

Abstract
Halogen bonding is an emerging noncovalent interaction for constructing supramolecular assemblies. Though similar to the more familiar hydrogen bonding, four primary differences between these two interactions make halogen bonding a unique tool for molecular recognition and the design of functional materials. First, halogen bonds tend to be much more directional than (single) hydrogen bonds. Second, the interaction strength scales with the polarizability of the bond-donor atom, a feature that researchers can tune through single-atom mutation. In addition, halogen bonds are hydrophobic whereas hydrogen bonds are hydrophilic. Lastly, the size of the bond-donor atom (halogen) is significantly larger than hydrogen. As a result, halogen bonding provides supramolecular chemists with design tools that cannot be easily met with other types of noncovalent interactions and opens up unprecedented possibilities in the design of smart functional materials.
This Account highlights the recent advances in the design of halogen-bond-based functional materials. Each of the unique features of halogen bonding, directionality, tunable interaction strength, hydrophobicity, and large donor atom size, makes a difference. Taking advantage of the hydrophobicity, researchers have designed small-size ion transporters. The large halogen atom size provided a platform for constructing all-organic light-emitting crystals that efficiently generate triplet electrons and have a high phosphorescence quantum yield. The tunable interaction strengths provide tools for understanding light-induced macroscopic motions in photoresponsive azobenzene-containing polymers, and the directionality renders halogen bonding useful in the design on functional supramolecular liquid crystals and gel-phase materials. Although halogen bond based functional materials design is still in its infancy, we foresee a bright future for this field. We expect that materials designed based on halogen bonding could lead to applications in biomimetics, optics/photonics, functional surfaces, and photoswitchable supramolecules.
Introduction
In recent years, halogen bonding has grown from a scientific curiosity to one of the most interesting noncovalent interactions for constructing supramolecular assemblies.1 According to the recently proposed IUPAC provisional recommendation,2 “A halogen bond occurs when there is evidence of a net attractive interaction between an electrophilic region associated with a halogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity”. This definition acknowledges the qualitative analogy between halogen bonding and the ubiquitous hydrogen bonding.3,4 The competition between halogen and hydrogen bonding in driving molecular recognition processes has been studied in crystal5,6 and biomolecular7 engineering, and in both cases it has been shown that it is possible for the former to outperform the latter under similar molecular environments.
The potential of halogen bonding, however, arises more from its differences, than from the similarities to hydrogen bonding. Herein we identify four such differences:
Directionality
Halogen bonding can be explained by the presence of a region of positive electrostatic potential, the so-called σ-hole, on the outermost surface of the halogen atom (X).8 The σ-hole is narrowly confined on the elongation of the R–X covalent bond axis, whereas the positive region of hydrogen bond donors is much more spread out over the surface of the hydrogen atom. Hence, halogen bonds tend to be more directional than (single) hydrogen bonds, with the typical R–X···Y (Y is any nucleophilic site) bond angle being closer to 180°.9
Tunability
The σ-hole strengthens with the electron withdrawing ability of the moiety the halogen atom is attached to.10 Importantly, it strengthens also with the polarizability of the halogen atom; hence, the bond strength develops in the order I > Br > Cl (fluorine atom can act as a halogen-bond donor only when attached to particularly strong electron-withdrawing groups11). Therefore, the halogen bond interaction strength can be tuned through single-atom mutation while hydrogen bond requires major changes into the residue the bond donor site is bound to.
Hydrophobicity
The presence of halogen atoms in a molecule increases its lipophilicity and hydrophobicity. Halogenated organic fragments, in particular perfluorocarbon residues, are less willing to mix with aqueous or polar solvents. In particular, it has been demonstrated that very polar solvents, for example, water, have little influence on the interaction energies and geometries of halogen-bonded adducts in solution.12 This is not the case for hydrogen-bonded adducts, which suffer much more from the competition with hydrogen bond-donor/acceptor solvents, as it is clear, for example, in anion transport applications (vide infra). Consequently, halogen bonding can be considered as a hydrophobic equivalent of the hydrophilic hydrogen bonding. This is a pivotal aspect especially in drug design, since many parameters such as absorption, biological barrier permeability, transport into organs and cells, and interaction with target molecules are controlled by the lipophilicity of the drug.13,14
Donor Atom Size
The van der Waals radii of iodine (bromine) and hydrogen are 1.98 Å (1.85 Å) and 1.20 Å, respectively.15 This may pose some steric limitations: for instance, halogen bonding between DNA base pairs is most stable using bromine rather than iodine, due to more favorable size/polarizability combination of the former.16 On the other hand, the bare size of halogen atoms may significantly alter the light-emitting properties of halogenated dyes by promoting singlet-to-triplet intersystem crossing.17
In this Account we pinpoint, through specific examples chosen from the past 2 years or so, that the above-mentioned characteristics of halogen bonding, high directionality, tunable interaction strength, hydrophobicity, and relatively large atom size, provide unique possibilities in constructing new classes of functional supramolecular materials. The examples chosen range from halogen-bond-driven ion transporters and phosphorescent cocrystals to photoresponsive materials and supramolecular gels. This Account should not be seen as a comprehensive review of halogen-bonded supramolecular materials, several such reviews have been published in the past years.18−21 We rather think that the chosen examples are good representatives of the future perspectives of this noncovalent interaction, the full potential of which is still far from being fully harnessed.
Halogen-Bond-Mediated Anion Transport
During the past decade, halogen bonding has been widely used to drive molecular self-assembly in the solid state. But halogen bonding can be strong enough to drive molecular recognition in solution as well.22,23 This area has grown rapidly in the past few years, driven by the possibility of using halogen bonding for rational drug design and catalysis.24−26 In particular, utilization of halogen bonding in anion recognition is a timely topic,27−31 which has also been recently reviewed.32,33 Anions are involved in a range of important biological, environmental, and chemical processes and their selective binding, extraction, and separation are frequently invoked as potential solutions to a number of fundamental and applied problems. It is thus not surprising that the design of host systems for anion recognition is a growing field of research, motivated by the need for new and selective anion receptors that function under competitive conditions, especially in light of their potential for biological activity.
Recent progress has shown the great potential of halogen bonding in driving transmembrane anion transport processes in aqueous solution, thanks to the hydrophobicity of halogen bond-donors.34−37
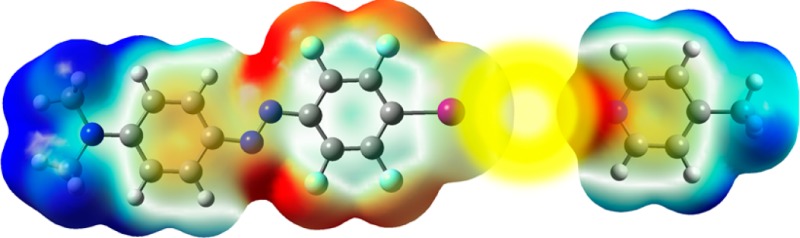
Physiologically, ion transporters carry the role of creating and maintaining intracellular ion concentration gradients. Such gradients act as energy sources to drive metabolic processes and they also control electrical signaling in nerves, muscles, and synapses. The need for synthetic anion transporters lies on one hand in genetic disease treatment, and on the other hand in niche technological applications such as sensing and separation processes.38 Anion transport can be based on various dynamic noncovalent interactions such as hydrogen bonding, ion pairing, or anion−π interactions, working best with weaker interactions than the strong ones used for efficient anion binding.39 This was nicely brought out in the recent work on calix[4]arene single-atom mutation series (Figure 1), designed to dissect the individual contributions to anion transport by halogen bonds, hydrogen bonds, and anion−π interactions.34 This was the first report suggesting that anion transport can be achieved with the contribution of halogen bonding.
Figure 1.
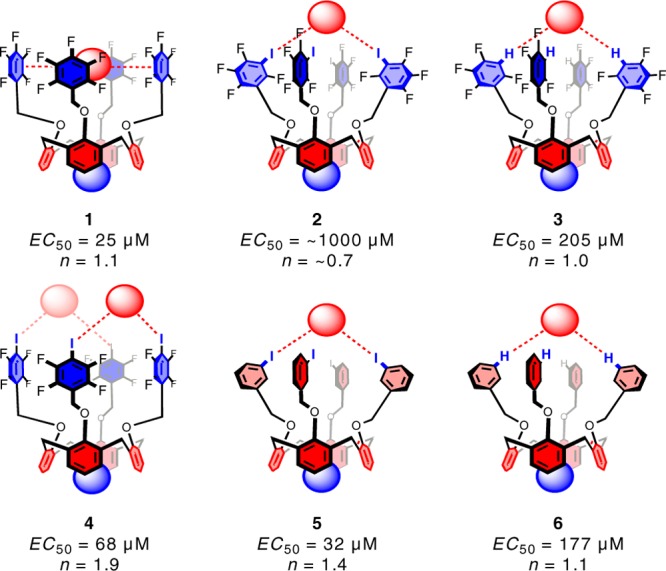
Ditopic ion transport systems made to study anion−π interactions and halogen bonding at work, with effective concentrations (EC50) and Hill coefficients (n) to summarize the transport activities. Red colors indicate electron-rich regions, and blue colors electron-poor regions. Red balls indicate anions, and blue balls tetramethylammonium cations. The dashed red lines illustrate possible anion−π (1), halogen-bonding (2, 4, 5), or hydrogen-bonding (3, 6) interactions between anions and transporters. Reprinted with permission from ref (34).
Within the series 1–6, the nature and strength of anion binding varied systematically without global structural changes. 1, 2, and 3 interact through anion−π, halogen, and hydrogen bonding, respectively. The system 1 was a rather good transporter, whereas 2, based on strong halogen bonding, was practically inactive. By replacing the strong halogen bonds with weaker hydrogen bonds, the activity was partially restored. If the halogen bonding was made weaker (5), the activity was similar to that in 1, highlighting that stronger binders are not the best transporters, and at the same time demonstrating halogen bonding at work in anion transport.
In a follow-up study, the same researchers provided unequivocal proof for halogen-bond-mediated transmembrane anion transport and designed the smallest anion transporters reported.35 A series of fluorinated candidates (Figure 2), selected for their ability to engage in strong, medium, or weak halogen bonds with anions were investigated with respect to their anion-transport activity, in comparison with hexafluorobenzene 19 as a homologous probe for anion−π interactions, pentafluorophenol 20 as a probe for hydrogen bonding, and diiodopyridinium 23 as a probe for symport.
Figure 2.
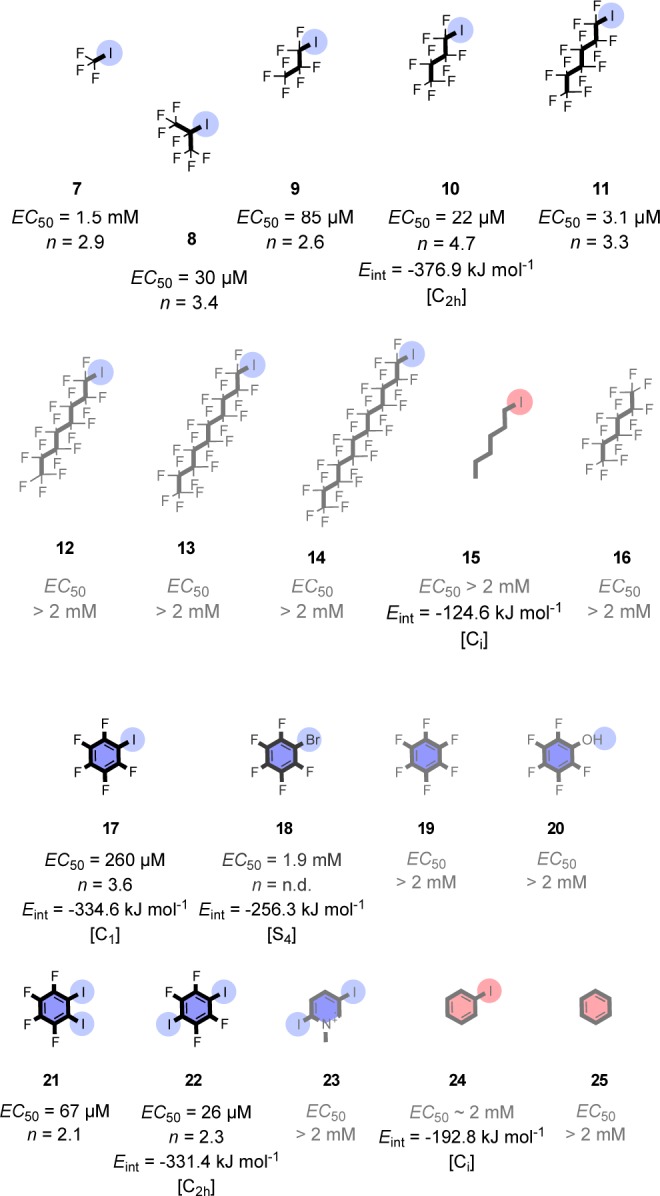
Candidates tested for halogen-bond-based anion transport. Effective concentrations (EC50) and Hill coefficients (n) are given to summarize transport activities in comparison to computed binding energies (Eint) of selected complexes. Reproduced with permission from ref (35).
The highest activity was observed for iodoperfluorohexane 11, while longer or shorter perfluoroalkyl chains reduced the anion transport ability. This is because short perfluoroalkyl chains have lower partition efficiencies in the membrane, while longer chains probably give rise to aggregates that prevent the anions to cross the membrane. Inactivity with defluorinated iodohexane 15 and deiodinated perfluorohexane 16 demonstrated that the activity of iodoperfluorohexane 11 originates from halogen bonding.
Transporters based on aromatic scaffolds showed exactly the same trend: the moderate activity of iodopentafluorobenzene 17 tailed off in perfluorinated bromobenzene 18, defluorinated iodobenzene 24, and benzene 25. The divalent diiodoarenes 21 and 22 were the most active aromatic transporters due to the increased halogen-bond donor moieties. Hexafluorobenzene 19, pentafluorophenol 20, and diiodopyridinium 23, in turn, were inactive, the latter two possibly because of their excessively hydrophilic nature.
The halogen-bond-mediated anion transport is surprisingly clean and efficient, given the small size and poor sophistication of the explored systems. The most interesting aspect arising from these studies is that anion transporters based on halogen bonds can be very small, as a consequence of the intrinsic high hydrophobicity of halogen bond-donors. Even iodotrifluoromethane 7, with one carbon atom only and a boiling point of −22 °C, can transport anions across lipid bilayer membranes without any membrane destruction or leakage. This is the smallest organic ion transporter ever reported.
Conversely, small hydrogen bond-donor transporters, such as short chain aliphatic alcohols, cannot be used for this purpose because of their hydrophilicity. On the other hand, long-chain alcohols disrupt lipid membranes and cannot be used either. For these reasons, typically, hydrogen bond donor moieties are supported on bulky molecular platforms in order to decrease water solubility of the transporters.
Phosphorescent Cocrystals Activated through Halogen Bonding
Organic phosphorescent materials bear immense potential for applications in, for example, organic optoelectronics, solid-state lighting, and sensing/imaging.40−42 The materials exhibiting high-quantum-efficiency phosphorescence are largely based on organometallics;43 purely organic, metal-free materials with efficient phosphorescence emission are rare.44,45 This matter was recently addressed through an ingenious materials design,46 demonstrating that organic crystals showing halogen bonds may open a pathway toward bright all-organic phosphors emitting at wavelengths ranging from blue to green.
The material is based on the aromatic aldehyde compound shown in Figure 3, used as a triplet-generating chromophore. In solution, the compound exhibits weak fluorescence and the triplet emission is inactive. In crystal form, the situation changes dramatically, and the compound exhibits bright green phosphorescence at around 500 nm with emission quantum yield of 2.9% (Figure 4). The enhanced phosphorescence emission in the crystalline state is explained as follows.
Figure 3.
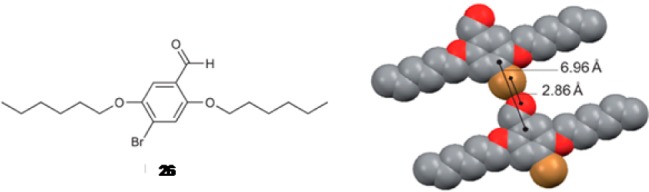
Left: Chemical structure of the compound 26. Right: Schematic depiction of the crystal packing of 26, highlighting the carbonyl oxygen–bromine distance defining the halogen bond. It is believed that this contact is responsible for the phosphorescence observed from crystals of 26. The van der Waals radii of Br and O are 1.85 and 1.52 Å, respectively. Reproduced with permission from ref (46).
Figure 4.
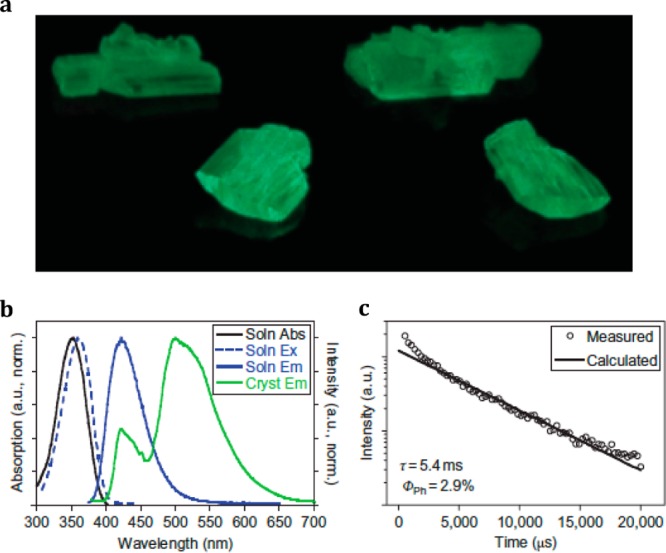
(a) Photograph of crystals of 26 under 365 nm irradiation. (b) Normalized UV–vis absorption (black solid line) and emission (blue solid line) of 26 in chloroform. Green solid line: photoluminescence emission (weak fluorescence and strong phosphorescence) of crystals of 26. (c) Time-resolved measurement of emission at 530 nm from 26 crystals. Phosphorescence lifetime and quantum yield are given in the inset. Reproduced with permission from ref (46).
Single-crystal X-ray diffraction studies revealed that C=O···Br halogen bonding drives the self-assembly of the compound in the solid state (Figure 3, right), which leads to a combination of two effects that promote triplet emission. Most importantly, the close proximity of the bromine atom to the carbonyl oxygen promotes singlet-to-triplet conversion through so-called heavy atom effect.47 Furthermore, the short C=O···Br bond is suggested to prevent nonradiative decay paths, hence further increasing the phosphorescence quantum yield. By diluting the chromophore in a nonemissive but structurally similar host compound to prevent self-quenching, emission quantum yield as high as 55% was reported.46 When bromine was substituted with iodine, no phosphorescence was detected, which was attributed to inferior size matching between the iodine atom and the aldehyde group. Later on, phosphorescent cocrystals based on C–I···π halogen bonding between 1,4-diiodotetrafluorobenzene and various conjugated π-electron donor systems have been constructed.48−50
The above examples comprise the first studies on halogen-bond-based crystal engineering for the design of light-emitting materials, in which the halogen atoms play a dual role as (i) electron acceptors and (ii) heavy-atom perturbers to enhance the triplet emission. Size matters: it has been reported using halogenated monohydroxyl corroles that the intersystem crossing rate undergoes a 60-fold increase passing from fluorine to iodine.17
Apart from providing new design principles for all-organic phosphors, these effects render halogenated dye molecules of potential interest as photosensitizers for photodynamic therapy.51 The utility of halogen atoms in directing the self-assembly of functional dye molecules is naturally by no means limited to promoting triplet emission; halogen-bond-based stilbene-containing cocrystal design allows for tuning fluorescence in the solid state,52 and supramolecular stacking of fluorescent squaraine dyes, driven by dispersive halogen–arene interactions, was reported last year.53
Halogen-Bond-Based Photoresponsive Materials
The term “photoresponsive” is rather ambiguous, as the interaction between light and matter can take various forms. More specifically, herein we refer to azobenzene-containing materials and the photoinduced effects that are triggered by photoisomerization of azobenzene.54−56 As we illustrate through two examples, halogen bonding, due to its high directionality and tunable interaction strength, may play an important role as a design tool for constructing photoresponsive supramolecular assemblies, bearing potential for advancing fundamental understanding on the photoinduced motions in azobenzene-containing polymers,57 as well as for designing low-molecular-weight materials with enhanced response to optical fields.58
One of the most striking examples of isomerization-triggered motions in azobenzene-containing materials (oftentimes polymers) is the light-induced surface patterning.54,59 In short, when a thin film of such material is irradiated with, for example, a sinusoidal interference pattern with spatially varying polarization/intensity profile, the sample surface deforms and forms a replica of the incident interference pattern. The resulting surface-relief patterns form through light-initiated mass migration of the azobenzene-containing material over distances of up to several micrometers. To give a few examples, such patterns are of interest in nanopatterning and optical-field nanoimaging.60−63 However, several aspects of the light-induced surface patterning phenomenon remain poorly understood.
Halogen bonding (or halogenated chromophores) provides us with unique tools to study the effect of strength and type of the noncovalent bond between the azobenzene units and the host polymer matrix on the light-induced surface patterning process. This has been demonstrated by complexing the azobenzenes 27a–c shown in Figure 5a with poly(4-vinylpyridine), which acted as a bond-accepting polymer host matrix for all the three molecules. What is unique about this series is that it allows controlling the halogen bond interaction strength (27a vs 27b; 5.1 and 3.5 kcal mol–1, respectively, based on DFT calculations57) as well as the nature of the interaction (27b vs 27c; halogen bonding vs hydrogen bonding, both of similar strength) without significantly altering the electronic properties (absorption spectrum, dipole moment) of the dyes, which are mainly determined by the electron-withdrawing tetrafluorobenzene ring. Hence with this series we aimed at decoupling the role of chromophore–polymer interaction from other effects that potentially influence the surface patterning. Such task can only be accomplished using halogen bonding.
Figure 5.
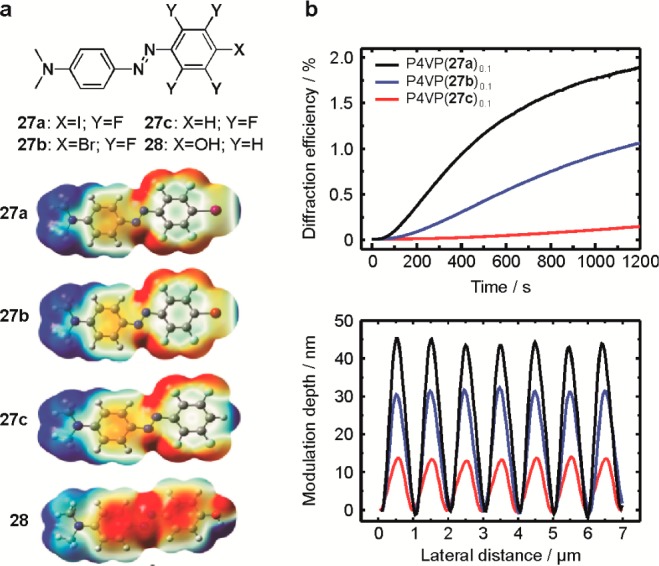
(a) Chemical structures (top) and computed plots of electrostatic potential surfaces (bottom) of the azobenzene derivatives 27a–c and 28. Electrostatic potentials range from −0.03 au (red) to 0.03 au (blue). Computational level: PBE0/6-311++G**; see ref (57) for further details. (b) Comparison between the first-order diffraction efficiency evolution upon inscription of the surface-relief grating (top), and the surface profiles measured with atomic orce microscope after the grating inscription, for thin films of P4VP(27a–c)0.1. The azobenzene concentration x = 0.1 depicts that, on average, every 10th polymer repeat unit is occupied by a chromophore. Reproduced with permission from ref (57).
As followed by in situ light-diffraction measurements and ex-situ atomic-force microscope observation (Figure 5b), the grating formation developed in the order 27c < 27b < 27a. This fact allowed us to conclude that (i) in the halogen-bonded complexes (cf. 27a and 27b) the surface patterning efficiency increases with the polymer–dye interaction strength, and (ii) the directionality of the noncovalent interaction (which we associate with the rigidity of the dye–polymer junction) promotes efficient light-induced surface patterning (cf. 27b and 27c; this statement was further supported by comparison between 27a and 28). So, halogen bonding seems to enhance the light-induced surface patterning in azo-containing polymers, and equally important, provides us with fundamental tools that are unforeseen in more conventional polymer systems.57
Halogen bonding also offers interesting prospects in designing photofunctional liquid crystals (LCs) from nonliquid-crystalline building blocks.58 Upon complexing 27a with a nonmesogenic stilbazole module (Figure 6), the resulting supramolecule 29 exhibited a monotropic nematic LC phase upon cooling from the isotropic phase, with an isotropic-to-nematic phase-transition temperature of 404.8 K and a mesophase temperature range of ca. 18 K. Neither of the constituents contains any flexible chains that are conventionally used in designing small-molecule liquid-crystalline complexes,64,65 which has important consequences for the optical response of the complex. Due to the high-temperature LC phase,66 initially isotropic spin-coated thin films of the azobenzene–stilbazole complex can be uniaxially aligned by irradiation with polarized light with high order parameter of molecular alignment (0.55).58 Furthermore, the lack of flexible spacers together with the presence of the dimethylamino group in the azobenzene module67,68 enhance the surface patterning efficiency of this complex. This is illustrated in Figure 6, which shows a grating with modulation depth of 600 nm (Figure 6b), inscribed on a thin film with starting thickness of 250 nm (Figure 6a). The fact that the modulation depth is more than twice the initial film thickness indicates that all the material is removed from the troughs of the grating, which in turn renders this material potentially interesting for nanostructuring applications.
Figure 6.
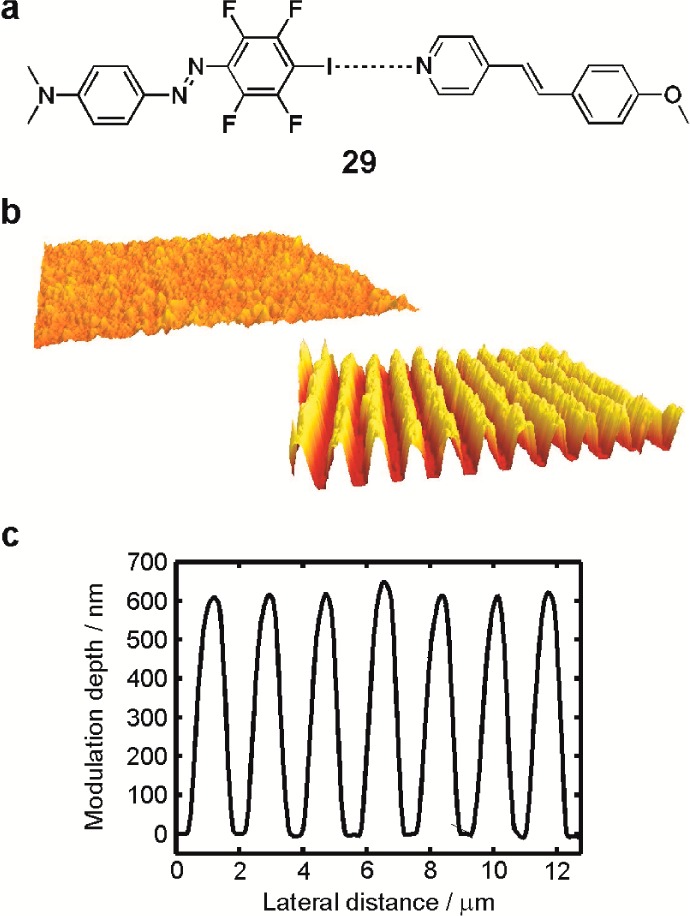
(a) Chemical structure of the liquid-crystalline azobenzene–stilbazole complex 29. (b) Upon irradiating with an interference pattern of 488 nm laser beams, a spin-coated film of 29 underwent pronounced light-induced mass transport, yielding a high-modulation-depth surface-relief grating. (c) Surface profile of the grating shown in (b). Reproduced with permission from ref (58).
The combination of efficient photoalignment and surface patterning is rather unique among the photoresponsive small-molecule complexes,65,66,69,70 and is made possible by the halogen-bond-based materials design: when 27a was replaced with 28, which forms a stronger but less directional interaction with the stilbazole-based bond acceptor, the complex exhibited no LC phase and was not photoresponsive at all.
Halogen-Bond-Triggered Supramolecular Gelation
Low-molecular-weight supramolecular gel-phase materials, in which the gelation is driven by self-assembly of small-molecule building blocks, are attracting increasing interest for applications as diverse as regenerative medicine, drug delivery, controlled crystal growth, and as responsive optical/electronic materials.71,72 The fibrous networks that provide gels with their solidlike rheological properties arise from complex hierarchical self-assembly, often initiated by hydrogen-bond-directed growth of one-dimensional fibrils. The dynamic self-association that nucleates the gel formation is very sensitive to competing noncovalent interactions, for example, anion binding or metal coordination.73,74 Hence, such interactions can be used to control the gel strength or, in extreme cases to inhibit (or alternatively to “turn on”), the gelation process altogether.75 If the interplay between the different intermolecular interactions involved is properly understood, they can, in principle, be harnessed to produce “smart” gel-phase materials with controllable properties. Recently, halogen bonding has been added to the family of supramolecular interactions for controlling gelation.76
The bis(urea) compounds 30, 31, and 33, shown in Figure 7, are nongelators as free ligands in polar organic solvents or organic solvent–water mixtures. This is because the formation of the urea α-tape motif responsible for gelation is inhibited by competitive pyridyl–urea hydrogen bonding (30), steric constraints (31), or, likely, iodo–carbonyl halogen bonding (33). Conversely, an equimolar solution of 30 and 32 (as well as 31 and 32) was found to gelate polar organic solvent–water mixtures. As shown by the single-crystal structure of the 1:1 complex between 30 and 32 (Figure 7, bottom), such halogen bond-induced turn-on of supramolecular gelation can be attributed to the halogen bond sequestration of the pyridyl groups by the diiodotetrafluorobenzene molecule, which prevents the gel-inhibiting pyridyl–urea interactions.
Figure 7.
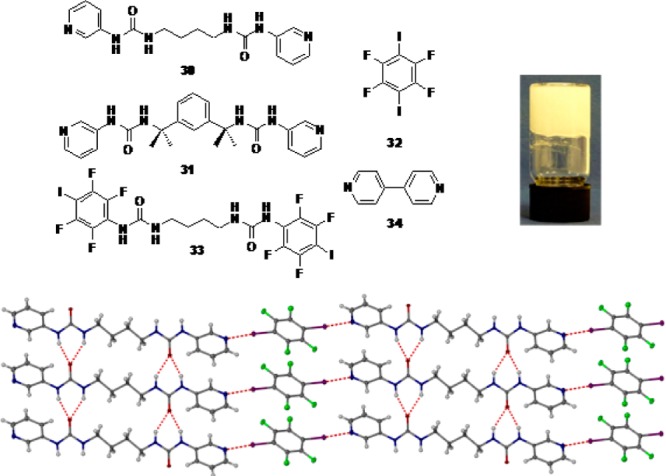
Top: Chemical formulas of the compounds used to obtain supramolecular low-molecular-weight gels (left). An example gel is shown on top right. Bottom: X-ray crystal structure of 30–32 cogel, showing the anticipated gel-forming urea-tape interaction and the halogen-bond cross-links involving the pyridyl groups. Reproduced with permission from ref (76).
The generality of the role of halogen bonding in turning on supramolecular gelation was confirmed by the reversed system involving 33 and 34, in which the bis(urea) and perfluoroaryliodide moieties were within the same compound (33) and the supramolecular cross-linking was done, instead, with 4,4′-bipyridine. Such complex resulted in a network of fibres gelling DMSO/water solutions (Figure 8, left). Finally, even cocrystallization of 30 and 33 in 1:1 ratio provided a supramolecular gel consisting of an intricate network of thin, homogeneous fibres showing some coalignment on the tens-of-micrometers scale, linked by a more disordered secondary mesh of material. These very thin fibres are consistent with the optical transparency of the gel (Figure 8, right).
Figure 8.

Scanning electron micrographs of dried cogels of 33–34 (on the left) and 33–30. Reproduced with permission from ref (76).
The results described above were the first demonstration that halogen bonding can play a role in driving the self-assembly of gel-phase materials. In fact, halogen bonding is sufficiently strong to induce gelation even in polar media, interfering with competing gel-inhibitory hydrogen-bond interactions in a manner similar to metal coordination by bis(pyridyl urea) gelators.77 Thus, halogen bond donors may substitute for metal ions when pyridyl-urea-based gelators are used, freeing the urea groups to form fibrils, and hence gels. The addition of halogen bonding into the toolbox of noncovalent interactions for controlling supramolecular gelation provides new opportunities in the development of this class of sophisticated functional materials, offering the possibility of manipulating the properties of supramolecular gels and the functional devices made thereof.
Conclusions and Perspectives
In this Account, each of the unique characteristics inherent to halogen bonding, directionality, tunable interaction strength, hydrophobicity, and large donor atom size, is shown to bring about or enhance functionalities that are absent without these characteristic features. Due to its hydrophobic nature, small-sized ion-transport systems can be designed based on halogen bonding.35 The large size of halogen atoms allows designing all-organic light-emitting crystals with efficient triplet generation and high phosphorescence quantum yield.46 The tunable interaction strength provides tools for understanding light-induced motions in halogen-bond-based azobenzene-containing photoresponsive polymers,57 and the directionality renders halogen bonding useful in the design of functional supramolecular liquid crystals58 as well as supramolecular gels.76
All of the above-described examples have been published in the course of the last 2 years, demonstrating that using halogen bonding in functional materials design is a vibrant new field of research. They serve to demonstrate the diversity and the potential that halogen bonding has to offer in this field, which may impact various other disciplines in the context of, for example, biomimetics, optics/photonics, functional surfaces, and photoswitchable supramolecules.
Hopefully, the highlighted examples will trigger a significant amount of further research. There is plenty of room for halogen bonding in functional materials design, and the future is wide open.
Acknowledgments
P.M. gratefully acknowledges the financial contribution from the European Research Council under the project FOLDHALO, Starting Grant ERC-2012-StG_20111012 (Grant Agreement Number 307108).
Biographies
Arri Priimagi earned his Ph.D. Degree in 2009 from the Department of Applied Physics, Helsinki University of Technology (nowadays know as Aalto University), Finland. After a two-year postdoctoral fellowship with Prof. Atsushi Shishido at Tokyo Institute of Technology, he returned to Aalto University where he presently works as a postdoctoral fellow. His main research interests involve the development of polymeric and supramolecular materials for optics and photonics and the design of light-responsive self-assemblies.
Gabriella Cavallo obtained her Ph.D. in Chemistry in 2006 at the University of Salerno. After two years at a chemical company, she joined the NFMLab at the Politecnico di Milano in 2008 as a postdoctoral researcher, where she became assistant professor in 2012. Her research interest is the synthesis of fluorinated compounds for recognition processes and molecular materials.
Pierangelo Metrangolo became full professor at the Politecnico di Milano in 2011. He is also visiting professor at the VTT – Technical Research Centre of Finland. He has also been EU Fellow at the University of Toulouse (France, 2001), visiting Professor at the University of York (UK, 2005) and at the University of Jyväskylä (Finland, 2006). He is on the Editorial Board of CrystEngComm (RSC) and Topic Editor of Crystal Growth & Design (ACS). His awards include the 2005 “G. Ciamician” medal of the Division of Organic Chemistry of the Italian Chemical Society. From 2013, he is European Research Council grantee (StG). His research interests include crystal engineering, supramolecular and biomimetic materials.
Giuseppe Resnati became full professor at the Politecnico di Milano in 2001. He was NATO fellow at the University of Clemson (Clemson, SC, 1990), and visiting scientist at the Université Paris XI (France, 1993) and Nagoya University (Japan, 2001). He is in the Editorial Board of the Journal of Fluorine Chemistry and member of the Academy of Europe. His awards include the 2008 Research Prize of the Division of Organic Chemistry of the Italian Chemical Society and the 2010 European Lecturship in Chemical Sciences Award of the RSC. His research interests are in fluorine chemistry and self-assembly processes.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Metrangolo P.; Resnati G.. Halogen Bonding: Fundamentals and Applications (Stucture and Bonding); Springer: Heidelberg, 2010. [Google Scholar]
- Desiraju G. R.; Ho P. S.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P. A.; Resnati G.; Rissanen K.. Definition of the halogen bond. Pure Appl. Chem. 2013, 85, doi: 10.1351/PAC-REC-12-05-10. [Google Scholar]
- Arunan E.; Desiraju G. R.; Klein R. A.; Sadlej J.; Scheiner S.; Alkorta I.; Clary D. C.; Crabtree R. H.; Dannenberg J. J.; Hobza P.; Kjaergaard H. G.; Legon A. C.; Mennucci B.; Nesbitt D. J. Definition of the hydrogen bond. Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar]
- Metrangolo P.; Neukirch H.; Pilati T.; Resnati G. Halogen bonding based recognition processes: a world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [DOI] [PubMed] [Google Scholar]
- Corradi E.; Meille S. V.; Messina M. T.; Metrangolo P.; Resnati G. Halogen bonding versus hydrogen bonding in driving self-assembly processes. Angew. Chem., Int. Ed. 2000, 39, 1782–1786. [DOI] [PubMed] [Google Scholar]
- Aakeröy C. B.; Fasulo M.; Schultheiss N.; Desper J.; Moore C. Structural competition between hydrogen bonds and halogen bonds. J. Am. Chem. Soc. 2007, 129, 13772–13773. [DOI] [PubMed] [Google Scholar]
- Voth A. R.; Hays F. A.; Ho P. S. Directing macromolecular conformation through halogen bonds. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6188–6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T.; Hennemann M.; Murray J. S.; Politzer P. Halogen bonding: the sigma hole. J. Mol. Model. 2007, 13, 291–296. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T. Halogen bonding: an electrostatically driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [DOI] [PubMed] [Google Scholar]
- Riley K. E.; Murray J. S.; Fanfrlík J.; Řezáč J.; Solá R. J.; Concha M. C.; Ramos F. M.; Politzer P. Halogen bond tunability I: the effects of aromatic fluorine substitution on the strengths of halogen-bonding interactions involving chlorine, bromine, and iodine. J. Mol. Model. 2011, 17, 3309–3318. [DOI] [PubMed] [Google Scholar]
- Metrangolo P.; Murray J. S.; Pilati T.; Politzer P.; Resnati G.; Terraneo G. The fluorine atom as a halogen bond donor, viz. a positive site. Cryst. Eng. Comm. 2011, 13, 6593–6596. [Google Scholar]
- Lu Y.; Li H.; Zhu X.; Zhu W.; Liu H. How does halogen bonding behave in solution? a theoretical study using implicit solvation model. J. Phys. Chem. A 2011, 115, 4467–4475. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Wang Y.; Zhu W. Nonbonding interactions of organic halogens in biological systems: inplications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. [DOI] [PubMed] [Google Scholar]
- Auffinger P.; Hays F. A.; Westhof E.; Ho P. S. Halogen bonds in biological molecules. Proc. Nat. Acad. Sci. U.S.A. 2004, 101, 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar]
- Parker A. J.; Stewart J.; Donald K. J.; Parish C. A. Halogen bonding in DNA base pairs. J. Am. Chem. Soc. 2012, 134, 5165–5172. [DOI] [PubMed] [Google Scholar]
- Shi L.; Liu H. Y.; Shen H.; Hu J.; Zhang G. L.; Wang H.; Ji L. N.; Chang C. K.; Jiang H. F. Fluorescence properties of halogenated mono-hydroxyl corroles: the heavy-atom effects. J. Porphyrins Phthalocyanines 2009, 13, 1221–1226. [Google Scholar]
- Meyer F.; Dubois P. Halogen bonding at work: recent applications in synthetic chemistry and materials science. CrystEngComm 2013, 15, 3058–3071. [Google Scholar]
- Rissanen K. Halogen bonded supramolecular complexes and networks. CrystEngComm. 2008, 10, 1107–1113. [Google Scholar]
- Fourmigué M. Halogen bonding: recent advances. Curr. Opin. Solid State Mater. Sci. 2009, 13, 36–45. [Google Scholar]
- Politzer P.; Murray J. S. Halogen bonding: an interim discussion. ChemPhysChem 2013, 14, 278–294. [DOI] [PubMed] [Google Scholar]
- Beale T. M.; Chudzinski M. G.; Sarwar M. G.; Taylor M. S. Halogen bonding in solution: thermodynamics and applications. Chem. Soc. Rev. 2013, 42, 1667–1680. [DOI] [PubMed] [Google Scholar]
- Erdélyi M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [DOI] [PubMed] [Google Scholar]
- Bruckmann A.; Pena M. A.; Bolm C. Organocatalysis through halogen-bond activation. Synlett 2008, 6, 900–902. [Google Scholar]
- Coulembier O.; Meyer F.; Dubois P. Controlled room temperature ROP of L-lactide by ICl3: a simple halogen-bonding catalyst. Polym. Chem. 2010, 1, 434–437. [Google Scholar]
- Walter S. M.; Kniep F.; Herdtweck E.; Huber S. M. Halogen-bond-induced activation of a carbon-heteroatom bond. Angew. Chem., Int. Ed. 2011, 50, 7187–7191. [DOI] [PubMed] [Google Scholar]
- Spence G. T.; Beer P. D. Expanding the scope of the anion templated synthesis of interlocked structures. Acc. Chem. Res. 2013, 46, 571. [DOI] [PubMed] [Google Scholar]
- Kilah N. L.; Wise M. D.; Beer P. D. Crystallographic implications for the design of halogen bonding anion receptors. Cryst. Growth Des. 2011, 11, 4565–4571. [Google Scholar]
- Sarwar M. G.; Dragisić B.; Dimitrijević E.; Taylor M. S. Halogen bonding between anions and iodoperfluoroorganics: solution-phase thermodynamics and multidentate-receptor design. Chem.—Eur. J. 2013, 19, 2050–2058. [DOI] [PubMed] [Google Scholar]
- Cavallo G.; Biella S.; Lü J.; Metrangolo P.; Pilati T.; Resnati G.; Terraneo G. Halide anion-templated assembly of di- and triiodoperfluorobenzenes into 2D and 3D supramolecular networks. J. Fluorine Chem. 2010, 131, 1165–1172. [Google Scholar]
- Raatikainen K.; Cavallo G.; Metrangolo P.; Resnati G.; Rissanen K.; Terraneo G. In the pursuit of efficient anion-binding organic ligands based on halogen bonding. Cryst. Growth Des. 2013, 13, 871–877. [Google Scholar]
- Cavallo G.; Metrangolo P.; Pilati T.; Resnati G.; Sansotera M.; Terraneo G. Halogen bonding: a general route in anion recognition and coordination. Chem. Soc. Rev. 2010, 39, 3772–3783. [DOI] [PubMed] [Google Scholar]
- Metrangolo P.; Pilati T.; Terraneo G.; Biella S.; Resnati G. Anion coordination and anion-templated assembly under halogen bonding control. CrystEngComm 2009, 11, 1187–1196. [Google Scholar]
- Vargas Jentzsch A.; Emery D.; Mareda J.; Metrangolo P.; Resnati G.; Matile S. Ditopic ion transport systems: anion- π interactions and halogen bonds at work. Angew. Chem., Int. Ed. 2011, 50, 11675–11678. [DOI] [PubMed] [Google Scholar]
- Vargas Jentzsch A.; Emery D.; Mareda J.; Nayak S. K.; Metrangolo P.; Resnati G.; Sakai N.; Matile S. Transmembrane anion transport mediated by halogen-bond donors. Nat. Commun. 2012, 3, 905. [DOI] [PubMed] [Google Scholar]
- Vargas Jentzsch A.; Matile S. Transmembrane halogen-bonding cascades. J. Am. Chem. Soc. 2013, 135, 5302–5303. [DOI] [PubMed] [Google Scholar]
- Vargas Jentzsch A.; Hennig A.; Mareda J.; Matile S.. Synthetic ion transporters that work with anion-π interactions, halogen bonds, and anionmacrodipole interactions. Acc. Chem. Res. 2013, DOI: 10.1021/ar400014r. [DOI] [PubMed] [Google Scholar]
- Gale P. A. From anion receptors to transporters. Acc. Chem. Res. 2011, 44, 216–226. [DOI] [PubMed] [Google Scholar]
- Behr J.-P.; Kirch M.; Lehn J.-M. Carrier-mediated transport through bulk liquid membranes: dependence of transport rates and selectivity on carrier properties in a diffusion-limited process. J. Am. Chem. Soc. 1985, 107, 241–246. [Google Scholar]
- Baldo M. A.; Lamansky S.; Burrows P. E.; Thompson M. E.; Forrest S. R. Very high-efficiency green organic light-emitting devices based on electrophosphorescence. Appl. Phys. Lett. 1999, 75, 4–6. [Google Scholar]
- D’Andrade B. W.; Forrest S. R. White organic light-emitting devices for solid-state lighting. Adv. Mater. 2004, 16, 1585–1595. [Google Scholar]
- Zhang G.; Palmer G. M.; Dewhirst M. W.; Fraser C. L. A dual-emissive-materials design concept enables tumour hypoxia imaging. Nat. Mater. 2009, 8, 747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y.; Park S. Y. Phosphorescent iridium(III) complexes: toward high phosphorescence quantum efficiency through ligand control. Dalton Trans. 2009, 8, 1267–1282. [DOI] [PubMed] [Google Scholar]
- Yuan W. Z.; Shen X. Y.; Zhao H.; Lam J. W. Y.; Tang L.; Lu P.; Wang C.; Liu Y.; Wang Z.; Zheng Q.; Sun J. Z.; Ma Y.; Tang B. Z. Crystallization-induced phosphorescence of pure organic luminogens at room temperature. J. Phys. Chem. C 2010, 114, 6090–6099. [Google Scholar]
- Hirata S.; Totani K.; Zhang J.; Yamashita T.; Kaji H.; Marder S. R.; Watanabe T.; Adachi C.. Efficient persistent room temperature phosphorescence in organic amorphous materials under ambient conditions. Adv. Funct. Mater. 2013, DOI: 10.1002/adfm.201203706. [DOI] [Google Scholar]
- Bolton O.; Lee K.; Kim H.-J.; Lin K. Y.; Kim J. Activating efficient phosphorescence from purely organic materials by crystal design. Nat. Chem. 2011, 3, 205–210. [DOI] [PubMed] [Google Scholar]
- Turro N. J.Modern Molecular Photochemistry; University Science Books: Sausalito, CA, 1991. [Google Scholar]
- Gao H. Y.; Zhao X. R.; Wang H.; Pang X.; Jin W. J. Phosphorescent cocrystals assembled by 1,4-diiodotetrafluorobenzene and fluorene and its heterocyclic analogues based on C-I−π halogen bonding. Cryst. Growth Des. 2012, 12, 4377–4387. [Google Scholar]
- Shen Q. J.; Pang X.; Zhao X. R.; Gao H. Y.; Sun H.-L.; Jin W. J. Phosphorescent cocrystals constructed by 1,4-diiodotetrafluorobenzene and polyaromatic hydrocarbons based on C-I−π halogen bonding and other assisting weak interactions. CrystEngComm 2012, 14, 5027–5034. [Google Scholar]
- Gao H. Y.; Shen Q. J.; Zhao X. R.; Yan X. Q.; Pang X.; Jin W. J. Phosphorescent co-crystal assembled by 1,4-diiodotetrafluorobenzene with carbazole based on C-I−π halogen bonding. J. Mater. Chem. 2012, 22, 5336–5343. [Google Scholar]
- Gorman A.; Killoran J.; O’Shea C.; Kenna T.; Gallagher W. M.; O’Shea D. F. In vitro demonstration of the heavy-atom effect for photodynamic therapy. J. Am. Chem. Soc. 2004, 126, 10619–10631. [DOI] [PubMed] [Google Scholar]
- Yan D.; Delori A.; Lloyd G. A.; Friscic T.; Day G. M.; Jones W.; Lu J.; Wei M.; Evans D. G.; Duan X. A cocrystal strategy to tune the luminescent properties of stilbene-type organic solid-state materials. Angew. Chem., Int. Ed. 2011, 50, 12483–12486. [DOI] [PubMed] [Google Scholar]
- Mayerhöffer U.; Würthner F. Halogen-arene interactions assist in self-assembly of dyes. Angew. Chem., Int. Ed. 2012, 51, 5615–5619. [DOI] [PubMed] [Google Scholar]
- Natansohn A.; Rochon P. Photoinduced motions in azo-containing polymers. Chem. Rev. 2002, 102, 4139–4175. [DOI] [PubMed] [Google Scholar]
- Ikeda T. Photomodulation of liquid crystal orientations for photonic applications. J. Mater. Chem. 2003, 13, 2037–2057. [Google Scholar]
- Mahimwalla Z.; Yager K. G.; Mamiya J.; Shishido A.; Priimagi A.; Barrett C. J. Azobenzene photomechanics: prospects and potential applications. Polym. Bull. 2012, 69, 967–1006. [Google Scholar]
- Priimagi A.; Cavallo G.; Forni A.; Gorynsztejn-Leben M.; Kaivola M.; Metrangolo P.; Milani R.; Shishido A.; Pilati T.; Resanti G.; Terraneo G. Halogen bonding versus hydrogen bonding in driving self-assembly and performance of light-responsive supramolecular polymers. Adv. Funct. Mater. 2012, 22, 2572–2579. [Google Scholar]
- Priimagi A.; Saccone M.; Cavallo G.; Shishido A.; Pilati T.; Metrangolo P.; Resnati G. Photoalignment and surface-relief grating formation are efficiently combined in low-molecular-weight halogen-bonded complexes. Adv. Mater. 2012, 24, OP345–OP352. [DOI] [PubMed] [Google Scholar]
- Viswanathan N. K.; Kim D. Y.; Bian S.; Williams J.; Liu W.; Li L.; Samuelson L.; Kumar J.; Tripathy S. K. Surface relief structures on azo polymer films. J. Mater. Chem. 1999, 9, 1941–1955. [Google Scholar]
- Hubert C.; Ryumantseva A.; Lerondel G.; Grand J.; Kostcheev S.; Billot L.; Vial A.; Bachelot R.; Royer P. Near-field photochemical imaging of noble metal nanostructures. Nano Lett. 2005, 5, 615–619. [DOI] [PubMed] [Google Scholar]
- Lee S.; Kang H. S.; Park J.-K. Directional photofluidization lithography: micro/nanostructural evolution by photofluidic motions of azobenzene materials. Adv. Mater. 2012, 24, 2069–2103. [DOI] [PubMed] [Google Scholar]
- Kravchenko A.; Shevchenko A.; Ovchinnikov V.; Priimagi A.; Kaivola M. Optical interference lithography using azobenzene-functionalized polymers for micro- and nanopatterning of silicon. Adv. Mater. 2011, 23, 4174–4177. [DOI] [PubMed] [Google Scholar]
- Ambrosio A.; Marrucci L.; Barbone F.; Roviello A.; Maddalena P. Light-induced spiral mass transport in azo-polymer films under vortex-beam illumination. Nat. Commun. 2012, 3, 989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loc Nguyen H.; Horton P. N.; Hursthouse M. B.; Legon A. C.; Bruce D. W. Halogen bonding: a new interaction for liquid crystal formation. J. Am. Chem. Soc. 2004, 126, 16–17. [DOI] [PubMed] [Google Scholar]
- Zakrevskyy Y.; Stumpe J.; Faul C. F. J. A supramolecular approach to optically anisotropic materials: photosensitive ionic self-assembly complexes. Adv. Mater. 2006, 18, 2133–2136. [Google Scholar]
- Kreger K.; Wolfer P.; Audorff H.; Kador L.; Stingelin-Stutzmann N.; Smith P.; Schmidt H. W. Stable holographic gratings with small-molecular trisazobenzene derivatives. J. Am. Chem. Soc. 2010, 132, 509–516. [DOI] [PubMed] [Google Scholar]
- You F.; Paik M. Y.; Häckel M.; Kador L.; Kropp D.; Schmidt H. W.; Ober C. K. Control and suppression of surface relief gratings in liquid-crystalline perfluoroalkyl-azobenzene polymers. Adv. Funct. Mater. 2006, 16, 1577–1581. [Google Scholar]
- Vapaavuori J.; Valtavirta V.; Alasaarela T.; Mamiya J.; Priimagi A.; Shishido A.; Kaivola M. Efficient surface structuring and photoalignment of supramolecular polymer-azobenzene complexes through rational chromophore design. J. Mater. Chem. 2011, 21, 15437–15441. [Google Scholar]
- Nakano H.; Takahashi T.; Kadota T.; Shirota Y. Formation of a surface relief grating using a novel azobenzene-based photochromic amorphous molecular material. Adv. Mater. 2002, 14, 1157–1160. [Google Scholar]
- Ishow E.; Lebon B.; He Y.; Wang X.; Bouteiller L.; Galmiche L.; Nakatani K. Structural and photoisomerization cross studies of polar photochromic monomeric glasses forming surface relief gratings. Chem. Mater. 2006, 18, 1261–1267. [Google Scholar]
- Hirst A. R.; Escuder B.; Miravet J. F.; Smith D. K. High-tech applications of self-assembling supramolecular nanostructured gel-phase materials: from regenerative medicine to electronic devices. Angew. Chem., Int. Ed. 2008, 47, 8002–8018. [DOI] [PubMed] [Google Scholar]
- Foster J. A.; Piepenbrock M. O. M.; Lloyd G. O.; Clarke N.; Howard J. A. K.; Steed J. W. Anion-switchable supramolecular gels for controlling pharmaceutical crystal growth. Nat. Chem. 2010, 2, 1037–1043. [DOI] [PubMed] [Google Scholar]
- Lloyd G. O.; Steed J. W. Anion tuning of supramolecular gel properties. Nat. Chem. 2009, 1, 437–442. [DOI] [PubMed] [Google Scholar]
- Piepenbrock M. O. M.; Lloyd G. O.; Clarke N.; Steed J. W. Metal- and anion-binding supramolecular gels. Chem. Rev. 2010, 110, 1960–2004. [DOI] [PubMed] [Google Scholar]
- Steed J. W. Anion-tuned supramolecular gels: a natural evolution from urea supramolecular chemistry. Chem. Soc. Rev. 2010, 39, 3686–3699. [DOI] [PubMed] [Google Scholar]
- Meazza L.; Foster J. A.; Fucke K.; Metrangolo P.; Resnati G.; Steed J. W. Halogen-bonding-triggered supramolecular gel formation. Nat. Chem. 2012, 5, 42–47. [DOI] [PubMed] [Google Scholar]
- Byrne P.; Lloyd G. O.; Applegarth L.; Anderson K. M.; Clarke N.; Steed J. W. Metal-induced gelation in dipyridyl ureas. New J. Chem. 2010, 34, 2261–2274. [Google Scholar]