

Abstract

We report DNA binding studies of the dinuclear ruthenium ligand [{Ru(phen)2}2tpphz]4+ in enantiomerically pure forms. As expected from previous studies of related complexes, both isomers bind with similar affinity to B-DNA and have enhanced luminescence. However, when tested against the G-quadruplex from human telomeres (which we show to form an antiparallel basket structure with a diagonal loop across one end), the ΛΛ isomer binds approximately 40 times more tightly than the ΔΔ, with a stronger luminescence. NMR studies show that the complex binds at both ends of the quadruplex. Modeling studies, based on experimentally derived restraints obtained for the closely related [{Ru(bipy)2}2tpphz]4+, show that the ΛΛ isomer fits neatly under the diagonal loop, whereas the ΔΔ isomer is unable to bind here and binds at the lateral loop end. Molecular dynamics simulations show that the ΔΔ isomer is prevented from binding under the diagonal loop by the rigidity of the loop. We thus present a novel enantioselective binding substrate for antiparallel basket G-quadruplexes, with features that make it a useful tool for quadruplex studies.
Introduction
Although it has been known for five decades that guanine-rich nucleic acids can form four-stranded structures, research into quadruplex DNA has rapidly escalated in recent years. One reason for continued interest is the demonstration that telomeres can and do fold into quadruplex structures in vivo.1−3 Shortening of telomeres on chromosomal replication is considered to be a major cause of senescence, and cancer cells have been shown to generate an immortal phenotype by upregulating telomerase.4,5 The activity of telomerase is inhibited by the presence of G-quadruplexes,6 leading to the possibility of novel anticancer agents that work by binding to and stabilizing such quadruplexes.
A second reason for interest is the observation that quadruplexes are found not only in telomeres but also in other parts of the genome. Typically they are found in upstream promoters7−9 and in some cases have been shown to perform a regulatory function on downstream genes.10−13 Quadruplexes are also formed by RNA and again are likely to have regulatory roles on translation.14 For all these reasons, there is considerable interest in finding small molecules that bind to quadruplexes and stabilize them and that could act as markers for their presence.
Over the past few years, it has become abundantly clear that guanine-rich sequences can fold into quadruplexes in many different ways.15 A given sequence can also fold differently depending on solution conditions, including counterions (potassium or sodium), molecular crowding,16−18 and dehydration.19 A case in point is the human telomere sequence, HTS, d[AG3(TTAG3)3], which has been observed in several conformations.20−27 It appears that such behavior is common.28 This plasticity makes it all the more important to identify small molecules that bind specifically to particular conformations and stabilize them, especially if the function and dysfunction of quadruplexes in normal and abnormal cellular function are to be delineated.29
Despite this importance, there is little detailed crystallographic or NMR data on ligand–HTS quadruplex structures.28,30,31 Of relevance to this work, there is only one report on metal complexes.32 Only four X-ray structures involving an intramolecular quadruplex have been reported, all of which involve the all-parallel conformer with the ligand end-stacking on terminal G-tetrads.33−36 Although, as outlined above, telomere sequences can take up a range of topologies, virtually all the other reported structures also involve ligands bound to all-parallel conformers, comprising tetramolecular or bimolecular quadruplexes.37−41 Indeed, given this paucity of data and the range of potential telomeric conformer targets, it has been suggested that the design of small molecules to stabilize G-quadruplexes should also be directed toward ligands that selectively target antiparallel and hybrid type G-quadruplex folding topologies.27 The structural data obtained for small molecules bound to non all-parallel quadruplex conformers indicate that these telomeric structures could be targeted through specific interactions. For example the crystallographic structures of disubstituted aminoalkylamidoacridine derivatives bound to the dimeric antiparallel G-quadruplex formed from the Oxytricha nova telomere sequence d(G4T4G4) reveal that while these structures display the expected end-stacking interaction, they also feature a second distinctive motif: the acridine moiety “threads” through the T4 diagonal loop.42,43
As part of a program to develop luminescent metal complexes as sequence and structure specific DNA binding substrates,44−46 we have studied the quadruplex binding properties of dinuclear ruthenium(II) complexes containing the tetrapyrido[3,2-a:2′,3′-c:3″,2″-h:2‴,3‴-j]phenazine (tpphz) ligand. Although the central tpphz ligand in such complexes is planar, the octahedral coordination geometry about the ruthenium centers gives rise to a “dumbbell” structure with bulky, and chiral, “stoppers”. These studies have revealed that both [{Ru(bipy)2}2tpphz]4+, 1, and [{Ru(phen)2}2tpphz]4+, 2, (where bipy = 2,2′-bipyridine, phen = 1,10-phenanthroline), Figure 1, bind to quadruplex DNA with high affinities (>107 M–1).47
Figure 1.

(A) Structures of complexes studied. (B) The two possible enantiomers of each of the metal centers in 1 and 2. (C) Schematic of the two diastereomers of 1 and 2 relevant to this study: left ΛΛ; right ΔΔ.
Both these complexes display a “DNA light-switch” effect, being essentially nonemissive in aqueous solution until DNA binding induces a several orders of magnitude increase in their Ru → tpphz 3MLCT-based luminescence. Uniquely, the emission and binding affinities of 1 and 2 are sensitive to DNA structure. While groove binding to all duplexes produces a relatively weak emission at >675 nm, binding to quadruplexes produces more complex emission changes. Intense blue-shifted luminescence (∼630 nm) and high affinity binding is observed only when the complex binds to antiparallel quadruplex structures containing external diagonal loops at least three bases in length. The presence of shorter lateral loops limits binding affinities by several orders of magnitude and results in negligible emission.48 This difference in luminescence output means that, despite the only modest selectivity in binding affinities, quadruplex structures can be detected in the presence of duplex DNA. Indeed, this concept has been illustrated by recently reported cell studies with these complexes.49
Fascinatingly, in cellulo studies reveal that, while 1 is only taken up by fixed cells, 2 is actively transported into live cells.49,50 Confocal microscopy studies confirm that 2 is a selective luminescent stain for heterochromatin. Furthermore, 2 displays distinctive noncolocalized multiple emission peaks, whose wavelengths are consistent with those obtained through in vitro studies, indicating that the complex is an in cellulo probe of DNA structure.49
Until now, these studies have used racemic mixtures of complexes 1 and 2. However, recently the Qu group have shown that the ΛΛ-enantiomer of a nonemissive dinuclear nickel(II) triple helicate complex displays a strong binding preference for specific quadruplex structures over duplex DNA,51,52 while Sugiyama and co-workers have demonstrated that a chiral helicene macrocycle can enantioselectively recognize quadruplex DNA.53 Furthermore, recent spectroscopic,54,55 crystallographic,56−58 and NMR59 studies on mono- and dinuclear RuII(dppz) (dppz = dipyrido[3,2-a:2′,3′-c]phenazine) systems with duplex DNA have also illustrated the importance of chirality in such interactions. In light of these studies, we discuss the DNA binding preferences of enantiopure samples of 2 and also report NMR-based studies designed to delineate the structural details of quadruplex binding by 1 and 2, followed by further rationalization of the results based on molecular dynamics (MD) simulations in water.
Results and Discussion
Spectroscopic Binding Studies
In previous studies, we have found that the luminescent binding response of rac-1 and rac-2 to duplex and quadruplex DNA is effectively identical. In both cases, binding to duplex DNA produces a 60-fold increase in emission, while binding to quadruplex produces a >150 times increase in blue-shifted emission. Furthermore comparisons of Kb values for rac-1 and rac-2 revealed they are almost identical within experimental error.47 In the study reported herein, the interaction of enantiomerically pure complexes ΔΔ-2 and ΛΛ-2 with duplex and quadruplex DNA was investigated through luminescence titrations using calf thymus DNA (CT-DNA) (Supporting Information Figure S01) and the human telomere sequence (HTS) d[AG3(TTAG3)3] (Figure 2).
Figure 2.

Typical data for the luminescence response of ΛΛ-2, ΔΔ-2, and an unresolved diastereomeric mixture of 2 to the progressive addition of the unimolecular HTS quadruplex d[AG3(TTAG3)3]. Conditions: 10 mM KH2PO4/K2HPO4, 1 mM K2EDTA, 200 mM KCl, pH 7.0, 298 K, [complex] = 7 μM. Lines show fitted Kb.
To aid comparisons, data for rac-2 in these conditions are also included. The CD spectrum of the HTS sequence in uncrowded K+ solutions confirmed that it adopts an antiparallel basket conformation (Supporting Information Figure S02), which is consistent with previous observations by Renčiuk et al.26 NMR and CD spectra of HTS in 100 mM NaCl are similar to those reported by Wang and Patel20 and indicate that the structure remains an antiparallel basket.
Characteristics for the interaction of CT-DNA with either enantiomer in aqueous buffer solutions were found to be very similar. Both show a very similar increase in steady-state luminescence, which is identical within experimental error to the changes observed for an unresolved diastereomeric mixture (see Supporting Information). Indeed, fits of the data to standard binding models lead to estimates of binding affinities that are almost identical to those previously reported for the unresolved mixture (Kb = 4.40 × 106 M–1), although it does appear that the binding affinity of ΛΛ-2 is slightly higher than that of ΔΔ-2 (Table 1).
Table 1. Estimated Binding Affinities for Different Diastereomers of 2 with Duplex and Quadruplex DNAa.
| complex | Kb(CT-DNA); M–1 | Kb(HTS); M–1 |
|---|---|---|
| ΛΛ-2 | 6.73 × 106 | 1.16 × 107 |
| ΔΔ-2 | 1.99 × 106 | 2.95 × 105 |
| unresolved 2b | 6.68 × 106 | 1.77 × 107 |
Calculated errors in estimates of Kb ≈ ±20%.
Binding constants for diastereomeric mixture are apparent Kbs.
By contrast, titrations of enantiomerically pure 2 with HTS produced clear differences in the luminescent response of ΛΛ-2 and ΔΔ-2. Addition of HTS to ΛΛ-2 led to increases in emission that were around 20% larger than those observed for an unresolved diastereomeric mixture. More fascinatingly, ΔΔ-2 displayed a much smaller emission response than either ΛΛ-2 or the unresolved solution mixture: at binding saturation, the steady-state emission intensity of ΔΔ-2 is 6-fold less than that of ΛΛ-2 (Figure 2). Because the binding-induced light-switch effect observed for these complexes is due to transfer from a polar bulk aqueous environment into a less polar binding environment, these data indicate that bound ΔΔ-2 is much more solvent accessible than ΛΛ-2, thus implying structural differences in the binding complexes with HTS for the two diastereomers. Fits of the luminescence changes to a simple one set of identical binding sites model offers further evidence to support this hypothesis, as the binding affinity of ΛΛ-2 is around 40 times higher than that of ΔΔ-2 (Table 1). The diastereomeric mixture contains 25% ΛΛ-2, 25% ΔΔ-2, and 50% ΛΔ-2, yet its emission intensity is roughly 85% the intensity of pure ΛΛ-2. This is almost exactly the value expected if ΛΔ-2 has the same emission intensity as ΛΛ-2. The results therefore suggest that ΛΔ-2 binds in the same way as ΛΛ-2, i.e., that for binding to HTS, Δ chirality is possible at one end of the ligand but not both.
As discussed before, addition of 2 (as a racemic mixture or as pure enantiomers) gives rise to hypochromic and bathochromic shifts in UV–vis spectra, indicative of stacking of the aromatic rings against DNA base pairs. A stacking mode of binding is also indicated by the strongly enhanced luminescence and the blue-shift of approximately 30 nm, which we have shown only occurs when the ligand is strongly shielded from solvent.48 Shielding to this extent can only arise when the ligand is covered by quadruplex loops: in other words, it implies stacking over a tetrad plane and shielding by loops, rather than groove binding. Furthermore, the markedly greater increase in luminescence for bound ΛΛ-2 implies significantly better shielding from solvent for this isomer. Further evidence for this hypothesis was obtained by NMR studies
NMR Binding Studies
To provide structural insights into the effects observed in our optical studies, the binding of 1 and 2 to HTS was further investigated using a combination of NMR spectroscopy and simulated annealing coupled with restrained molecular dynamics simulations for structure determination. Assignments of the folded quadruplex before addition of any complex are given in the Supporting Information (Table S1).
On addition of ΔΔ-2 to HTS, severe line broadening of DNA signals was observed by NMR, mainly from the lateral loop end (red dots, bottom end of Figure 3). This implies that the ligand is binding at this end and causing structural perturbations concomitant with binding, which occur on a time scale in the millisecond range. Unfortunately, this is a common observation in studies of DNA/ligand interactions and it makes structure determination of the complexes difficult by severely reducing the information content of spectra. By contrast, addition of the more tightly binding ΛΛ-2 produced less severe relaxation-induced broadening although enough to abolish intermolecular NOEs. Chemical shift changes occurred at both ends of the quadruplex, and intramolecular NOEs were broadened and lost at both ends. It therefore appears that whereas ΔΔ-2 binds mainly at the lateral loop end, ΛΛ-2 binds at both ends. Taken together with the luminescence data, the implication is that binding of ΛΛ-2 at the diagonal loop end (top end of Figure 3) is accompanied by a high degree of shielding of the ligand from solvent. This in turn implies that the ligand is stacked onto the tetrad underneath the diagonal loop.
Figure 3.
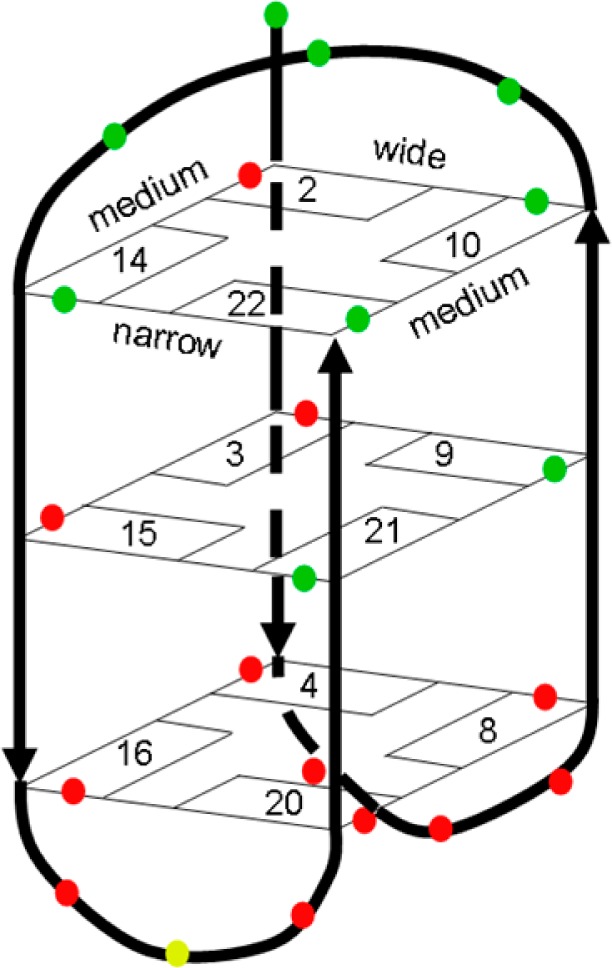
The antiparallel basket formed by HTS. The colors show the residues still present (green), missing (red), and possible exchange doublet (yellow), upon addition of ΔΔ-2.
All attempts to alter the solution conditions so as to bring back intermolecular NOEs were unsuccessful. We therefore carried out titrations with 1. The absorption and emission response of this complex to HTS binding is very similar to that of 2: in particular, the enhancement and blue-shifting of its luminescence are very similar, indicating analogous modes of binding.48 These studies were carried out on a diastereomeric mixture as this had the advantage that both putative binding sites could be potentially investigated in a single experiment.
Addition of 1 to HTS gave rise to chemical shift changes at both ends of the quadruplex. There was broadening of signals throughout, which led to a general reduction in the intensities of intramolecular NOEs, although the largest effects were seen at the lateral loop end (Table 2). However, gratifyingly, a large number of new intermolecular NOEs could be seen to the ligand (Figure 4).
Table 2. Intermolecular NOE Crosspeaks and the Corresponding DNA Residues Identified for the Interaction of 1 with HTS.
| complex signal | DNA signal | assigned DNA atom |
|---|---|---|
| One-Loop End | ||
| 7.79 | 4.38 | G10 H5′ |
| 7.79 | 2.89 | G14 H2′ |
| 7.79 | 2.74 | G14 H2″ |
| 7.80 | 5.65 | T11 H1′ |
| 7.80 | 6.30 | G10 H1′ |
| 7.76 | 6.31 | G10 H1′ |
| 7.74 | 3.31 | G22 H2′ |
| 7.55 | 2.40 | G22 H2″ |
| 6.95 | 5.58 | T12 H1′ |
| 6.95 | 1.14 | T12 H2′ |
| 6.29 | 5.46 | G2 H1′ |
| 8.36 | 2.61 | G9 H2″ |
| 8.36 | 2.94 | G9 H2′ |
| Two-Loop End | ||
| 8.36 | 2.51 | G8 H2″ |
| 8.36 | 3.08 | G8 H2′ |
| 8.11 | 1.91 | T5 CH3 |
| 6.93 | 1.30 | T18 H2′ |
| 6.93 | 2.07 | T17 CH3 |
| 6.93 | 4.04 | T6 H5′* |
| 8.22 | 2.83 | A19 H2′ |
Figure 4.

Selected sections of spectra showing the new crosspeaks identified upon addition of a diastereomeric mixture of 1 to the HTS sequence.
In free solution, NMR resonances from the four symmetry-related positions of 1 have identical chemical shifts. However, in the presence of HTS, most signals were split into four, in some cases with fairly large chemical shift changes. The increased complexity made it impossible to assign the ligand signals in the complex to individual positions and meant that, although we were able to observe 20 intermolecular NOEs in the complex, we were only able to assign the nucleotide signals (Table 2).
Analysis of the NOEs reveals that they are not compatible with a single structure for the complex, because 13 derive from contacts at the diagonal loop end and seven from contacts at the lateral loop end. On the basis of the discussion above, this is not surprising. In particular, by analogy with 2, we expect ΔΔ-1 to bind mainly at the lateral loop end and ΛΛ-1 (and probably also ΛΔ-1) mainly at the diagonal loop end. NOE intensities are compatible with this expectation. As anticipated, the NOEs at the diagonal loop end are consistent with 1 binding under the diagonal loop.
NMR-Based Structures of Complexes
The experimentally observed NOEs from the lateral loop end were used to calculate structures for both ΛΛ-1 and ΔΔ-1 bound at the lateral loop end using restrained simulated annealing. The structure generated for ΔΔ-1 is shown in Figure 5 and has no violations of the NOE constraints greater than 0.5 Å.
Figure 5.
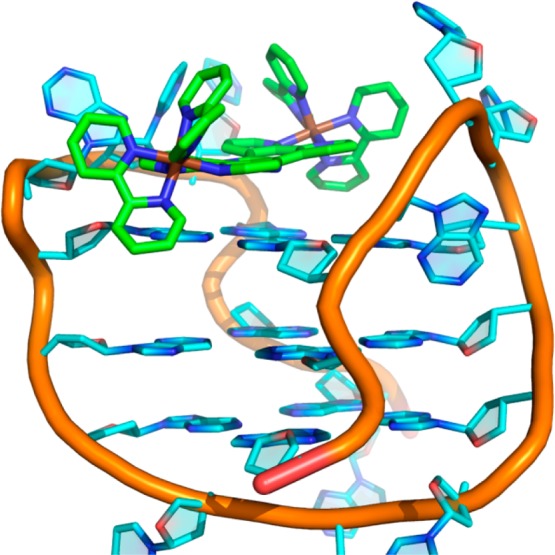
Molecular dynamics-based structure of ΔΔ-1 bound to the lateral loop end of HTS, generated using experimentally observed NOEs for the interaction.
While the tpphz ligand stacks on top of the tetrad bases, this interaction is reinforced by electrostatics: the positively charged ruthenium centers are located at the edge of the tetrad close to the anionic phosphate backbone. There is little perturbation to the quadruplex structure, with slight movement of the lateral loops to accommodate the ligand. Bases in the loops partially shield the tpphz rings, as expected from the luminescence. There is little direct contact between the bipy ligands and the quadruplex, and therefore both the ΛΛ and ΔΔ complexes bind in a similar way, with similar energies.
The observed NOEs were also used to calculate a structure for the complex bound at the diagonal loop end. The bound ΛΛ-1 structure is shown in Figure 6. The bipy rings fit neatly against the phosphate backbone making close van der Waals contact. The diagonal loop holds the tpphz in place and shields it from solvent, as expected from the luminescence data.
Figure 6.

Molecular dynamics-based structure of ΛΛ-1 bound to the diagonal loop end of HTS, generated using experimentally observed NOEs for the interaction. Left: Illustrating the bipy ring at front left parallel to the DNA backbone. Right: Showing how the 5′ end of the DNA backbone (front center) moves to avoid the ligand.
By contrast, attempts to use the same NOEs to calculate a structure with ΔΔ-1 bound at the same site result in a very high-energy state (roughly 10 times higher). In this structure, one end of the ligand is able to fit straightforwardly, by displacing the terminal nucleotide of the quadruplex DNA chain (Figure 7). However, in this calculation, the other end of the ligand does not displace the DNA backbone. Instead, the calculation generates a number of physically impossible solutions, of which the lowest energy is shown in Figure 7: the phosphate backbone passes through the middle of one of the aromatic bipy rings. The other solution is to displace one bipy ligand completely away from the tpphz plane. Structures have been calculated for the ΛΛ and ΔΔ isomers of 2 in the same way and show similar features (Supporting Information Figures S04 and S05). These calculations imply that the experimental NOE data are compatible with the ΛΛ isomer binding at the diagonal loop end but not with the ΔΔ isomer binding at the same site. This result is thus in agreement with the conclusion reached above, that both isomers bind at the lateral loop end, but only the ΛΛ isomer (and probably the ΛΔ isomer also) binds at the diagonal loop end. To investigate this issue further, unconstrained molecular dynamics simulations were carried out in a TIP3P water model.
Figure 7.
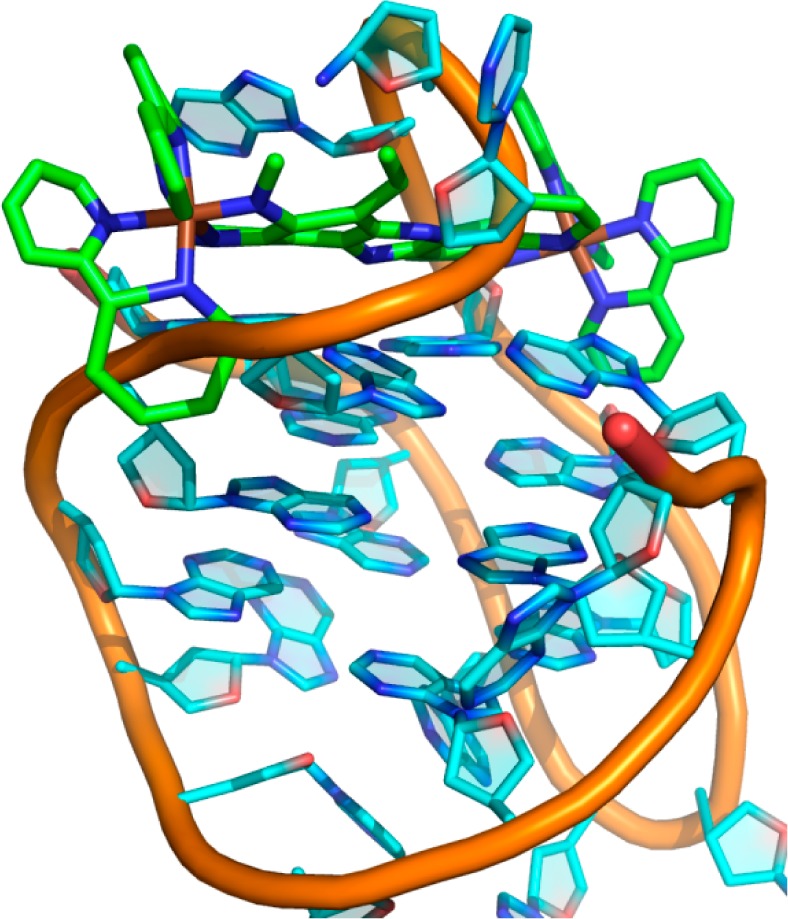
Molecular dynamics-based structure of ΔΔ-1 bound to the diagonal loop end of HTS, generated using experimentally observed NOEs for the interaction.
Unconstrained MD Simulations
To evaluate the impact of ΛΛ-1 and ΔΔ-1 complexes on the quadruplex structure when binding under the diagonal loop, molecular dynamics simulations were performed on free HTS and its association with ΛΛ-1 and ΔΔ-1. For free HTS, three independent simulations were undertaken using the structures presented in Figures 5–7 (henceforth denoted A, B, and C) as starting conformations. These results were then compared with simulations of the associations formed between the quadruplex and both diastereomers ΛΛ-1 and ΔΔ-1, labeled ΛΛ-1-HTS and ΔΔ-1-HTS, respectively. For the ΛΛ-1-HTS simulation, the NMR-based structure shown in Figure 6 was used as starting point, while for simulation of ΔΔ-1-HTS, the starting structure was generated from ΛΛ-1-HTS by replacing ΛΛ-1 with ΔΔ-1, superimposing the two ruthenium atoms and the tpphz bridging ligand. Thus, only the spatial arrangement of the two Ru(bipy)2 moieties was changed, in agreement with the respective diastereomers in the octahedral metal coordination sphere. Further molecular dynamics simulation details, together with an extended discussion, are provided in Supporting Information.
Simulations A, B, and C of the free HTS show that the G-tetrads yield low root mean-square deviations (RMSD), calculated relative to the starting structures, consistent with a minor conformational rearrangement experienced by these subunits throughout the 50 ns of simulation time, which is to be expected because they are held in place by hydrogen bonds. In contrast, the diagonal loops are more mobile (see Supporting Information Figure S06) showing higher RMSD values. Overall, the three independent simulations sampled significant HTS conformational space as suggested by the representative conformations of simulations A, B, and C, which have slightly different structures with cross RMSD values collected in Supporting Information Table S03 between 2.71 and 3.88 Å. We then went on to look at the interaction of HTS with the metal complex using the same method.
Figure 8 shows the RMSD values over the simulation time (using the unrelaxed starting structures as reference) for both the ΛΛ-1-HTS and ΔΔ-1-HTS associations. In both cases, after an initial jump due to the geometry relaxation using the ideal force field parameters, the values tend to stabilize. Focusing on the RMSD values for ΛΛ-1-HTS, the G-tetrad and the HTS structure typically stabilize after the first five ns while the values for the diagonal loop oscillate. This loop mobility is assigned to the presence of ΛΛ-1, as it was not observed in simulations A, B, and C of free HTS. Apart from this increased loop mobility, the antiparallel basket is as stable as free HTS was in simulations A, B, and C, so unsurprisingly the RMSDs between the representative conformations of ΛΛ-1-HTS and simulations A, B, and C are within the variability found for the calculated cross RMSDs obtained in these three simulations (see Supporting Information Table S03).
Figure 8.
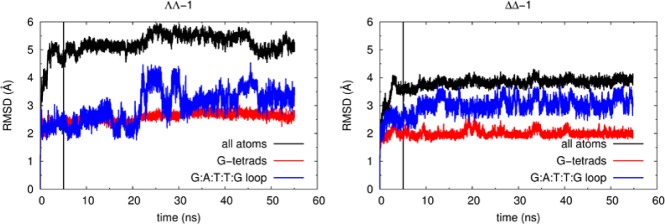
Variation of the RMSD values throughout the course of the MD simulations for ΛΛ-1-HTS (left) and ΔΔ-1-HTS (right). The vertical black line marks the separation between the equilibration and collection simulation stages.
By contrast, for ΔΔ-1-HTS, all the RMSD values converge very quickly, including those associated with the diagonal loop. This indicates that complex ΔΔ-1 is able to fit under the loop without causing major G-DNA conformational changes. Indeed, the representative conformation is comparable to the representative conformation of the ΛΛ-1-HTS simulation (with a RMSD value of only 1.55 Å) and the representative frames of the free HTS simulationsm A, B, and C, with cross RMSD values ranging from 1.54 to 2.81 Å. This indicates that the observed experimental recognition of only ΛΛ-1 under the diagonal loop is not caused by the intrinsic loop cavity shape or size because both isomers are able to fit into the cavity.
Analyzing the representative conformations of ΛΛ-1-HTS and ΔΔ-1-HTS represented in Figure 9, both ruthenium complexes bind under the diagonal loop with the tpphz ligand stacking over the top G-tetrad. However, some differences are evident. Complex ΛΛ-1 adopts a diagonal arrangement over the guanine bases and under the loop whereas in ΔΔ-1, the stacking of the tpphz ligand is more localized over two guanines of only one-half of the G-tetrad.
Figure 9.

Representative snapshots of simulations ΛΛ-1-HTS (top, magenta) and ΔΔ-1-HTS (bottom, aquamarine). Side and top views are presented on the left and center, respectively. Right pictures represent the complexes stacked over the top G-quartet.
To clarify this point, a surface representation was constructed for the position occupied by the tpphz ligand (excluding the hydrogen atoms) over a 50 ns collection period for the HTS association with each of the diastereomers (Figure 10). These surfaces clearly show a striking difference: in ΛΛ-1-HTS, the tpphz ligand is able to “float” over the G-tetrad interacting with the four guanines. In contrast, in ΔΔ-1-HTS, the tpphz ligand remains “locked” over two guanines of the G-tetrad throughout the entire simulation time. Because tpphz is present in both dinuclear complexes, the difference in dynamic behavior must be caused by the stereochemistry of the bipy ligands in the octahedral Ru(II) coordination spheres. This is consistent with the observation that in ΛΛ-1-HTS the diagonal loop appears to be more flexible, oscillating over the middle of both Ru(bipy)2 moieties. On the other hand, in ΔΔ-1-HTS, the loop is more rigid and locks the ΔΔ-1 complex movement over the G-tetrad.
Figure 10.
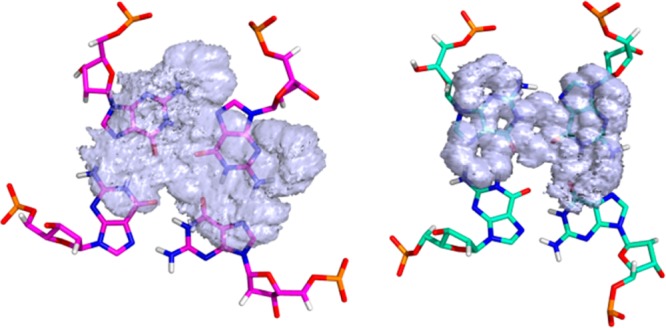
Surface (isovalue = 1) representing the histogram of positions occupied by the tpphz atoms (excluding hydrogen) over the 50 ns collection period in ΛΛ-1-HTS (left) and ΔΔ-1-HTS (right).
The above results suggest that, although both ΛΛ-1 and ΔΔ-1 complexes can fit under the loop, sufficient HTS loop conformational freedom to permit the complex entrance into the prefolded G-DNA structure is only present for ΛΛ-1. In contrast, the stereochemistry of ΔΔ-1 appears to induce increased rigidity on the loop, preventing complex entrance into the loop arch. In other words, ΔΔ-1 does not bind under the diagonal loop because the loop arch does not fulfill the complex’s “stereochemical requirements”.
As mentioned above, the simulated annealing structure proposed for the binding of ΔΔ-1 to the diagonal loop end of HTS corresponds to an impossible solution (Figure 7), which could also be rationalized by our unconstrained MD simulations in water. If one calculates the expected NOEs (not shown) from the representative conformations of simulations ΛΛ-1-HTS and ΔΔ-1-HTS (or from the relaxed structures obtained by Molecular Mechanics minimization), the protons that contact the ligand are quite different, meaning that the interaction of ΛΛ-1 and ΔΔ-1 under the loop results in clearly distinct NOEs. Therefore, the experimental interactions (presumably generated only by the “ΛΛ-1-HTS” interaction) are not suitable as restraints to generate a hypothetical “ΔΔ-1-HTS” association with this binding mode.
Conclusions
We have shown that ΛΛ-2 binds mainly at the diagonal loop end of HTS, stabilized by a good steric fit under the loop. By contrast, ΔΔ-2 binds >40-fold more weakly and almost entirely at the other end. Both isomers also bind to B-DNA, but the affinity of ΛΛ-2 is greater for HTS than for B-DNA and, importantly, when bound to HTS the luminescence is more intense and is blue-shifted from ∼675 to ∼630 nm. Studies with other ligands that bind to quadruplex DNA have shown that compounds that bind strongly to one conformation also stabilize that conformation, as expected from thermodynamic arguments.10,52,60−62 We have previously shown49,50 that 2 is actively taken up by cells, and confocal images show that it generates punctate images centered mainly in the heterochromatin, as might be expected for a probe that highlights G-quadruplexes. It also has a wavelength and intensity of emission that is different for different quadruplex structures.48 ΛΛ-2 is therefore a useful tool for specifically stabilizing and imaging antiparallel basket structures with a diagonal loop. This study also confirms that the antiparallel structure of HTS can be selectively targeted. In comparison with the use of antibodies to detect quadruplexes,63−65 small molecules like complex 2 and its analogues are much simpler to rationally design and potentially have wider applicability as they can be used directly on living cells. Furthermore, and as this study indicates, because such systems can be made to target specific features of a quadruplex, they can be made specific to individual quadruplex structures. Consequently, with the structural information obtained by this study, we are exploring this potential for enhanced specificity through informed design of derivatives of 2. Although this work is intended to image antiparallel basket quadruplexes in living cells, it could subsequently be extended into molecular tools for selectively stabilizing quadruplexes.
Experimental Section
Ligands were synthesized using published methods. Ru(phen)2Cl2·2H2O and Ru(bipy)2Cl2·2H2O were prepared following a standard literature method.64 The racemic mixtures of the tpphz complexes were prepared by methods we have previously described.47 Enantiomers of 2 were prepared by the route first described by MacDonnell and Bodige.66 Characterization data for these complexes were identical to the original reports.
The HTS oligonucleotide d[AG3(TTAG3)3] was purchased from Eurogentec Biotechnology (Southampton, UK), purified by HPLC, and used without further annealing. Samples for UV–vis and luminescence titrations were in 10 mM KH2PO4/K2HPO4 and 1 mM K2EDTA in 50–200 mM KCl (pH 7.0, 298 K), in which DNA was added to 10 μM ligand, while samples for NMR were prepared in 50 mM NaCl, pH 7, using 300 mM DNA at 298 K. Titrations were conducted with DNA and ligand of the same order of concentration as 1/Kb, shown to be the optimum values for obtaining accurate binding constants.67 The optimum temperature for observing NOEs to 1 was 283 K. CD spectra were recorded on a Jasco J-810 spectrophotometer using a Peltier variable temperature controller: 100 nm/min from 200 to 320 nm. CD melting experiments gave a melting temperature of 63.3 °C. Luminescence measurements were carried out on a Hitachi F-4500 fluorescence spectrophotometer using a 1 cm path length. UV–vis titrations were carried out using a Cary 3 Bio. Binding affinities were obtained by fitting using Origin 7.0 software to a standard one-set-of-binding-sites model.
NMR experiments were carried out on Bruker Avance 800, 600, and 500 spectrometers. Assignments were made using COSY, TOCSY, and NOESY spectra (mixing times 60 or 90 ms for TOCSY, 100 ms for NOESY), supported by 1H–31P HSQC (Supporting Information Table S01). Spectra were analyzed using FELIX (Felix NMR, Inc., San Diego, CA).
Structure calculations were carried out using Xplor with the parallhdg.dna parameters. Calculations used simulated annealing over 8000 steps from 2000 to 100 K, using standard square-well potentials for NOEs. All calculations imposed planarity and hydrogen bonding in the tetrads plus planarity in tpphz and bipy polypyridyl ligands and octahedral geometry around the ruthenium. The force field included van der Waals and electrostatic terms. Threading of the ligand into the complex was accomplished by starting with almost zero van der Waals radii and increasing the radius in a geometric progression during the simulated annealing. For each complex, only the intermolecular NOEs relevant to that end were included, all specified as ambiguous NOEs to any ligand proton, with an upper limit of 5 Å. Convergence was improved by including restraints to position the tpphz ring close to the relevant tetrad plane. NOEs for the ΛΛ-1-HTS and ΔΔ-1-HTS complexes were calculated by numerical integration of the Solomon equations.68
Unconstrained MD simulations were carried out using the AMBER ff99bsc0 set of parameters and charges for DNA69 combined with General Amber Force Field (GAFF) parameters70 with extra terms71,72 and RESP charges73 for both ΔΔ-1 and ΛΛ-1 complexes. These simulations were undertaken with the pmemd.cuda AMBER executable, able to accelerate explicit solvent Particle Mesh Ewald (PME)74,75 calculations through the use of GPUs with the new Single Precision Fixed Point (SPFP) model.76
Acknowledgments
We thank the EPSRC for a Ph.D. studentship for T.W. through the White Rose Life Science DTC. P.J.C. thanks FCT for the postdoctoral grant SFRH/BPD/27082/2006. V.F. acknowledges the funding from QREN-FEDER through the Operational Program Competitiveness Factors—COMPETE and National Funds through the FCT under project PTDC/QUI-QUI/101022/2008.
Glossary
Abbreviations Used
- HTS
human telomere sequence
- tpphz
tetrapyrido[3,2-a:2′,3′-c:3″,2″-h:2‴,3‴-j]phenazine
- bipy
2,2′-bipyridine
- phen
1,10-phenanthroline
- dppz
dipyrido[3,2-a:2′,3′-c]phenazine
- MD
molecular dynamics
- CD
circular dichroism
- CT-DNA
calf thymus DNA
- NOE
nuclear Overhauser effect
Supporting Information Available
Details of MD; luminescent titrations with CT-DNA; circular dichroism spectrum of HTS quadruplex; titration with ΔΔ-2; NMR assignments of the HTS quadruplex; models of ΛΛ-2 and ΔΔ-2 bound to HTS. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and chemical shift data have been deposited at the PDB with the entry codes 2mcc (ΔΔ-1 complex) and 2mco (ΛΛ-1 complex) and also at BMRB with the entry codes 19435 and 19448, respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Gellert M.; Lipsett M. N.; Davies D. R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. U. S. A. 1962, 48, 2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D.; Gilbert W. Formation of parallel 4-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [DOI] [PubMed] [Google Scholar]
- Paeschke K.; Simonsson T.; Postberg J.; Rhodes D.; Lipps H. J. Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nature Struct. Mol. Biol. 2005, 12, 847–854. [DOI] [PubMed] [Google Scholar]
- Kim N. W.; Piatyszek M. A.; Prowse K. R.; Harley C. B.; West M. D.; Ho P. L. C.; Coviello G. M.; Wright W. E.; Weinrich S. L.; Shay J. W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Counter C. M.; Hahn W. C.; Wei W. Y.; Caddle S. D.; Beijersbergen R. L.; Lansdorp P. M.; Sedivy J. M.; Weinberg R. A. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 14723–14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler A. M.; Williamson J. R.; Cech T. R.; Prescott D. M. Inhibition of telomerase by G-quartet DNA structures. Nature 1991, 350, 718–720. [DOI] [PubMed] [Google Scholar]
- Huppert J. L.; Balasubramanian S. G-Quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A.; Halder K.; Halder R.; Yadav V. K.; Rawal P.; Thakur R. K.; Mohd F.; Sharma A.; Chowdhury S. Genome-wide computational and expression analyses reveal G-quadruplex DNA motifs as conserved cis-regulatory elements in human and related species. J. Med. Chem. 2008, 51, 5641–5649. [DOI] [PubMed] [Google Scholar]
- Du Z.; Zhao Y.; Li N. Genome-wide analysis reveals regulatory role of G4 DNA in gene transcription. Genome Res. 2008, 18, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui-Jain A.; Grand C. L.; Bearss D. J.; Hurley L. H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J. X.; Dexheimer T. S.; Chen D.; Carver M.; Ambrus A.; Jones R. A.; Yang D. Z. An intramolecular G-quadruplex structure with mixed parallel/antiparallel G-strands formed in the human BCL-2 promoter region in solution. J. Am. Chem. Soc. 2006, 128, 1096–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogoi S.; Xodo L. E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirude P. S.; Okumus B.; Ying L.; Ha T.; Balasubramanian S. Single-molecule conformational analysis of G-quadruplex formation in the promoter DNA duplex of the proto-oncogene C-kit. J. Am. Chem. Soc. 2007, 129, 7484–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S.; Bugaut A.; Huppert J. L.; Balasubramanian S. An RNA G-quadruplex in the 5 ′ UTR of the NRAS proto-oncogene modulates translation. Nature Chem. Biol. 2007, 3, 218–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider S. M.; Neidle S.; Parkinson G. N. A structural analysis of G-quadruplex/ligand interactions. Biochimie 2011, 93, 1239–1251. [DOI] [PubMed] [Google Scholar]
- Xue Y.; Kan Z.-Y.; Wang Q.; Yao Y.; Liu J.; Hao Y.-H.; Tan Z. Human telomeric DNA forms parallel-stranded intramolecular G-quadruplex in K+ solution under molecular crowding condition. J. Am. Chem. Soc. 2007, 129, 11185–11191. [DOI] [PubMed] [Google Scholar]
- Heddi B.; Anh Tuan P. Structure of human telomeric DNA in crowded solution. J. Am. Chem. Soc. 2011, 133, 9824–9833. [DOI] [PubMed] [Google Scholar]
- Dhakal S.; Cui Y.; Koirala D.; Ghimire C.; Kushwaha S.; Yu Z.; Yangyuoru P. M.; Mao H. Structural and mechanical properties of individual human telomeric G-quadruplexes in molecularly crowded solutions. Nucleic Acids Res. 2013, 41, 3915–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. C.; Buscaglia R.; Chaires J. B.; Lane A. N.; Trent J. O. Hydration is a major determinant of the G-quadruplex stability and conformation of the human telomere 3 ′ sequence of d(AG3(TTAG3)3). J. Am. Chem. Soc. 2010, 132, 17105–17107. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Patel D. J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure 1993, 1, 263–282. [DOI] [PubMed] [Google Scholar]
- Parkinson G. N.; Lee M. P. H.; Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [DOI] [PubMed] [Google Scholar]
- Ambrus A.; Chen D.; Dai J. X.; Bialis T.; Jones R. A.; Yang D. Z. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006, 34, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu K. N.; Phan A. T.; Kuryavyi V.; Lacroix L.; Patel D. J. Structure of the human telomere in K+ solution: an intramolecular (3 + 1) G-quadruplex scaffold. J. Am. Chem. Soc. 2006, 128, 9963–9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J.; Carver M.; Punchihewa C.; Jones R. A.; Yang D. Structure of the hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007, 35, 4927–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. W.; Amrane S.; Bouaziz S.; Xu W.; Mu Y.; Patel D. J.; Luu K. N.; Phan A. T. Structure of the human telomere in K+ solution: a stable basket-type G-quadruplex with only two G-tetrad layers. J. Am. Chem. Soc. 2009, 131, 4301–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renčiuk D.; Kejnovská I.; Školáková P.; Bednářová K.; Motlová J.; Vorlíčková M. Arrangements of human telomere DNA quadruplex in physiologically relevant K+ solutions. Nucleic Acids Res. 2009, 37, 6625–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänsel R.; Löhr F.; Foldynová-Trantírková S.; Bamberg E.; Trantírek L.; Dötsch V. The parallel G-quadruplex structure of vertebrate telomeric repeat sequences is not the preferred folding topology under physiological conditions. Nucleic Acids Res. 2011, 39, 5768–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidle S. The structures of quadruplex nucleic acids and their drug complexes. Curr. Opin. Struct. Biol. 2009, 19, 239–250. [DOI] [PubMed] [Google Scholar]
- Georgiades S. N.; Abd Karim N. H.; Suntharalingam K.; Vilar R. Interaction of metal complexes with G-quadruplex DNA. Angew. Chem., Int. Ed. 2010, 49, 4020–4034. [DOI] [PubMed] [Google Scholar]
- Ou T.-M.; Lu Y.-J.; Tan J.-H.; Huang Z.-S.; Wong K.-Y.; Gu L.-Q. G-Quadruplexes: targets in anticancer drug design. ChemMedChem 2008, 3, 690–713. [DOI] [PubMed] [Google Scholar]
- Xu Y. Chemistry in human telomere biology: structure, function and targeting of telomere DNA/RNA. Chem. Soc. Rev. 2011, 40, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Campbell N. H.; Abd Karim N. H.; Parkinson G. N.; Gunaratnam M.; Petrucci V.; Todd A. K.; Vilar R.; Neidle S. Molecular basis of structure–activity relationships between salphen metal complexes and human telomeric DNA quadruplexes. J. Med. Chem. 2012, 55, 209–222. [DOI] [PubMed] [Google Scholar]
- Parkinson G. N.; Cuenca F.; Neidle S. Topology conservation and loop flexibility in quadruplex–drug recognition: Crystal structures of inter- and intramolecular telomeric DNA quadruplex–drug complexes. J. Mol. Biol. 2008, 381, 1145–1156. [DOI] [PubMed] [Google Scholar]
- Bazzicalupi C.; Ferraroni M.; Bilia A. R.; Scheggi F.; Gratteri P. The crystal structure of human telomeric DNA complexed with berberine: an interesting case of stacked ligand to G-tetrad ratio higher than 1:1. Nucleic Acids Res. 2013, 41, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collie G. W.; Promontorio R.; Hampel S. M.; Micco M.; Neidle S.; Parkinson G. N. Structural basis for telomeric G-quadruplex targeting by naphthalene diimide ligands. J. Am. Chem. Soc. 2012, 134, 2723–2731. [DOI] [PubMed] [Google Scholar]
- Micco M.; Collie G. W.; Dale A. G.; Ohnmacht S. A.; Pazitna I.; Gunaratnam M.; Reszka A. P.; Neidle S. Structure-based design and evaluation of naphthalene diimide G-quadruplex ligands as elomere targeting agents in pancreatic cancer cells. J. Med. Chem. 2013, 56, 2959–2974. [DOI] [PubMed] [Google Scholar]
- Fedoroff O. Y.; Salazar M.; Han H. Y.; Chemeris V. V.; Kerwin S. M.; Hurley L. H. NMR-based model of a telomerase-inhibiting compound bound to G-quadruplex DNA. Biochemistry 1998, 37, 12367–12374. [DOI] [PubMed] [Google Scholar]
- Gavathiotis E.; Heald R. A.; Stevens M. F. G.; Searle M. S. Drug recognition and stabilisation of the parallel-stranded DNA quadruplex d(TTAGGGT)4 containing the human telomeric repeat. J. Mol. Biol. 2003, 334, 25–36. [DOI] [PubMed] [Google Scholar]
- Clark G. R.; Pytel P. D.; Squire C. J.; Neidle S. Structure of the first parallel DNA quadruplex–drug complex. J. Am. Chem. Soc. 2003, 125, 4066–4067. [DOI] [PubMed] [Google Scholar]
- Parkinson G. N.; Ghosh R.; Neidle S. Structural basis for binding of porphyrin to human telomeres. Biochemistry 2007, 46, 2390–2397. [DOI] [PubMed] [Google Scholar]
- Martino L.; Virno A.; Pagano B.; Virgilio A.; Di Micco S.; Galeone A.; Giancola C.; Bifulco G.; Mayol L.; Randazzo A. Structural and thermodynamic studies of the interaction of distamycin a with the parallel quadruplex structure [d(TGGGGT)]4. J. Am. Chem. Soc. 2007, 129, 16048–16056. [DOI] [PubMed] [Google Scholar]
- Haider S. M.; Parkinson G. N.; Neidle S. Structure of a G-quadruplex–ligand complex. J. Mol. Biol. 2003, 326, 117–125. [DOI] [PubMed] [Google Scholar]
- Campbell N. H.; Patel M.; Tofa A. B.; Ghosh R.; Parkinson G. N.; Neidle S. Selectivity in ligand recognition of G-quadruplex loops. Biochemistry 2009, 48, 1675–1680. [DOI] [PubMed] [Google Scholar]
- Metcalfe C.; Thomas J. A. Kinetically inert transition metal complexes that reversibly bind to DNA. Chem. Soc. Rev. 2003, 32, 215–224. [DOI] [PubMed] [Google Scholar]
- Gill M. R.; Thomas J. A. Ruthenium(II) polypyridyl complexes and DNA—from structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012, 41, 3179–3192. [DOI] [PubMed] [Google Scholar]
- Waywell P.; Gonzalez V.; Gill M. R.; Adams H.; Meijer A. J. H. M.; Williamson M. P.; Thomas J. A. Structure of the complex of [Ru(tpm)(dppz)py]2+ with a B-DNA oligonucleotide: a single-substituent binding switch for a metallo-intercalator. Chem.—Eur. J. 2010, 16, 2407–2417. [DOI] [PubMed] [Google Scholar]
- Rajput C.; Rutkaite R.; Swanson L.; Haq I.; Thomas J. A. Dinuclear monointercalating Ru-II complexes that display high affinity binding to duplex and quadruplex DNA. Chem.—Eur. J. 2006, 12, 4611–4619. [DOI] [PubMed] [Google Scholar]
- Wilson T.; Williamson M. P.; Thomas J. A. Differentiating quadruplexes: binding preferences of a luminescent dinuclear ruthenium(II) complex with four-stranded DNA structures. Org. Biomol. Chem. 2010, 8, 2617–2621. [DOI] [PubMed] [Google Scholar]
- Gill M. R.; Garcia-Lara J.; Foster S. J.; Smythe C.; Battaglia G.; Thomas J. A. A ruthenium(II) polypyridyl complex for direct imaging of DNA structure in living cells. Nature Chem. 2009, 1, 662–667. [DOI] [PubMed] [Google Scholar]
- Tian X.; Gill M. R.; Canton I.; Thomas J. A.; Battaglia G. Live cell luminescence imaging as a function of delivery mechanism. ChemBioChem 2011, 12, 548–551. [DOI] [PubMed] [Google Scholar]
- Yu H.; Wang X.; Fu M.; Ren J.; Qu X. Chiral metallo-supramolecular complexes selectively recognize human telomeric G-quadruplex DNA. Nucleic Acids Res. 2008, 36, 5695–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Geng J.; Feng L.; Ren J.; Qu X. Chiral metallo-supramolecular complexes selectively induce human telomeric G-quadruplex formation under salt-deficient conditions. Chem.—Eur. J. 2011, 17, 8209–8215. [DOI] [PubMed] [Google Scholar]
- Shinohara K.; Sannohe Y.; Kaieda S.; Tanaka K.; Osuga H.; Tahara H.; Xu Y.; Kawase T.; Bando T.; Sugiyama H. A chiral wedge molecule inhibits telomerase activity. J. Am. Chem. Soc. 2010, 132, 3778–3782. [DOI] [PubMed] [Google Scholar]
- Andersson J.; Lincoln P. Stereoselectivity for DNA threading intercalation of short binuclear ruthenium complexes. J. Phys. Chem. B 2011, 115, 14768–14775. [DOI] [PubMed] [Google Scholar]
- Johansson J. R.; Wang Y.; Eng M. P.; Kann N.; Lincoln P.; Andersson J. Bridging ligand length controls AT selectivity and enantioselectivity of binuclear ruthenium threading intercalators. Chem.—Eur. J. 2013, 19, 6246–6256. [DOI] [PubMed] [Google Scholar]
- Hall J. P.; O’Sullivan K.; Naseer A.; Smith J. A.; Kelly J. M.; Cardin C. J. Structure determination of an intercalating ruthenium dipyridophenazine complex which kinks DNA by semiintercalation of a tetraazaphenanthrene ligand. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 17610–17614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niyazi H.; Hall J. P.; O’Sullivan K.; Winter G.; Sorensen T.; Kelly J. M.; Cardin C. J. Crystal structures of Λ-Ru(phen)2dppz2+ with oligonucleotides containing TA/TA and AT/AT steps show two intercalation modes. Nature Chem. 2012, 4, 621–628. [DOI] [PubMed] [Google Scholar]
- Song H.; Kaiser J. T.; Barton J. K. Crystal structure of Δ-Ru(bpy)2dppz2+ bound to mismatched DNA reveals side-by-side metalloinsertion and intercalation. Nature Chem. 2012, 4, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Reymer A.; Persson C.; Kazimierczuk K.; Brown T.; Lincoln P.; Nordén B.; Billeter M. Initial DNA interactions of the binuclear threading intercalator Λ,Λ-[m-bidppz(bipy)4Ru24+: an NMR study with d(CGCGAATTCGCG)2. Chem.—Eur. J. 2013, 19, 5401–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seenisamy J.; Rezler E. M.; Powell T. J.; Tye D.; Gokhale V.; Joshi C. S.; Siddiqui-Jain A.; Hurley L. H. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J. Am. Chem. Soc. 2004, 126, 8702–8709. [DOI] [PubMed] [Google Scholar]
- Shi S.; Geng X. T.; Zhao J.; Yao T. M.; Wang C. R.; Yang D. J.; Zheng L. F.; Ji L. N. Interaction of [Ru(bpy)2(dppz)]2+ with human telomeric DNA: preferential binding to G-quadruplexes over i-motif. Biochimie 2010, 92, 370–377. [DOI] [PubMed] [Google Scholar]
- Sun D.; Liu Y.; Liu D.; Zhang R.; Yang X.; Liu J. Stabilization of G-quadruplex DNA, inhibition of telomerase activity and live cell imaging studies of chiral ruthenium(II) complexes. Chem.—Eur. J. 2012, 18, 4285–4295. [DOI] [PubMed] [Google Scholar]
- Biffi G.; Tannahill D.; McCafferty J.; Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nature Chem. 2013, 5, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffitzel C.; Berger I.; Postberg J.; Hanes J.; Lipps H. J.; Pluckthun A. In vitro generated antibodies specific for telomeric guanine–quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8572–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan B. P.; Salmon D. J.; Meyer T. J. Mixed phosphine 2,2′-bipyridine complexes of ruthenium. Inorg. Chem. 1978, 17, 3334–3341. [Google Scholar]
- MacDonnell F. M.; Bodige S. Efficient stereospecific syntheses of chiral ruthenium dimers. Inorg. Chem. 1996, 35, 5758–&. [Google Scholar]
- Granot J. Determination of dissociation constants of 1:1 complexes from NMR data. Optimization of the experimental setup by statistical analysis of simulated experiments. J. Magn. Reson. 1983, 55, 216–224. [Google Scholar]
- Williamson M. P. Guidelines for the design of kinetic NOE experiments from computer simulation. Magn. Reson. Chem. 1987, 25, 356–361. [Google Scholar]
- Perez A.; Marchan I.; Svozil D.; Sponer J.; Cheatham T. E. III; Laughton C. A.; Orozco M. Refinenement of the AMBER force field for nucleic acids: Improving the description of a/g conformers. Biophys. J. 2007, 92, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. M.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Brandt P.; Norrby T.; Akermark E.; Norrby P. O. Molecular mechanics (MM3*) parameters for ruthenium(II)–polypyridyl complexes. Inorg. Chem. 1998, 37, 4120–4127. [DOI] [PubMed] [Google Scholar]
- Moret M.-E.; Tavernelli I.; Rothlisberger U. Combined QM/MM and classical molecular dynamics study of Ru(bpy)32+ in water. J. Phys. Chem. B 2009, 113, 7737–7744. [DOI] [PubMed] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W. D.; Kollman P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Le Grand S.; Goetz A. W.; Walker R. C. SPFP: Speed without compromise: a mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.