determining the probability of each biological function/disease or pathway being assigned by chance. The build function of IPA allows for generating pathways that can complete the data analysis by showing interactions of identified proteins with a specific group of molecules.
Western blotting
Fifty micrograms of each cardiac LV lysate were loaded onto a 4–20% gradient, 10 or 18% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked and immunoblotted with diluted primary antibodies to sarcolemmal membrane-associated protein (SLMAP) (1:1000, sc-100957, Santa Cruz, Biotechnology Inc., Santa Cruz, CA, USA), HSPB1 (anti-HSP27, 1:1000, sc1049 Santa Cruz, Biotechnology), CRYAB (1:2000, Thermo Fisher Scientific, Illkirch, France), NDRG2 (1:5000, ab72140, Abcam, Paris, France), ERp29 (1:2500, ab11420, Abcam), huntingtin (HTT) (1:1000, clone 1HU-4C8, MAB2166, Millipore), and finally incubated with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit secondary antibodies, as appropriate. Visualization by chemiluminescence detection was carried out according to the manufacturer's instructions (ECL kit, GE Healthcare). Equal protein loading for LV lysates was assessed by stripping blot and reprobing with an anti-calsequestrin antibody (CSQ; 1: 2500, PA1-913, Affinity BioReagents, Golden, CO, USA). Quantitation of digitized images of immunoblots was done using ImageJ software (http://rsb.info.nih.gov/nih-image/about.html.). The intensity of immunoreactive bands was normalized to that of calsequestrin. Data are expressed as percentages of the respective control (means ± SEM).
Coimmunoprecipitation
Immunoprecipitation (IP) buffer (50 mmol/L TRis-HCl pH7.4, 100 mmol/L NaCl, 15 mol/L EDTA, 1% Triton × 100) supplemented with a complete cocktail of inhibitor proteases (fast Sigma Aldrich Chimie, Saint-Quentin-Fallavier, France) was added to 200 μg of LV lysates to a final volume of 0.5 mL. The LV lysates were depleted in immunoglobulins by incubation with protein A- and protein G- Sepharose magnetic beads for 1 h at 4°C. Using Pierce Crosslink magnetic IP/Co-IP kit (Thermo Scientific, Illkirch, France), 5 μg of anti-HSP27 antibody (sc1049) were crosslinked onto protein A/G magnetic beads according to the manufacturer's instructions. The cleared lysates were incubated with HSP27 antibody-crosslinked beads on a rotator overnight at 4°C, then the beads were collected with a magnetic stand and nonbound sample was removed and saved for analysis. Following three washes of beads with IP-buffer, 100 μL of elution buffer was added and incubated under rotation for 5 min at room temperature. The beads were magnetically separated and the pH of elution buffer containing the IP-target antigen was adjusted by adding 1 mol/L Tris, pH 7.5. Laemmli loading buffer was added to the eluted protein. Immunoprecipitated protein samples were fractioned on 3–8% NuPAGE Tris-Acetate gel (Life technologies, Saint Aubin, France), then transferred to nitrocellulose membrane. After blocking, the membrane was incubated with primary antibodies to anti-HTT, 1:1000 or anti- IMMT, 1:5000. After 3 washes, the membranes were probed, respectively, with HRP-linked anti-mouse or -rabbit secondary antibodies. Co-IP protein was detected by chemiluminescence. Films were digitized and quantitated using ImageJ.
Statistical analysis
Results were presented as means ± SEM. To determine the effects of surgery, gender, or genotype and their interaction on expressed identified protein in LV, statistical differences were determined using ANOVAs followed by the FDR correction method (Benjamini and Hochberg, 1995) for multiple comparisons, P ≤ 0.05 indicates statistical significance.
Results
TAC induces pathological LV hypertrophy leading to congestive HF
Gravimetric data are reported in Table 2. A significant gender difference in myocardial mass was observed in mice of both genotypes (W: wild type; K: FKBP12.6 overexpressing mice) without TAC, which was higher in male than in female mice (32 and 48%, respectively). Thirty days following surgery, heart weight to tibia length ratios were significantly increased in W and K mice with TAC compared with sham-operated mice (Table 2). Moreover, the presence of pulmonary edema (defined as lung weight to tibia length ratio in TAC mice > mean value in sham group + 3 SD) was used as the criterion to classify TAC mice. Accordingly, two groups were defined as noncongested mice (C) and as mice with congestive heart failure (H) (Fig. 2A). As expected, the hypertrophic response to TAC was more important in H than in C mice (Fig. 2B).
Table 2.
Gravimetric data
| Female | Male | ||||||
|---|---|---|---|---|---|---|---|
| Genotype | Sham | TAC | Sham | TAC | |||
| FWS | FWC | FWH | MWS | MWC | MWH | ||
| W | Mice (n) | 4 | 4 | 4 | 4 | 4 | 4 |
| BW (g) | 20 ± 1 | 22 ± 1 | 22 ± 1 | 31 ± 1 | 28 ± 0.2 | 28.4 ± 1.4 | |
| LVW (mg) | 79 ± 3 | 116 ± 6* | 154 ± 9* | 112 ± 4† | 158 ± 4* | 171 ± 2* | |
| HW/TL (mg/mm) | 6.6 ± 0.3 | 8.8 ± 0.4* | 13 ± 1*$ | 8.4 ± 0.2 | 12 ± 0.4* | 13.0 ± 0.4* | |
| Lu W/TL (mg/mm) | 8.6 ± 0.5 | 8.8 ± 0.3 | 24 ± 4*$ | 9.1 ± 0.3 | 9.5 ± 0.6 | 18.1 ± 2.4*$ | |
| K | FKS | FKC | FKH | MKS | MKC | MKH | |
| Mice (n) | 4 | 4 | 4 | 4 | 4 | 4 | |
| BW (g) | 20 ± 1 | 23 ± 1 | 22 ± 1 | 28 ± 2 | 28 ± 1 | 25 ± 3 | |
| LVW(mg) | 73 ± 4 | 127 ± 10* | 179 ± 2* | 110 ± 10† | 154 ±15 | 181 ± 18* | |
| HW/TL (mg/mm) | 6.4 ± 0.4 | 9.9 ± 0.8* | 14 ± 0.3*$ | 8.5 ± 0.4 | 11 ± 1* | 13.9 ± 1.6*$ | |
| Lu W/TL (mg/mm) | 8.5 ± 0.5 | 9.9 ± 0.3* | 21 ± 3*$ | 8.7 ± 0.4 | 9.3 ± 0.2* | 14.4 ± 3.4*$ | |
Data are mean ± SEM. W, wild-type mice; K, FKBP12.6 overexpressing mice; S, sham-operated; TAC, thoracic aortic constriction; F, female; M, male; C, noncongested mice; H, mice with congestive heart failure; BW, body weight; LVW; left ventricle weight; HW/TL, ratio heart weight to tibia length; Lu/TL, ratio lung weight to tibia length. *P < 0.05 TAC versus sham; $P < 0.05 C versus H mice; †P < 0.05 female versus male.
Figure 2.
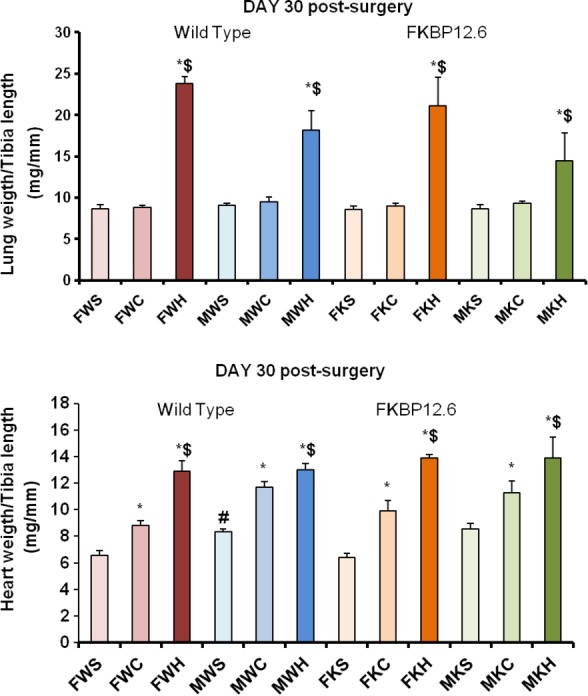
Characterization of the animal model. At 30 day postsurgery, thoracic aortuc constriction (TAC) induced pathological left ventricular hypertrophy and congestive HF in wild-type mice (W) and in FKBP12.6 overexpressing mice (K) of both genders (M for male; F for female). Sham-operated mice (S) were also studied at 30 days. (A), histograms showing the ratio of lung weight to tibia length in groups where pulmonary edema index was used to classify TAC mice into noncongested mice (C) or mice with congestive heart failure (H). (B), hypertrophic responses to TAC, (heart weight to tibia length as hypertrophy index) for mice of both genotypes and genders. Data are means ± SEM; *P < 0.05 TAC compared with Sham; $P < 0.05, mice with lung congestion (H) compared with mice without lung congestion (C).
Functional remodeling
Echocardiographic data are reported in Table 3. No difference was observed in stenotic jet indicating a similar degree of aortic constriction in all TAC mice (Table 3). Marked increases in interventricular septum (IVSTD) and posterior wall thickness (PWTD) at end diastole were found in female failing hearts of both genotypes. A significant genotype difference in the hypertrophic response to TAC was also observed. Male fKbp12.6 overexpressing TAC (MKT) mice exhibited a greater increase in IVSTD and PWTD than MWT mice. This increase was more pronounced in the MKH group. As expected, TAC was associated with alterations in LV function. In TAC mice, ejection fraction (EF) was reduced in both genders and genotypes, indicating systolic dysfunction. A genotype difference in the increase in LV end diastolic diameter (LVEDD) was also found in female mice, failing hearts being dilated in FK mice only. Early diastolic velocity of the mitral annulus (Ea) was decreased in all banded mice, whatever the gender or the genotype, indicating altered diastolic function. A gender difference was also observed in the TAC-induced decrease in aortic outflow (30 and 20% in male and female mice, respectively, P < 0.05). Thus, both genders of W and K mice developed LV hypertrophy following TAC accompanied with systolic and diastolic LV dysfunction.
Table 3.
Echocardiographic data
| Female | Male | ||||||
|---|---|---|---|---|---|---|---|
| Genotype | Sham | TAC | Sham | TAC | |||
| W | FWS | FWC | FWH | MWS | MWC | MWH | |
| Mice (n) | 4 | 5 | 3 | 5 | 4 | 4 | |
| BW (g) | 23 ± 1 | 21 ± 2 | 21 ± 1 | 25 ± 3 | 25 ± 2 | 25 ±1 | |
| HR (bpm) | 423 ± 38 | 420 ± 60 | 419 ± 38 | 456 ± 48 | 450 ± 50 | 435 ± 24 | |
| IVSTD (mm) | 0.57 ± 0.10 | 0.74 ± 0.07* | 0.97 ± 0.07*$ | 0.61 ± 0.02 | 0.63 ± 0.18 | 0.68 ± 0.15 | |
| PWTD (mm) | 0.51 ± 0.06 | 0.80 ± 0.15* | 1.12 ± 0.24*$ | 0.59 ± 0.10 | 0.77 ± 0.20* | 0.76 ± 0.22 | |
| LVmass (mg) | 80.8 ± 9.4 | 140 ± 38* | 211 ± 50* | 99.9 ± 13.2 | 134.4 ± 20.7 | 164.6 ± 52.3 | |
| LVEDD (mm) | 4.3 ± 0.2 | 4.5 ± 0.4 | 4.5 ± 0.1 | 4.5 ± 0.4 | 4.8 ± 0.6 | 5.2 ± 0.1 | |
| FS% | 44 ± 7 | 32 ± 5* | 28 ± 5* | 39 ± 8 | 32 ± 2 | 22 ± 5* | |
| EF% | 82 ± 6 | 68 ± 7* | 63 ± 8* | 77 ± 9 | 68 ± 3 | 52 ± 10* | |
| Spw (cm/sec) | 3.1 ± 0.7 | 2.3 ± 0.1* | 2.4 ± 0.5 | 3.1 ± 0.3 | 2.9 ± 0.4 | 2.3 ± 0.2*$ | |
| Ea(cm/sec) | 4.7 ± 0.5 | 3.3 ± 1.0* | 3.2 ± 0.2* | 4.6 ± 0.6 | 3.7 ± 0.6 | 2.9 ± 0.4*$ | |
| E/Ea | 0.2 ± 0.0 | 0.4 ± 0.1* | 0.3 ± 0.0 | 0.2 ± 0.0 | 0.3 ± 0.0* | 0.4 ± 0.0*$ | |
| Aortic outflow (m/sec) | 1.0 ± 0.2 | 0.8 ± 0.1 | 0.8 ± 0.1* | 1.3 ± 0.2 | 1.0 ± 0.2 * | 0.9 ± 0.0*$ | |
| Stenotic jet velocity(m/sec) | 4.5 ± 0.9 | 5.0 ± 0.2 | 4.4 ± 0.9 | 4.3 ± 1.1 | |||
| Velocity ratio | 5.5 ± 1.0 | 6.5 ± 0.4 | 4.4 ± 1.4 | 4.9 ± 1.4 | |||
| K | FKS | FKC | FKH | MKS | MKC | MKH | |
| Mice (n) | 6 | 5 | 4 | 6 | 3 | 3 | |
| BW (g) | 24 ± 3 | 23 ± 2 | 22 ± 1 | 25 ± 2 | 24 ± 1 | 22 ± 2 | |
| HR (bpm) | 432 ± 18 | 417 ± 52 | 424 ± 33 | 450 ± 67 | 392 ± 49 | 359 ± 30 | |
| IVSTD (mm) | 0.58 ± 009 | 0.84 ± 0.08* | 0.85 ± 0.14* | 0.53 ± 0.07 | 0.71 ± 0.06 | 0.87 ± 0.04* | |
| PWTD (mm) | 0.59 ± 0.14 | 0.95 ± 0.18* | 1.03 ± 0.09*$ | 0.53 ± 0.08 | 140.6 ± 25.6* | 0.95 ± 0.06*$ | |
| LVmass (mg) | 87.3 ± 27.0 | 165.7 ± 23.8* | 205.9 ± 12.2* | 88.9 ± 12.2 | 4.8 ± 0.3 | 188 ± 41* | |
| LVEDD (mm) | 4.2 ± 0.3 | 4.5 ± 0.3 | 4.9 ± 02*$ | 4.6 ± 0.3 | 29 ± 3* | 4.8 ± 0.7 | |
| FS% | 40 ± 5 | 37 ± 7 | 30 ± 4* | 40 ± 6 | 65 ± 5* | 26 ± 5* | |
| EF% | 78 ± 6 | 74 ± 8 | 65 ± 6* | 77 ± 6 | 2.2 ± 0.2* | 60 ± 9* | |
| Spw (cm/sec) | 2.8 ± 0.3 | 2.4 ± 8 | 2.4 ± 0.4 | 3.0 ± 0.6 | 4.0 ± 0.8 | 1.5 ± 0.3* | |
| Ea(cm/sec) | 4.3 ± 0.4 | 3.2 ± 0.7* | 3.0 ± 0.6* | 4.0 ± 0.8 | 0.2 ± 0.1 | 2.1 ± 0.7*$ | |
| E/Ea | 0.2 ± 0.0 | 0.3 ± 0.1* | 0.3 ± 0.0 * | 0.3 ± 0.1 | 1.0 ± 0.0 | 0.4 ± 0.1*$ | |
| Aortic outflow (m/sec) | 1.0 ± 0.1 | 0.9 ± 0.1* | 0.8 ± 0.1* | 1.3 ± 0.2 | 3.8 ± 01 | 1.1 ± 0.0*$ | |
| Stenotic jet velocity (m/sec) | – | 4.4 ± 0.4 | 4.5 ± 0.4 | – | 3.7 ± 0.1 | 4.1 ± 1.3 | |
| Velocity ratio | – | 5.0 ± 0.7 | 5.8 ± 0.9 | – | 3.9 ± 1.2 | ||
W, wild-type mice; K, FKBP12.6 overexpressing mice; F, female; M, male; S, sham-operated; TAC, thoracic aortic constriction; C, noncongested mice; H mice with congestive heart failure; BW, body weight; TL, tibial length; HR, heart rate; IVSTD, interventricular septum thickness; PWTD, posterior wall thickness; LVEDD, LV end diastolic diameter; LV, left ventricle mass; EF, LV ejection fraction; Spw, maximum systolic velocity of posterior wall; E/Ea, peak velocity of early mitral inflow/early diastolic velocity of the mitral annulus; Velocity ratio, stenotic jet velocity/LV outflow velocity. Data are mean ± SEM. *P < 0.05, TAC versus sham; $P < 0.05, C versus H; †P < 0.05, female versus male.
2D-DIGE differential proteomic analysis
Forty eight cardiac LV protein extracts, representing the 12 experimental groups (FWS [Female wild type sham-operated], FWC [Female wild type noncongested], FWH [Female wild type congestive heart failure], FKS [Female FKBP12.6 overexpressing Sham-operated], FKC [Female FKBP12.6 overexpressing noncongested], FKH [Female FKBP12.6 overexpressing congestive heart failure], MWS [Male wild type sham-operated], MWC [Male wild type noncongested], MWH [Male wild type congestive heart failure], MKS [Male FKBP12.6 overexpressing sham-operated], MKC [Male FKBP12.6 overexpressing noncongested], and MKH [Male FKBP12.6 overexpressing congestive heart failure]), were analyzed by 2D-DIGE technology. A representative set of 2D-DIGE gel images is shown in Figure 3A, and reveals the reproducibility of resolved protein maps, allowing the detection of 1610 ± 140 spots per gel, and 1312 ± 106 spots matched for the 24 2D-DIGE gels of the experiment.
Figure 3.

Mouse cardiac LV 2D-DIGE analysis. (A) Representative spot maps of each experimental group analyzed by 2D-DIGE. Protein extracts from cardiac left ventricles were prepared, labeled, and separated by 2D-DIGE, as described in Supplemental methods. (B) Differentially expressed protein spots, identified by DeCyder software, were identified by mass spectrometry (numbered/circled spots). Protein expression data were filtered by the following criteria: false discovery rate (FDR) and 1.2-fold difference in abundance. FDR correction was applied as a multiple testing correction method to keep the overall error rate as low as possible and P-value less than 0.05 in at least one of the following comparisons; MWH versus FWH; MWC versus FWC; MWS versus FWS; MKS versus FKS; MKH versus FKH; MKC versus FKC, FKH versus FWH; MKH versus MWH;FKC versus FWC; MKC versus MWC; FKS versus FWS; MKS versusMWS, FWC versus FWS; MWC versus MWS;FKC versusFKS; MKC versus MKS; FWH versus FWS; MWH versus MWS; FKH versus FKS; MKH versus MKS. (C) Principal component analysis (b>PCA) performed from the protein spots detected and matched. The score plot shows experimental maps.
In order to assess significant differential expression as a result of gender, genotype, and TAC, multiple group-to-group comparisons were performed (see Fig. 3B) using the DeCyder biological variation analysis (BVA) module. We identified a total of 96 spots differentially expressed by mass spectrometry (Table 4).
Table 4.
MS/MS data
| Exp. | Theo. | Mass spectrometry | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spot number | Protein name | Uniprot ID | pI | Mw | pI | Mw | Number of unique identified peptides in MSMS / in MS + MSMS | Total ion score | Total ion score | Sequence coverage (%) |
| 4 | ATP synthase subunit d, mitochondrial | Q9DCX2 | 5.5 | 18 | 5.5 | 19 | 8 | 612 | 116 | 60 |
| 6 | Ferritin heavy chain | P09528 | 5.6 | 17 | 5.5 | 21 | 1 | 18 | 18 | 5 |
| 7 | Ferritin heavy chain | P09528 | 5.6 | 18 | 5.5 | 21 | 2 | 40 | 21 | 10 |
| 8 | Alpha-crystallin B chain | P23927 | 6.6 | 19 | 6.8 | 20 | 4 | 125 | 37 | 30 |
| 9 | Glutathione S-transferase Mu 7 | Q80W21 | 6.8 | 21 | 6.3 | 26 | 4 | 235 | 83 | 23 |
| 10 | Glutathione S-transferase Mu 5 | P48774 | 6.7 | 22 | 6.8 | 27 | 10 | 617 | 106 | 49 |
| 12 | Glutathione S-transferase omega-1 | O09131 | 6.5 | 27 | 6.9 | 27 | 2 | 40 | 24 | 10 |
| 17 | Glutathione S-transferase Mu 5 | P48774 | 6.3 | 22 | 6.8 | 27 | 7 | 284 | 76 | 35 |
| 18 | Guanylate kinase | Q64520 | 6.3 | 19 | 6.1 | 22 | 2 | 44 | 22 | 9 |
| 20 | Thioredoxin-dependent peroxide reductase, mitochondrial | P20108 | 5.8 | 20 | 7.2 | 28 | 3 | 119 | 70 | 14 |
| 21 | Endoplasmic reticulum protein ERp29 | P57759 | 5.9 | 25 | 5.9 | 29 | 1 | 16 | 16 | 3 |
| 23 | Heat shock protein beta-1 | P14602 | 5.7 | 22 | 6.1 | 23 | 6 | 311 | 110 | 32 |
| 28 | Heat shock protein beta-1 | P14602 | 5.3 | 22 | 6.1 | 23 | 5 | 308 | 111 | 28 |
| 30 | 14-3-3 protein beta/alpha | Q9CQV8 | 4.6 | 24 | 4.8 | 28 | 11 | 84 | 55 | 47 |
| 31 | 14-3-3 protein gamma | P61982 | 4.6 | 25 | 4.8 | 28 | 10 | 97 | 54 | 42 |
| 32 | 14-3-3 protein zeta/delta | P63101 | 4.6 | 24 | 4.7 | 28 | 10 | 301 | 85 | 49 |
| 35 | Tropomyosin alpha-3 chain | P21107 | 4.6 | 29 | 4.7 | 33 | 1 | 22 | 22 | 3 |
| 39 | Annexin A5 | P48036 | 4.7 | 33 | 4.8 | 36 | 11 | 748 | 117 | 50 |
| 43 | Ubiquinone biosynthesis protein COQ9, mitochondrial | Q8K1Z0 | 5.0 | 30 | 5.6 | 35 | 7 | 479 | 118 | 31 |
| 44 | Chloride intracellular channel protein 1 | Q9Z1Q5 | 5.1 | 27 | 5.1 | 27 | 6 | 287 | 64 | 34 |
| 45 | Sarcolemmal membrane-associated protein | Q3URD3 | 5.0 | 38 | 5.2 | 67 | 2 | 60 | 39 | 2 |
| 46 | Sarcolemmal membrane-associated protein | Q3URD3 | 5.0 | 38 | 5.2 | 67 | 7 | 286 | 66 | 9 |
| 47 | Microtubule-associated protein RP/EB family member 2 | Q8R001 | 5.2 | 33 | 5.2 | 37 | 2 | 50 | 29 | 6 |
| 48 | F-actin-capping protein subunit beta | P47757 | 5.4 | 31 | 5.5 | 31 | 2 | 43 | 26 | 7 |
| 51 | Electron transfer flavoprotein subunit alpha, mitochondrial | Q99LC5 | 6.7 | 33 | 8.6 | 35 | 8 | 600 | 109 | 37 |
| 53 | PDZ and LIM domain protein 1 | O70400 | 6.5 | 36 | 6.4 | 36 | 2 | 74 | 38 | 7 |
| 58 | Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, mitochondrial | O35459 | 6.3 | 31 | 7.6 | 36 | 7 | 404 | 92 | 25 |
| 60 | Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, mitochondrial | O35459 | 6.0 | 31 | 7.6 | 36 | 2 | 95 | 61 | 8 |
| 62 | Malate dehydrogenase, cytoplasmic | P14152 | 5.9 | 34 | 6.2 | 36 | 6 | 377 | 91 | 21 |
| 63 | Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase | Q9DBB8 | 6.1 | 39 | 8.5 | 48 | 4 | 150 | 54 | 13 |
| 65 | Leukocyte elastase inhibitor A | Q9D154 | 6.0 | 42 | 5.9 | 43 | 5 | 151 | 39 | 15 |
| 70 | L-lactate dehydrogenase B chain | P16125 | 5.8 | 33 | 5.7 | 37 | 8 | 563 | 115 | 30 |
| 71 | Isocitrate dehydrogenase | Q9D6R2 | 5.7 | 39 | 6.3 | 40 | 8 | 486 | 111 | 27 |
| 72 | [Protein ADP-ribosylarginine] hydrolase-like protein 1 | Q8BGK2 | 5.7 | 41 | 5.6 | 40 | 8 | 430 | 66 | 26 |
| 74 | Serpin B6 | Q60854 | 5.7 | 44 | 5.5 | 43 | 1 | 117 | 117 | 5 |
| 75 | L-lactate dehydrogenase B chain | P16125 | 5.6 | 37 | 5.7 | 37 | 6 | 369 | 104 | 19 |
| 78 | Isocitrate dehydrogenase | Q9D6R2 | 5.4 | 39 | 6.3 | 40 | 8 | 374 | 66 | 27 |
| 85 | Adenosine kinase | P55264 | 5.8 | 47 | 5.8 | 40 | 8 | 454 | 81 | 25 |
| 89 | Protein NDRG2 | Q9QYG0 | 4.6 | 48 | 5.2 | 41 | 1 | 27 | 27 | 3 |
| 93 | Calreticulin | P14211 | 4.3 | 70 | 4.3 | 48 | 4 | 159 | 72 | 12 |
| 96 | Serine protease inhibitor A3K | P07759 | 4.4 | 68 | 5.1 | 47 | 3 | 138 | 55 | 12 |
| 101 | Alpha-1-antitrypsin 1-4 | Q00897 | 4.6 | 63 | 5.2 | 46 | 1 | 25 | 25 | 4 |
| 102 | Alpha-1-antitrypsin 1-3 | Q00896 | 4.6 | 63 | 5.2 | 46 | 4 | 152 | 77 | 16 |
| 103 | Alpha-1-antitrypsin 1-1 | P07758 | 4.7 | 62 | 5.4 | 46 | 6 | 268 | 74 | 25 |
| 104 | Protein disulfide-isomerase | P09103 | 4.7 | 61 | 4.8 | 57 | 7 | 479 | 117 | 23 |
| 105 | Alpha-1-antitrypsin 1-3 | Q00896 | 4.7 | 62 | 5.2 | 46 | 7 | 380 | 99 | 29 |
| 106 | Alpha-1-antitrypsin 1-1 | P07758 | 4.8 | 61 | 5.4 | 46 | 7 | 402 | 105 | 26 |
| 107 | Alpha-1-antitrypsin 1-2 | P22599 | 4.8 | 61 | 5.3 | 46 | 7 | 410 | 114 | 22 |
| 110 | Liver carboxylesterase N | P23953 | 4.4 | 75 | 5.1 | 61 | 1 | 27 | 27 | 2 |
| 115 | ATP synthase subunit beta, mitochondrial | P56480 | 4.9 | 56 | 5.2 | 56 | 12 | 903 | 112 | 33 |
| 116 | ATP synthase subunit beta, mitochondrial | P56480 | 4.9 | 55 | 5.2 | 56 | 12 | 1156 | 124 | 33 |
| 119 | Dynactin subunit 2 | Q99KJ8 | 5.2 | 54 | 5.1 | 44 | 5 | 189 | 52 | 15 |
| 120 | Desmin | P31001 | 5.3 | 56 | 5.2 | 54 | 3 | 75 | 42 | 5 |
| 121 | 60 kDa heat shock protein, mitochondrial | P63038 | 5.3 | 56 | 5.9 | 61 | 8 | 424 | 91 | 21 |
| 122 | 60 kDa heat shock protein, mitochondrial | P63038 | 5.3 | 56 | 5.9 | 61 | 8 | 763 | 164 | 23 |
| 123 | Desmin | P31001 | 5.2 | 56 | 5.2 | 54 | 13 | 1049 | 142 | 33 |
| 124 | Desmin | P31001 | 5.2 | 56 | 5.2 | 54 | 13 | 848 | 108 | 32 |
| 125 | Desmin | P31001 | 5.1 | 56 | 5.2 | 54 | 12 | 1017 | 128 | 30 |
| 126 | Desmin | P31001 | 5.1 | 57 | 5.2 | 54 | 12 | 818 | 132 | 28 |
| 127 | Vimentin | P20152 | 5.1 | 58 | 5.2 | 54 | 12 | 716 | 90 | 27 |
| 128 | Actin, alpha skeletal muscle | P68134 | 5.2 | 68 | 5.2 | 42 | 3 | 146 | 69 | 10 |
| 130 | NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial | Q91VD9 | 5.1 | 74 | 5.5 | 80 | 11 | 564 | 92 | 22 |
| 132 | NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial | Q91VD9 | 5.1 | 74 | 5.5 | 80 | 4 | 77 | 22 | 8 |
| 133 | Kelch repeat and BTB domain-containing protein 10 | Q9ER30 | 5.0 | 70 | 5.0 | 68 | 6 | 206 | 41 | 11 |
| 134 | Kelch repeat and BTB domain-containing protein 10 | Q9ER30 | 5.0 | 70 | 5.0 | 68 | 3 | 57 | 24 | 4 |
| 135 | Protein-glutamine gamma-glutamyltransferase 2 | P21981 | 4.9 | 75 | 5.0 | 77 | 3 | 132 | 57 | 5 |
| 136 | Protein-glutamine gamma-glutamyltransferase 2 | P21981 | 4.9 | 75 | 5.0 | 77 | 3 | 103 | 43 | 4 |
| 143 | Collagen alpha-1(VI) chain | Q04857 | 5.1 | 102 | 5.2 | 108 | 3 | 97 | 43 | 3 |
| 144 | Collagen alpha-1(VI) chain | Q04857 | 5.1 | 100 | 5.2 | 108 | 5 | 170 | 50 | 6 |
| 148 | Major vault protein | Q9EQK5 | 5.5 | 83 | 5.4 | 96 | 8 | 369 | 73 | 14 |
| 154 | Serum albumin | P07724 | 5.7 | 68 | 5.8 | 69 | 14 | 1301 | 148 | 28 |
| 156 | Serum albumin | P07724 | 5.8 | 69 | 5.8 | 69 | 2 | 46 | 23 | 4 |
| 168 | Moesin | P26041 | 6.5 | 72 | 6.2 | 68 | 3 | 92 | 43 | 3 |
| 169 | Serotransferrin | Q921I1 | 6.6 | 75 | 6.9 | 77 | 1 | 28 | 28 | 1 |
| 170 | Moesin | P26041 | 6.6 | 72 | 6.2 | 68 | 5 | 182 | 66 | 7 |
| 173 | Moesin | P26041 | 6.7 | 71 | 6.2 | 69 | 2 | 79 | 51 | 3 |
| 177 | Propionyl-CoA carboxylase beta chain, mitochondrial | Q99MN9 | 6.6 | 59 | 7.2 | 58 | 2 | 98 | 60 | 4 |
| 179 | Carboxylesterase 3 | Q8VCT4 | 6.4 | 59 | 6.2 | 62 | 2 | 24 | 40 | 5 |
| 182 | Fibrinogen beta chain | Q8K0E8 | 6.3 | 59 | 6.7 | 55 | 4 | 159 | 67 | 9 |
| 184 | Dihydropyrimidinase-related protein 2 | O08553 | 6.3 | 65 | 6.0 | 62 | 6 | 229 | 54 | 15 |
| 185 | Aldehyde dehydrogenase, mitochondrial | P47738 | 6.3 | 55 | 7.5 | 57 | 4 | 161 | 59 | 8 |
| 186 | Lipoamide acyltransferase component of branched-chain alpha-keto acid dehydrogenase complex, mitochondrial | P53395 | 6.3 | 54 | 8.9 | 53 | 4 | 141 | 40 | 10 |
| 187 | T-complex protein 1 subunit beta | P80314 | 6.2 | 57 | 6.0 | 57 | 5 | 188 | 60 | 13 |
| 188 | Adenylyl cyclase-associated protein 2 | Q9CYT6 | 6.2 | 61 | 6.0 | 53 | 1 | 23 | 23 | 1 |
| 190 | Aldehyde dehydrogenase, mitochondrial | P47738 | 6.1 | 54 | 5.7 | 57 | 4 | 41 | 14 | 8 |
| 191 | Sarcalumenin | Q7TQ48 | 6.3 | 55 | 4.4 | 99 | 7 | 459 | 119 | 18 |
| 193 | Selenium-binding protein 1 | P17563 | 6.0 | 56 | 5.9 | 52 | 11 | 746 | 129 | 26 |
| 197 | EH domain-containing protein 4 | Q9EQP2 | 6.6 | 66 | 6.3 | 61 | 8 | 319 | 67 | 17 |
| 199 | Septin-2 | P42208 | 6.3 | 44 | 6.1 | 41 | 5 | 293 | 85 | 18 |
| 203 | Short/branched chain specific acyl-CoA dehydrogenase, mitochondrial | Q9DBL1 | 6.4 | 42 | 8.0 | 47 | 4 | 160 | 66 | 13 |
| 204 | Acyl-coenzyme A thioesterase 2, mitochondrial | Q9QYR9 | 6.4 | 46 | 6.9 | 50 | 4 | 145 | 51 | 13 |
| 205 | Acyl-coenzyme A thioesterase 2, mitochondrial | Q9QYR9 | 6.4 | 46 | 6.9 | 50 | 5 | 375 | 102 | 15 |
| 206 | Elongation factor Tu | Q8BFR5 | 6.4 | 48 | 7.2 | 49 | 9 | 450 | 78 | 33 |
| 209 | Isovaleryl-CoA dehydrogenase | Q9JHI5 | 6.6 | 44 | 8.5 | 46 | 14 | 513 | 131 | 44 |
| 212 | Creatine kinase M-type | P07310 | 6.7 | 45 | 6.6 | 43 | 6 | 352 | 73 | 29 |
| 213 | Isocitrate dehydrogenase | O88844 | 6.8 | 46 | 6.4 | 47 | 8 | 376 | 81 | 22 |
| 214 | Beta-enolase | P21550 | 6.7 | 50 | 6.7 | 47 | 12 | 906 | 130 | 43 |
| 219 | Mitochondrial inner membrane protein | Q8CAQ8 | 6.7 | 69 | 6.2 | 84 | 7 | 455 | 98 | 12 |
| 328 | Catechol O-methyltransferase | O88587 | 5.0 | 29 | 5.5 | 29 | 1 | 28 | 28 | 4 |
| 331 | Heat shock protein beta-1 | P14602 | 5.8 | 22 | 6.1 | 23 | 6 | 283 | 75 | 32 |
Gene name and accession number according to SwissProt.Experimental (E) Isoelectric point (pI) and molecular weight (Mw). Theoretical (T) Isoelectric point (pI) and molecular weight (Mw).
Principal component analysis, performed on all spots detected is illustrated in Figure 3C and revealed patterns that clearly and orderly segregated the 12 groups. We have circled the sham-operated (S), noncongested (C) mice and mice with congestive heart failure (H) suggesting that the C group was intermediate between S and H.
Gender-related differential expression profile of mouse LV
Two-dimensional differential in-gel electrophoresis analysis revealed that 13 spots showed differential expression between male and female mice listed in Table 5. This comparison highlighted two proteins downregulated in males (Table 5), related to distinct functions, that is, xenobiotic detoxification (ESTN: liver carboxyl esterase N) and alcohol metabolism (ALDH2: aldehyde dehydrogenase, mitochondrial). Eleven spots (corresponding to eight proteins) were upregulated in male mice. Among these were all isoforms of alpha-1-antitrypsin 1 (A1AT1, A1AT2, A1AT3, and A1AT4), the fibrinogen beta chain (FIBB), protein disulfide-isomerase (PDIA1), and calreticulin (CALR).
Table 5.
Gender comparison
| M versus F | ||||||
|---|---|---|---|---|---|---|
| Spot number | Protein name | Uniprot accession | Accession number | 1-ANOVA | P-value | FC |
| 93 | Calreticulin | CALR_MOUSE | P14211 | 2,9E-06 | 3,6E-11 | 2,9 |
| 96 | Serine protease inhibitor A3K | SPA3K_MOUSE | P07759 | 2,4E-04 | 1,4E-06 | 1,8 |
| 101 | Alpha-1 -antitrypsin 1-4 | A1AT4_MOUSE | Q00897 | 9,5E-06 | 1,9E-09 | 3,2 |
| 102 | Alpha-1-antitrypsin 1-3 | A1AT3_MOUSE | Q00896 | 1,2E-06 | 5,8E-11 | 2,7 |
| 103 | Alpha-1-antitrypsin 1-1 | A1AT1_MOUSE | P07758 | 7,7E-08 | 1,3E-11 | 2,0 |
| 105 | Alpha-1-antitrypsin 1-3 | A1AT3_MOUSE | Q00896 | 4,0E-08 | 2,2E-12 | 1,8 |
| 106 | Alpha-1-antitrypsin 1-1 | A1AT1_MOUSE | P07758 | 3,6E-05 | 2,4E-09 | 1,7 |
| 107 | Alpha-1-antitrypsin 1-2 | A1AT2_MOUSE | P22599 | 6,0E-07 | 2,0E-08 | 2,0 |
| 110 | Liver carboxylesterase N | EST1C_MOUSE | P23953 | 2,9E-01 | 8,2E-03 | −1,7 |
| 182 | Fibrinogen beta chain | FIBB_MOUSE | Q8K0E8 | 1,5E-04 | 2,0E-07 | 1,4 |
| 185 | Aldehyde dehydrogenase, mitochondrial | ALDH2_MOUSE | P47738 | 2,5E-05 | 2,2E-04 | −1,3 |
| 190 | Aldehyde dehydrogenase, mitochondrial | ALDH2_MOUSE | P47738 | 5,0E-07 | 2,4E-05 | −1,4 |
| 104 | Protein disulfide-isomerase | PDIA1_MOUSE | P09103 | 5,8E-08 | 2,2E-04 | 1,4 |
FC, Fold change; using DeCyder software, between normalized spot volume between male (M) and female (F) mouse samples.
Genotype-related differential expression profile
Proteomic analysis revealed 11 identified proteins that were differentially expressed between wild type (W) and FKBP12.6-overexpressing mice (K) which are listed in Table 6. A set of differentially expressed proteins (14-3-3 G, ADK, GSTM5, GSTM7, ILEUA, KBTBA, MARE2, and SBP1 characterized the genotype difference [Table 6]). Among the upregulated proteins (Table 6), a marked upregulation of the detoxification enzymes, glutathione S-transferases (GSTM5 and GSTM7), was observed in K mice. SBP1, an antioxidant protein, was also upregulated in MKH mice. The cytoskeletal protein (MARE2: microtubule-associated protein RP/EB family member2) and the leukocyte elastase inhibitor (ILEUA) were downregulated in K mice whereas others such as PDZ and LIM domain protein1 (PDLI1 = PDLIM1) were simultaneously increased (Table 6).
Table 6.
Genotype comparison
| K versus W | ||||||
|---|---|---|---|---|---|---|
| Spot number | Protein name | Uniprot accession | Accession number | 1-ANOVA | P-value | FC |
| 9 | Glutathione S-transferase Mu 7 | GSTM7_MOUSE | Q80W21 | 1,2E-05 | 3,2E-04 | 1,4 |
| 10 | Glutathione S-transferase Mu 5 | GSTM5_MOUSE | P48774 | 2,4E-05 | 1,4E-08 | 4,0 |
| 17 | Glutathione S-transferase Mu 5 | GSTM5_MOUSE | P48774 | 5,1E-05 | 1,4E-08 | 5,4 |
| 31 | 14-3-3 protein gamma | 1433G_MOUSE | P61982 | 2,5E-07 | 3,2E-04 | 1,2 |
| 47 | Microtubule-associated protein RP/EB family member 2 | MARE2_MOUSE | Q8R001 | 3,1E-06 | 3,8E-05 | −1,3 |
| 53 | PDZand LIM domain protein 1 | PDLI1_MOUSE | 070400 | 6,4E-06 | 1,5E-02 | 1,3 |
| 65 | Leukocyte elastase inhibitor A | ILEUA_MOUSE | Q9D154 | 5,2E-05 | 8,9E-02 | −1,4 |
| 85 | Adenosine kinase | ADK_MOUSE | P55264 | 3,7E-07 | 3,2E-04 | 1,3 |
| 133 | Kelch repeat and BTB domain-containing protein 10 | KBTBA_MOUSE | Q9ER30 | 6,8E-05 | 6,6E-03 | 1,3 |
| 191 | Sarcalumenin | SRCA_MOUSE | Q7TQ48 | 7,2E-04 | 3,2E-04 | 1,3 |
| 193 | Selenium-binding protein 1 | SBP1_MOUSE | P17563 | 4,0E-08 | 1,2E-03 | 1,3 |
FC, Fold change; using DeCyder software, between normalized spot volume between wild-type (W) and FKBP12.6-overexpressing (K) mouse samples.
Differential expression profiles of TAC-induced pathological LVH
At 30 days postsurgery, chronic PO induced LVH associated with an altered protein expression pattern (Table 7). We selected a few proteins differentially expressed and identified by 2D-DIGE for immunoblotting. Figure 4B–E show representative Western blot analyses of SLMAP (Fig. 4B), NDRG2 (Fig. 4C), endoplasmic reticulum stress-associated proteins (ERP29, Fig. 4D), and alpha-crystallinB chain (CRYAB, Fig. 4E) expression in C and H mice (from four individuals in each group) and CSQ as a control for protein loading (Fig. 4A). H mice exhibited an obvious upregulation of SLMAP and CRYAB. TAC induced a significant increase in NDRG2 protein level in MWH whereas no significant change was observed in MKH mice (Fig. 4C). TAC was also associated with an increased level of ERP 29 (Fig. 4D). These upregulations of the selected proteins were consistent with 2D-DIGE results.
Table 7.
Differentially expressed and identified cardiac left ventricular spots in response to TAC in mice
| (C, H) versus S | C versus S | H versus S | H versus C | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spot number | Protein name | Uniprot accession | Accession number | 1-ANOVA | P-Value | FC | P-Value | FC | P-Value | FC | P-Value | FC |
| A: Spots with fold change not significantly different between H and C groups | ||||||||||||
| 6 | Ferritin heavy chain | FRIH_MOUSE | P09528 | 1,1E-02 | 6,9E-06 | 1,8 | 3,1E-03 | 1,8 | 7,6E-05 | 1,7 | 5,3E-01 | −1,0 |
| 8 | Alpha-crystallin B chain | CRYAB_MOUSE | P23927 | 5,6E-03 | 2,8E-05 | 2,0 | 2,2E-02 | 1,8 | 2,8E-05 | 2,2 | 1,9E-01 | 1,3 |
| 9 | Glutathione S-transferase Mu 7 | GSTM7_MOUSE | Q80W21 | 1,2E-05 | 1,3E-04 | −1,4 | 1,2E-02 | −1,3 | 1,4E-03 | −1,4 | 3,4E-01 | −1,1 |
| 18 | Guanylate kinase | KGUA_MOUSE | Q64520 | 1,4E-05 | 3,4E-07 | 1,5 | 4,3E-03 | 1,4 | 4,4E-06 | 1,5 | 2,3E-01 | 1,1 |
| 23 | Heat shock protein beta-1 | HSPB1_MOUSE | P14602 | 6,4E-06 | 2,5E-06 | 2,1 | 5,9E-03 | 1,9 | 5,5E-06 | 2,3 | 1,6E-01 | 1,2 |
| 28 | Heat shock protein beta-1 | HSPB1_MOUSE | P14602 | 2,3E-03 | 6,0E-05 | 1,7 | 2,2E-02 | 1,5 | 8,1E-05 | 1,9 | 1,4E-01 | 1,3 |
| 43 | Ubiquinone biosynthesis protein COQ9 | COQ9_MOUSE | Q8K1Z0 | 1,1E-06 | 1,6E-06 | −1,3 | 8,8E-03 | −1,2 | 1,9E-06 | −1,4 | 4,5E-03 | −1,1 |
| 53 | PDZ and LIM domain protein 1 | PDLI1_MOUSE | 070400 | 6,4E-06 | 5,8E-06 | 1,5 | 1,7E-02 | 1,4 | 5,0E-05 | 1,6 | 1,3E-01 | 1,1 |
| 70 | L-lactate dehydrogenase B chain | LDHB_MOUSE | P16125 | 2,1E-04 | 6,3E-06 | −1,3 | 1,4E-02 | −1,2 | 1,6E-06 | −1,3 | 4,0E-02 | −1,1 |
| 71 | Isocitrate dehydrogenase [NAD] subunit alpha | IDH3A_MOUSE | Q9D6R2 | 1,6E-04 | 1,6E-03 | −1,2 | 5,7E-02 | −1,2 | 2,1E-02 | −1,2 | 3,1E-01 | 1,1 |
| 72 | Protein ADP-ribosylarginine] hydrolase-like protein 1 | ARHL1_MOUSE | Q8BGK2 | 1,2E-07 | 2,7E-08 | 1,4 | 3,8E-05 | 1,4 | 2,2E-06 | 1,4 | 4,3E-01 | −1,0 |
| 75 | L-lactate dehydrogenase B chain | LDHB_MOUSE | P16125 | 5,0E-03 | 2,8E-05 | −1,4 | 4,4E-02 | −1,3 | 7,0E-05 | −1,5 | 5,5E-02 | −1,1 |
| 85 | Adenosine kinase | ADK_MOUSE | P55264 | 3,7E-07 | 6,0E-03 | 1,2 | 2,5E-01 | 1,2 | 1,0E-02 | 1,3 | 3,0E-01 | 1,1 |
| 119 | Dynactin subunit 2 | DCTN2_MOUSE | Q99KJ8 | 2,4E-03 | 2,2E-05 | 1,3 | 1,7E-02 | 1,3 | 1,2E-04 | 1,4 | 1,7E-01 | 1,1 |
| 128 | Actin. alpha skeletal muscle | ACTS_MOUSE | P68134 | 4,8E-03 | 1,8E-03 | 1,2 | 2,4E-02 | 1,2 | 3,1E-03 | 1,3 | 1,2E-01 | 1,1 |
| 154 | Serum albumin | ALBU_MOUSE | P07724 | 1,5E-04 | 2,0E-04 | 1,3 | 5,7E-02 | 1,3 | 1,4E-03 | 1,4 | 3,3E-01 | 1,1 |
| 185 | Aldehyde dehydrogenase | ALDH2_MOUSE | P47738 | 2,5E-05 | 2,4E-03 | −1,2 | 2,8E-01 | −1,2 | 2,3E-03 | −1,3 | 1,3E-01 | −1,1 |
| 187 | T-complex protein 1 subunit beta | TCPB_MOUSE | P80314 | 1,3E-02 | 3,7E-05 | 1,5 | 2,9E-02 | 1,4 | 6,7E-04 | 1,5 | 4,2E-01 | 1,0 |
| 193 | Selenium-binding protein 1 | SBP1_MOUSE | P17563 | 4,0E-08 | 5,8E-05 | −1,4 | 6,4E-02 | −1,3 | 1,1E-05 | −1,5 | 5,9E-02 | −1,2 |
| 209 | Isovaleryl-CoA dehydrogenase | IVD_MOUSE | Q9JHI5 | 5,2E-04 | 6,3E-06 | −1,3 | 3,8E-02 | −1,2 | 7,0E-07 | −1,4 | 1,2E-01 | −1,2 |
| B: Spots with fold change significantly different between H and C groups | ||||||||||||
| 4 | ATP synthase subunit delta | ATP5H_MOUSE | Q9DCX2 | 1,3E-02 | 5,2E-04 | −1,4 | 1,6E-01 | −1,2 | 3,3E-04 | −1,6 | 5,0E-02 | −1,3 |
| 7 | Ferritin heavy chain | FRIH_MOUSE | P09528 | 3,9E-03 | 5,1E-05 | −1,5 | 5,7E-02 | −1,3 | 4,6E-06 | −1,7 | 5,0E-02 | −1,3 |
| 12 | Long-chain specific acyl-CoA dehydrogenase | ACADL_MOUSE | P51174 | 9,3E-04 | 1,6E-04 | −1,3 | 1,4E-01 | −1,2 | 2,5E-05 | −1,4 | 2,9E-02 | −1,2 |
| 20 | Thioredoxin-dependent peroxide reductase | PRDX3_MOUSE | P20108 | 6,4E-06 | 2,6E-06 | −1,4 | 1,9E-02 | −1,2 | 2,5E-08 | −1,5 | 3,9E-03 | −1,3 |
| 21 | Endoplasmic reticulum resident protein 29 | ERP29_MOUSE | P57759 | 5,3E-03 | 2,9E-04 | 1,4 | 7,4E-02 | 1,2 | 1,3E-04 | 1,6 | 2,7E-02 | |
| 30 | 14-3-3 protein beta/alpha | 1433B_MOUSE | Q9CQV8 | 3,2E-04 | 2,9E-04 | 1,2 | 2,7E-01 | 1,1 | 2,7E-06 | 1,3 | 6,0E-04 | 1,2 |
| 32 | 14-3-3 protein zeta/delta | 1433Z_MOUSE | P63101 | 2,3E-05 | 2,8E-05 | 1,2 | 6,0E-02 | 1,1 | 2,4E-07 | 1,3 | 1,1E-03 | 1,2 |
| 35 | Tropomyosin alpha-3 chain | TPM3_MOUSE | P21107 | 1,4E-04 | 1,7E-05 | 1,4 | 5,9E-02 | 1,2 | 1,1E-08 | 1,6 | 3,7E-03 | 1,3 |
| 39 | Annexin A5 | ANXA5_MOUSE | P48036 | 6,0E-07 | 1,5E-07 | 1,4 | 3,8E-03 | 1,3 | 6,2E-09 | 1,6 | 1,6E-04 | 1,3 |
| 44 | Chloride intracelular channel protein 1 | CLIC1_MOUSE | Q9Z1 Q5 | 5,6E-04 | 1,4E-05 | 1,3 | 6,2E-02 | 1,2 | 8,1E-07 | 1,4 | 1,5E-03 | 1,2 |
| 45 | Sarcolemmal membrane-associated protein | SLMAP_MOUSE | Q3URD3 | 2,5E-07 | 3,7E-08 | 1,6 | 1,2E-03 | 1,4 | 2,8E-08 | 1,8 | 1,1E-03 | |
| 46 | Sarcolemmal membrane-associated protein | SLMAP_MOUSE | Q3URD3 | 5,8E-08 | 1,5E-09 | 1,8 | 2,9E-04 | 1,6 | 5,8E-09 | 2,0 | 7,2E-04 | 1,3 |
| 48 | F-actin-capping protein subunit beta | CAPZB_MOUSE | P47757 | 7,7E-08 | 5,6E-07 | 1,4 | 8,8E-03 | 1,3 | 1,0E-08 | 1,6 | 6,3E-03 | 1,2 |
| 51 | Electron transfer flavoprotein subunit alpha | ETFA_MOUSE | Q99LC5 | 1,9E-03 | 4,3E-05 | −1,3 | 7,1E-02 | −1,2 | 4,6E-05 | −1,4 | 1,9E-02 | −1,2 |
| 58 | Delta (3,5)-Delta (2,4)-dienoyl-CoAisomerase | ECH1_MOUSE | 035459 | 4,0E-08 | 3,7E-08 | −1,7 | 1,8E-03 | −1,4 | 2,3E-10 | −2,0 | 3,1E-04 | −1,4 |
| 60 | Delta (3,5)-Delta (2,4)-dienoyl-CoAisomerase | ECH1_MOUSE | 035459 | 5,8E-08 | 1,1E-07 | −1,7 | 3,9E-03 | −1,5 | 2,2E-08 | −2,0 | 7,0E-04 | −1,3 |
| 62 | Malate dehydrogenase | MDHC_MOUSE | P14152 | 2,3E-03 | 1,8E-05 | −1,3 | 3,6E-02 | −1,2 | 9,8E-06 | −1,4 | 2,7E-02 | −1,2 |
| 63 | Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase | DHDH_MOUSE | Q9DBB8 | 2,0E-02 | 1,1E-03 | −1,3 | 1,9E-01 | −1,2 | 6,7E-04 | −1,5 | 2,3E-02 | −1,3 |
| 65 | Leukocyte elastase inhibitor A | ILEUA_MOUSE | Q9D154 | 5,2E-05 | 1,4E-02 | 1,4 | 6,5E-01 | 1,1 | 1,5E-03 | 1,8 | 1,8E-02 | −1,6 |
| 74 | Serpin B6 | SPB6_MOUSE | Q60854 | 6,4E-06 | 2,8E-06 | 1,4 | 1,8E-02 | 1,2 | 1,3E-06 | 1,5 | 1,7E-03 | 1,2 |
| 78 | Isocitrate dehydrogenase [NAD] subunit alpha | IDH3A_MOUSE | Q9D6R2 | 1,1E-05 | 1,8E-03 | −1,3 | 5,9E-01 | −1,1 | 2,7E-06 | −1,6 | 6,2E-04 | −1,5 |
| 89 | Protein NDRG2 | NDRG2_MOUSE | Q9QYG0 | 4,1E-03 | 1,1E-02 | 1,3 | 6,9E-01 | 1,1 | 1,7E-03 | 1,5 | 1,8E-02 | 1,4 |
| 104 | Protein disulfide-isomerase | PDIA1_MOUSE | P09103 | 5,8E-08 | 3,1E-04 | 1,4 | 1,8E-01 | 1,2 | 4,5E-05 | 1,5 | 9,7E-03 | 1,3 |
| 115 | ATP synthase subunit beta | ATPB_MOUSE | P56480 | 1,5E-03 | 6,7E-06 | −1,4 | 1,7E-02 | −1,3 | 2,5E-05 | −1,5 | 2,9E-02 | -1,2 |
| 116 | ATP synthase subunit beta | ATPB_MOUSE | P56480 | 2,6E-05 | 2,8E-07 | −1,4 | 5,0E-03 | −1,3 | 2,4E-07 | −1,5 | 1,6E-03 | -1,2 |
| 120 | Desmin | DESM_MOUSE | P31001 | 2,7E-05 | 2,8E-05 | 1,4 | 6,7E-02 | 1,2 | 4,5E-08 | 1,6 | 7,4E-04 | 1,4 |
| 121 | 60 kDa heat shock protein | CH60_MOUSE | P63038 | 2,5E-07 | 5,7E-05 | −1,2 | 1,4E-01 | −1,1 | 8,3E-09 | −1,3 | 5,3E-06 | −1,2 |
| 122 | 60 kDa heat shock protein | CH60_MOUSE | P63038 | 5,2E-05 | 1,5E-05 | −1,3 | 6,1E-02 | −1,2 | 1,7E-07 | −1,5 | 2,5E-04 | −1,3 |
| 123 | Vimentin | VIME_MOUSE | P20152 | 3,1E-07 | 7,1E-07 | 1,6 | 4,3E-03 | 1,4 | 7,1E-10 | 1,9 | 9,2E-04 | 1,4 |
| 124 | Desmin | DESM_MOUSE | P31001 | 1,5E-04 | 8,8E-05 | 1,4 | 1,2E-01 | 1,2 | 1,2E-07 | 1,6 | 1,6E-03 | 1,4 |
| 125 | Vimentin | VIME_MOUSE | P20152 | 3,3E-06 | 4,7E-06 | 1,5 | 1,7E-02 | 1,3 | 2,1E-08 | 1,7 | 1,1E-03 | 1,4 |
| 126 | Vimentin | VIME_MOUSE | P20152 | 4,9E-06 | 3,0E-06 | 1,4 | 1,4E-02 | 1,2 | 1,7E-08 | 1,5 | 3,0E-03 | 1,3 |
| 127 | Vimentin | VIME_MOUSE | P20152 | 1,9E-04 | 5,8E-05 | 1,4 | 1,2E-01 | 1,2 | 2,4E-07 | 1,6 | 2,0E-03 | 1,3 |
| 130 | NADH-ubiquinone oxidoreductase 75 kDa subunit | NDUS1_MOUSE | Q91VD9 | 5,2E-04 | 1,6E-04 | −1,3 | 1,8E-01 | −1,2 | 2,8E-05 | −1,5 | 7,0E-04 | −1,3 |
| 132 | NADH-ubiquinone oxidoreductase 75 kDa subunit | NDUS1_MOUSE | Q91VD9 | 4,8E-03 | 1,3E-04 | −1,3 | 1,4E-01 | −1,2 | 5,0E-05 | −1,5 | 4,1E-03 | −1,2 |
| 133 | Kelch repeat and BTB domain-containing protein 10 | KBTBA_MOUSE | Q9ER30 | 6,8E-05 | 2,4E-04 | 1,3 | 1,4E-01 | 1,2 | 3,4E-04 | 1,5 | 2,5E-02 | 1,2 |
| 134 | Kelch repeat and BTB domain-containing protein 10 | KBTBA_MOUSE | Q9ER30 | 2,2E-04 | 5,8E-05 | 1,2 | 7,3E-02 | 1,2 | 1,8E-05 | 1,3 | 1,5E-02 | 1,2 |
| 135 | Protein-glutamine gamma-glutamyltransferase 2 | TGM2_MOUSE | P21981 | 9,6E-06 | 2,8E-07 | 1,8 | 2,8E-03 | 1,5 | 3,5E-08 | 2,0 | 8,8E-03 | 1,4 |
| 136 | Protein-glutamine gamma-glutamyltransferase 2 | TGM2_MOUSE | P21981 | 6,6E-06 | 6,9E-07 | 1,6 | 2,8E-03 | 1,4 | 2,4E-07 | 1,9 | 7,7E-03 | 1,3 |
| 143 | Collagen alpha-l (VI) chain | C06A1_MOUSE | Q04857 | 7,4E-05 | 1,4E-05 | 1,7 | 5,7E-02 | 1,4 | 1,2E-07 | 2,0 | 2,0E-03 | 1,5 |
| 144 | Collagen alpha-1 (VI) chain | C06A1_MOUSE | Q04857 | 3,6E-03 | 6,2E-02 | 1,6 | 2,3E-01 | 1,3 | 7,6E-05 | 2,0 | 9,7E-03 | 1,5 |
| 148 | Major vault protein | MVP_MOUSE | Q9EQK5 | 2,8E-07 | 1,5E-09 | 1,7 | 3,8E-05 | 1,5 | 2,1E-09 | 1,8 | 2,8E-02 | 1,2 |
| 168 | Moesin | MOES_MOUSE | P26041 | 5,3E-04 | 1,7E-03 | 1,3 | 3,5E-01 | 1,1 | 1,4E-04 | 1,5 | 4,1E-03 | 1,4 |
| 169 | Serotransferrin | TRFE_MOUSE | Q921I1 | 3,4E-02 | 3,9E-02 | 1,2 | 9,1E-01 | 1,0 | 1,1E-02 | 1,3 | 2,8E-02 | 1,3 |
| 170 | Moesin | MOES_MOUSE | P26041 | 1,5E-02 | 7,8E-04 | 1,3 | 2,6E-01 | 1,1 | 4,4E-05 | 1,4 | 6,8E-03 | 1,3 |
| 173 | Moesin | MOES_MOUSE | P26041 | 4,5E-03 | 1,2E-03 | 1,4 | 3,5E-01 | 1,2 | 1,0E-05 | 1,6 | 4,3E-03 | 1,4 |
| 177 | Propionyl-CoA carboxylase beta chain, mitochondrial | PCCB_MOUSE | Q99MN9 | 5,8E-08 | 1,5E-07 | −1,9 | 4,2E-03 | −1,5 | 5,9E-10 | −2,3 | 2,3E-03 | −1,5 |
| 179 | Carboxylesterase 1D | CES3_MOUSE | Q8VCT4 | 1,2E-07 | 6,9E-06 | −2,0 | 5,1E-02 | −1,6 | 1,7E-06 | −2,5 | 4,0E-05 | −1,6 |
| 184 | Dihydropyrimidinase-related protein 2 | DPYL2_MOUSE | 008553 | 1,9E-04 | 1,5E-05 | 1,3 | 4,2E-02 | 1,2 | 9,6E-07 | 1,4 | 2,9E-03 | 1,2 |
| 186 | Lipoamide acyltransferase component of branched-chain alpha-keto acid dehydrogenase complex | ODB2_MOUSE | P53395 | 9,1E-06 | 3,4E-07 | −1,3 | 8,9E-03 | −1,2 | 2,3E-10 | −1,5 | 7,2E-03 | −1,2 |
| 188 | Adenylyl cyclase-associated protein 2 | CAP2_MOUSE | Q9CYT6 | 4,5E-04 | 1,7E-04 | 1,4 | 1,6E-01 | 1,2 | 1,7E-04 | 1,7 | 6,2E-04 | 1,4 |
| 197 | EH domain-containing protein 4 | EHD4_MOUSE | Q9EQP2 | 1,4E-04 | 4,8E-02 | 1,2 | 8,7E-01 | 1,0 | 6,7E-04 | 1,4 | 8,3E-03 | 1,4 |
| 199 | Septin-2 | SEPT2_MOUSE | P42208 | 1,1E-05 | 6,9E-06 | 1,5 | 2,5E-02 | 1,3 | 3,5E-08 | 1,7 | 1,1E-03 | 1,4 |
| 203 | Short/branched chain specific acyl-CoA dehydrogenase | ACDSB_MOUSE | Q9DBL1 | 1,3E-03 | 5,4E-03 | −1,2 | 3,3E-01 | −1,1 | 8,6E-03 | −1,3 | 2,3E-02 | −1,2 |
| 204 | Acyl-coenzyme Athioesterase 2 | ACOT2_MOUSE | Q9QYR9 | 3,1E-05 | 1,6E-05 | −1,3 | 4,4E-02 | −1,2 | 2,4E-08 | −1,6 | 4,0E-04 | −1,3 |
| 205 | Acyl-coenzyme Athioesterase 2 | ACOT2_MOUSE | Q9QYR9 | 5,3E-05 | 2,1E-05 | −1,3 | 3,6E-02 | −1,2 | 2,4E-07 | −1,5 | 6,2E-04 | −1,3 |
| 206 | Elongation factor Tu | EFTU_MOUSE | Q8BFR5 | 1,1E-02 | 2,7E-03 | −1,3 | 5,6E-01 | −1,1 | 7,5E-05 | −1,6 | 1,1E-03 | −1,5 |
| 212 | Creatine kinase M-type | KCRM_MOUSE | P07310 | 9,3E-04 | 1,6E-04 | −1,3 | 1,4E-01 | −1,2 | 2,5E-05 | −1,4 | 2,9E-02 | −1,2 |
| 213 | Isocitrate dehydrogenase [NADP] | IDHC_MOUSE | 088844 | 7,2E-03 | 4,8E-03 | −1,3 | 5,5E-01 | −1,1 | 2,0E-04 | −1,5 | 5,9E-03 | −1,4 |
S, sham-operated mice; (C, H), TAC mice; C, noncongested mice; H, mice with congestive heart failure; FC, fold change; bold values, fold change not statistically valid (P > 0.05).
Figure 4.

Validation of a few selected 2D DIGE proteins. (A), representative immunoblots of SLMAP, NDRG2, ERP29, and CRYAB expression levels in all experimental groups. Western blot of calsequestrin (CSQ) was used as a control of protein loading, and to normalize the densitometric data of each protein expression. (B–E), respective histograms of levels of SLMAP, NDRG2, ERP29, and CRYAB proteins (are expressed as fold) increases in TAC mouse groups (C and H) relative to their respective shams (S), in the FKBP12.6 overexpressing (K) group compared with wild-type mice (W) of both genders (M or F). Data are mean ± sem. *P < 0.05, TAC versus sham; $P < 0.05, C versus. H; #P < 0.05, female versus male.
Bioinformatic analysis of proteomic finding
Ingenuity pathway analysis allowed us to place the differential proteomic findings into a biological context. After giving information concerning their location within the cell, identified proteins are subdivided into slices based on cellular functions (Table 7) or involvement in canonical pathways. The building of networks shows how an identified molecule can participate in a given biological pathway. According to the presence of pulmonary edema, mice with congestive failing hearts (H) or without lung congestion (noncongested mice, C) were compared with their respective sham-operated mice (S). Figures 5 and 6 show the IPA networks obtained from data related to the following two TAC-induced differential protein expression profiles; first, between C mice and S mice (Fig. 5A) and second, between H mice and S mice (Fig. 5B).
Figure 5.

TAC-induced specific molecular signatures. Graphical representation of the most significant protein interaction networks using ingenuity pathway analysis (IPA) of differentially expressed identified proteins. The upregulated proteins are marked in red and the downregulated in green. The nodes represent proteins that are connected with one or several arrows; the solid arrows represent direct interactions and the dotted arrows indirect interactions. (A), the network shows the difference between noncongested (C) and nonfailing hearts (S), (B), the network demonstrates the difference between congestive failing hearts (H) and nonfailing hearts (S), (see Table 7). (C) Common bio-signature of TAC-induced pathological LVH: an IPA analysis shows that the TAC induces similar alterations in protein content in C and H mice. Upregulated identified proteins are depicted in red and downregulated identified proteins in green. Solid lines indicate direct interaction or regulation, and dashed lines indirect relationships.
Figure 6.
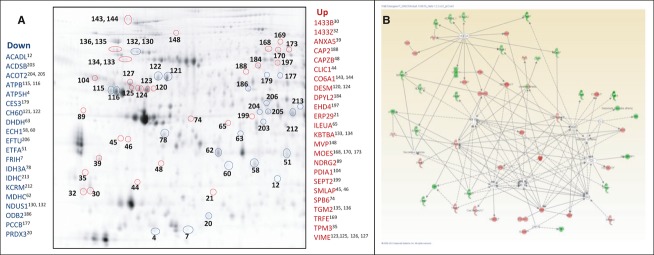
Comparative differentially expressed and identified protein spots (H vs. C). (A) 2D-DIGE analysis revealed 59 spots differentially expressed in HF analysis. Thirty-four spots were upregulated (surrounded in red) and 25 spots were downregulated (surrounded in blue) in the FKBP12.6 overexpressing mouse group compared with wild-type mice. (B) The networks demonstrate the difference between congestive failing hearts (H) and noncongestive hearts (C), upregulated (red), and downregulated proteins (green) where direct interactions or regulation (solid lines), and indirect relationships (dashed lines) are shown. (C). Here is the list of differentially expressed identified spot proteins in H mice.
When H mice were compared with C mice, they exhibited a specific molecular signature (Fig. 6A and B) not found in C mice.
Similarities of PO-induced changes in C and H mice
Compared to sham mice, 26 identified spots were differentially expressed in approximatively the same range for C and H mice, and were not significantly different between H and C groups. Out of these 26 spots, 12 spots were upregulated and eight spots were downregulated in TAC mice (Table 7A). In TAC mice, a similar expression pattern (Fig. 5C) was observed for proteins related to the electron transport chain (ETFA: electron transfer flavoprotein subunit alpha; COQ9: ubiquinone biosynthesis protein COQ9), and to the citric acid cycle (IDH3A). Most of these key enzymes displayed various degrees of downregulation. As expected, alpha skeletal actin (ACTS) was upregulated in TAC mice. Serum albumin was also increased in TAC mice. A significant upregulation of major vault protein (MVP, also known as lung resistance-related protein), SLMAP, PDZ and LIM domain protein1 (PDLI1 = PDLIM1) was observed in all TAC mice.
Molecular signature of TAC-induced congestive failing mouse heart (H)
Interestingly, within the group of TAC mice, the molecular signature of those with congestive HF (H) differed from that of mice without pulmonary edema (C) (Fig. 5A–B). Indeed, a different expression pattern was seen between the two phenotypes. Of 59 differentially expressed proteins, 34 were upregulated and 23 were downregulated in H mice relative to C mice (Fig. 6A and Table 7B). Decreases in ferritin heavy chain (FRIH = FTH1), 60 kDa heat shock protein (CH60 = HSPD1), and peroxiredoxin 3 (PRDX3) were observed. The downregulation in the mitochondrial proteins (Acyl-coenzyme A thioesterase2 [ACOT2], carboxylesterase 3 [CES3], a component of branched-chain alpha-keto acid dehydrogenase [ODB2], propionyl-CoA carboxyl beta chain [PCCB]) associated with metabolic energy processor playing a role in the tricarboxylic acid cycle (isocitrate dehydrogenase: IDHC) or the respiratory chain (ATP synthase subunit d: ATP5H) were also observed.
Most of the protein upregulations occurring in H mice concerned cytoskeletal remodeling (F-actin-capping protein subunit beta: CAPZB; desmin [DSM = DES]; dihydropyrimidinase-related protein2 [DPYL2]; microtubule-associated protein RP/EB family member2 [MARE2]; moesin [MOES = MSN]; septine 2 [SEPT2]; tropomyosin alpha3-chain [TPM3]; vimentin [VIM]). Moreover, proteins involved in antioxidative mechanisms were also upregulated in H mice. In addition, the stress-responsive glutathione S-transferase omega-1 (GSTO-1) and proteins of calcium homeostasis, like annexin A5 (ANXA5), were also higher in congestive failing hearts. Protein-glutamine gamma-glutamyltransferase 2 (TGM2), known to interact with a number of different substrates and to play a role in the response to injury, is associated with congestive HF. On the one hand, the intracellular serpin (leukocyte elastase inhibitor A [ILEUA]) protein level was higher in H than in C mice; on the other hand, we found an increase in serpin B6 (SPB6). In addition, several proteins: adenylyl cyclase–associated protein2 (Cap2), chloride intracellular channel protein1 (CLIC1), EH domain-containing protein 4 (EHD4), Kelch repeat and BTB domain-containing protein 10 (KBTBA), NDRG2 and endoplasmic reticulum protein ERp29 (ERP29) were upregulated in failing LVs.
Validation of cardiac HTT expression and one of its direct interactions by immunoblotting
Ingenuity pathway analysis revealed that HTT is one of the most interconnected nodes involving 23 differentially identified proteins in C mice (Fig. 5A) and related directly or indirectly with still more proteins in H mice (Fig. 5B). In order to estimate the predictive involvement of HTT in HF (Fig. 7A), the Western blot analysis illustrated in Figure 7B shows that TAC is associated with a significant increase in the expression level of HTT in H mice whatever the gender or genotype. Here, we showed that the HSPB1 protein expression level was upregulated in TAC mice. These results were consistent with those of 2D-DIGE. Coimmunoprecipitation experiments (with the Hsp27 antibody) show a physical interaction between HSPB1 and HTT in mouse hearts (Fig. 7C), which confirmed the IPA predictive direct interaction HTT/HSPB1.
Figure 7.

Predictive emerging role of huntingtin in heart failure. (A) In order to predict how the cross-talk among proteins takes part in the progression of heart failure, by using IPA software we organized identified proteins in congestive failing heart (H) into different networks. It appears that one major network, in which huntingtin protein (HTT) was found, gathers proteins involved in mitochondrial function and cytoskeletal remodeling. (B) Western blotting analysis validates the expression of HTT and HSPB1 in mouse heart of both strains after TAC. (C). Coimmunoprecipitation of HTT with endogenous HSPB1 (Hsp27). The anti-Hsp27 antibody was used to immunoprecipitate HSPB1 from mouse heart lysates. Immunoprecipitates (IP) separated by SDS-PAGE electrophoresis (3–8% Tris-Acetate) and probed with an anti-HTT. Lane 1, brain lysate; lane 2, MagicMark™ XP Western protein standard in the range of 20–220 kDa; lane 3, HSP27 immunoprecipitate with Protein A/G sepharose magnetic beads in the absence of lysate; lane 4, lysate precipitated with Protein A/G beads lacking anti-Hsp27 antibody; lanes: 5-9, respective flow-through of LV lysates immunoprecipitated with anti-HSP27; lanes10-14, eluates of LV lysates immunoprecipitated with anti-HSP27 linked to Protein A/G sepharose (lanes 10-11, IP from two individual H mouse LV; lanes12-13, IP from two individual C mouse LV; lane 14, IP from S mouse LV). HTT was coimmunoprecipitated with endogenous HSPB1 in cardiac lysates.
Discussion
The present study deals with the integrative physiology of the effects of TAC at day 30 in mice overexpressing FKBP12.6 and their wild-type controls of both genders. As expected, TAC induced LVH. Despite no difference in stenotic jets indicating a similar degree of aortic constriction in all TAC mice studied, two distinct TAC-induced LVH responses were observed: without lung pathology, considered as likely compensatory LVH, (C) or with pulmonary edema, considered as congestive heart failure (H). Following TAC, mice of both genotypes exhibited similar systolic and diastolic LV dysfunction. These observations were associated with pronounced changes in the pattern of protein expression. The proteomic approaches used here allowed separation of proteins only within the range of approximately 10–150 kDa and between pH ranges of 4–7. Therefore, we were able to analyze here a substantial part of, but not the entire, LV proteome.
The major new findings are that (1) gender differences exist in plasma inflammatory response proteins; (2) there are genotype differences in the detoxifying enzymes; (3) differential comparative proteomic and bioinformatic analysis allowed us to distinguish molecular changes occurring in both C and H mice as a bio-signature of the early stage of failure initiation, then drastic changes in the abundance of mitochondrial proteins and antioxidant stress proteins in H mice that could be a signature characterizing the late end stage of HF; 4) according to the networks built using the IPA software, pathway analysis identified the HTT signaling node as a potential mediator of mitochondrial changes (Fig. 7) upon TAC-induced congestive HF.
Gender molecular signature
In humans and mice, several groups reported that males exhibit higher α1-antitrypsin expression than females (Zabel et al. 2002; Regitz-Zagrosek 2006; Diedrich et al. 2007). Consistent with these previous reports, although serum contaminations of our myocardial LV samples cannot be totally excluded, we found that the cardiac protein level of the four isoforms of alpha1-antitrypsin and fibrinogen beta chain were more highly expressed in male than in female mice, whether submitted to TAC or not. Cardiac α1-antitrypsin expression might confer multiple protective roles associated with its antiinflammatory and immunomodulatory properties (Janciauskiene et al. 2011). Calreticulin, a multifunctional Ca2+-buffering chaperone, is involved in multiple cell processes (Michalak et al. 1999; Papp et al. 2009). TAC increased its expression in both genders and genotypes and male mouse hearts of both genotypes expressed higher calreticulin levels than female hearts. Mice overexpressing cardiac calreticulin exhibited a decreased systolic function and chamber dilation (Nakamura et al. 2001). Accordingly, it can be suggested that the TAC-induced upregulation of calreticulin plays a role in systolic function in our mouse model.
It is generally accepted that the mitochondria can serve as a source of NO-based cell signals that may originate independently of NO synthase activity (Chen et al. 2005). Mitochondrial ALDH2 (mtALDH2) is the main enzyme involved in acetaldehyde oxidation and in NO formation (Daiber et al. 2009; Song et al. 2011). The cardioprotective role of mtALDH2 is well known from studies in knockout mice (ALDH2-/- mice), who show marked vascular dysfunction (Wenzel et al. 2008). In addition, it is tempting to speculate that the lower cardiac expression of mtADLH2, found here in males, might result in less NO production. There is ample evidence that in PO-induced LVH and end-stage HF, the substrate preference switches from free fatty acids (FFA) to glucose and is associated with a downregulation of enzymes involved in FFA metabolism (Ventura-Clapier et al. 2002; Mettauer et al. 2006; Regitz-Zagrosek et al. 2010). Liver carboxylesterase N (ESTN) although predominantly expressed in liver, is also expressed at lower levels in the heart (Islam et al. 1999; Diczfalusy et al. 2001). We found lower levels of ESTN in male than in female hearts, indicative of a gender difference in fatty acid metabolism.
Given all these concomitant changes, male and female mouse hearts differed in terms of Ca2+-homeostasis, energy metabolism, signaling, and stress responses, but the outcome of such differences still remains unclear. Furthermore, in whole LV tissue it is difficult to define whether all differential protein expression occurred exclusively in the cardiomyocytes, as other cell types could also contribute.
Genotype molecular signature
Mice overexpressing FKBP12.6 differed markedly from wild-type mice; they exhibited very high levels of detoxification enzymes such as glutathione S-transferases (GST Mu5 and GST Mu7) and SBP1. Based on these findings, we may speculate that K mice handle oxidative stress differently, compared with WT mice. Interestingly, we found higher adenosine kinase (ADK) expression in K mouse hearts. ADK, a key enzyme in the purine salvage pathway, prevents toxic levels of adenosine building up within the cell (Kulkarni et al. 1998). This suggests that transgenic mice may also manage the adenosine pathway differently. We also observed a genotype difference in PDZ and LIM domain protein, with a small upregulation in K mice. These proteins may act as adapters between kinases and the cytoskeleton (Kotaka et al. 2001; Kadrmas and Beckele 2004) and colocalize with α-actinin at sites of actin anchorage, such as the intercalated disks of cardiac muscle cells. In this context, we can speculate that K mice may also present differences in the regulation of actin structure and dynamics. Besides this, MAPRE2 is one of the three microtubule-associated proteins that regulate microtubule functions and dynamics (Abiatari et al. 2009). Thus, the downregulation of MAPRE2 levels in K mice might alter the dynamics of cell morphology. However, addressing the functions of MAPRE2 in the heart awaits the generation of a mouse knockout model.
TAC induced two molecular bio-signatures of pathological LVH
Thoracic aortic constriction causes chronic PO leading to pathological LVH and its progression to HF is accompanied with increases in cell size and surface area, and in protein synthesis. TAC also induced pulmonary edema in a subgroup of mice (H) of both genders and genotypes. Both C and H mice shared similar TAC-induced changes in a set of common proteins, either up- or downregulated, indicative of molecular events occurring at the early stage of HF.
Molecular bio-signature of the early stage of progression to HF
We found that TAC induced an increase in two essential enzymes involved in purine metabolism, that is, ADK and KGUA (GUK1), each exerting its action through different cell signaling pathways in a dependent manner via their cellular localization. More recently, it was shown that ADK acts as an important mediator of adenosine attenuation of cardiomyocyte hypertrophy (Fassett et al. 2011) and also as a homeostatic bioenergetic network regulator adenosine (Shen et al. 2011). Here, TAC induced higher ADK protein levels in K hearts than in respective controls, and it is tempting to speculate that ADK might be a potential target for modulating the cardiac injury level in the early stage of HF in K mice. Besides these, KGUA catalyzes the phosphorylation of GMP to GDP and is also implicated in the regulation of the supply of guanine nucleotide to cell signaling pathways. Moreover, GMP-induced KGUA conformational changes are sufficient to convert KGUA to a membrane-associated guanylate kinase (MAGUK) involved in stabilizing cell–cell adhesion (Johnston et al. 2011). These protein dynamics raise the question of whether such neofunctionalization can occur in vivo in PO hearts. However, it is still unclear how the heart manages the balance between adenosine and guanine nucleotide availability.
Here, the MVP level was upregulated in C mice and further increased in failing mice (H). Despite the diverse conditions characterized by the upregulation of MVP, such as chemotherapy resistance (Scheffer et al. 2000), new roles have been recently assigned to MVP, including the association with insulin-like growth factor-1, hypoxia-inducible factor-1 alpha, and the major DNA repair machineries (Lara et al. 2001). Although the function of MVP is still largely unknown in the heart, in view of our observations, we may consider MVP as a potential prognostic factor associated with the progression to HF.
We have also identified two isoforms of SLMAP in mouse hearts of both genotypes. SLMAP plays a potential role in organizing the excitation–contraction (E–C) coupling apparatus of the cardiomyocyte and resides at distinct subcellular locations (Guzzo et al. 2005). Mice overexpressing cardiac SLMAP exhibit less response to an isoproterenol challenge, and their altered cardiac function was associated with diminished expression of Ca2+-handling proteins of SR, such as RyR2 and SERCA2a (Nader et al. 2012). Together these observations and our previous report showing downregulated Ca2+-handling proteins (Prévilon et al. 2011), suggest that the presently observed TAC-induced SLMAP upregulation plays a potential role in the impaired systolic and diastolic function in mouse hearts of both genotypes. As SLMAP level was higher in congestive failing hearts, it might be associated with diastolic dysfunction. We may suggest that SLMAP could qualify as a biomarker for the transition to HF.
Among the downregulated proteins observed in TAC mouse hearts of both genotypes were two glycolytic enzymes (ENOB [beta-enolase] and LDHB [L-lactate dehydrogenase B chain]). A decrease in ENOB was previously reported in rat pressure-overloaded hearts (Keller et al. 1995). Besides their glycolytic functions, they have also been shown to be suitable auxiliary proteins for DNA biology (Popanda et al. 1998). Concomitant alterations in given metabolic pathways were reflected by net changes in enzymes leading to mitochondrial dysfunction. Together, altered ACADS and ETFA levels involved in mitochondrial fatty acid beta-oxidation, and ATPB in the proton transport during oxidative phosphorylation, might contribute to decrease oxidative production of ATP, but also potentially increase production of reactive oxygen species (ROS). The decrease in IDH3A would impair the tricarboxylic acid cycle and the decrease in ubiquinone biosynthesis protein COQ9 would ensure a deficit in lipophilic antioxidant and therefore may affect the respiratory electron transfer chain. Taken together, these observations confirmed that mitochondrial machinery is altered in TAC-induced pathological LVH. Multiple changes in mitochondrial oxidative stress protein expression are determinant for the progression to HF whatever the genotype. Indeed, HF is frequently associated with energetic impairment (De Sousa et al. 2002; Neubauer 2007; Ingwall 2009; Turer et al. 2010).
Molecular bio-signature of the end stage of HF
Multiple pathological changes have been reported at different levels in failing hearts. The impairment of EF and the increase in LVEDD, reflecting chamber dilatation, are key changes at the organ level.
At the tissue level, our previous work showed less TAC-induced LV fibrotic response in K mice, with a predominant perivascular but also interstitial fibrosis (Prévilon et al. 2011), potentially impairing oxygen diffusion. Here, the noticeably enhanced collagen type 6 (Co6A) expression level in MWH may explain, at least in part, diastolic dysfunction whereas MKT mice with a preserved diastolic function exhibited a smaller increase in CO6A. Thus, it is conceivable that the fibrotic response may result in a chronic hypoxic stress in congestive failing hearts. In agreement with recent data showing cardiomyocyte NDRG2 expression (Sun et al. 2011) and upregulation by hypoxia-induced stress (Wang et al. 2008), the present upregulation of NDRG2 might be attributed to the hypoxic environment. Recently, its adenoviral-mediated overexpression attenuated liver fibrosis (Yang et al. 2011). Elucidation of the biological function of NDRG2 in HF may provide a promising strategy for the treatment of myocardial fibrosis. Protein–glutamine gamma-glutamyl transferase (TGM2) functions as an extracellular matrix stabilizer (Deasey et al. 2013) and its Ca2+-dependent cross-linking activity has been implicated in many fibrotic diseases (Tovar-Vidales et al. 2011). Therefore, we can speculate that enhanced TGM2 expression levels participate in TAC-induced cardiac fibrosis although additional experimental studies are needed to assess its precise involvement in HF.
At the cell level, changes in cytoskeletal structure in LVH and HF have also been reported (Periasamy et al. 2008; Dhalla et al. 2009; Palazzuoli and Nuti 2010). Here, we observed the upregulation of several cytoskeletal proteins which might contribute to organize a network of proteins combining structural and signaling functions. Among them, desmin is well known to provide connections between the different organelles (nucleus, mitochondrion, sarcolemma) and influences their localization and function. Desmin is increased in human failing hearts and causes a loss of myocyte cross-striation (Heling et al. 2000). The increment in desmin observed here in H mice may also result from changes in function of its chaperone molecule, αB-crystallin (CryAB). Recently, molecular chaperones or heat shock proteins (HSPB6, HSPB1, CryAB) have been considered as multifunctional protective agents, their actions being implicated in intracellular protein quality control (QPR) (Kumarapeli et al. 2010) and in maintaining muscle integrity (Edwards et al. 2011) and contractile function (Fan and Kranias 2011). Besides these cardioprotective roles, CryAB suppressed the hypertrophy induced by short-term PO in the heart (Kumarapeli et al. 2008). Consistent with this, we also found higher levels of expression of CryAB, HSPB6, and HSPB1 that might alter QPR in congestive failing hearts.
A significant number of reports also support the notion that mitochondrial dysfunction plays a critical role in the pathogenesis of HF (O'Rourke et al. 2001; Russel et al. 2005; Ventura-Clapier2009; Hollander et al. 2011). Mitochondrial oxidative stress is also important in TAC-induced HF (Bugger et al. 2010; Dai et al. 2012). Here, in both genotypes, TAC-induced congestive failing hearts exhibited an aggravated downregulation of distinct mitochondrial protein sets which might cause an imbalance in energy supply and an increase in ROS production, affecting mitochondrial function through peroxidation. For instance, the overexpression of PRDX3, a mitochondrial antioxidant, prevents LV remodeling and failure after myocardial infarction in mice (Matsushima et al. 2006). Needless to say, the decrease in PRDX3 and heat shock protein (CH60) observed here might be detrimental for mitochondria, suggesting that congestive failing hearts may be more sensitive to oxidative stress. Although mitochondrial-targeted antioxidants have proved protective in various animal models of disease, they still await positive clinical trials (Griffiths 2012).
Our bioinformatic analysis highlighted a network in which huntingtin protein was found to be at the center of a cluster of mitochondrial proteins (Fig. 7A), but its precise function in heart disease remains to be elucidated. Here, we validate by Western blot analysis that TAC-induced cardiac HTT expression level was upregulated in HF. In addition, the IAP-derived interaction of the HTT with HSPB1 in heart was confirmed by coimmunoprecipitation experiments. However, we did not verify all interactomes with HTT. In order to better understand the involvement of HTT in the progression to HF, further functional experiments are required. When mutant huntingtin protein is expressed only in mouse cardiomyocytes (Pattison et al. 2008), the animals develop HF whereas knocking down huntingtin expression in mice (Zeitling et al. 1995) and in zebrafish (Lumsden et al. 2007) is embryonically lethal, resulting in defects in all three germ layers and in iron utilization, respectively. The notion that huntingtin protein may damage neurons by directly interfering with mitochondrial function in Huntington's disease (Cattaneo and Calabresi 2002), allows us to suggest that such a similar chain of events may occur in HF. Huntingtin protein appeared as a key node between mitochondrial dysfunction and cytoskeletal remodeling (TMG2). It is tempting to hypothesize that the increase in TGM2 and the dependence on the degree of the HTT polyglutamine expansion might contribute to more susceptibility to apopain cleavage leading to cytotoxic effects and likely to result in accelerated dysfunction in cellular energy metabolism. Speculatively, we suggest that huntingtin protein will become a new potential target for pathogenic mechanisms involved in HF progression.
However, mitochondria are also involved in intracellular Ca2+ handling. Mitochondrial Ca2+-transport is important in the generation of ROS and in the opening of the mitochondrial permeability transition pore, a factor potentially involved in HF (Griffiths 2012).
As a consequence of oxidative stress, mitochondrial alterations and functional impairment might lead to Ca2+ leak which, in turn, could directly interfere with the regulation of endoplasmic reticulum (ER) Ca2+-cycling. Thus, the ER might be overloaded with Ca2+ in congestive HF. A disturbance of Ca2+-homeostasis and overexpression of normal and/or incorrectly folded proteins interfere with ER function (Minamino and Kitakaze 2010). We also confirmed an increase in both ER stress proteins (ERP29, GRP78). Indeed, a marked increase in GRP78 involved in activation of the unfolded protein response (UPR) has been associated with the pathophysiology of human HF (Dally et al. 2009) and in mouse PO hearts (Okada et al. 2004). Another chaperone, HSP90, involved in UPR and regulation of apoptotic signaling through interaction with the proteins involved in the degradation process (Patterson and Cyr 2002), was also increased in failing hearts. These findings reflected an ER stress that might impair the UPR in H mice. Like the ER, the SR which is considered as a reservoir for Ca2+ release via the RyR2 channel during systolic contraction, then for Ca2+ capture via SERCA2a during relaxation, can also be stressed, either by altered SR Ca2+-homeostasis and/or by altered proteins which trigger cardiac dysfunction. A recent report associated ER stress with a reduced abundance of SERCA2a (Liu et al. 2011). We previously showed that altered Ca2+-handling proteins were associated with altered systolic and diastolic LV function in TAC mice (Prévilon et al. 2011). If the lumen of ER and SR is functionally well connected, the downregulation of SERCA2a might thus result in a reduced Ca2+ store in ER of congestive failing hearts. However, it remains to be demonstrated that ER and SR share a common Ca2+ store. Indeed, the concept that excessive levels of ROS/RNS (reactive nitrogen species) (Fassett et al. 2011; Tsutsui et al. 2011) and abnormal Ca2+-handling (Bers 2008) contribute to the development of contractile dysfunction in congestive HF is well accepted. ROS/RNS, normally produced in the heart, promote endogenous reversible RyR2-S-nitrosylation and S-glutathionylation (Donoso et al. 2011). Lastly, GSTO1 and ion chloride channel (CLIC1) bind to RyR channels (Donoso et al. 2011; Dulhunty et al. 2011). Both binding sites are on the clamp region of RyR2, a region that undergoes significant structural changes with channel opening and which bind another important regulator, FKBP12.6 (Donoso et al. 2011). Especially, the dissociation of FKBP12.6 from RyR2 caused Ca2+ leak via RyR2 and has been implicated in phenotypic changes in HF (Marks et al., 2002; Huang et al. 2006). We found that GSTO1 and CLIC1 protein levels were upregulated in failing hearts from mice of both genotypes. Thus, in such a context, these proteins might also modulate RyR2 activity in order to maintain low levels of Ca2+ leak during diastole, which might prevent arrhythmia and sudden cardiac death. Galfré et al.(2012) also highlighted some controversy over the functional effects of FKBP12.6 as the only channel stabilizer of RyR2. Furthermore, among the multiple molecular partners carefully orchestrating Ca2+-homeostasis, we also found an enhanced ANXA5 protein expression level in H mice, in agreement with its increased expression in failing human hearts, and its relation to systolic dysfunction in hypertensive patients (Camors et al. 2005, 2006; Ravassa et al. 2007). It also acted as a regulatory factor of Ca2+-handling proteins when it formed a complex with the Na+/Ca2+ -exchanger. Therefore, it is tempting to speculate that ANXA5 might contribute to impaired systolic function in mouse congestive HF.
Conclusion
This study demonstrates distinct changes in the pathological LVH protein profile in response to TAC. The overexpression of FKBP12.6 is not sufficient to confer a sustained protection against chronic systolic overload. Compelling evidence supports a relationship between SR/ER and mitochondria which likely involve several Ca2+ and ROS-sensitive factors in the pathophysiology of end-stage HF. We speculate that huntingtin protein will become a new potential target for pathogenic mechanisms involved in HF progression. In TAC mice, huntingtin protein upregulation was related with a remarkable downregulation of proteins involved in energy metabolism. Although our understanding of the failing heart proteome has progressed in recent years, many important issues are still unresolved. As a consequence of PO, multiple cross-talks between organs can occur differently; therefore an integrative pathophysiology at the body level remains also to be explored. Nevertheless, in order to identify the pathways and to prove their mediating role, a large number of further cell biology and animal experiments are required that will provide us with new targets for drug discovery and therapeutic intervention.
Acknowledgments
This study was funded by the Institut National de la Santé et de la Recherche Médicale (Inserm), Université Paris Diderot (Paris 7), and by EU FP6 grant LSHM-CT-2005-018,833, EUGeneHeart. Miresta Prévilon was supported by a scholarship of the Ministère de l'Enseignement Supérieur et de la Recherche. We thank M. Osborne-Pellegrin for her help in editing the manuscript.
Conflict of Interest
None declared.
References
- Abiatari I, Gillen S, DeOliveira T, Klose T, Bo K, Giese NA, et al. The microtubule-associated protein MAPRE2 is involved in perineural invasion of pancreatic cancer cells. Int. J. Oncol. 2009;35:1111–1116. doi: 10.3892/ijo_00000426. doi: 10.3892/ijo_00000426. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Benjamini Y, Hochberg Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J. Educ. Behav. Stat. 2000;25:60–83. [Google Scholar]
- Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008;70:29–49. doi: 10.1146/annurev.physiol.70.113006.100455. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- Bugger H, Scwharzer M, Chen D, Schrepper A, Amorin PA, Schoeppe M, et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overloaded-induced heart failure. Cardiovasc. Res. 2010;85:376–384. doi: 10.1093/cvr/cvp344. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- Camors E, Monceau V, Charlemagne D. Annexins and Ca2+-handling in the heart. Cardiovasc. Res. 2005;65:793–802. doi: 10.1016/j.cardiores.2004.11.010. doi: 10.1016/j.cardiores.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Camors E, Charue D, Trouvé P, Monceau V, Loyer X, Russo-Marie F, et al. Association of annexin A5 with Na+/Ca2+ exchanger and caveolin-3 in non failing and failing human heart. J. Mol. Cell. Cardiol. 2006;4:47–55. doi: 10.1016/j.yjmcc.2005.08.009. doi: 10.1016/j.yjmcc.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Calabresi P. Mutant huntingtin goes straight to the heart. Nat. Neurosci. 2002;5:711–712. doi: 10.1038/nn0802-711. doi: 10.1038/nn0802-711. [DOI] [PubMed] [Google Scholar]
- Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, et al. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin biactivation. Proc. Natl. Acad. Sci. USA. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, et al. Mitochondrial proteome remodelling in pressure overloaded-heart failure: the role of mitochondrial oxidative stress. Cardiovasc. Res. 2012;93:79–88. doi: 10.1093/cvr/cvr274. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A, Oelze M, Wenzel P, Wickramanayake JM, Schuhmacher S, Jansen T, et al. Nitrate tolerance as a model of vascular dysfunction: roles for mitochondrial aldehyde dehydrogenase and mitochondrial oxidative stress. Pharmacol. Rep. 2009;61:33–48. doi: 10.1016/s1734-1140(09)70005-2. PMID:19307691. [DOI] [PubMed] [Google Scholar]
- Dally S, Monceau V, Corvazier E, Bredoux R, Raies A, Bobe R, et al. Compartimentalized expression of three novel sarco/endoplasmic reticulum Ca2+ ATPase 3 isoforms including the switch to ER stress, SERCA3f, in nonfailing and failing human heart. Cell Calcium. 2009;45:144–154. doi: 10.1016/j.ceca.2008.08.002. doi: 10.1016/j.ceca.2008.08.002. [DOI] [PubMed] [Google Scholar]
- De Sousa E, Lechene P, Fortin D, N'Guessan B, Belmadani S, Bigard X, et al. Cardiac and skeletal muscle energy metabolism in heart failure: beneficial effects of voluntary activity. Cardiovas Res. 2002;56:260–268. doi: 10.1016/s0008-6363(02)00540-0. doi: 10.1016/S0008-6363(02)00540-0. [DOI] [PubMed] [Google Scholar]
- Deasey S, Shanmugasundaram S, Nurminskaya M. Tissue-specific response to loss of transglutaminase 2. Amino Acids. 2013;44:179–187. doi: 10.1007/s00726-011-1183-9. doi: 10.1007/s00726-011-1183-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalla NS, Saini-Chohan HK, Rodriguez-Leyva D, Elimban V, Dent MR, Tappia PS. Subcelluar remodeling may induce cardiac dysfunction in congestive heart failure. Cardiovasc. Res. 2009;81:429–438. doi: 10.1093/cvr/cvn281. doi: 10.1093/cvr/cvn281. [DOI] [PubMed] [Google Scholar]
- Diczfalusy MA, Björkhem I, Einarsson C, Hillebrant CG, Alexon SHE. Characterization of enzymes involved formation of ethyl esters of long-chain fatty acids in humans. J. Lipid Res. 2001;42:1025–1032. PMID:11441128. [PubMed] [Google Scholar]
- Diedrich M, Tadic J, Mao L, Wacker NebrichG, Hetzer R, Regitz-Zagrosek V, et al. Heart expression related to age and sex in mice and humans. Int. J. Mol. Med. 2007;20:865–874. PMID:17982696. [PubMed] [Google Scholar]
- Donoso P, Sanchez G, Bull R, Hildalgo C. Modulation of cardiac ryanodine activity by ROS and RNS. Front. Biosci. 2011;16:553–567. doi: 10.2741/3705. PMID:21196188. [DOI] [PubMed] [Google Scholar]
- Dulhunty A, Hewawasam R, Liu D, Casarotto MG, Board PG. Regulation of the cardiac muscle ryanodine receptor by glutathione transferases. Drug Metab. Rev. 2011;43:236–252. doi: 10.3109/03602532.2010.549134. doi: 10.3109/03602532.2010.549134. [DOI] [PubMed] [Google Scholar]
- Edwards HV, Cameron RT, Baillie GS. The emerging role of HSP20 as a multifunctional protective agent. Cell. Signal. 2011;23:1447–1454. doi: 10.1016/j.cellsig.2011.05.009. doi: 10.1016/j.cellsig.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Fan GC, Kranias EG. Small heat shock protein 20 (HspB6) in cardiac hypertrophy and failure. J. Mol. Cell. Cardiol. 2011;51:574–577. doi: 10.1016/j.yjmcc.2010.09.013. doi: 10.1016/j.yjmcc.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassett JT, Hu X, Xu X, Lu Z, Zhang P, Chen Y, et al. Adenosine kinase regulation of cardiomyocyte hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H1722–H1732. doi: 10.1152/ajpheart.00684.2010. doi: 10.1152/ajpheart.00684.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galfré E, Pitt SJ, Venturi E, Sitsapesan M, Zaccai NR, Tsaneva-Atanasova K, et al. FKBP12 activates the cardiac ryanodine receptor Ca2+-release channel and is antagonised by FKBP12.6. PLoS ONE. 2012;7:e31956. doi: 10.1371/journal.pone.0031956. doi: 10.1371/journal.pone.0031956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellen B, Fernandez-Velasco M, Briec F, Vinet L, LeQuang K, Rouet-Benzineb P, et al. Conditional FKBP12.6 over-expression in mouse cardiac myocytes prevents triggered ventricular tachycardia through specific alterations in excitation–contraction coupling. Circulation. 2008;117:1778–1786. doi: 10.1161/CIRCULATIONAHA.107.731893. doi: 10.1161/CIRCULATIONAHA.107.731893. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ. Mitochondria and heart disease. Adv. Exp. Med. Biol. 2012;2012:249–267. doi: 10.1007/978-94-007-2869-1_11. doi: 10.1007/978-94-007-2869-1_11. [DOI] [PubMed] [Google Scholar]
- Guzzo RM, Salih M, Moore ED, Tuana BS. Molecular properties of cardiac tail-anchored membrane protein SLMAP are consistent with structural role in arrangement of excitation-contraction coupling apparatus. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H1810–H1819. doi: 10.1152/ajpheart.01015.2004. doi: 10.1152/ajpheart.01015.2004. [DOI] [PubMed] [Google Scholar]
- Harrar Y, Bellini C, Faure JD. FKBPs: at the crossroads of folding and transduction. Trends Plant Sci. 2001;6:426–431. doi: 10.1016/s1360-1385(01)02044-1. PMID:11544132. [DOI] [PubMed] [Google Scholar]
- Heling A, Zimmermann R, Kostin MaenoY, Hein S, Devaux B, Bauer E, et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Cir. Res. 2000;86:846–853. doi: 10.1161/01.res.86.8.846. doi: 10.1161/01.RES.86.8.846. [DOI] [PubMed] [Google Scholar]
- Hollander JM, Baseler WA, Dabkowski ER. Proteomic remodeling of mitochondria in heart failure. Congest. Heart Fail. 2011;17:262–268. doi: 10.1111/j.1751-7133.2011.00254.x. doi: 10.1111/j.1751-7133.2011.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Shan J, Reiken S, Wehrens XH, Marks AR. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc. Natl. Acad. Sci. USA. 2006;103:3456–3461. doi: 10.1073/pnas.0511282103. doi: 10.1073/pnas.0511282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009;81:412–419. doi: 10.1093/cvr/cvn301. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MR, Waheed A, Shah GN, Tomatsu S, Sly WS. Human egasyn binds beta-glucuronidase but neither the esterase active site of egasyn nor the C-terminus of beta-glucuronidase is involved in their interaction. Arch. Biochem. Biophys. 1999;372:53–61. doi: 10.1006/abbi.1999.1449. PMID:10562416. [DOI] [PubMed] [Google Scholar]
- Janciauskiene SM, Bais R, Koczulla R, Vogelmeier C, Köhnlein T, Welte T. The discovery of α1-antitrypsin and its role in health and disease. Respir. Med. 2011;105:1129–1139. doi: 10.1016/j.rmed.2011.02.002. doi: 10.1016/j.rmed.201.02.002. [DOI] [PubMed] [Google Scholar]
- Jeyakumar LH, Ballester L, Chenug DS, McIntyre JO, Chang P, Olivey HE, et al. FKBP binding characteristics of cardiac microsomes from diverse vertebrates. Biochem. Biophys. Res. Commun. 2001;281:979–986. doi: 10.1006/bbrc.2001.4444. doi: 10.1006/bbrc.2001.4444. [DOI] [PubMed] [Google Scholar]
- Johnston CA, Whitney DS, Volkman BF, Doe CQ, Prehoda KE. Conversion of the enzyme guanylate kinase into a mitotic –spindle orientating protein by a single mutation that inhibits GMP-Induced closing. Proc. Natl. Sci. Acad. USA. 2011;108:E973–E978. doi: 10.1073/pnas.1104365108. doi: 10.1073/pnas.1104365108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadrmas JL, Beckele MC. The Lim domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 2004;5:920–931. doi: 10.1038/nrm1499. doi: 10.1038/nrm1499. [DOI] [PubMed] [Google Scholar]
- Keller A, Rouzeau JD, Farhadian F, Wisnewsky C, Marotte F, Lamandé N, et al. Differential expression of alpha- and beta-enolase genes during rat heart development and hypertrophy. Am. J. Physiol. 1995;269:H1843–H1851. doi: 10.1152/ajpheart.1995.269.6.H1843. PMID:8594891. [DOI] [PubMed] [Google Scholar]
- Kotaka M, Lau YM, Cheung K-K, Lee SMY, Li HY, Chan WY, et al. Elfin is expressed during early heart development. J. Cell. Biochem. 2001;83:463–472. doi: 10.1002/jcb.1244. doi: 10.1002/jcb.1244. [DOI] [PubMed] [Google Scholar]
- Kulkarni JS, Przywara DA, Wakade TD, Wakade AR. Adenosine induces apoptosis by inhibiting mRNA and protein synthesis in chick embryonic sympathetic neurons. Neurosci. Lett. 1998;248:187–190. doi: 10.1016/s0304-3940(98)00369-3. PMID: 9654340. [DOI] [PubMed] [Google Scholar]
- Kumarapeli AR, Su H, Huang W, Tang M, AZheng H, Horak KM, et al. Apha B-crystallin suppresses pressure overload cardiac hypertrophy. Cir. Res. 2008;103:1473–1482. doi: 10.1161/CIRCRESAHA.108.180117. doi: 10.1161/CIRCRESAHA.108.180117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumarapeli AR, Horak K, Wang X. Protein quality control in protection against systolic overload cardiomyopathy: the long term role of small heat shock proteins. Am. J. Transl. Res. 2010;2:390–401. PMID:20733949. [PMC free article] [PubMed] [Google Scholar]
- Lam E, Martin MM, Timerman AP, Sabers C, Fleischer S, Lukas T, et al. A novel FK506 binding protein can mediate the immunosuppressive effects of FK506 and is associated with the cardiac ryanodine receptor. J. Biol. Chem. 1995;270:26511–26522. doi: 10.1074/jbc.270.44.26511. doi: 10.1074/jbc.270.44.26511. [DOI] [PubMed] [Google Scholar]
- Lara PC, Pruschy M, Zimmermann M, Henriquez-Hernandez LA. MVP and vaults: a role in the radiation response. Radiat. Oncol. 2001;6:148. doi: 10.1186/1748-717X-6-148. doi: 10.1186/1746-717X-6-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XH, Zhang ZY, Andersson KB, Husberg C, Enger UH, Raeder MG, et al. Cardyomycyte-specific disruption of Serca2 in adult mice causes sarco(endo)plasmic reticulum stress and apoptosis. Cell Calcium. 2011;49:201–207. doi: 10.1016/j.ceca.2010.09.009. doi: 10.1016/j.ceca.2010.09.009. [DOI] [PubMed] [Google Scholar]
- Lumsden AL, Henshall TL, Dayan S, Lardelli MT, Richards RI. Hungtingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum. Mol. Genet. 2007;16:1905–1920. doi: 10.1093/hmg/ddm138. doi: 10.1093/hmg/ddm138. [DOI] [PubMed] [Google Scholar]
- Marks AR. Immunophilin modulation of calcium channel gating. Methods. 1996;9:177–187. doi: 10.1006/meth.1996.0024. PMID:8812665. [DOI] [PubMed] [Google Scholar]
- Marouga R, David S, Hawkins E. The development of the DIGE system: 2D fluorescence gel analysis technology. Anal. Bioanal. Chem. 2005;382:669–678. doi: 10.1007/s00216-005-3126-3. PMID:15900442. [DOI] [PubMed] [Google Scholar]
- Marks AR, Reiken S, Marx SO. Progression of Heart Failure: is Protein Kinase A Hyperphosphorylation of the Ryanodine Receptor a Contributing Factor? Circulation. 2002;105:272–275. [PubMed] [Google Scholar]
- Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006;113:1779–1786. doi: 10.1161/CIRCULATIONAHA.105.582239. doi: 10.1161/CIRCULATIONAHA.105.582239. [DOI] [PubMed] [Google Scholar]
- Mettauer B, Zoll J, Garnier A, Ventura-Clapier R. Heart failure: a model of cardiac and skeletal muscle energetic failure. Pflugers Arch. 2006;452:653–666. doi: 10.1007/s00424-006-0072-7. doi: 10.1007/s00424-006-0072-7. [DOI] [PubMed] [Google Scholar]
- Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin:one protein, one gene, many functions. Biochem. J. 1999;344(Pt2):281–292. PMID:10567207. [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Kitakaze M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010;48:1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- Nader M, Westendorp B, Hawari O, Salih M, Stewart AFR, Leenen FHH, et al. Tail-anchored membrane protein SLMAP is a novel regulator of cardiac function at the sarcoplasmic reticulum. Am. J. Physiol. Heart Circ. Physiol. 2012;302:H1138–H1145. doi: 10.1152/ajpheart.00872.2011. doi: 10.1152/ajpheart.00872.2011. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Robertson M, Liu G, Dickie P, Nakamura K, Gou JQ, et al. Complete heartblock and sudden death in mice overexpressing calreticulin. J. Clin. Invest. 2001;107:1245–1253. doi: 10.1172/JCI12412. doi: 10.1172/JCI12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer S. The failing heart-An engine out of fuel. N. Engl. J. Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- O'Rourke B, Foster J, Van Eyk DB. Mitochondrial protein phosphorylation as a regulatory modality: implications for mitochondrial dysfunctions in heart failure. Congest. Heart Fail. 2001;17:269–282. doi: 10.1111/j.1751-7133.2011.00266.x. doi: 10.1111/j.1751-7133.2011.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzuoli A, Nuti R. Heart failure: pathophysiology and clinical picture. Contrib. Nephrol. 2010;2010:1–10. doi: 10.1159/000313714. doi: 10.1159/000313714. [DOI] [PubMed] [Google Scholar]
- Papp S, Dziak E, Opas M. Embryonic stem cell-derived cardiomyogenesis: a novel role for calreticulin as a regulator. Stem Cells. 2009;27:1507–1515. doi: 10.1002/stem.85. doi: 10.1002/stem.85. [DOI] [PubMed] [Google Scholar]
- Patterson C, Cyr D. Welcome to machine: a cardiologist's introduction to protein folding and degradation. Circulation. 2002;106:2741–2746. doi: 10.1161/01.cir.0000041145.30519.6b. doi: 10.1161/01.CIR.0000041145.30519.6B. [DOI] [PubMed] [Google Scholar]
- Pattison JS, Sanbe A, Maloyan A, Osinka H, Klevistsky R, Robbins J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation. 2008;117:2743–2751. doi: 10.1161/CIRCULATIONAHA.107.750232. doi: 10.1161/CIRCULATIONAHA.107.750232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy M, Paul ML, Janssen PML. Molecular basis of diastolic dysfunction. Heart Fail. Clin. 2008;4:13–21. doi: 10.1016/j.hfc.2007.10.007. doi: 10.1016/j.hfc.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popanda O, Fox G, Thielmann H. Modulation of DNA polymerase α, δ and ε by lactate dehydrogenase and 3-phosphoglycerate kinase. Biochim. Biophys. Acta. 1998;1397:102. doi: 10.1016/s0167-4781(97)00229-7. PMID:9545551. [DOI] [PubMed] [Google Scholar]
- Prestle J, Janssen PM, Janssen AP, Zeitz O, Lehnart SE, Bruce L, et al. Over-expression of FK506-binding protein FKBP12.6 in cardiomyocytes reduces ryanodine receptor-mediated Ca2+ leak from the sarcoplasmic reticulum and increases contractility. Circ. Res. 2001;88:188–194. doi: 10.1161/01.res.88.2.188. doi: 10.1161/01.RES.88.2.188. [DOI] [PubMed] [Google Scholar]
- Prévilon M, Pezet M, Dachez C, Mercadier JJ, Rouet-Benzineb P. Sequential alterations in Akt, GSK3b, and calcineurin signalling in the mouse left ventricle after thoracic aortic constriction. Can. J. Physiol. Pharmacol. 2010;88:1093–1101. doi: 10.1139/y10-087. doi: 10.1139/y10-087. [DOI] [PubMed] [Google Scholar]
- Prévilon M, Pezet M, Semprez F, Mercadier JJ, Rouet-Benzineb P. FKBP12.6 mice display temporal gender differences in cardiac Ca2+-signalling phenotype upon chronic pressure overload. Can. J. Physiol. Pharmacol. 2011;89:769–782. doi: 10.1139/y11-075. doi: 10.1139/Y11-075. [DOI] [PubMed] [Google Scholar]
- Ravassa S, Gonzalez A, Lopez B, Beaumont J, Querejeta R, Larman M, et al. Upregulation of myocardial annexin A5 in hypertensive heart disease: association with systolic dysfunction. Eur. Heart J. 2007;28:2785–2791. doi: 10.1093/eurheartj/ehm370. doi: 10.1093/eurheartj/ehm370. [DOI] [PubMed] [Google Scholar]
- Regitz-Zagrosek V. Therapeutic implications of the gender-specific aspects of cardiovascular disease. Nat. Rev. Drug Discov. 2006;5:425–438. doi: 10.1038/nrd2032. doi: 10.1038/nrd2032. [DOI] [PubMed] [Google Scholar]
- Regitz-Zagrosek V, Oertelt-Prigione S, Seeland U, Hetzer R. Sex and gender difference in myocardial hypertrophy and heart failure. Circ. J. 2010;74:1265–1273. doi: 10.1253/circj.cj-10-0196. doi: org/10.1253/circj.CJ-10-0196. [DOI] [PubMed] [Google Scholar]
- Russel LK, Finck BN, Kelly DP. Mouse models of mitochondrial dysfunction and heart failure. J. Mol. Cell. Cardiol. 2005;38:81–91. doi: 10.1016/j.yjmcc.2004.10.010. doi: 10.1016/j.yjmcc.2004.10010. [DOI] [PubMed] [Google Scholar]
- Scheffer GL, Schroeijers AB, Izquierdo MA, Wiemer EA, Scheper RJ. Lung resistance-related protein/major vault protein and vaults in multidrug-resistant cancer. Curr. Opin. Oncol. 2000;12:550–556. doi: 10.1097/00001622-200011000-00007. PMID:11085454. [DOI] [PubMed] [Google Scholar]
- Schreiber SL. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- Shen HY, Lusardi TA, Williams-Karnesky RL, Lan JQ, Poulsen DJ, Boison D. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J. Cereb. Blood Flow Metab. 2011;31:1648–1659. doi: 10.1038/jcbfm.2011.30. doi: 10.1038/jcbfm.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song BJ, Abdelmegeed MA, Yoo SH, Kim BJ, Jo SA, Jo I, et al. Post-translational modifications of mitochondrial aldehyde dehydrogenase and biomedical implications. J. Proteomics. 2011;74:2691–2702. doi: 10.1016/j.jprot.2011.05.013. doi: 10.1016/j.jprot.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Shen L, Sun X, Tong G, Sun D, Han T, et al. Variation of NDRG2 and c-Myc expression in rat heart during the acute stage of ischemia/reperfusion injury. Histochem. Cell Biol. 2011;135:27–35. doi: 10.1007/s00418-010-0776-9. doi: 10.1007/s00414-010-0776-9. [DOI] [PubMed] [Google Scholar]
- Tovar-Vidales T, Clark AF, Wordinger RJ. Focus on molecules: transglutaminase 2. Exp. Eye Res. 2011;93:2–3. doi: 10.1016/j.exer.2010.06.006. doi: 10.1016/j.exer.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H2181–H2190. doi: 10.1152/ajpheart.00554.2011. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- Turer AT, Cr Malloy, Newgard CB, Podgoreanu MV. Energetics and metabolism in the failing heart: important but poorly understood. Curr. Opin. Clin. Nutr. Metab. Care. 2010;13:458–465. doi: 10.1097/MCO.0b013e32833a55a5. doi: 10.1097/MCO.0b013e32833a55a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R. Exercice training, energy metabolism, and heart failure. Appl. Physiol. Nutr. Metab. 2009;34:336–339. doi: 10.1139/h09-013. doi: 10.1139/h09-013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R, Veksler E, De Sousa V. Metabolic myopathy in heart failure. News Physiol. Sci. 2002;17:191–196. doi: 10.1152/nips.01392.2002. PMID:2270955. [DOI] [PubMed] [Google Scholar]
- Vinet L, Pezet M, Bito V, Briec F, Biesmans L, Rouet-Benzineb P, et al. Cardiac FKBP12.6 over expression protects against triggered ventricular tachycardia in pressure overload mouse hearts. Basic Res. Cardiol. 2012;107:1–14. doi: 10.1007/s00395-012-0246-8. doi: 10.1007/s00395-012-0246-8. [DOI] [PubMed] [Google Scholar]
- Viswanathan S, Unlu M, Minden JS. Two-dimensional difference gel electrophoresis. Nat. Protoc. 2006;1:1351–1358. doi: 10.1038/nprot.2006.234. doi: 10.1038/nprot.2006.234. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu N, Yao L, Li F, Zhang J, Deng Y, et al. NDRG2 is a new HIF-1 target gene necessary for hypoxia-induced apoptosis in A549 cells. Cell. Physiol. Biochem. 2008;21:239–250. doi: 10.1159/000113765. doi: 10.1159/000113765. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. PMID:12837242. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Müller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M, et al. ALDH-2 deficiency increases cardiovascular oxidative stress–evidence for indirect antioxidative properties. Biochem. Biophys. Res. Commun. 2008;367:137–143. doi: 10.1016/j.bbrc.2007.12.089. doi: 10.1016/j.bbrc.2007.12.089. [DOI] [PubMed] [Google Scholar]
- Xiao J, Tian X, Jones PP, Bolstad J, Kong H, Wang R, et al. Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J. Biol. Chem. 2007;282:34828–34838. doi: 10.1074/jbc.M707423200. doi: 10.1074/jbc.M707423200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, et al. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature. 2002;416:334–338. doi: 10.1038/416334a. doi: 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- Yang J, Zheng J, Wu L, Shi M, Zhang H, Wang X, et al. NDRG2 ameliorates hepatic fibrosis by inhibiting the TGF-b1/Smad pathway and altering the MMP2/TIMP2 ratio in rats. PLoS ONE. 2011;6:e27710. doi: 10.1371/journal.pone.0027710. doi: 10.1371/journal.pone.0027710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel C, Chamrad DC, Priller J, Woodman B, Meyer HE, Bates GP, et al. Alterations in the mouse and human proteome caused by Huntington's disease. Mol. Cell. Proteomics. 2002;1:366–375. doi: 10.1074/mcp.m200016-mcp200. doi: 10.1074/mcp.M200016-MCP200. [DOI] [PubMed] [Google Scholar]
- Zeitling S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadys A. Increased apoptosis and early embryonic lethality in mice nullizygous for Huntington's gene homologue. Nat. Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]